

Abstract
A liver transplant patient was admitted with cholangitis, for which meropenem therapy was started. Initial cultures showed a carbapenem-susceptible (CS) Escherichia coli strain, but during admission, a carbapenem-resistant (CR) E. coli strain was isolated. Analysis of the outer membrane protein profiles showed that both CS and CR E. coli lacked the porins OmpF and OmpC. Furthermore, PCR and sequence analysis revealed that both CS and CR E. coli possessed blaCTX-M-15 and blaOXA-1. The CR E. coli strain additionally harbored blaCMY-2 and demonstrated a >15-fold increase in β-lactamase activity against nitrocefin, but no hydrolysis of meropenem was detected. However, nitrocefin hydrolysis appeared strongly inhibited by meropenem. Furthermore, the CMY-2 enzyme demonstrated lower electrophoretic mobility after its incubation either in vitro or in vivo with meropenem, indicative of its covalent modification with meropenem. The presence of the acyl-enzyme complex was confirmed by mass spectrometry. By transformation of the CMY-2-encoding plasmid into various E. coli strains, it was established that both porin deficiency and high-level expression of the enzyme were needed to confer meropenem resistance. In conclusion, carbapenem resistance emerged by a combination of elevated β-lactamase production and lack of porin expression. Due to the reduced outer membrane permeability, only small amounts of meropenem can enter the periplasm, where they are trapped but not degraded by the large amount of the β-lactamase. This study, therefore, provides evidence that the mechanism of “trapping” by CMY-2 β-lactamase plays a role in carbapenem resistance.
INTRODUCTION
Until a few years ago, carbapenem resistance in Escherichia coli and Klebsiella pneumoniae was rare. In recent reports, however, genes such as KPC, NDM, VIM, IMP, and OXA, which encode carbapenemases, have been described (1, 2). The expression of these enzymes in E. coli can result in carbapenem resistance. Occasionally, E. coli or K. pneumoniae isolates resistant to carbapenem antibiotics are detected without the aforementioned carbapenemases. The explanation for this phenomenon is the presence of two mechanisms of resistance operating in unison, i.e., the presence of large quantities of chromosomal or plasmid-encoded β-lactamases together with decreased permeability of the outer membrane (3–5). The extreme imbalance between the number of antibiotic molecules entering the bacterial cell and the high quantities of β-lactamase present in the periplasm is used to explain the resistance to the carbapenems.
This explanation, however, is controversial, as both Mammeri et al. (6) and Queenan et al. (7) have demonstrated that AmpC and CTX-M enzymes have no or little hydrolytic activity toward carbapenems. Nevertheless, these authors concluded that the increased MICs for carbapenems were able to be explained by the low-but-not-zero hydrolysis rate of the antibiotics by the AmpC enzymes (8). Another possible mechanism that might be involved is trapping, which involves complex formation between antibiotics and β-lactamases to prevent the antibiotics from reaching their targets (9–12). However, this mechanism is controversial for β-lactam antibiotics except moxalactam, for which trapping has been accepted as a mechanism of resistance (13, 14). Additionally, the trapping mechanism has been demonstrated for ceftazidime and a mutated TEM β-lactamase (15). Recently, a covalent acyl-enzyme complex of imipenem with AmpC β-lactamase has been demonstrated by crystallography. The structure revealed that the electrophilic acyl center of imipenem was not bound in the oxyanion hole of the enzyme but was displaced and therefore escaped hydrolysis (16).
In the present study, we report the clinical and microbiological characteristics associated with carbapenem resistance of an E. coli isolate that was selected in vivo by a meropenem-containing regimen and provide evidence for the mechanism of trapping by a plasmid-encoded CMY-2 β-lactamase.
MATERIALS AND METHODS
Patient and isolates.
A 22-year-old female received a liver transplant in September 2007 at the Erasmus University Medical Center, Rotterdam, the Netherlands. The transplantation procedure was complicated by intra-abdominal infection with a multiresistant extended-spectrum-β-lactamase (ESBL)-producing E. coli strain for which she received treatment with meropenem. Shortly thereafter, she suffered from substantial intra-abdominal bleeding due to the rupture of a mycotic aortic aneurysm. A vascular prosthesis was positioned, and the patient was discharged in December 2007. Since infection of the vascular prosthesis was anticipated, meropenem was continued until spring 2009. In July 2009, the patient was readmitted with acute liver failure and cholangitis and meropenem was restarted and continued for 7 days until adequate drainage of the biliary tract was achieved. Initial cultures showed a carbapenem-susceptible E. coli isolate. However, at later stages of the hospitalization, several E. coli isolates with changed susceptibility patterns were obtained from abdominal specimens (Table 1).
Table 1.
Susceptibility of different E. coli isolates to cephalosporins and carbapenems
| Drug | MIC (μg/ml) for each isolate (date of isolation, specimen type)a |
||||||
|---|---|---|---|---|---|---|---|
| EC-1 (14/9/2007, urine) | EC-2 (28/7/2009, bile) | EC-3 (30/7/2009, abdomen) | EC-8 (11/8/2009, abdomen) | EC-9 (12/11/2009, abdomen) | EC-11 (30/7/2009, urine) | EC-13 (23/10/2009, abdomen) | |
| Cefotaxime | ≥32 | ≥32 | ≥32 | ≥32 | ≥32 | 4 | 4 |
| Ceftazidime | 16 | 32 | 32 | ≥256 | ≥256 | 32 | 32 |
| Ceftriaxone | ≥32 | ≥32 | ≥32 | ≥32 | ≥32 | 2 | 2 |
| Cefepime | 2 | ≥16 | ≥16 | ≥16 | ≥16 | 0.5 | 0.5 |
| Imipenem | 0.125 | 0.125 | 0.125 | ≥32 | ≥32 | 0.125 | 0.125 |
| Meropenem | 0.012 | 0.125 | 0.125 | ≥32 | ≥32 | 0.012 | 0.012 |
The susceptibility (MIC in μg/ml) of the isolates was determined by Etest. The dates of isolation are formatted as day/month/year.
Identification and susceptibility testing.
Patient isolates were identified to the species level with Vitek 2 (Vitek AMS; bioMérieux Vitek Systems Inc., Hazelwood, MO). Susceptibilities to antimicrobial drugs were determined with Vitek 2 using AST-N140 (Vitek AMS) and confirmed by Etest (AB bioMérieux, Solna, Sweden) according to the manufacturer's instructions. Susceptibility tests and the applied modified Hodge test were performed according to the 2010 CLSI guidelines (17).
PFGE.
The isolates were typed by pulsed-field gel electrophoresis (PFGE) as described previously (18). Cluster designation was based on isolates showing approximately 80% or greater relatedness, which corresponds to the “possibly related (4 to 6 bands difference)” criterion of Tenover et al. (19).
OMP analysis.
Outer membrane protein (OMP) profiles were studied by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and confirmed by immunoblotting. Briefly, cell envelopes were isolated from bacteria grown overnight at 37°C in L broth or, when indicated, in a phosphate-limited medium (20, 21). Cells were disrupted by ultrasonication, and Sarkosyl (2% end concentration) was added to the lysates to dissolve the inner membrane proteins (22). After 30 min of incubation at room temperature, the insoluble OMPs were collected by centrifugation for 30 min at 16,100 × g. Cell fractions were analyzed by SDS-PAGE as described previously (23) with 0.2% SDS and, to separate the porins OmpF and OmpC, 5 M urea in the running gel. Proteins were stained with Coomassie brilliant blue or transferred to nitrocellulose membranes by semidry electroblotting. The blots were incubated with a polyclonal antiserum that was raised against the E. coli porin PhoE and cross-reacts with the related porins OmpF and OmpC and, subsequently, with alkaline phosphatase-conjugated goat anti-rabbit IgG antiserum (BioSource International Inc., Camarillo, CA). The blots were then stained with 0.5 mg/ml 5-bromo-4-chloro-3-indolylphosphate and 0.1 mg/ml nitroblue tetrazolium (Sigma-Aldrich, St. Louis, MO) in 100 mM NaHCO3 and 1 mM MgCl2 (pH 9.8) until color developed.
Characterization of β-lactamases.
PCRs were used as previously described to amplify several β-lactamase genes coding for ESBLs (blaCTX-M, blaTEM, and blaSHV), plasmid-encoded AmpCs (blaCMY, blaFOX, blaMOX, blaDHA, blaLAT, blaACT, and blaMIR), and oxacillinases (24–29). Additionally, we tested for the carbapenemase genes blaVIM, blaIMP, and blaKPC (30, 31). Sequencing of PCR products was performed using a 3100 ABI Prism genetic analyzer (Applied Biosystems, Foster City, CA) with an additional primer for the CTX-M-1 group (28). For sequencing of the CMY-2 gene, primers described by Bauernfeind et al. were used (32). Analysis and comparisons of nucleotide and amino acid sequence data were carried out using MegAlign software (DNAStar Inc., Madison, WI).
Transformation.
Plasmids were isolated from the carbapenem-resistant patient isolates EC-8 and EC-9 (Table 1) and used to transform E. coli strain BL21(DE3) (Novagen), which produces OmpF as the only porin, and an ompF mutant derivative of this strain, designated CE1536 (33). Transformation was done by electroporation selecting for resistance to 100 μg/ml ampicillin.
Analysis of β-lactamase activity in periplasmic fractions.
β-Lactamase activity in periplasmic extracts was determined using nitrocefin (Calbiochem, Merck KGaA, Darmstadt, Germany) as a chromogenic substrate (34). First, periplasmic fractions were isolated from bacteria exponentially growing in L broth. The bacteria were harvested and converted to spheroplasts (35), which were removed by centrifugation for 1 min at 16,000 × g. The supernatant was used as the periplasmic extract. Appropriate dilutions of the extract in 1 ml of 10 mM HEPES and 5 mM MgCl2 (pH 7.2) were incubated at room temperature with 50 μM nitrocefin, and the initial rate of nitrocefin cleavage was measured by the change in optical density at 486 nm (OD486). A change in the OD486 of 1 corresponds to the degradation of 68.1 nmol nitrocefin, and the measured activity was calculated back to the β-lactamase activity of 108 cells. Inhibition studies were performed on periplasmic extracts incubated for 1 min with 0.005, 0.05, 0.5, 5, and 50 μM meropenem (AstraZeneca) prior to the addition of nitrocefin and subsequent measurement of β-lactamase activity. Preincubation of periplasmic extracts with cephaloridine (Sigma-Aldrich, St. Louis, MO) at 50 and 500 μM served as a control in this assay. To study the degradation of meropenem, periplasmic extracts were incubated in 1 ml of 10 mM HEPES and 5 mM MgCl2 (pH 7.2) with 50 nmol/ml of meropenem, and the opening of the β-lactam ring was measured during 10 min at 300 nm using a Unicam UV1 spectrometer. The degradation of cephaloridine measured at 260 nm was used as a control. In an alternative assay, the microiodometric method described by Zimmermann and Rosselet was used (36). Again, the degradation of cephaloridine was measured as a control.
For zymography, samples of periplasmic fractions were analyzed by semi-native SDS-PAGE without SDS in the gel (37). The sample buffer contained 0.1% of SDS, no β-mercaptoethanol, and 10% sucrose instead of glycerol, and the samples were not boiled before electrophoresis. After electrophoresis, the gels were rinsed twice for 15 min in 10 mM HEPES and 5 mM MgCl2 (pH 7.2) containing 1% bovine serum albumin. β-Lactamase activity was detected by incubating the gel for 15 min in 10 mM HEPES and 5 mM MgCl2 (pH 7.2) containing 0.1 mM nitrocefin. The result was photographed immediately.
Mass spectrometry.
Periplasmic extracts were either incubated or not with 1 mM meropenem and separated on 10% NU-PAGE gels (Invitrogen) under nonreducing conditions according to the manufacturer's recommendations. The gel was stained with Coomassie brilliant blue R250. Selected bands were excised, digested in gel with trypsin, and analyzed on an AB Sciex 45800 MALDI TOF/TOF, all as described previously (38). Mass spectra were searched using the Mascot engine against the Swiss-Prot 57.11 database. For each peptide mass fingerprint search, the mass tolerance was set to 0.05 Da. One missed tryptic cleavage was allowed. The mass tolerance for database searching with tandem mass spectrometry (MS/MS) spectra was set to 0.3 Da. The mass of tryptic peptides of the AmpC β-lactamase of the resistant strains was predicted using the PeptideMass program at http://web.expasy.org/peptide_mass/.
In vivo modification of AmpC β-lactamase with meropenem.
To study whether meropenem is bound in vivo by AmpC β-lactamase, bacteria were grown at 37°C in L broth to an OD660 of 0.6. Then meropenem was added at various concentrations and incubation was continued at 37°C without aeration for 20 min. No cell lysis was observed in this time period. The cells were collected by centrifugation and resuspended in the same volume of 0.9% NaCl. A periplasmic extract derived from 5 × 107 cells of strain BL21(DE3) overproducing TEM β-lactamase from the multicopy plasmid pET11 (Invitrogen) was added per ml of washed cells to capture any remaining free meropenem in the solution. After incubation for 5 min at room temperature, cells were collected by centrifugation and washed with 0.9% NaCl. The cells were then converted to spheroplasts (35), which were lysed by freeze-thawing followed by sonication for 10 s. Cell debris was removed by centrifugation for 30 min in a microcentrifuge, and the soluble fraction was analyzed by SDS-PAGE on gels containing 8% acrylamide and 5 M urea to obtain optimal separation of the CMY-2 enzyme and the acyl-enzyme adduct. Western blotting was performed as described above using a rabbit polyclonal antiserum raised against AmpC from Enterobacter cloacae (Aviva Systems Biology, San Diego, CA) at a 1:500 dilution, which was extensively preabsorbed with a whole-cell lysate of E. coli K-12 strain DH5α.
RESULTS
Characterization of the clinical E. coli isolates.
During hospitalization of the patient, various multiresistant E. coli strains were isolated, and the susceptibility data are shown in Table 1. The most remarkable result was that isolates EC-8 and EC-9 showed MICs for imipenem and meropenem of ≥32 μg/ml. The genetic similarity of the different isolates was investigated by PFGE, which identified three clusters of E. coli. The carbapenem-resistant isolates EC-8 and EC-9 belonged to cluster 1, EC-2 and EC-3 belonged to cluster 2, and EC-11 and EC-13 belonged to cluster 3 (Fig. 1). Isolates within each one of the clusters had >80% similar PFGE profiles. Interestingly, isolates from cluster 2 exhibited >60% similarity of PFGE profiles to cluster 1, suggesting that isolates from clusters 1 and 2 were related (Fig. 1). The remaining isolate (EC-1) was not related to isolates from clusters 1, 2, or 3. These genotypic differences demonstrate that the patient was colonized/infected by different E. coli strains during hospital admission, suggesting that subsequent exposure to different antibiotics has probably selected the different isolates with their corresponding resistance mechanism(s).
Fig 1.
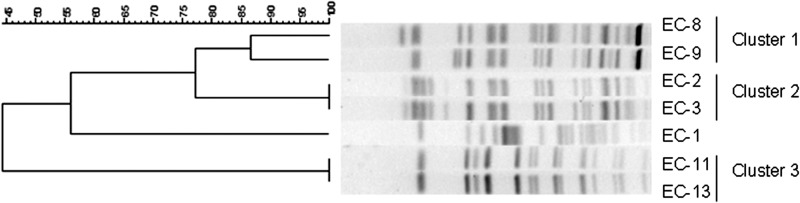
PFGE profiles of E. coli isolates. Genetic similarity was determined by using the Dice similarity index. The carbapenem-resistant isolates EC-8 and EC-9 cluster together.
Identification of β-lactamases.
To investigate the involvement of carbapenemases in the resistance phenotype, the resistant isolates EC-8 and EC-9 were subjected to the Hodge test, which was negative (data not shown). Additionally, all isolates were tested by PCR for the presence of prevalent β-lactamases, such as VIMs, IMPs, KPCs, TEMs, SHVs, CTX-Ms, and OXAs. Isolates EC-11 and EC-13 harbored TEM-1 and SHV-genes, while EC-1, EC-2, EC-3, EC-8, and EC-9 also contained CTX-M and OXA genes; these genes were subsequently identified via sequencing as CTX-M-15 and OXA-1, respectively. None of the isolates were positive for blavim, blaimp, or blakpc.
Analysis of the OMP profiles.
On its own, the presence (and probable expression) of the identified β-lactamase genes does not explain the resistance to carbapenem antibiotics. However, it is known that reduced outer membrane permeability contributes to the resistance of isolates expressing ESBL and/or AmpC enzymes to carbapenem antibiotics (39, 40). The permeability of the outer membrane to β-lactam antibiotics is determined largely by the presence of a class of abundant, channel-forming OMPs, designated porins (41). E. coli K-12 strains generally produce two porins, OmpF and OmpC, when grown under routine laboratory conditions. To study possible differences in the porin profiles, OMPs from the various isolates were subjected to SDS-PAGE. In the carbapenem-susceptible isolates EC-1, EC-11, and EC-13, OmpF and OmpC were readily detected (Fig. 2A). The carbapenem-resistant isolates EC-8 and EC-9, but also the carbapenem-susceptible isolates EC-2 and EC-3, did not express OmpF and OmpC. The absence of these porins was confirmed by Western blotting (Fig. 2B). Another porin, PhoE, is expressed in E. coli K-12 during growth under phosphate limitation (42, 43). Hence, we also investigated the expression of this porin in the carbapenem-susceptible and -resistant isolates. PhoE was detected in OMP preparations of all strains after their growth in a phosphate-limited medium (results not shown). Since both the carbapenem-resistant isolates EC-8 and EC-9 and the carbapenem-susceptible isolates EC-2 and EC-3 lacked OmpF and OmpC, we concluded that porin deficiency on its own was not able to explain the resistance of strains EC-8 and EC-9 to carbapenems.
Fig 2.

SDS-PAGE gel (A) and Western blot (B) of outer membrane proteins of E. coli isolates. (A) SDS-PAGE of OMP profiles of carbapenem-susceptible (EC-1 to EC-3, EC-11, and EC-13) and -resistant (EC-8 and EC-9) isolates. The lane marked K12 shows the OMP profile of an E. coli K-12 control strain expressing both OmpC and OmpF. The lane marked M contains molecular mass marker proteins. Arrows indicate the positions of OmpC, OmpF, and OmpA. (B) Western blot of OMP profiles incubated with anti-porin antibodies.
Identification of CMY-2 β-lactamase in the resistant isolates.
Although the PCR experiments described above revealed a similar set of β-lactamase genes in both clusters of strains, they did not exclude differences in enzyme activity.
To test this possibility, β-lactamase activity was determined in periplasmic extracts of the different isolates using nitrocefin as the substrate. In comparison to the carbapenem-susceptible isolates EC-2 and EC-3, the resistant isolates EC-8 and EC-9 demonstrated a >15-fold-higher β-lactamase activity (data not shown). To identify the enzyme responsible for the increased β-lactamase activity, cell extracts were analyzed by zymography. The zymogram revealed, besides a 28-kDa band present in all isolates examined, a very prominent band of 35 kDa present only in isolates EC-8 and EC-9 (Fig. 3A). The corresponding protein was very abundant and was readily detected in whole-cell lysates on Coomassie brilliant blue-stained gels, and it was the most prominent band in periplasmic extracts of the resistant strains (Fig. 3B). Mass spectrometry performed on the excised 35-kDa band revealed a protein with homology to an AmpC β-lactamase of Citrobacter freundii (accession number P05193) with 15 matching peptides (E value, 0.00026). Subsequent PCR and sequencing of a PCR product obtained with ampC-specific primers confirmed the presence of a CMY-2 β-lactamase. The possibility that the CMY-2 β-lactamase has carbapenemase activity was spectrophotometrically examined using meropenem as the substrate. No hydrolysis of meropenem was observed. In control experiments, with cephaloridine as the substrate, a hydrolysis rate of 110 nmol/min/108 cells was observed. These data were confirmed in a microiodometric assay in which no hydrolysis of meropenem was detected either, while cephaloridine was hydrolyzed at a rate of 26 nmol/min/108 cells. Thus, in spite of the difference in β-lactamase activity between the susceptible isolates EC-2 and EC-3 and the carbapenem-resistant isolates EC-8 and EC-9, this β-lactamase activity did not result in the measurable hydrolysis of meropenem.
Fig 3.

Zymogram and SDS-PAGE gel revealing expression of β-lactamase in the carbapenem-resistant E. coli isolates. (A) The zymogram was obtained by incubating the gel, on which periplasmic proteins from isolates EC-8, EC-9, EC-2, and EC-3 were separated, with nitrocefin as a β-lactamase substrate. A predominant band of 35 kDa with β-lactamase activity is detected only in the carbapenem-resistant isolates EC-8 and EC-9. A weaker band of 28 kDa is found in all four isolates. (B) SDS-PAGE profiles of whole-cell lysates of the same isolates as in panel A and of periplasmic extracts. The lane marked M contains molecular mass marker proteins; their molecular mass (in kDa) is indicated.
CMY-2 covalently binds meropenem.
As an alternative resistance mechanism, we next considered the possibility that the carbapenem antibiotics are irreversibly bound (“trapped”) by the CMY-2 β-lactamase produced in the meropenem-resistant isolates. Such a mechanism was first assessed by preincubating periplasmic extracts of the resistant strains with meropenem at various concentrations and subsequently determining the remaining β-lactamase activity by measuring the hydrolysis of nitrocefin. The rate of nitrocefin hydrolysis, which was 558 nmol/min/108 cells in the absence of meropenem, drastically decreased in the presence of meropenem. An ∼50% inhibition of nitrocefin hydrolysis was observed after preincubation with meropenem at a concentration 1,000-fold lower than that of nitrocefin (Fig. 4). No hydrolysis of nitrocefin was detected when meropenem was used at equimolar concentrations. Preincubation of the periplasmic extract with various concentrations of the cleavable β-lactamase substrate cephaloridine only marginally decreased the rate of nitrocefin hydrolysis; at an equimolar cephaloridine concentration, the rate of nitrocefin hydrolysis was reduced by only 18% (data not shown). Together, these results demonstrate that meropenem binds the β-lactamase and thereby inhibits its activity. If meropenem were slowly hydrolyzed by the CMY-2 β-lactamase, prolonged preincubation of the enzyme with meropenem would have resulted in a slow recovery of nitrocefin hydrolysis activity. However, no such recovery of enzyme activity was observed after 1 h of preincubation at any of the meropenem concentrations tested (results not shown). This result confirms that the CMY-2 β-lactamase binds meropenem but does not hydrolyze it at any biologically significant rate.
Fig 4.

Inhibition of β-lactamase activity by meropenem. Periplasmic extracts of isolate EC-8 were incubated for 1 min with various concentrations of meropenem as indicated. Subsequently, the remaining β-lactamase activity was determined using nitrocefin as a β-lactamase substrate. β-Lactamase activity after preincubation without meropenem is set at 100%.
To substantiate the irreversible binding of meropenem to the CMY-2 enzyme, periplasmic extracts, either incubated or not with meropenem, were analyzed by SDS-PAGE. The results showed a slight increase in the electrophoretic mobility of the ∼35-kDa CMY-2 band after preincubation with meropenem (Fig. 5). This shift in electrophoretic mobility indicates the covalent modification of the β-lactamase by meropenem. To further confirm the covalent binding of meropenem to the enzyme, the 35-kDa bands were excised from the gel and digested with trypsin and the resulting peptides were identified by mass spectrometry. In the sample of the protein that was not incubated with meropenem, a peptide with a mass of 2,369.04 Da that was absent from the sample incubated with meropenem was detected (Fig. 6A). The mass of this peptide corresponds to the calculated mass of 2,369.21 Da of the peptide containing the active-site serine residue of the enzyme. In the sample treated with meropenem, a peptide that was absent in the sample not treated with meropenem was observed at 2,708.23 Da (Fig. 6B). The mass of this fragment is consistent with that of the tryptic peptide containing the active-site serine, modified with meropenem after removal of an acetaldehyde group (Fig. 6C). These results confirm the hypothesis that CMY-2 is trapping meropenem in an inactivated acyl-enzyme complex.
Fig 5.
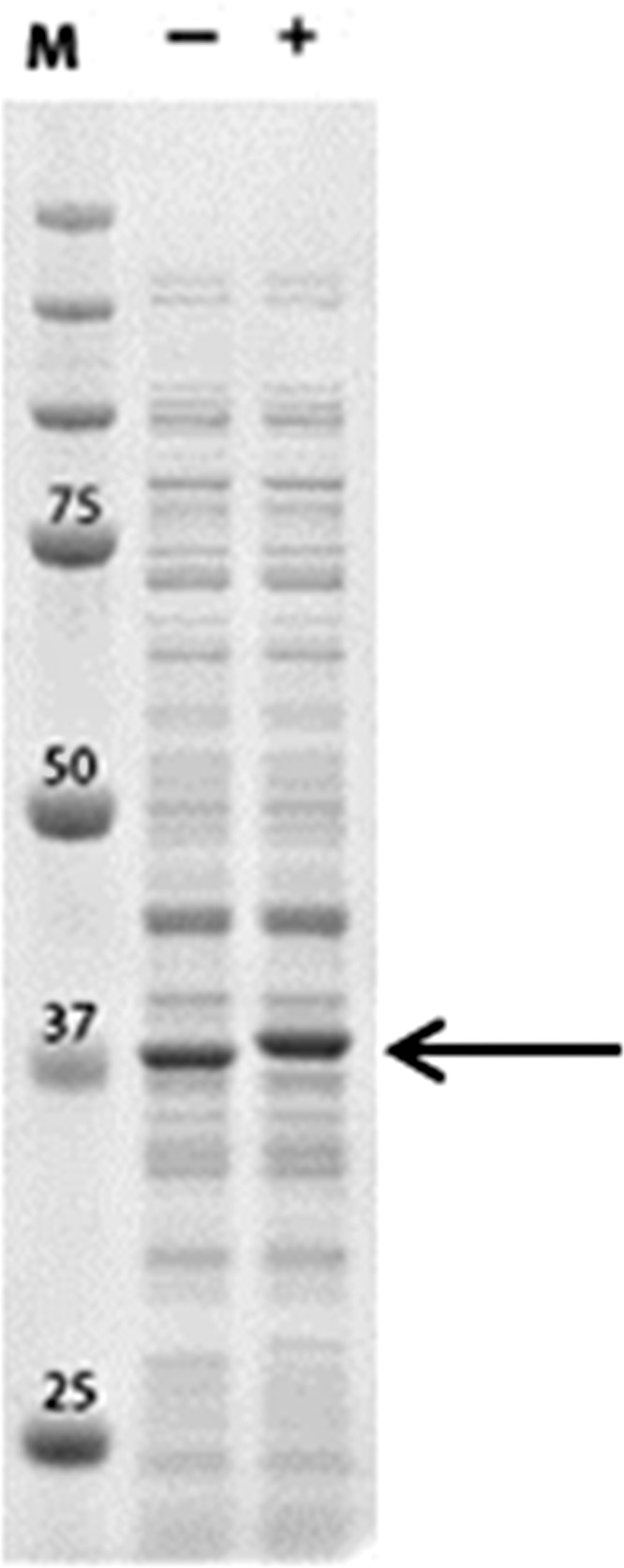
Covalent modification of CMY-2 with meropenem revealed by SDS-PAGE. Periplasmic extract of EC-8 was either incubated (+) or not (−) with 1 mM meropenem and analyzed by SDS-PAGE after boiling of the samples. The plasmid-encoded CMY-2 is visible as a predominant band indicated by the arrow. The lane marked M contains molecular mass marker proteins; the molecular mass of the most relevant bands is indicated (in kDa).
Fig 6.

Mass spectra of tryptic peptides of the CMY-2 enzyme after incubation in the absence or presence of meropenem. (A) Mass spectra (range, 2,200 to 2,800) of tryptic peptides of CMY-2 either preincubated (lower spectrum) or not (top spectrum) with meropenem. The peak at a mass of 2,369.04 (arrow) corresponds to the calculated mass of 2,369.21 Da of the peptide ADIANNHPVTQQTLFELGSVSK, containing the active-site serine (in bold) of the CMY-2 enzyme. (B) Mass spectra (range, 2,650 to 2,900) of tryptic peptides of CMY-2 either preincubated (lower panel) or not (top panel) with meropenem. The calculated mass of the CMY-2 tryptic peptide containing the active-site serine (2,369.21 Da) substituted with meropenem (383.46 Da) after removal of an acetaldehyde group (44.05 Da) (see panel C) is 2,708.62 Da, consistent with the peak at a mass of 2,708.23 Da that was found in our experiments in the sample incubated with meropenem (arrow). (C) Proposed chemical mechanisms for the BlaC-catalyzed reaction with meropenem resulting in a covalent acyl-enzyme adduct (53). Reprinted from reference 53 with permission from AAAS.
Both CMY-2 and porin deficiency are required to confer meropenem resistance.
To investigate whether the CMY-2 β-lactamase of strains EC-8 and EC-9 is encoded on a plasmid and whether the porin deficiency of these strains contributes to the observed carbapenem resistance, plasmid DNA was isolated from these strains and used to electroporate the laboratory E. coli strain BL21(DE3) and its porin-deficient mutant derivative, CE1536. SDS-PAGE and zymography revealed that the ampicillin-resistant transformants obtained abundantly produced the 35-kDa CMY-2 β-lactamase but not the 28-kDa enzyme that was also detected in the zymograms of periplasmic extracts of EC-8 and EC-9 (data not shown), demonstrating that the CMY-2 gene is encoded on a plasmid that does not contain the gene for the 28-kDa enzyme. PCR analysis confirmed the presence of the CMY-2 gene as the only β-lactamase present in the transformants. As in the case of the original isolates EC-8 and EC-9, no degradation of meropenem was demonstrated in periplasmic extracts of the transformants of BL21(DE3) and CE1536, while the high nitrocefin-hydrolyzing capacity of these extracts was effectively inhibited by preincubation with low concentrations of meropenem (data not shown). Subsequently, the susceptibility of the recombinants to carbapenems and cephalosporins was investigated (Table 2). The expression of the CMY-2 β-lactamase in strain BL21(DE3) did not substantially affect the MICs for imipenem and meropenem but increased the MICs to cefotaxime, ceftazidime, and, to a lesser extent, cefepime (Table 2). However, introduction of the CMY-2-encoding plasmid into the porin-deficient strain CE1536 dramatically increased the MICs to imipenem and meropenem (Table 2). These results demonstrate that both the expression of the CMY-2 β-lactamase and the absence of porins are required to confer full resistance to meropenem and imipenem.
Table 2.
Susceptibility of E. coli strain BL21(DE3), its porin-deficient derivative CE1536, and their transformants containing the CMY-2-encoding plasmid from isolate EC-8 for several antibioticsa
| Strain | MIC (μg/ml) |
||||
|---|---|---|---|---|---|
| Imipenem | Meropenem | Ceftazidime | Cefotaxime | Cefepime | |
| BL21(DE3) | 0.125 | 0.016 | 0.125 | 0.004 | ≤0.032 |
| BL21(DE3) CMY-2 | 0.25 | 0.016 | ≥256 | ≥32 | <0.25 |
| CE1536 | 0.125 | 0.012 | 0.25 | 0.032 | ≤0.032 |
| CE1536 CMY-2 | ≥32 | ≥32 | ≥256 | ≥32 | ≥16 |
The susceptibility (MIC in μg/ml) of the isolates was determined by Etest.
Trapping of meropenem by CMY-2 β-lactamase in the periplasm.
Next we investigated whether trapping of meropenem by CMY-2 β-lactamase can also be demonstrated in vivo in the bacterial periplasm. Various concentrations of meropenem were added to intact cells of E. coli BL21(DE3) and its porin-deficient derivative, CE1536, both containing the CMY-2-encoding plasmid. After incubation for 20 min, the binding of meropenem to the CMY-2 enzyme was analyzed by SDS-PAGE of the soluble fraction of cell lysates and Western blotting. In the wild-type strain, the CMY-2 β-lactamase was readily modified even at the lowest concentration of meropenem tested, as appeared from the detection of the slower-migrating acyl-enzyme adduct on the blots (Fig. 7A). In contrast, the acyl-enzyme adduct was detected in the porin-deficient mutant only after incubation with much higher concentrations of meropenem. These experiments demonstrated that meropenem covalently modifies CMY-2 β-lactamase also in its native periplasmic environment and confirmed that the presence of porins facilitates the transport of meropenem across the outer membrane. Curiously, a fraction of the β-lactamase molecules in the wild-type strain escaped modification by meropenem even at the highest concentration tested (Fig. 7A). At present, we do not have a clear explanation for this observation.
Fig 7.
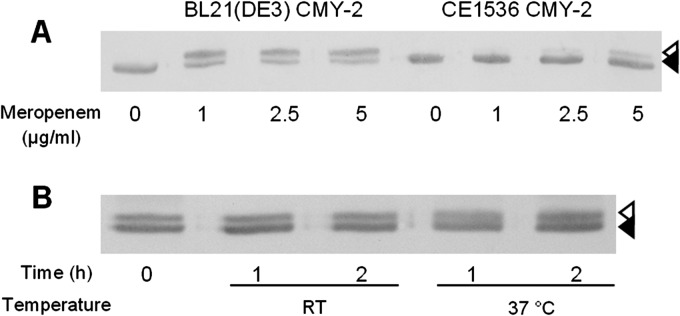
Covalent modification of CMY-2 β-lactamase in vivo and stability of the complex. (A) Cells of strain BL21(DE3) and its porin-deficient mutant, CE1536, both carrying the CMY-2-encoding plasmid, were incubated for 20 min with meropenem at the concentrations indicated. Then the cells were extensively washed and the soluble fraction was isolated and analyzed by SDS-PAGE and Western blotting with AmpC-specific antiserum. (B) BL21(DE3) cells carrying the CMY-2-encoding plasmid were incubated for 20 min with 1 μg/ml meropenem, after which the cells were washed extensively. Then the soluble fraction was isolated and incubated for the indicated time periods at room temperature (RT) or 37°C before analysis by SDS-PAGE and Western blotting with AmpC-specific antiserum. The CMY-2 β-lactamase and its covalently modified product are indicated by filled and open arrowheads, respectively.
To test the stability of the acyl-enzyme complexes formed in vivo, BL21(DE3) cells carrying the CMY-2-encoding plasmid were incubated with meropenem, after which the isolated, soluble fractions of the cells were incubated for various time periods at room temperature or at 37°C before analysis by SDS-PAGE and Western blotting. No hydrolysis of the acyl-enzyme adduct was observed, even after 2 h of incubation (Fig. 7B), again confirming that the CMY-2 β-lactamase does not hydrolyze meropenem at a biologically significant rate.
DISCUSSION
Despite several alarming reports describing the increase of acquired carbapenemases, i.e., NDM and KPC in the United Kingdom and India, resistance to carbapenems in E. coli is still rare in Western Europe. However, due to the worldwide spread of CTX-M-positive E. coli strains, the selection pressure imposed by carbapenems increases, as carbapenem treatment is often the only option left. Carbapenems are also considered first-line treatment for serious infections due to E. coli expressing chromosomal or plasmid-encoded AmpC β-lactamases. As it already had been shown that the patient in our case was colonized with CTX-M-positive E. coli strains, i.e., isolates EC-1, EC-2, and EC-3, we initially concluded that the resistance of isolates EC-8 and EC-9 may be due to the expression of CTX-M enzymes in combination with decreased permeability of the outer membrane (39, 40). Subsequently, however, we demonstrated the presence of the additional plasmid-encoded CMY-2 β-lactamase in EC-8 and EC-9 in accordance with recent literature that describes a significant increase in Enterobacteriaceae carrying plasmids with an ampC gene in Spain (44), Norway (45), United Kingdom (46), Canada (47, 48), and the United States (49). The most commonly reported plasmid-encoded AmpC enzyme in all countries is the CMY-2 variant, which was detected mainly in isolates of E. coli.
The phenotypes of isolates with a plasmid-encoded AmpC vary depending on the specific AmpC expressed. In most cases, such isolates demonstrate reduced susceptibility to 3rd-generation cephalosporins and cefoxitin (50). However, such isolates stay susceptible to carbapenem antibiotics because chromosomal and plasmid-encoded AmpCs are not or are barely able to hydrolyze these antibiotics (6, 7, 16). In E. coli and K. pneumoniae isolates with reduced susceptibility to carbapenems, the increased MICs observed have been suggested to result from a combination of decreased outer membrane permeability and extreme production of β-lactamase (39, 40). As porins are the main pathway for passage of β-lactam antibiotics across the outer membrane, decreased expression of porins plays an important role in development of resistance (51). When the permeability of the outer membrane is decreased, the number of drug molecules entering the periplasm is restricted and overproduction of β-lactamase results in an enzyme excess. Consequently, the relatively few drug molecules that cross the outer membrane are unlikely to escape from enzymes to exert their antibacterial effect. The magnitude of this effect depends on the specific β-lactamases present and their expression level. The effect of outer membrane permeability on the susceptibility to carbapenem antibiotics is clearly demonstrated in our study. Introduction of the plasmid encoding the CMY-2 β-lactamase in the porin-producing strain BL21(DE3) hardly affected the MICs for imipenem and meropenem, while the MICs for cephalosporins were increased. However, introduction of the plasmid into the porin-deficient strain CE1536 dramatically increased the MICs for imipenem and meropenem to ≥32 μg/ml. Also, the MIC for cefepime was much more affected upon introduction of the plasmid into the porin-deficient strain compared to the parental strain (Table 2).
Extreme overproduction of CMY-2, as demonstrated by the pronounced protein band detected by SDS-PAGE in whole-cell lysates (Fig. 3B), in combination with low influx is required for carbapenem resistance (Table 2). But even this combination of traits does not fully explain the high MICs for imipenem and meropenem, as these carbapenems, in spite of their high affinity for AmpC-type β-lactamases, show extremely low hydrolysis rates (6, 7). Therefore, it had been suggested that resistance arises by the formation of a stable acyl-enzyme intermediate (11, 12, 52). Thus, due to the high affinity of the drug for the enzyme, the drug would be “trapped” and unable to reach its target.
The proposed “trapping hypothesis” for β-lactams has remained controversial for a long time due to the insensitivity of β-lactamase assays and the failure to consider all periplasmic determinants of enzyme function (10, 14).
A first line of evidence for the trapping mechanism was provided by Beadle and Stoichet (16) using crystallography, which showed a structural basis for the very poor hydrolysis of the acyl-enzyme adduct formed upon interaction of an AmpC with a carbapenem. Our experiments are consistent with the trapping hypothesis in that we did not detect any hydrolysis of meropenem in periplasmic extracts of isolates EC-8 and EC-9. In addition, by performing competition/inhibition experiments, we were able to demonstrate indirectly the high affinity of the CMY-2 β-lactamase for meropenem. Preincubation with small amounts of meropenem was sufficient to fully inhibit the hydrolysis of nitrocefin by the enzyme. Importantly, no recovery of nitrocefin-hydrolyzing activity was observed after incubation of the periplasmic extracts for prolonged periods with concentrations of meropenem that fully or partially inhibited the β-lactamase, confirming the absence of meropenem-hydrolyzing activity at biologically relevant time scales. In control experiments, large amounts of the hydrolysable substrate cephaloridine only marginally inhibited the hydrolysis of nitrocefin. Additional evidence for the covalent binding of meropenem to CMY-2 was obtained by SDS-PAGE, which revealed a decreased electrophoretic mobility of the β-lactamase after incubation with meropenem. The evidence for “trapping” was further substantiated by mass spectrometry, which showed the disappearance from the spectrum of the tryptic fragment containing the active-site serine residue after incubation of the enzyme with meropenem. Concomitantly, a new fragment was detected in the spectrum, the mass of which corresponded to that of the peptide containing the active-site serine modified with the expected acyl adduct. Importantly, we were able to demonstrate that the enzyme-acyl adduct is also formed in vivo in the native periplasmic environment of the CMY-2 β-lactamase when intact bacterial cells are incubated with meropenem and is therefore not an in vitro artifact. The acyl-enzyme adduct formed appeared to be very stable, as no sign of deacylation was observed during 2 h of incubation, i.e., during several bacterial generations. Hence, any hydrolysis that still might occur at a very low and, in our assays, undetectable rate cannot be biologically relevant and significantly contribute to the observed resistance level. Previously, a mutant form of the TEM-1 β-lactamase was suggested to trap ceftazidime in the periplasm. By binding this third-generation cephalosporin with high affinity and no observable deacylation of the formed adduct, this mutant enzyme was shown to confer drastically increased resistance to the antibiotic (15). Similarly, the high expression level of CMY-2, the high affinity of the CMY-2 enzyme for meropenem (7), and the unobservable deacylation rate, combined with the low influx of the antibiotic in porin-deficient strains, explain the high resistance level of the patient isolates EC-8 and EC-9 by a trapping mechanism.
It is likely that the porin-deficient mutants resulted from the long-term treatment of the patient with meropenem after a complicated liver transplantation in 2007 and that carbapenem resistance emerged by stepwise accumulation of multiple drug resistance determinants. Emergence of carbapenem-resistant E. coli during carbapenem therapy is particularly bothersome since carbapenem therapy is often used as a first-line choice in patients with infections caused by AmpC- or ESBL-producing bacteria. To prevent such a scenario, the use of a rapid and specific assay for the detection of a plasmid-encoded AmpC is highly recommended, especially because our results suggest that this mechanism of resistance is present and can spread unnoticed. It may even create a resistance problem that might be more important than acquired carbapenemases.
In conclusion, the carbapenem resistance of the E. coli isolates is explained by the decreased outer membrane permeability and the large amounts of the CMY-2 β-lactamase produced that trap the small amounts of carbapenem entering the periplasm into a biologically inactive complex.
ACKNOWLEDGMENTS
This project was in part supported financially by a research grant from the Dutch ZonMW organization.
We thank Nicole Lemmens, Marco Mol, Gisele Peirano, and Gregory Koningstein for their technical support of this study.
Footnotes
Published ahead of print 3 June 2013
REFERENCES
- 1. Nordmann P, Cuzon G, Naas T. 2009. The real threat of Klebsiella pneumoniae carbapenemase-producing bacteria. Lancet Infect. Dis. 9:228–236 [DOI] [PubMed] [Google Scholar]
- 2. Walsh TR, Toleman MA, Poirel L, Nordmann P. 2005. Metallo-β-lactamases: the quiet before the storm? Clin. Microbiol. Rev. 18:306–325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Doumith M, Ellington MJ, Livermore DM, Woodford N. 2009. Molecular mechanisms disrupting porin expression in ertapenem-resistant Klebsiella and Enterobacter spp. clinical isolates from the UK. J. Antimicrob. Chemother. 63:659–667 [DOI] [PubMed] [Google Scholar]
- 4. Liu Y-F, Yan JJ, Ko WC, Tsai SH, Wu JJ. 2008. Characterization of carbapenem-non-susceptible Escherichia coli isolates from a university hospital in Taiwan. J. Antimicrob. Chemother. 61:1020–1023 [DOI] [PubMed] [Google Scholar]
- 5. Yang Q, Wang H, Sun H, Chen H, Xu Y, Chen M. 2010. Phenotypic and genotypic characterization of Enterobacteriaceae with decreased susceptibility to carbapenems: results from large hospital-based surveillance studies in China. Antimicrob. Agents Chemother. 54:573–577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mammeri H, Guillon H, Eb F, Nordmann P. 2010. Phenotypic and biochemical comparison of the carbapenem-hydrolyzing activities of five plasmid-borne AmpC β-lactamases. Antimicrob. Agents Chemother. 54:4556–4560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Queenan AM, Shang W, Flamm R, Bush K. 2010. Hydrolysis and inhibition profiles of β-lactamases from molecular classes A to D with doripenem, imipenem, and meropenem. Antimicrob. Agents Chemother. 54:565–569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lakaye B, Dubus A, Lepage S, Groslambert S, Frére JM. 1999. When drug inactivation renders the target irrelevant to antibiotic resistance: a case story with β-lactams. Mol. Microbiol. 31:89–101 [DOI] [PubMed] [Google Scholar]
- 9. Gutmann L, Williamson R. 1983. A model system to demonstrate that β-lactamase-associated antibiotic trapping could be a potential means of resistance. J. Infect. Dis. 148:316–321 [DOI] [PubMed] [Google Scholar]
- 10. Livermore DM. 1985. Do β-lactamases ‘trap’ cephalosporins? J. Antimicrob. Chemother. 15:511–514 [DOI] [PubMed] [Google Scholar]
- 11. Sanders CC. 1984. Inducible β-lactamases and non-hydrolytic resistance mechanisms. J. Antimicrob. Chemother. 13:1–3 [DOI] [PubMed] [Google Scholar]
- 12. Then RL, Angehrn P. 1982. Trapping of nonhydrolyzable cephalosporins by cephalosporinases in Enterobacter cloacae and Pseudomonas aeruginosa as a possible resistance mechanism. Antimicrob. Agents Chemother. 21:711–717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Livermore DM. 1987. “Covalent trapping” and latamoxef resistance in β-lactamase-derepressed Pseudomonas aeruginosa. J. Antimicrob. Chemother. 20:7–13 [DOI] [PubMed] [Google Scholar]
- 14. Vu H, Nikaido H. 1985. Role of β-lactam hydrolysis in the mechanism of resistance of a β-lactamase-constitutive Enterobacter cloacae strain to expanded-spectrum β-lactams. Antimicrob. Agents Chemother. 27:393–398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Antunes NT, Frase H, Toth M, Mobashery S, Vakulenko SB. 2011. Resistance to third-generation cephalosporin ceftazidime by a deacylation-deficient mutant of the TEM β-lactamase by the uncommon covalent-trapping mechanism. Biochemistry 50:6387–6395 [DOI] [PubMed] [Google Scholar]
- 16. Beadle BM, Shoichet BK. 2002. Structural basis for imipenem inhibition of class C β-lactamases. Antimicrob. Agents Chemother. 46:3978–3980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Clinical and Laboratory Standards Institute 2010. Performance standards for antimicrobial susceptibility testing. 20th informational supplement. Clinical and Laboratory Standards Institute, Wayne, PA [Google Scholar]
- 18. Pitout JDD, Gregson DB, Campbell L, Laupland KB. 2009. Molecular characteristics of extended-spectrum-β-lactamase-producing Escherichia coli isolates causing bacteremia in the Calgary Health Region from 2000 to 2007: emergence of clone ST131 as a cause of community-acquired infections. Antimicrob. Agents Chemother. 53:2846–2851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tenover FC, Arbeit RD, Goering RV, Mickelsen PA, Murray BE, Persing DH, Swaminathan B. 1995. Interpreting chromosomal DNA restriction patterns produced by pulsed-field gel electrophoresis: criteria for bacterial strain typing. J. Clin. Microbiol. 33:2233–2239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Levinthal C, Singer ER, Fetherolf K. 1962. Reactivation and hybridization of reduced alkaline phosphatase. Proc. Natl. Acad. Sci. U. S. A. 48:1230–1237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tommassen J, van Tol H, Lugtenberg B. 1983. The ultimate localization of an outer membrane protein of Escherichia coli K-12 is not determined by the signal sequence. EMBO J. 2:1275–1279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lugtenberg B, Meijers J, Peters R, van der Hoek P, van Alphen L. 1975. Electrophoretic resolution of the “major outer membrane protein” of Escherichia coli K-12 into four bands. FEBS Lett. 58:254–258 [DOI] [PubMed] [Google Scholar]
- 23. Laemmli UK. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685 [DOI] [PubMed] [Google Scholar]
- 24. Hasman H, Mevius D, Veldman K, Olesen I, Aarestrup FM. 2005. β-Lactamases among extended-spectrum β-lactamase (ESBL)-resistant Salmonella from poultry, poultry products and human patients in The Netherlands. J. Antimicrob. Chemother. 56:115–121 [DOI] [PubMed] [Google Scholar]
- 25. Karisik E, Ellington MJ, Pike R, Warren RE, Livermore DM, Woodford N. 2006. Molecular characterization of plasmids encoding CTX-M-15 β-lactamases from Escherichia coli strains in the United Kingdom. J. Antimicrob. Chemother. 58:665–668 [DOI] [PubMed] [Google Scholar]
- 26. Mabilat C, Goussard S. 1993. PCR detection and identification of genes for extended-spectrum β-lactamases, p 553–559 In Persing DH, Smith TF, Tenover FC, White TJ. (ed), Diagnostic molecular microbiology: principles and applications. American Society for Microbiology, Washington, DC [Google Scholar]
- 27. Pérez-Pérez FJ, Hanson ND. 2002. Detection of plasmid-mediated AmpC β-lactamase genes in clinical isolates by using multiplex PCR. J. Clin. Microbiol. 40:2153–2162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Severin JA, Mertaniasih NM, Kuntaman K, Lestari ES, Purwanta M, Lemmens-Den Toom N, Duerink DO, Hadi U, van Belkum A, Verbrugh HA, Goessens WH, Study Group “Antimicrobial Resistance in Indonesia: Prevalence and Prevention” (AMRIN) 2010. Molecular characterization of extended-spectrum β-lactamases in clinical Escherichia coli and Klebsiella pneumoniae isolates from Surabaya, Indonesia. J. Antimicrob. Chemother. 65:465–469 [DOI] [PubMed] [Google Scholar]
- 29. Woodford N, Fagan EJ, Ellington MJ. 2006. Multiplex PCR for rapid detection of genes encoding CTX-M extended-spectrum β-lactamases. J. Antimicrob. Chemother. 57:154–155 [DOI] [PubMed] [Google Scholar]
- 30. Bradford PA, Bratu S, Urban C, Visalli M, Mariano N, Landman D, Rahal JJ, Brooks S, Cebular S, Quale J. 2004. Emergence of carbapenem-resistant Klebsiella species possessing the class A carbapenem-hydrolyzing KPC-2 and inhibitor-resistant TEM-30 β-lactamases in New York City. Clin. Infect. Dis. 39:55–60 [DOI] [PubMed] [Google Scholar]
- 31. Pitout JDD, Gregson DB, Poirel L, McClure JA, Le P, Church DL. 2005. Detection of Pseudomonas aeruginosa producing metallo-β-lactamases in a large centralized laboratory. J. Clin. Microbiol. 43:3129–3135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bauernfeind A, Stemplinger I, Ungwirth R, Giamarellou H. 1996. Characterization of the plasmidic β-lactamase CMY-2, which is responsible for cephamycin resistance. Antimicrob. Agents Chemother. 40:221–224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Renault M, Tommassen-van Boxtel R, Bos MP, Post JA, Tommassen J, Baldus M. 2012. Cellular solid-state nuclear magnetic resonance spectroscopy. Proc. Natl. Acad. Sci. U. S. A. 109:4863–4868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. O'Callaghan CH, Morris A, Kirby SM, Shingler AH. 1972. Novel method for detection of β-lactamases by using a chromogenic cephalosporin substrate. Antimicrob. Agents Chemother. 1:283–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Osborn MJ, Gander JE, Parisi E. 1972. Mechanism of assembly of the outer membrane of Salmonella typhimurium. Site of synthesis of lipopolysaccharide. J. Biol. Chem. 247:3973–3986 [PubMed] [Google Scholar]
- 36. Zimmermann W, Rosselet A. 1977. Function of the outer membrane of Escherichia coli as a permeability barrier to beta-lactam antibiotics. Antimicrob. Agents Chemother. 12:368–372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Voulhoux R, Bos MP, Geurtsen J, Mols M, Tommassen J. 2003. Role of a highly conserved bacterial protein in outer membrane protein assembly. Science 299:262–265 [DOI] [PubMed] [Google Scholar]
- 38. Roussel-Jazédé V, Jongerius I, Bos MP, Tommassen J, van Ulsen P. 2010. NalP-mediated proteolytic release of lactoferrin-binding protein B from the meningococcal cell surface. Infect. Immun. 78:3083–3089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Martínez-Martínez L, Pascual A, Hernández-Allés S, Alvarez-Díaz D, Suárez I, Tran J, Bendí VJ, Jacoby GA. 1999. Roles of β-lactamases and porins in activities of carbapenems and cephalosporins against Klebsiella pneumoniae. Antimicrob. Agents Chemother. 43:1669–1673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Poirel L, Héritier C, Spicq C, Nordmann P. 2004. In vivo acquisition of high-level resistance to imipenem in Escherichia coli. J. Clin. Microbiol. 42:3831–3833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Nikaido H. 2003. Molecular basis of bacterial outer membrane permeability revisited. Microbiol. Mol. Biol. Rev. 67:593–656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Janssen R, Verjans GM, Kusters JG, Tommassen J. 1995. Induction of the phoE promoter upon invasion of Salmonella typhimurium into eukaryotic cells. Microb. Pathog. 19:193–201 [DOI] [PubMed] [Google Scholar]
- 43. Tommassen J, Lugtenberg B. 1980. Outer membrane protein e of Escherichia coli K-12 is co-regulated with alkaline phosphatase. J. Bacteriol. 143:151–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Mata C, Miró E, Rivera A, Mirelis B, Coll P, Navarro F. 2010. Prevalence of acquired AmpC β-lactamases in Enterobacteriaceae lacking inducible chromosomal ampC genes at a Spanish hospital from 1999 to 2007. Clin. Microbiol. Infect. 16:472–476 [DOI] [PubMed] [Google Scholar]
- 45. Naseer U, Haldorsen B, Simonsen GS, Sundsfjord A. 2010. Sporadic occurrence of CMY-2-producing multidrug-resistant Escherichia coli of ST-complexes 38 and 448, and ST131 in Norway. Clin. Microbiol. Infect. 16:171–178 [DOI] [PubMed] [Google Scholar]
- 46. Woodford N, Reddy S, Fagan EJ, Hill RL, Hopkins KL, Kaufmann ME. 2007. Wide geographic spread of diverse acquired AmpC-type β-lactamases among Escherichia coli and Klebsiella spp. in the UK and Ireland. J. Antimicrob. Chemother. 59:102–105 [DOI] [PubMed] [Google Scholar]
- 47. Baudry PJ, Mataseje L, Zhanel GG, Hoban DJ, Mulvey MR. 2009. Characterization of plasmids encoding CMY-2 AmpC β-lactamases from Escherichia coli in Canadian intensive care units. Diagn. Microbiol. Infect. Dis. 65:379–383 [DOI] [PubMed] [Google Scholar]
- 48. Pitout JDD, Le PG, Moore KL, Church DL, Gregson DB. 2010. Detection of AmpC β-lactamases in Escherichia coli, Klebsiella spp., Salmonella spp., and Proteus mirabilis in a regional clinical microbiology laboratory. Clin. Microbiol. Infect. 16:165–170 [DOI] [PubMed] [Google Scholar]
- 49. Hanson ND, Moland ES, Hong SG, Propst K, Novak DJ, Cavalieri SJ. 2008. Surveillance of community-based reservoirs reveals the presence of CTX-M, imported AmpC, and OXA-30 β-lactamases in urine isolates of Klebsiella pneumoniae and Escherichia coli in a U.S. community. Antimicrob. Agents Chemother. 52:3814–3816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Pitout JDD, Gregson DB, Church DL, Laupland KB. 2007. Population-based laboratory surveillance for AmpC β-lactamase-producing Escherichia coli, Calgary. Emerg. Infect. Dis. 13:443–448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Nikaido H. 1994. Prevention of drug access to bacterial targets: permeability barriers and active efflux. Science 264:382–388 [DOI] [PubMed] [Google Scholar]
- 52. Pechère JC, Levesque R. 1983. β-lactamases: clinical and genetic perspectives. J. Antimicrob. Chemother. 12:529–534 [DOI] [PubMed] [Google Scholar]
- 53. Hugonnet JE, Tremblay LW, Boshoff HI, Barry CE, III, Blanchard JS. 2009. Meropenem-clavulanate is effective against extensively drug-resistant Mycobacterium tuberculosis. Science 323:1215–1219 [DOI] [PMC free article] [PubMed] [Google Scholar]