

Abstract
Meningococcal disease is characterized by a fast progression and a high mortality rate. Cell-penetrating peptides (CPPs), developed as vectors for cargo delivery into eukaryotic cells, share structural features with antimicrobial peptides. A screen identified two CPPs, transportan-10 (TP10) and model amphipathic peptide (MAP), with bactericidal action against Neisseria meningitidis. Both peptides were active in human whole blood at micromolar concentrations, while hemolysis remained negligible. Additionally, TP10 exhibited significant antibacterial activity in vivo. Uptake of SYTOX green into live meningococci was observed within minutes after TP10 treatment, suggesting that TP10 may act by membrane permeabilization. Apart from its bactericidal activity, TP10 suppressed inflammatory cytokine release from macrophages infected with N. meningitidis as well as from macrophages stimulated with enterobacterial and meningococcal lipopolysaccharide (LPS). Finally, incubation with TP10 reduced the binding of LPS to macrophages. This novel endotoxin-inhibiting property of TP10, together with its antimicrobial activity in vivo, indicates the possibility to design peptide-based therapies for infectious diseases.
INTRODUCTION
Cell-penetrating peptides (CPPs), such as Tat and penetratin (AntpHD43-58), are defined by their ability to translocate across cellular membranes in a nondisruptive manner. They have been developed for the delivery of cell-impermeable molecules into eukaryotic cells and are successfully employed as vehicles for pharmaceutical compounds in vivo (1–4). Depending on the CPP and its cargo, both endocytic as well as physically driven cellular uptakes have been reported, and the two routes may coexist (5, 6). The class of CPPs consists of peptides from diverse origins, including protein transduction domains responsible for membrane-translocating properties of native proteins but also chimeric and designed peptides (7). Characteristically, CPPs are short, cationic peptides that often have amphipathic properties and thus share features with antimicrobial peptides (AMPs) (5). In line with this similarity, some CPPs exhibit antimicrobial action toward bacteria, parasites, and fungi in vitro (8–11). Such antimicrobial activity is observed at higher concentrations than required for translocation across eukaryotic membranes.
AMPs are characterized by rapid and broad-spectrum antimicrobial activity and a strong interaction with microbial membranes (8). Antimicrobial activity results from a membrane-permeabilizing effect, the interaction with intracellular targets such as nucleic acids or proteins, or the disruption of cellular processes (12, 13). The interaction of AMPs with nonprotein targets, the coexistence of several killing mechanisms, and the presence of multiple peptides in vivo may contribute to the low level of AMP resistance (14, 15). Additionally, eukaryotic AMPs possess immunomodulatory functions or inhibit proinflammatory responses induced by microbial components (16–18). Such immunomodulatory activity has also been reported for the lantibiotic nisin Z and synthetic AMP derivates (19–21). These properties open up alternative approaches to combat microbial infections (15). The identification and development of novel antimicrobial agents are obvious aims in the battle against multiresistant bacteria. The therapeutic use of many AMPs may be limited by potential toxicity and reduced activity in vivo (15). Nevertheless, there are promising AMPs that have reached preclinical or clinical trials (22–24).
The Gram-negative diplococcus Neisseria meningitidis (meningococcus) is an obligate human pathogen and a major cause of bacterial meningitis and sepsis worldwide. Serogroups A, B, C, W, X, and Y are frequently associated with disease. Serotyping is based on the expression of a polysaccharide capsule, which ensures survival in the bloodstream by inhibiting phagocytosis and complement attack. The second major virulence factor, lipopolysaccharide (LPS) or endotoxin, is largely responsible for the devastating inflammatory response that characterizes systemic meningococcal disease (25).
In this study, we performed a screen of eight CPPs and identified two peptides with bactericidal action toward meningococci, namely, transportan-10 (TP10) and model amphipathic peptide (MAP). TP10 is a deletion analogue of the CPP transportan, a chimeric construct consisting of the N-terminal part of the neuropeptide galanin linked to the full-length wasp venom peptide mastoparan but without the toxic side effects of its parent peptide (26). The MAP used in this study is a variant of previously reported MAPs (27, 28), which are artificial model peptides designed for efficient membrane translocation. Both TP10 and MAP were found to be more potent than the human cathelicidin LL-37 and the peptide antibiotic polymyxin B (PMB), and we were able to demonstrate antimicrobial activities of both CPPs in human whole blood. TP10 was further studied in a mouse model and exhibited significant antimicrobial activity in vivo. Fluorescence microscopy of SYTOX green uptake into live meningococci suggested that TP10 exhibits membrane-permeabilizing activity. Finally, we describe an inhibitory effect of TP10 on inflammatory cytokine release induced by N. meningitidis infection or by purified meningococcal LPS.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
N. meningitidis serogroup A (Z6244), B (MC58), C (FAM20), and W (JB515) strains were grown on GC agar (Acumedia) plates containing 1% Kelloggs' supplement in a humidified environment at 37°C and 5% CO2 for 16 to 18 h. All in vitro assay mixtures were incubated at 37°C and 5% CO2.
Cell culture and cell viability assays.
Eukaryotic cells were maintained at 37°C and 5% CO2 in a humidified environment. RAW 264.7 murine macrophages (ATCC TIB-71) were cultured in Dulbecco's modified Eagle's medium (DMEM) with GlutaMAX and pyruvate (Invitrogen), supplemented with 10% heat-inactivated fetal bovine serum (FBS) (Sigma-Aldrich). Human monocytic THP1 cells (ATCC TIB-202) were cultured in RPMI 1640 medium with GlutaMAX (Invitrogen) and 10% FBS and differentiated with 100 ng/ml phorbol 12-myristate 13-acetate (Sigma-Aldrich). Cell viability after treatment with TP10 (0 to 128 μM), MAP (0 to 128 μM), or polymyxin B (0 to 512 μM) for 6 h was assessed in three independent experiments using MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] (Invitrogen) according to the manufacturer's recommendations. Formaldehyde-fixed cells were used as a blank. Triton X-100 (0.5%) was used as a positive control.
Peptide synthesis and purification.
Peptides (Table 1) were synthesized (SYRO multiple peptide synthesizer; MultiSynTech, Germany) on 9-fluorenylmethoxy carbonyl (Fmoc)-Rink amide-4-methylbenzhydrylamine resin (0.67 mmol/g; IRIS Biotech, Germany), using standard Fmoc solid-phase peptide synthesis (29). The peptide was cleaved by using 95% trifluoroacetic acid (TFA)–2.5% water–2.5% triisopropylsilane for 3 h and precipitated in diethyl ether. The obtained crude peptide was dried in a vacuum overnight. The peptide was purified by high-performance liquid chromatography (HPLC) on a Discovery C18 Supelco column (Sigma-Aldrich), using a gradient of acetonitrile-water containing 0.1% TFA. The identity of the purified product was verified by analytical HPLC and by a PerkinElmer prOTOFTM 2000 matrix-assisted laser desorption ionization–time of flight mass spectrometer (PerkinElmer, Sweden). The mass spectrum was acquired in the positive ion reflector mode by using α-cyano-4-hydroxycinnamic acid (Sigma-Aldrich) as a matrix (10 mg/ml, 7:3 acetonitrile:water, 0.1% TFA). The molarity of the peptide was determined based on dilutions of accurately weighed substances. CPPs were resuspended in phosphate-buffered saline (PBS) and stored at −20°C. LL-37 (Innovagen) was resuspended in 0.1% TFA. Magainin II and polymyxin B (Sigma-Aldrich) were resuspended in PBS.
Table 1.
Properties of the peptides included in this studya
| Peptide | Sequence | No. of aa | Net chargeb | Hydrophobic ratio (%)b | Origin | Reference(s) |
|---|---|---|---|---|---|---|
| Penetratin | RQIKIWFQNRRMKWKK-amide | 16 | +7 | 37 | Drosophila transcription factor Antennapedia | 61 |
| pVEC | LLIILRRRIRKQAHAHSL-amide | 18 | +7 | 50 | Murine vascular endothelial cadherin | 9, 40 |
| R41 | FILFILFILGGKHKHKHKHKHK-amide | 22 | +11 | 40 | Designed peptide | Unpublished |
| R8 | GPPRFPPRFPPRFPPRFPPRFP-amide | 22 | +5 | 22 | Design based on sequence of PR-39 | Unpublished |
| TP10 | AGYLLGKINLKALAALAKKIL-amide | 21 | +4 | 61 | Deletion analogue of transportan, a chimeric peptide | 26, 38, 39 |
| M918 | MVTVLFRRLRIRRASGPPRVRV-amide | 22 | +7 | 45 | Tumor suppressor protein p14ARF | 62 |
| YTA-4 | Acetyl-IAWVKAFIRKLRKGPLG-amide | 17 | +5 | 52 | Substrate of matrix metalloprotease 2 | 63 |
| MAP | KLALKALKALKAALKLA-amide | 17 | +5 | 70 | Designed model amphipathic peptide | 9, 27 |
| LL-37 | LLGDFFRKSKEKIGKEFKRIVQRIKDFLRNLVPRTES | 37 | +6 | 35 | Human cathelicidin | 64 |
Abbreviations: TP10, transportan-10; MAP, model amphipathic peptide; aa, amino acids.
As predicted by using the Antimicrobial Peptide Database website (http://aps.unmc.edu/AP/main.html) (65).
Bactericidal assays.
N. meningitidis FAM20 cells were resuspended in GC liquid and incubated with agitation. Survival was analyzed by plating of serial dilutions. The effects of CPPs, LL-37, and polymyxin B on FAM20 cells (105 CFU/ml) were screened at 30 μM for 6 h and compared to effects on PBS-treated controls. Screening was performed in ≥2 independent experiments. Concentration-dependent killing of FAM20 or serogroup A, B, or W cells (105 CFU/ml) was analyzed after incubation with 0 to 16 μM TP10, 0 to 32 μM MAP, or 0 to 1,024 μM polymyxin B for 6 h and performed ≥3 times in duplicate or triplicate. Time-dependent killing of FAM20 cells (105 CFU/ml) was analyzed at 0 to 60 min, 3 h, 6 h, and 12 h with TP10 (0 to 16 μM) or MAP (0 to 32 μM) in three independent experiments.
Hemolytic assay and bactericidal activity in human whole blood.
Venous blood was collected from healthy human volunteers in heparinized Vacutainer tubes (BD Bioscience). Heparin was chosen as the anticoagulant due to the low survival rate of bacteria in EDTA-treated blood. For hemolysis assays, TP10 or MAP (0 or 100 μM) was added to four parts of blood mixed with one part of PBS with or without bacteria and incubated for 3 h. Samples were diluted in PBS (1:25) and centrifuged for 5 min at 1,000 × g, and supernatants were read at 540 nm. The negative (PBS) and positive (1% Triton X-100) controls were set to 0% and 100% hemolysis, respectively. The experiment was performed three times.
For bactericidal assays in whole blood, N. meningitidis MC58 cells were resuspended in PBS and mixed with four parts of whole blood to obtain 105 CFU/ml. TP10 or MAP was added (0 to 100 μM) for 3 h. Blood killing was tested in ≥3 independent experiments. After incubation, samples were also assayed for hemolysis, as described above.
Mouse model.
The CD46 transgenic mouse strain expresses human CD46 in a human-like pattern and was described previously (30). Mice were challenged intraperitoneally (i.p.) with 109 CFU of MC58 cells in 100 μl of GC broth. Treatment was performed at 1 h postinfection by i.p. injection of 100 μl of TP10 in PBS (0.016 mg TP10/g) or PBS alone (≥15 mice per group). Bacteremia was examined immediately before and 1 h after TP10 treatment (corresponding to 1 h and 2 h after bacterial challenge) by plating serial dilutions of blood samples. Animal care and experiments were done in accordance with institutional guidelines and were approved by national ethical committees (identification number N376/11).
Live microscopy of fluorescent dye uptake.
Bacteria were grown to the logarithmic phase and added to glass-bottom plates (MatTek). SYTOX green (5 μM; Invitrogen) was added together with TP10 (0, 4, or 8 μM). Microscopy of live bacteria was performed at 37°C and 5% CO2 by using an inverted Zeiss Cell Observer microscope. Images were captured between 1 and 10 min after addition of TP10 at a ×1,000 magnification and with fixed exposure settings. TP10 and controls were treated and visualized sequentially. SYTOX green microscopy was performed in three independent experiments. A total of ≥10 samples was imaged for each concentration.
In vitro differentiation of human peripheral monocyte-derived macrophages.
Isolation of monocytes was performed essentially as previously described (31, 32). In brief, CD14+ monocytes were isolated from buffy coats by negative selection using RosetteSep human monocyte enrichment cocktail (0.5 ml/10 ml blood; StemCell Technologies, Vancouver, Canada) and separated on Ficoll-Paque Plus. Cells were cultured for 6 to 7 days in complete medium containing RPMI 1640 with GlutaMAX and 10% FBS. The medium was further supplemented with recombinant human macrophage colony-stimulating factor (M-CSF) (50 ng/ml; Immunotools) to differentiate monocytes into macrophages. Purity of isolated monocytes and differentiated macrophages was determined by CD14 and CD68 staining, respectively, followed by fluorescence-activated cell sorter (FACS) analysis.
Analysis of cytokine expression.
RAW 264.7 macrophages were infected with N. meningitidis FAM20 cells (105 CFU/ml) in DMEM–10% FBS. TP10 (0 or 16 μM) was added simultaneously, and cell supernatants were collected after 6 h. For stimulation of RAW 264.7 cells, THP1 cells, and primary macrophages with dead bacteria, FAM20 cells were heat inactivated in PBS at 56°C for 1 h. Heat-killed bacteria were added to cells (equaling 3 × 105 CFU/ml of live bacteria) simultaneously with TP10 (0 or 16 μM) for 6 h. For stimulation with LPS, RAW 264.7 cells were treated with LPS from Escherichia coli O111:B4 (0 to 200 ng/ml; Sigma-Aldrich) or purified L3-type LPS (0 to 10 ng/ml) and TP10 (0 or 16 μM) for 6 h. Meningococcal L3-type LPS was a kind gift from Tone Tønjum (Centre for Molecular Biology and Neuroscience, Department of Microbiology, Oslo University Hospital). Briefly, LPS was purified from proteinase K-treated, boiled bacterial cells. Tricine-SDS-PAGE gels were run for 17 h at a constant current of 20 mA and silver stained. LPS was isolated by the hot phenol-water extraction method and dissolved in 1 mM Tris (pH 7.5) (33). Tumor necrosis factor alpha (TNF-α) and interleukin-6 (IL-6) levels in cell supernatants were analyzed by enzyme-linked immunosorbent assays (ELISAs), using Invitrogen antibody pairs. Cytokine concentrations were calculated from A450 values by using four-parameter curve fitting. For all settings, three independent experiments were performed.
LPS-FITC binding to macrophages.
RAW 264.7 cells (5 × 105 cells/well) were incubated for 30 min at 37°C with fluorescein isothiocyanate (FITC)-coupled LPS from Escherichia coli O111:B4 (1 μg/ml; Sigma-Aldrich) with or without 16 μM TP10, LL-37, or magainin II and subsequently washed. Binding of LPS-FITC was analyzed by flow cytometry in three independent experiments. Results are expressed as relative binding of LPS compared to the control without peptide.
Statistical analysis.
Differences between multiple groups were analyzed by ANOVA (analysis of variance) followed by the Bonferroni posttest. Data from animal experiments were analyzed by using the Mann-Whitney U test. Statistical differences between ratios were analyzed after log transformation of the data. A P value below 0.05 was considered statistically significant. Statistical analysis was performed by using Graph Pad Prism 4 software.
RESULTS
Identification of peptides with antibacterial activity against N. meningitidis.
Antimicrobial peptides have been suggested as potential drugs against bacterial infections (15). However, their in vivo use has often been limited by poor activity under physiological conditions (34). In order to identify novel peptides with antibacterial and anti-inflammatory activity against N. meningitidis, we screened a set of peptides for the ability to inhibit meningococcal survival (see Table 1 for a list of peptides). Three peptides, i.e., TP10, YTA-4, and MAP, exhibited antimicrobial activity against N. meningitidis serogroup C strain FAM20 after 6 h of incubation at a concentration of 30 μM. The most potent peptides, TP10 and MAP, eradicated meningococci from a starting concentration of 105 CFU/ml and thus proved to be more potent than the human cationic AMP LL-37 and the peptide antibiotic polymyxin B (Fig. 1). Both TP10 and MAP caused killing of N. meningitidis in a dose-dependent manner (Fig. 2A and B). A >3-log reduction in viable bacteria compared to untreated controls was observed at 8 μM for TP10 (17.5 μg/ml) and at 16 μM for MAP (28.2 μg/ml) after 6 h of incubation (Fig. 2A and B). These concentrations are referred to as the inhibitory concentrations. In contrast, more than 64 μM polymyxin B (≥88.7 μg/ml) was required for a similar effect (Fig. 2C). Kinetic studies revealed that both TP10 and MAP were able to decrease bacterial viability within 10 min (Fig. 2D and E), a time span too short to allow for the growth of N. meningitidis. This indicates a bactericidal action of the peptides. The bactericidal concentrations of 16 μM TP10 and 32 μM MAP cleared an inoculum of 105 CFU/ml within 40 to 60 min, while 512 μM polymyxin B was required for a bactericidal effect (Fig. 2C). At half the bactericidal concentration, TP10 (8 μM) inhibited bacterial growth above the inoculum concentration (<105 CFU/ml) for up to 12 h of incubation, compared to >108 CFU/ml for untreated controls (Fig. 2D), while bacteria treated with 16 μM MAP were able to grow to roughly 108 CFU/ml within 12 h (Fig. 2E). The meningococcal capsule is involved in AMP resistance (35, 36), and its composition may thus impact the bactericidal activity of CPPs. However, both TP10 and MAP were active against all tested serogroups (A, B, C, and W), while a much higher concentration of polymyxin B was required for bactericidal activity (Table 2).
Fig 1.
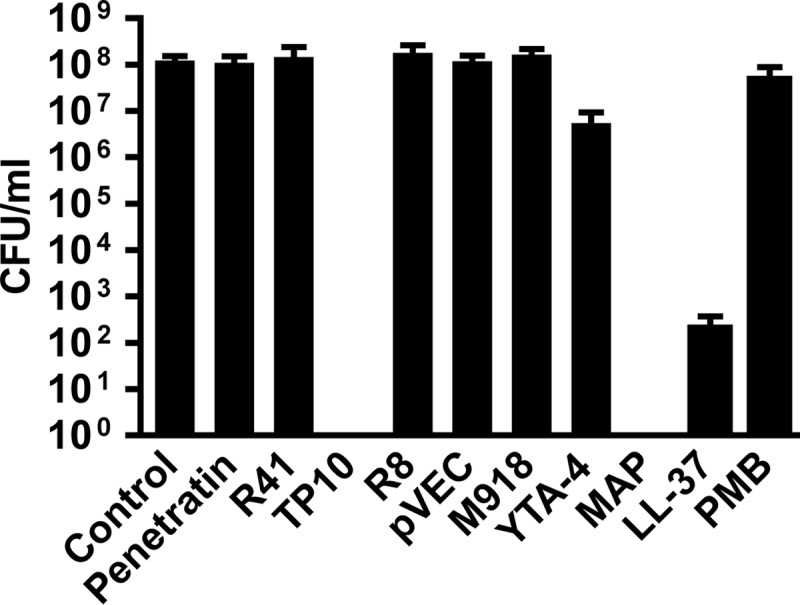
Identification of peptides with potent antimicrobial activity against N. meningitidis. N. meningitidis serogroup C strain FAM20 cells (105 CFU/ml) were incubated for 6 h with CPPs, LL-37, or polymyxin B (PMB) at a concentration of 30 μM in PBS or with PBS alone (control) in GC broth. LL-37 was suspended to 30 μM in 0.1% TFA. Treatment with 0.1% TFA alone (for LL-37) gave results equal to those with PBS alone (control). CFU were determined by plating of serial dilutions. Data are presented as means and standard deviations of ≥2 independent experiments. Peptides are listed in Table 1.
Fig 2.

Transportan-10 (TP10) and model amphipathic peptide (MAP) induce rapid and exponential killing. (A to C) Dose-dependent action of TP10, MAP, and polymyxin B (PMB) in GC broth. FAM20 cells (105 CFU/ml) were incubated for 6 h with TP10 (0, 4, 8, and 16 μM) (A), MAP (0, 8, 16, and 32 μM) (B), or PMB (0, 64, 128, 256, and 512 μM) (C). Bars represent the means and standard errors of ≥3 independent experiments. (D and E) Antimicrobial action of TP10 (D) and MAP (E) over time. FAM20 cells (105 CFU/ml) were incubated with TP10 (0, 4, 8, and 16 μM) or MAP (0, 8, 16, and 32 μM) in GC broth. Bacteria were plated after 0 to 12 h. Results are presented as means ± standard errors of three independent experiments. (F to H) Cell viability was assayed by using an MTT assay after incubation of RAW 264.7 macrophages with TP10 (0 to 128 μM) (F), MAP (0 to 128 μM) (G), or PMB (0 to 512 μM) (H) for 6 h. Formaldehyde-fixed samples (PFA) were used as the blank, and 0.5% Triton X-100 (TX) was used as the positive control. Cell viability was expressed as the absorbance ratio of TP10-treated cells to viable controls (PBS). Data are presented as means and standard errors of ≥3 independent experiments. Statistically significant differences are indicated by an asterisk (P < 0.05, determined analysis of variance followed by the Bonferroni posttest). PFA, paraformaldehyde.
Table 2.
Bactericidal concentrationsa of TP10, MAP, and polymyxin B for different meningococcal serogroups after 6 h of treatment
| Peptide | Bactericidal concn (μM) (concn in μg/ml) against serogroup |
|||
|---|---|---|---|---|
| A | B | C | W | |
| TP10 | 16 (35.0) | 8 (17.5) | 16 (35.0) | 32 (70.0) |
| MAP | ≥32 (56.4) | 32 (56.4) | 32 (56.4) | >32 (56.4) |
| Polymyxin B | 256 (354.7) | 128 (177.4) | 512 (709.4) | ≥512 (709.4) |
Defined as the concentrations causing eradication of all viable bacteria from an inoculum of 105 CFU/ml.
In order to assess peptide toxicity to eukaryotic cells, we performed MTT cell viability assays with RAW 264.7 macrophages treated with TP10, MAP, and polymyxin B for 6 h. Significant toxicity was not observed until concentrations of 64 μM TP10 and 128 μM MAP were used, equaling 8-fold inhibitory concentrations at 6 h (Fig. 2F and G). In contrast, polymyxin B exhibited toxic effects already at two times the inhibitory concentration (128 μM) (Fig. 2H).
Antimicrobial activity of transportan-10 and model amphipathic peptide in whole blood.
Next, the bactericidal activities of TP10 and MAP were assessed in human whole blood. Serogroup B strain MC58 was chosen for whole-blood assays, since FAM20 had a limited capacity to survive in human whole blood (data not shown). At 3 h of incubation with meningococci, both TP10 and MAP exhibited significant antimicrobial activity in human whole blood at concentrations of 50 and 100 μM (Fig. 3A). To exclude eukaryotic cell toxicity at these concentrations, red blood cell lysis was quantified after the treatment of human whole blood with TP10 or MAP. After 3 h of incubation at 100 μM, the peptides induced only negligible hemolysis (<2% for TP10 and <1% for MAP) (Fig. 3B). Hemolysis was not observed after incubation of blood with either peptide in the presence of bacteria (data not shown).
Fig 3.

Antimicrobial effect of TP10 in human whole blood and in vivo. (A) Survival of MC58 cells incubated with TP10 or MAP (0, 50, and 100 μM) in human whole blood for 3 h. Results are expressed as the mean survival ratios [(CFU after peptide incubation)/(CFU after control incubation)] and standard errors of three independent experiments. Statistically significant differences are indicated by an asterisk (P < 0.05, determined by analysis of variance with the Bonferroni posttest). (B) Human whole blood incubated with 100 μM TP10 or MAP for 3 h. Hemolysis was quantified by measuring the absorbance at 540 nm of supernatants from diluted and centrifuged samples. Triton X-100 (TX) (1%) was included as positive control. The absorbance value from the positive control was set to 100%. Results are presented as means and standard errors of three independent experiments. (C) Bacterial blood counts in CD46 transgenic mice infected i.p. with MC58 at a dose of 1 × 109 CFU. TP10 (0.016 mg/g mouse) or PBS (control) (n ≥ 15 per group) was injected i.p. at 1 h postinfection. Bacterial counts in blood (CFU/ml) were determined immediately before and 1 h after peptide treatment. Results are presented as the fold change in bacterial load (bacterial counts after treatment/bacterial counts before treatment). The lines indicate the median fold change in bacterial load for each group. Statistically significant differences are indicated by an asterisk (P < 0.05, determined by a Mann-Whitney U test on the log fold change).
Transportan-10 reduces bacteremia in mice.
The therapeutic potential of the peptides was further evaluated in a mouse model (30). Due to the toxicity of MAP observed in initial in vivo experiments (data not shown), all further studies were performed solely with TP10. Mice were infected intraperitoneally (i.p.) with serogroup B strain MC58 and treated with TP10 (0.016 mg/g mouse) i.p. at 1 h postinfection. The bacterial load in blood was determined immediately before and 1 h after TP10 treatment, and the fold change in bacteremia was calculated. TP10 treatment resulted in significantly decreased bacteremia levels compared to the control treatment (Fig. 3C). No adverse side effects were observed in infected mice upon i.p. administration of a single dose of TP10. In summary, these data show that TP10 exhibited significant antimicrobial activity in an in vivo model.
Impact of transportan-10 on membrane integrity of N. meningitidis.
Since TP10 was found to be superior to MAP with respect to bactericidal activity and suitability for in vivo use, we continued to characterize the activity of this peptide. The impact of TP10 on membrane integrity was studied by fluorescence microscopy of N. meningitidis in the presence of the cell-impermeable nucleic acid stain SYTOX green. SYTOX green is selectively taken up into cells with compromised membrane integrity and exhibits greatly enhanced fluorescence upon DNA binding. SYTOX green and TP10 were added simultaneously to log-phase bacteria, and microscopy images were captured from 1 min to 10 min. Upon addition of 8 μM TP10, SYTOX green-positive bacteria were detected as early as 1 min after treatment (Fig. 4). The SYTOX green intensity increased strongly over time, and SYTOX green was detectable in nearly all bacteria within 5 to 10 min. Following treatment of N. meningitidis with 4 μM TP10, SYTOX green-positive bacteria were detectable after ≤5 min, but not all bacteria of a given microcolony were permeabilized within 10 min. In control cells treated with 0 μM TP10, only a few SYTOX green-positive bacteria were detected. Taken together, these results suggest a membrane-disruptive action of TP10.
Fig 4.

Effects of TP10 on membrane integrity. Membrane integrity was analyzed by fluorescence microscopy of N. meningitidis in the presence of the cell-impermeable nucleic acid stain SYTOX green, which exhibits greatly enhanced fluorescence upon uptake and DNA binding. FAM20 cells were grown to log phase in GC broth. After the addition of SYTOX green (5 μM) and TP10 (0, 4, or 8 μM), bright-field and fluorescence images (SYTOX green) were captured at the indicated time points and with fixed exposure settings. Representative images are shown.
Transportan-10 decreases inflammatory cytokine secretion from infected macrophages.
In order to investigate whether the synthetic peptide TP10 had an effect on inflammatory cytokine secretion, murine RAW 264.7 macrophages were infected with live N. meningitidis cells for 6 h with or without TP10 at 16 μM. Infection with bacteria in the presence of peptide significantly reduced the release of TNF-α (Fig. 5A) and IL-6 (Fig. 5B). The observed decrease in inflammatory cytokine secretion upon TP10 treatment was found to be independent of antibacterial activity, as an equivalent decrease was detected after stimulation of macrophages with heat-killed bacteria (Fig. 5A and B). A similar reduction of inflammatory cytokine release after treatment with TP10 was also observed for human peripheral monocyte-derived macrophages stimulated with heat-killed N. meningitidis (Fig. 5C and D) and for monocytic THP1 cells (data not shown). MTT cell viability assays confirmed that TP10 did not exhibit cytotoxic effects on RAW 264.7 macrophages (Fig. 2F) or THP1 cells (data not shown) at the concentrations used in the cytokine assays. In primary macrophages, a slight but nonsignificant decrease in cell viability was observed at 16 μM TP10 (Fig. 5E); however, this cannot explain the inhibitory effect of TP10 on inflammatory cytokine release that was already observed at the lower concentration of 8 μM TP10. Taken together, these results demonstrate that TP10 decreases levels of inflammatory cytokines induced by both live and heat-killed N. meningitidis, suggesting that this effect is not based on its antimicrobial activity.
Fig 5.

TP10 decreases cytokine release from macrophages infected with N. meningitidis. (A and B) RAW 264.7 murine macrophages were infected with live or heat-killed FAM20 cells with or without TP10 (16 μM) for 6 h. Release of TNF-α (A) or IL-6 (B) was quantified by ELISA. Results are presented as means and standard errors of three independent experiments. (C and D) Human peripheral monocyte-derived macrophages were infected with heat-killed FAM20 cells and treated with 0, 8, and 16 μM TP10 for 6 h. Release of TNF-α (C) and IL-6 (D) was quantified by ELISA. One representative experiment from a total of two (8 μM) or four (16 μM) is shown. Bars represent means and standard deviations of triplicate wells. (E) Cell viability was assayed by using an MTT assay after incubation of human peripheral monocyte-derived macrophages with TP10 (0 to 32 μM) or 0.5% Triton X-100 (TX) for 6 h. Formaldehyde-fixed samples (PFA) were used as the blank. Cell viability was expressed as the absorbance ratio of TP10-treated cells to viable controls (0 μM TP10). Data are presented as means and standard errors of four independent experiments. Statistically significant differences are indicated by an asterisk (P < 0.05, determined by analysis of variance followed by the Bonferroni posttest).
Transportan-10 reduces both LPS-mediated inflammatory cytokine secretion and binding of LPS to macrophages.
LPS is one of the main inflammatory mediators in meningococcal disease and is related to the LPSs of other Gram-negative bacteria. When stimulating RAW 264.7 macrophages with up to 200 ng/ml of purified LPS from Escherichia coli for 6 h, levels of secretion of both TNF-α and IL-6 were significantly reduced in the presence of 16 μM TP10 (Fig. 6A and B). Next, the same experiment was performed with purified meningococcal LPS. The presence of 16 μM TP10 significantly decreased the secretion of TNF-α and IL-6 from RAW 264.7 cells stimulated with up to 10 ng/ml of meningococcal LPS (Fig. 6C and D). Therefore, we hypothesized that the decrease in cytokine release in the presence of TP10 could be linked to the amount of LPS bound to macrophages. To test this, we used FITC-coupled LPS and quantified LPS binding to RAW 264.7 cells in the presence or absence of TP10 by flow cytometry. The antimicrobial peptides LL-37, a known inhibitor of LPS binding, and magainin II, lacking such an LPS-inhibitory effect, served as positive and negative controls, respectively (37). We found that TP10 significantly decreased the binding of LPS to RAW 264.7 macrophages (Fig. 6E), corresponding to the effects of LL-37. These results demonstrate that TP10 decreases cytokine release induced by both E. coli and N. meningitidis LPSs and reduces the binding of LPS to RAW 264.7 macrophages.
Fig 6.

TP10 reduces LPS-induced cytokine release from macrophages. (A and B) RAW 264.7 cells were stimulated with 0, 20, or 200 ng/ml of purified E. coli LPS in the absence or presence of 16 μM TP10 for 6 h. TNF-α and IL-6 secretions were analyzed by ELISA. One representative experiment out of three is shown. Bars represent means and standard deviations of triplicate wells. (C and D) Similarly, RAW 264.7 cells were stimulated with 0, 1, or 10 ng/ml of purified N. meningitidis LPS with or without 16 μM TP10 for 6 h. Cytokine secretion was analyzed by ELISA. Bars represent means and standard errors of three independent experiments. (E) RAW 264.7 macrophages were incubated with LPS-FITC (1 μg/ml) in the absence (control) or presence of 16 μM TP10, LL-37, or magainin II. LPS binding was analyzed by flow cytometry. Data are presented as relative LPS binding compared to controls. Bars represent the mean ratios and standard errors of three independent experiments. Statistically significant differences are indicated by an asterisk (P < 0.05, determined by analysis of variance followed by the Bonferroni posttest).
DISCUSSION
In a search for peptides with antimicrobial activity against N. meningitidis, we identified two CCPs, TP10 and MAP, with antimicrobial activities superior to those of the human AMP LL-37 and the peptide antibiotic polymyxin B. TP10 as well as an analogue of the MAP used in this study were previously tested in vitro on Staphylococcus aureus, E. coli, and Plasmodium falciparum (9, 38, 39). In this work, we show that these peptides are active in human whole blood and that TP10 exhibits significant antimicrobial activity against N. meningitidis in a mouse model. Moreover, we report a novel activity of TP10, namely, the inhibition of inflammatory cytokine secretion induced by lipopolysaccharide (LPS).
Using fluorescence microscopy, we detected an uptake of the membrane-impermeable dye SYTOX green into meningococci treated with low concentrations of TP10 (4 and 8 μM) within 1 to 5 min. These results indicate that TP10 exhibits membrane-disruptive activity toward the Gram-negative species N. meningitidis. This is in agreement with the results of Nekhotiaeva et al. (39), who previously suggested such an activity regarding the Gram-positive bacterium S. aureus. The concentrations of TP10 required for membrane permeabilization (≥4 μM) and antimicrobial activity (≥8 μM) did not affect eukaryotic cell viability (MTT assays in Fig. 2F and 5E). Correspondingly, it was suggested that the uptake of certain CPPs into eukaryotic cells is achieved without or with only transient membrane permeabilization, while CPPs may selectively disrupt prokaryotic membranes, thus resembling membrane-active AMPs (8, 26, 40–42). Altogether, accumulating data suggest a dual effect of TP10: efficient cargo delivery into eukaryotic cells at submicromolar concentrations (43) and a membrane-disruptive activity toward microbial membranes at 4 to 8 μM.
In kinetic studies with 8 μM TP10, we observed an increase in bacterial viability between 3 and 6 h, although bacterial growth above the inoculum concentration of 105 CFU/ml was inhibited for up to 12 h (Fig. 2D). This partial recovery may result from bacterial adaptation, loss of peptide activity, or the emergence of resistant clones. It was previously argued that bacterial resistance to AMPs may occur at lower levels than antibiotic resistance (44). However, studies reported both the spontaneous occurrence of AMP-resistant mutants and the experimental evolution of AMP resistance (45–48). In our short-term studies, bacterial adaptation or potential resistance was not able to overcome the growth-inhibitory effect of 8 μM TP10 within 12 h.
AMPs have been suggested as potential future treatments of multidrug-resistant bacteria and other bacterial infections (15, 49). However, the in vivo use of many AMPs is limited by poor activity under physiological conditions as well as an undesired toxicity to eukaryotic cells at higher concentrations (34). TP10 and MAP exhibited significant activity against N. meningitidis in human whole blood. Higher concentrations of peptide were needed for bacterial killing in whole blood than in growth medium, presumably due to the high protein content and the presence of serum. In contrast to previous data on purified erythrocytes (50), hemolytic activity was not observed at antimicrobial concentrations of the peptides. The hemolysis assay (Fig. 3B) was performed by using heparinized venous blood, with a final heparin concentration of 17 U/ml. This concentration may potentially interfere with complement-mediated hemolysis (51). However, experiments with TP10 in EDTA-anticoagulated blood showed negligible hemolysis as well (data not shown). In RAW 264.7 cell cultures, toxic effects of TP10 were not observed until 8 times the inhibitory concentration was used, while for polymyxin B, no margin between toxic and inhibitory drug concentrations was observed.
In a mouse model, in which infection was established before treatment was initiated intraperitoneally, TP10 was able to significantly reduce the bacterial load compared to the bacterial load in mice given PBS alone. Sarko et al. (52) reported a good serum half-life of TP10 exceeding 72 h. The same study suggested a longer circulation time of TP10 than of other CPPs after peptide injection into rats. Other studies have shown that CPPs disperse easily throughout the body irrespective of the administration route (41). Injection of TP10 into mice resulted in an accumulation of the peptide in spleen and liver (52). This accumulation coincided with decreasing TP10 levels in the circulation, comparable to the clearance of other CPPs (52). We observed no adverse side effects of TP10 in mice infected with N. meningitidis. However, the injection of TP10 into uninfected controls caused transient impairment of activity and locomotion. Mice regained normal appearance and activity after 10 to 20 min, without signs of long-term side effects over 10 days of observation (data not shown).
In addition to its antimicrobial effect, TP10 was found to significantly decrease inflammatory cytokine release from macrophages infected with N. meningitidis or macrophages stimulated with purified meningococcal LPS. This effect correlated with reduced binding of LPS to TP10-treated macrophages and was unrelated to its bactericidal activity. Endotoxin-neutralizing activity has been described for certain AMPs and peptides derived from diverse LPS-binding proteins (53–56). Similar to TP10, the peptide antibiotic polymyxin B is able to inhibit the inflammatory action of LPS both in vitro and in vivo (57, 58). Regarding inhibition of meningococcal LPS, polymyxin B is less effective (59), a trend which we also observed for TP10. We found bactericidal concentrations of polymyxin B (128 to ≥512 μM [177.4 to ≥709.4 μg/ml]) to overlap concentrations at which toxicity for human cells was observed (128 μM [177.4 μg/ml]). Thus, TP10's advantage is that meningococci are sensitive to its antimicrobial action, while they are intrinsically resistant to nontoxic concentrations of polymyxin B (60).
Meningococcal disease is characterized by a rapid onset, a high mortality rate, and serious sequelae in survivors (25). Antibiotic treatment, together with intensive care, is often initiated late and, hence, ineffectively, and supplementary therapeutic strategies are needed. By combining rapid tissue penetration and good serum stability (41, 52) with LPS-inhibitory potential and in vivo antibacterial activity, peptides such as TP10 may provide a scaffold for the design of novel therapies.
ACKNOWLEDGMENTS
This work was supported by the Swedish Research Council, the Swedish Cancer Society, the Knut and Alice Wallenbergs Foundation, Ragnar Söderbergs Stiftelse, and Torsten Söderbergs Stiftelse.
We thank the Electron Microscopy Center (EMC) at the Department of Materials and Environmental Chemistry (MMK), Stockholm University, for the use of their scanning electron microscope equipment as well as Kjell Jansson (MMK) for his technical expertise. We thank Tone Tønjum (Centre for Molecular Biology and Neuroscience, Department of Microbiology, Oslo University Hospital) for kindly providing purified meningococcal LPS and Sushil Kumar Pathak (Department of Molecular Biosciences, The Wenner-Gren Institute, Stockholm University) for help with isolation of monocytes.
Footnotes
Published ahead of print 20 May 2013
REFERENCES
- 1. Fonseca SB, Pereira MP, Kelley SO. 2009. Recent advances in the use of cell-penetrating peptides for medical and biological applications. Adv. Drug Deliv. Rev. 61:953–964 [DOI] [PubMed] [Google Scholar]
- 2. Lehto T, Simonson OE, Mäger I, Ezzat K, Sork H, Copolovici DM, Viola JR, Zaghloul EM, Lundin P, Moreno PM, Mae M, Oskolkov N, Suhorutsenko J, Smith CE, Andaloussi SE. 2011. A peptide-based vector for efficient gene transfer in vitro and in vivo. Mol. Ther. 19:1457–1467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Uchida T, Kanazawa T, Kawai M, Takashima Y, Okada H. 2011. Therapeutic effects on atopic dermatitis by anti-RelA siRNA with functional peptides, Tat and AT1002. J. Pharmacol. Exp. Ther. 338:443–450 [DOI] [PubMed] [Google Scholar]
- 4. Yin L, Ahmad R, Kosugi M, Kawano T, Avigan D, Stone R, Kharbanda S, Kufe D. 2010. Terminal differentiation of chronic myelogenous leukemia cells is induced by targeting of the MUC1-C oncoprotein. Cancer Biol. Ther. 10:483–491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Henriques ST, Melo MN, Castanho MA. 2006. Cell-penetrating peptides and antimicrobial peptides: how different are they? Biochem. J. 399:1–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Raagel H, Saalik P, Pooga M. 2010. Peptide-mediated protein delivery—which pathways are penetrable? Biochim. Biophys. Acta 1798:2240–2248 [DOI] [PubMed] [Google Scholar]
- 7. Mae M, Langel U. 2006. Cell-penetrating peptides as vectors for peptide, protein and oligonucleotide delivery. Curr. Opin. Pharmacol. 6:509–514 [DOI] [PubMed] [Google Scholar]
- 8. Splith K, Neundorf I. 2011. Antimicrobial peptides with cell-penetrating peptide properties and vice versa. Eur. Biophys. J. 40:387–397 [DOI] [PubMed] [Google Scholar]
- 9. Palm C, Netzereab S, Hallbrink M. 2006. Quantitatively determined uptake of cell-penetrating peptides in non-mammalian cells with an evaluation of degradation and antimicrobial effects. Peptides 27:1710–1716 [DOI] [PubMed] [Google Scholar]
- 10. Park N, Yamanaka K, Tran D, Chandrangsu P, Akers JC, de Leon JC, Morrissette NS, Selsted ME, Tan M. 2009. The cell-penetrating peptide, Pep-1, has activity against intracellular chlamydial growth but not extracellular forms of Chlamydia trachomatis. J. Antimicrob. Chemother. 63:115–123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jung HJ, Park Y, Hahm KS, Lee DG. 2006. Biological activity of Tat (47-58) peptide on human pathogenic fungi. Biochem. Biophys. Res. Commun. 345:222–228 [DOI] [PubMed] [Google Scholar]
- 12. Brogden KA. 2005. Antimicrobial peptides: pore formers or metabolic inhibitors in bacteria? Nat. Rev. Microbiol. 3:238–250 [DOI] [PubMed] [Google Scholar]
- 13. Nicolas P. 2009. Multifunctional host defense peptides: intracellular-targeting antimicrobial peptides. FEBS J. 276:6483–6496 [DOI] [PubMed] [Google Scholar]
- 14. Peschel A, Sahl HG. 2006. The co-evolution of host cationic antimicrobial peptides and microbial resistance. Nat. Rev. Microbiol. 4:529–536 [DOI] [PubMed] [Google Scholar]
- 15. Hancock RE, Sahl HG. 2006. Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies. Nat. Biotechnol. 24:1551–1557 [DOI] [PubMed] [Google Scholar]
- 16. Zughaier SM, Svoboda P, Pohl J, Stephens DS, Shafer WM. 2010. The human host defense peptide LL-37 interacts with Neisseria meningitidis capsular polysaccharides and inhibits inflammatory mediators release. PLoS One 5:e13627. 10.1371/journal.pone.0013627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Grigat J, Soruri A, Forssmann U, Riggert J, Zwirner J. 2007. Chemoattraction of macrophages, T lymphocytes, and mast cells is evolutionarily conserved within the human alpha-defensin family. J. Immunol. 179:3958–3965 [DOI] [PubMed] [Google Scholar]
- 18. Bowdish DM, Davidson DJ, Scott MG, Hancock RE. 2005. Immunomodulatory activities of small host defense peptides. Antimicrob. Agents Chemother. 49:1727–1732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kindrachuk J, Jenssen H, Elliott M, Nijnik A, Magrangeas-Janot L, Pasupuleti M, Thorson L, Ma S, Easton DM, Bains M, Finlay B, Breukink EJ, Sahl HG, Hancock RE. 2013. Manipulation of innate immunity by a bacterial secreted peptide: lantibiotic nisin Z is selectively immunomodulatory. Innate Immun. 19:315–327 [DOI] [PubMed] [Google Scholar]
- 20. Scott MG, Dullaghan E, Mookherjee N, Glavas N, Waldbrook M, Thompson A, Wang A, Lee K, Doria S, Hamill P, Yu JJ, Li Y, Donini O, Guarna MM, Finlay BB, North JR, Hancock RE. 2007. An anti-infective peptide that selectively modulates the innate immune response. Nat. Biotechnol. 25:465–472 [DOI] [PubMed] [Google Scholar]
- 21. Nijnik A, Madera L, Ma S, Waldbrook M, Elliott MR, Easton DM, Mayer ML, Mullaly SC, Kindrachuk J, Jenssen H, Hancock RE. 2010. Synthetic cationic peptide IDR-1002 provides protection against bacterial infections through chemokine induction and enhanced leukocyte recruitment. J. Immunol. 184:2539–2550 [DOI] [PubMed] [Google Scholar]
- 22. Rubinchik E, Dugourd D, Algara T, Pasetka C, Friedland HD. 2009. Antimicrobial and antifungal activities of a novel cationic antimicrobial peptide, omiganan, in experimental skin colonisation models. Int. J. Antimicrob. Agents 34:457–461 [DOI] [PubMed] [Google Scholar]
- 23. Yeung AT, Gellatly SL, Hancock RE. 2011. Multifunctional cationic host defence peptides and their clinical applications. Cell. Mol. Life Sci. 68:2161–2176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lipsky BA, Holroyd KJ, Zasloff M. 2008. Topical versus systemic antimicrobial therapy for treating mildly infected diabetic foot ulcers: a randomized, controlled, double-blinded, multicenter trial of pexiganan cream. Clin. Infect. Dis. 47:1537–1545 [DOI] [PubMed] [Google Scholar]
- 25. Stephens DS, Greenwood B, Brandtzaeg P. 2007. Epidemic meningitis, meningococcaemia, and Neisseria meningitidis. Lancet 369:2196–2210 [DOI] [PubMed] [Google Scholar]
- 26. Soomets U, Lindgren M, Gallet X, Hallbrink M, Elmquist A, Balaspiri L, Zorko M, Pooga M, Brasseur R, Langel U. 2000. Deletion analogues of transportan. Biochim. Biophys. Acta 1467:165–176 [DOI] [PubMed] [Google Scholar]
- 27. Oehlke J, Scheller A, Wiesner B, Krause E, Beyermann M, Klauschenz E, Melzig M, Bienert M. 1998. Cellular uptake of an alpha-helical amphipathic model peptide with the potential to deliver polar compounds into the cell interior non-endocytically. Biochim. Biophys. Acta 1414:127–139 [DOI] [PubMed] [Google Scholar]
- 28. Magzoub M, Graslund A. 2004. Cell-penetrating peptides: from inception to application. Q. Rev. Biophys. 37:147–195 [DOI] [PubMed] [Google Scholar]
- 29. Coin I, Beyermann M, Bienert M. 2007. Solid-phase peptide synthesis: from standard procedures to the synthesis of difficult sequences. Nat. Protoc. 2:3247–3256 [DOI] [PubMed] [Google Scholar]
- 30. Johansson L, Rytkonen A, Wan H, Bergman P, Plant L, Agerberth B, Hokfelt T, Jonsson AB. 2005. Human-like immune responses in CD46 transgenic mice. J. Immunol. 175:433–440 [DOI] [PubMed] [Google Scholar]
- 31. Hertzen E, Johansson L, Kansal R, Hecht A, Dahesh S, Janos M, Nizet V, Kotb M, Norrby-Teglund A. 2012. Intracellular Streptococcus pyogenes in human macrophages display an altered gene expression profile. PLoS One 7:e35218. 10.1371/journal.pone.0035218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hertzen E, Johansson L, Wallin R, Schmidt H, Kroll M, Rehn AP, Kotb M, Morgelin M, Norrby-Teglund A. 2010. M1 protein-dependent intracellular trafficking promotes persistence and replication of Streptococcus pyogenes in macrophages. J. Innate Immun. 2:534–545 [DOI] [PubMed] [Google Scholar]
- 33. Westphal O, Jann JK. 1965. Bacterial lipopolysaccharide: extraction with phenol-water and furthers applications of the procedure. Methods Carbohydr. Chem. 5:83–91 [Google Scholar]
- 34. Brogden NK, Brogden KA. 2011. Will new generations of modified antimicrobial peptides improve their potential as pharmaceuticals? Int. J. Antimicrob. Agents 38:217–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Jones A, Geörg M, Maudsdotter L, Jonsson AB. 2009. Endotoxin, capsule, and bacterial attachment contribute to Neisseria meningitidis resistance to the human antimicrobial peptide LL-37. J. Bacteriol. 191:3861–3868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Spinosa MR, Progida C, Tala A, Cogli L, Alifano P, Bucci C. 2007. The Neisseria meningitidis capsule is important for intracellular survival in human cells. Infect. Immun. 75:3594–3603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Rosenfeld Y, Papo N, Shai Y. 2006. Endotoxin (lipopolysaccharide) neutralization by innate immunity host-defense peptides. Peptide properties and plausible modes of action. J. Biol. Chem. 281:1636–1643 [DOI] [PubMed] [Google Scholar]
- 38. Arrighi RB, Ebikeme C, Jiang Y, Ranford-Cartwright L, Barrett MP, Langel U, Faye I. 2008. Cell-penetrating peptide TP10 shows broad-spectrum activity against both Plasmodium falciparum and Trypanosoma brucei brucei. Antimicrob. Agents Chemother. 52:3414–3417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Nekhotiaeva N, Elmquist A, Rajarao GK, Hallbrink M, Langel U, Good L. 2004. Cell entry and antimicrobial properties of eukaryotic cell-penetrating peptides. FASEB J. 18:394–396 [DOI] [PubMed] [Google Scholar]
- 40. Elmquist A, Lindgren M, Bartfai T, Langel Ü. 2001. VE-cadherin-derived cell-penetrating peptide, pVEC, with carrier functions. Exp. Cell Res. 269:237–244 [DOI] [PubMed] [Google Scholar]
- 41. Vives E, Schmidt J, Pelegrin A. 2008. Cell-penetrating and cell-targeting peptides in drug delivery. Biochim. Biophys. Acta 1786:126–138 [DOI] [PubMed] [Google Scholar]
- 42. Dunkin CM, Pokorny A, Almeida PF, Lee HS. 2011. Molecular dynamics studies of transportan 10 (tp10) interacting with a POPC lipid bilayer. J. Phys. Chem. B 115:1188–1198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kilk K, El-Andaloussi S, Jarver P, Meikas A, Valkna A, Bartfai T, Kogerman P, Metsis M, Langel U. 2005. Evaluation of transportan 10 in PEI mediated plasmid delivery assay. J. Control. Release 103:511–523 [DOI] [PubMed] [Google Scholar]
- 44. Zasloff M. 2002. Antimicrobial peptides of multicellular organisms. Nature 415:389–395 [DOI] [PubMed] [Google Scholar]
- 45. Leszczynska K, Namiot D, Byfield FJ, Cruz K, Zendzian-Piotrowska M, Fein DE, Savage PB, Diamond S, McCulloch CA, Janmey PA, Bucki R. 2013. Antibacterial activity of the human host defence peptide LL-37 and selected synthetic cationic lipids against bacteria associated with oral and upper respiratory tract infections. J. Antimicrob. Chemother. 68:610–618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Pranting M, Negrea A, Rhen M, Andersson DI. 2008. Mechanism and fitness costs of PR-39 resistance in Salmonella enterica serovar Typhimurium LT2. Antimicrob. Agents Chemother. 52:2734–2741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Perron GG, Zasloff M, Bell G. 2006. Experimental evolution of resistance to an antimicrobial peptide. Proc. Biol. Sci. 273:251–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Samuelsen O, Haukland HH, Jenssen H, Kramer M, Sandvik K, Ulvatne H, Vorland LH. 2005. Induced resistance to the antimicrobial peptide lactoferricin B in Staphylococcus aureus. FEBS Lett. 579:3421–3426 [DOI] [PubMed] [Google Scholar]
- 49. Pathan FK, Venkata DA, Panguluri SK. 2010. Recent patents on antimicrobial peptides. Recent Pat. DNA Gene Seq. 4:10–16 [DOI] [PubMed] [Google Scholar]
- 50. Saar K, Lindgren M, Hansen M, Eiriksdottir E, Jiang Y, Rosenthal-Aizman K, Sassian M, Langel U. 2005. Cell-penetrating peptides: a comparative membrane toxicity study. Anal. Biochem. 345:55–65 [DOI] [PubMed] [Google Scholar]
- 51. Logue GL. 1977. Effect of heparin on complement activation and lysis of paroxysmal nocturnal hemoglobinuria (PNH) red cells. Blood 50:239–247 [PubMed] [Google Scholar]
- 52. Sarko D, Beijer B, Garcia Boy R, Nothelfer EM, Leotta K, Eisenhut M, Altmann A, Haberkorn U, Mier W. 2010. The pharmacokinetics of cell-penetrating peptides. Mol. Pharm. 7:2224–2231 [DOI] [PubMed] [Google Scholar]
- 53. Ding JL, Li P, Ho B. 2008. The Sushi peptides: structural characterization and mode of action against Gram-negative bacteria. Cell. Mol. Life Sci. 65:1202–1219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Gough M, Hancock RE, Kelly NM. 1996. Antiendotoxin activity of cationic peptide antimicrobial agents. Infect. Immun. 64:4922–4927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Larrick JW, Hirata M, Balint RF, Lee J, Zhong J, Wright SC. 1995. Human CAP18: a novel antimicrobial lipopolysaccharide-binding protein. Infect. Immun. 63:1291–1297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Mookherjee N, Brown KL, Bowdish DM, Doria S, Falsafi R, Hokamp K, Roche FM, Mu R, Doho GH, Pistolic J, Powers JP, Bryan J, Brinkman FS, Hancock RE. 2006. Modulation of the TLR-mediated inflammatory response by the endogenous human host defense peptide LL-37. J. Immunol. 176:2455–2464 [DOI] [PubMed] [Google Scholar]
- 57. Corrigan JJ, Jr, Bell BM. 1971. Comparison between the polymyxins and gentamicin in preventing endotoxin-induced intravascular coagulation and leukopenia. Infect. Immun. 4:563–566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Stokes DC, Shenep JL, Fishman M, Hildner WK, Bysani GK, Rufus K. 1989. Polymyxin B prevents lipopolysaccharide-induced release of tumor necrosis factor-alpha from alveolar macrophages. J. Infect. Dis. 160:52–57 [DOI] [PubMed] [Google Scholar]
- 59. Baldwin G, Alpert G, Caputo GL, Baskin M, Parsonnet J, Gillis ZA, Thompson C, Siber GR, Fleisher GR. 1991. Effect of polymyxin B on experimental shock from meningococcal and Escherichia coli endotoxins. J. Infect. Dis. 164:542–549 [DOI] [PubMed] [Google Scholar]
- 60. Tzeng YL, Ambrose KD, Zughaier S, Zhou X, Miller YK, Shafer WM, Stephens DS. 2005. Cationic antimicrobial peptide resistance in Neisseria meningitidis. J. Bacteriol. 187:5387–5396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Derossi D, Joliot AH, Chassaing G, Prochiantz A. 1994. The third helix of the Antennapedia homeodomain translocates through biological membranes. J. Biol. Chem. 269:10444–10450 [PubMed] [Google Scholar]
- 62. El-Andaloussi S, Johansson HJ, Holm T, Langel Ü. 2007. A novel cell-penetrating peptide, M918, for efficient delivery of proteins and peptide nucleic acids. Mol. Ther. 15:1820–1826 [DOI] [PubMed] [Google Scholar]
- 63. Lindgren M, Rosenthal-Aizman K, Saar K, Eiriksdottir E, Jiang Y, Sassian M, Ostlund P, Hallbrink M, Langel U. 2006. Overcoming methotrexate resistance in breast cancer tumour cells by the use of a new cell-penetrating peptide. Biochem. Pharmacol. 71:416–425 [DOI] [PubMed] [Google Scholar]
- 64. Sorensen O, Cowland JB, Askaa J, Borregaard N. 1997. An ELISA for hCAP-18, the cathelicidin present in human neutrophils and plasma. J. Immunol. Methods 206:53–59 [DOI] [PubMed] [Google Scholar]
- 65. Wang G, Li X, Wang Z. 2009. APD2: the updated antimicrobial peptide database and its application in peptide design. Nucleic Acids Res. 37:D933–D937. 10.1093/nar/gkn823 [DOI] [PMC free article] [PubMed] [Google Scholar]