

Abstract
Antivirulence strategies targeting bacterial behavior, such as adhesion and biofilm formation, are expected to exert low selective pressure and have been proposed as alternatives to biocidal antibiotic treatments to avoid the rapid occurrence of bacterial resistance. Here, we tested this hypothesis using group 2 capsule polysaccharide (G2cps), a polysaccharidic molecule previously shown to impair bacterium-surface interactions, and we investigated the nature of bacterial resistance to a nonbiocidal antibiofilm strategy. We screened an Escherichia coli mutant library for an increased ability to form biofilm in the presence of G2cps, and we identified several mutants displaying partial but not total resistance to this antibiofilm polysaccharide. Our genetic analysis showed that partial resistance to G2cps results from multiple unrelated mutations leading to modifications in surface physicochemical properties that counteract the changes in ionic charge and Lewis base properties induced by G2cps. Moreover, some of the identified mutants harboring improved biofilm formation in the presence of G2cps were also partially resistant to other antibiofilm molecules. This study therefore shows that alterations of bacterial surface properties mediate only partial resistance to G2cps. It also experimentally validates the potential value of nonbiocidal antibiofilm strategies, since full resistance to antibiofilm compounds is rare and potentially unlikely to arise in clinical settings.
INTRODUCTION
Rapid emergence of resistance to antibiotics acquired through mutations or horizontal gene transfer constitutes an increasingly common cause of therapeutic failure when treating bacterial infections (1, 2). Antibiotic resistance may also result from acquisition of the high antibiotic tolerance displayed by bacterial biofilm communities growing on the surface of contaminated medical implants (3, 4). While elimination of already formed biofilms remains challenging, a number of preventive strategies using a bactericidal or bacteriostatic coating with antibiotic or antimicrobial peptides, as well as nonspecific antiseptics, such as silver, zinc, or cupric oxides, have been reported to limit bacterial colonization on catheter surfaces (5, 6). These approaches, however, are also associated with problematic selection of multiresistant bacterial pathogens (7).
Several alternative nonbiocidal strategies that specifically target molecular events leading to biofilm formation and the onset of virulence factors have been proposed (8, 9). These approaches include antagonistic interference with bacterial communication signaling (10), inhibition of cyclic di-GMP-dependent biofilm switch (11), inhibition of signal transduction systems inducing biofilm formation (12), and prevention of adhesin assembly, hindering microbial attachment (13). Another promising approach uses inhibition of bacterial initial adhesion by surface-active compounds impairing bacterial attachment to surfaces (14). Alongside synthetic molecules that affect wettability and related surfactant properties, surfactants are also naturally produced by a wide variety of microorganisms (15). These molecules are active under physiological conditions; they are biodegradable and contribute to population dynamics by reducing the adhesion of competing microbes (16–18). Since biosurfactants target behavior rather than bacterial fitness, they are expected to exert milder evolutionary selective pressure and therefore are less likely to contribute to the selection of resistant mutants (8). Hence, biosurfactants represent an attractive antibiofilm strategy; however, the validity of these assumptions remains untested.
In the present study, we sought to determine whether mutants resistant to antiadhesion polysaccharide could arise by screening a transposon library of biofilm-forming Escherichia coli mutants and looking for those mutants able to adhere to and form biofilm, despite the presence of group 2 capsule polysaccharide (G2cps). G2cps is a hydrophilic and negatively charged polysaccharide polymer produced by most extraintestinal E. coli strains and previously shown to impair surface adhesion of both Gram-negative and Gram-positive bacteria by a still unknown mechanism (19). While we did not identify any mutant displaying full resistance to G2cps, partial resistance to G2cps arose from multiple unrelated mutations that led to modifications in physicochemical surface charge properties, counteracting the antibiofilm effect of G2cps and other antibiofilm compounds. This study thus provides insight into potential mechanisms of resistance to antibiofilm molecules and supports the hypothesis that prophylactic use of nonbiocidal antiadhesion compounds could represent a valuable approach to preventing pathogen surface colonization in clinical settings.
MATERIALS AND METHODS
Bacterial strains, plasmids, and growth conditions.
The bacterial strains and plasmids used in this study are listed in Table S1 in the supplemental material. All experiments were performed in 0.4% glucose M63B1 minimal medium (M63B1-glu) at 37°C. All liquid cultures were agitated. Antibiotics were added when required at the following concentrations: chloramphenicol (Cm) at 25 μg/ml and kanamycin (Km) at 50 μg/ml. Anhydrotetracycline (aTc) was used as an inducer for the KmRExTET cassette (described in reference 20) at a concentration of 50 ng/ml (20).
G2cps extract and antibiofilm supernatant preparations.
Overnight cultures of E. coli CFT073 ΔmchB unable to produce biocidal microcin that could interfere with the G2cps effect, along with the iai44, Ec094, iai73, and H19 E. coli natural isolates grown in M63B1-glu, were centrifuged for 10 min at 8,000 rpm and 4°C and filtered through a 0.45-μm-pore-size filter. Supernatant containing G2cps was further concentrated by precipitation with 3 volumes of cold 100% ethanol and dialyzed against deionized water (10-kDa cassettes; Pierce, Rockford, IL). The purity of the G2cps-containing extract was verified by purification by anion-exchange chromatography, followed by sizing chromatography and gas-phase chromatography to analyze the extract composition, as described in reference 19. The total amounts of neutral sugars were quantified by phenol-sulfuric acid methods using glucose as a standard (21).
Biofilm inhibition assay and biofilm quantification.
Overnight cultures were adjusted to an optical density at 600 nm (OD600) of 0.05 in 100 μl in 96-well polyvinyl chloride (PVC) microtiter plates (Falcon; Becton, Dickinson Labware, Oxnard, CA) in the presence or absence of 30 μg/ml G2cps extract and the supernatant of iai44, Ec094, iai73, or H19 diluted 1:1 in M63B1-glu, 12 μg/ml of surfactin (Sigma-Aldrich), or 0.00013% Tween 80 (Sigma-Aldrich). Biofilms were left to grow for 16 h at 37°C, revealed, and quantified as previously described (19).
Polymyxin B MIC determination and biofilm susceptibility assay.
The MIC value of polymyxin B (Sigma-Aldrich) was determined by dilution in M63B1-glu, as previously described (22). The MIC of polymyxin B for wild-type (WT) strain TG1-c was determined to be 2 ng/ml. Biofilms were formed for 24 h as described above. Unattached and planktonic bacteria were first removed from biofilms preformed for 24 h, and wells were then filled with 100 μl of M63B1 containing 4, 8, or 15 ng/ml of polymyxin B. After 24 h of incubation at 37°C, the polymyxin B susceptibility of the treated biofilm population versus that of a nontreated biofilm was determined by CFU counts.
Genetic analysis of mutants partially resistant to G2cps.
It has recently been reported that transfer of the mariner transposon (Tn) carried by Tn-psc189Km and the Mu prophage present in E. coli S17-1 λpir results in double mutagenesis of the recipient strain (23). Thus, mutants partially resistant to the G2cps extract were transduced to the wild type in order to verify that their resistance was due to the Tn insertion only (the transduction mutants are listed in Table S1 in the supplemental material). The presence of the Mu phophage was then verified by PCR using primers Mu-5 and Mu-3 (the sequences are listed in Table S2 in the supplemental material).
Determination of Tn and Mu insertion sites involved a first round of PCR using a primer specific for the right end of the Tn (pscIR2Km) or either the right or left end of Mu (MuR200-5 or MuL200-3, respectively) and an arbitrary primer (ARBN1 or ARBN6). A second PCR was then performed on the product from the first PCR using a primer specific to the rightmost end of the Tn (pscIR2Kmbis) or the right or left end of Mu (MuR100-5 or MuL100-3, respectively) and a primer identical to the 5′ end of the arbitrary primer (ARBbis) (see Table S2 in the supplemental material) (24). Homology searches were performed using the Colibri server (http://genolist.pasteur.fr/colibri/), and data on the prediction of gene function and protein location were collected from the EcoCyc database (http://ecocyc.org).
Deletion mutants were generated either by transduction from mutants belonging to the Keio Collection (25) or by the bacteriophage λ red linear DNA gene inactivation method using the three-step PCR procedure (26, 27; see http://www.pasteur.fr/recherche/unites/Ggb/3SPCRprotocol.html). Overexpression mutants were realized by addition of the aTc-inducible cassette KmRExTet upstream from the selected gene using the three-step PCR procedure (20). All mutants realized in this study are listed in Table S1 in the supplemental material, and primers used to either delete or overexpress genes presented in this study are listed in Table S2 in the supplemental material. When necessary, the resistance conferred by the Km cassette either by the use of Tn-psc189Km or after replacement of a gene deleted by the KmFRT cassette was removed after transformation of the pCP20 plasmid and excision of the cassette after FLP recombination target (FRT) recombination sequences (28). All constructs were checked by PCR with specific primers (see Table S2 in the supplemental material).
Electrophoretic mobility measurement.
The OD600 of bacteria grown overnight in M63B1-glu was adjusted to about 0.03 in water, allowing conductivity of 0.20 to 0.23 mS/cm. Electrophoretic mobility was measured at ∼20°C and pH 7 with an automated laser zetameter (Zetaphoremètre II; CAD Instrumentations, Paris, France). The results were based on an automated video analysis of about 200 bacteria per measure under an electric field of 50 V.
MATS method.
The microbial adhesion to solvents (MATS) method is based on comparison of microbial cell affinity to a polar solvent and microbial cell affinity to a nonpolar solvent (29). The polar solvent can be an electron acceptor or an electron donor, but both solvents must have similar van der Waals surface tension components. The following pairs of solvents were used: the first pair was chloroform, an electron acceptor solvent, and hexadecane, a nonpolar solvent, and the second pair was ethyl acetate, a strong electron donor solvent, and decane, a nonpolar solvent. Because of the surface tension properties of these solvents, differences between results obtained with chloroform and hexadecane and results obtained with ethyl acetate and decane indicated that there were electron donor-electron acceptor interactions at the bacterial cell surface and revealed hydrophobic and hydrophilic properties. The initial OD600 (ODi) of bacteria grown overnight in M63B1-glu was adjusted to 0.9 in 1.5 ml. The microbial suspension was vortexed for 2 min with 0.25 ml of a solvent. This high concentration of electrolyte was used to avoid charge interference by a masking cell charge, because some solvent droplets, especially hexadecane, become negatively charged in aqueous suspensions. The mixture was allowed to stand for 15 min to ensure that the two phases were completely separated before a sample (1 ml) was carefully removed from the aqueous phase and the final OD600 (ODf) was determined. Microbial adhesion to each solvent was calculated as [(ODi − ODf)/ODi] × 100 and is presented as a percentage.
Statistical analysis.
Each experiment was performed at least three times. For each experiment, means were calculated from three samples. The mean from at least three individual experiments was graphically represented, as was the standard deviation. Student's t tests were performed for all experiments. The level of significance is shown in each figure.
RESULTS
Screening for E. coli mutants able to form biofilm in the presence of antiadhesion G2cps.
To study the potential occurrence of resistance to bacterial surface-active compounds, we chose to identify bacterial mutants able to form biofilm, in spite of the presence of antiadhesion group 2 capsular polysaccharide (G2cps), a previously described broad-spectrum antibiofilm molecule produced by most uropathogenic and E. coli strains from the B2 phylogenetic group (Fig. 1A) (19). We screened a library of 11,000 Tn insertion mutants in E. coli K-12 TG1-c (30), a Cm-resistant biofilm-forming E. coli K-12 strain expressing the F conjugative pilus (31). Using a G2cps concentration (30 μg/ml) leading to a 75% reduction in TG1-c biofilm formation in microtiter plates (Fig. 1AB), we did not obtain any mutant with full resistance to G2cps. We nevertheless selected 26 mutants, named A to Z, that were partially resistant to G2cps and that formed significantly more biofilm than WT strain TG1-c in the presence of G2cps (Fig. 1C). However, use of increasing concentrations of purified G2cps still totally impaired their adhesion (data not shown). Among these 26 mutants, 9 of them (C, D, E, K, L, M, P, R, and Z mutants) produced 15 to 30% more biomass than WT strain TG1-c in the presence of G2cps (Fig. 1C). To exclude the possibility that the increased resistance to G2cps observed was due to improved adhesion of the mutants, we compared the biofilm-forming capacities of the WT and its 26 corresponding partially resistant mutants in the absence of extracted G2cps. Biofilm quantification showed that 17 out of the 26 identified mutants formed 10 to 45% more biofilm than the WT, whereas 9 mutants displayed biomass in amounts equivalent to the amount for the WT or reduced compared to the amount for the WT (see Fig. S1A in the supplemental material). To take this heterogeneity into account, we determined, for each mutant, the ratio between the biomass formed with and without G2cps and we identified 16 mutants (0.145% of the 11,000 transposon insertion mutants) displaying significantly higher partial resistance to the antibiofilm activity of G2cps than the WT (see Fig. S1B in the supplemental material). Among these 16 remaining mutants, we chose to focus our study on the seven mutants (C, D, E, L, M, P, and Z) which displayed neither a planktonic nor a biofilm growth defect compared to the growth of WT strain TG1-c (data not shown; see Fig. S1A in the supplemental material) while displaying resistance to G2cps (Fig. 1C).
Fig 1.
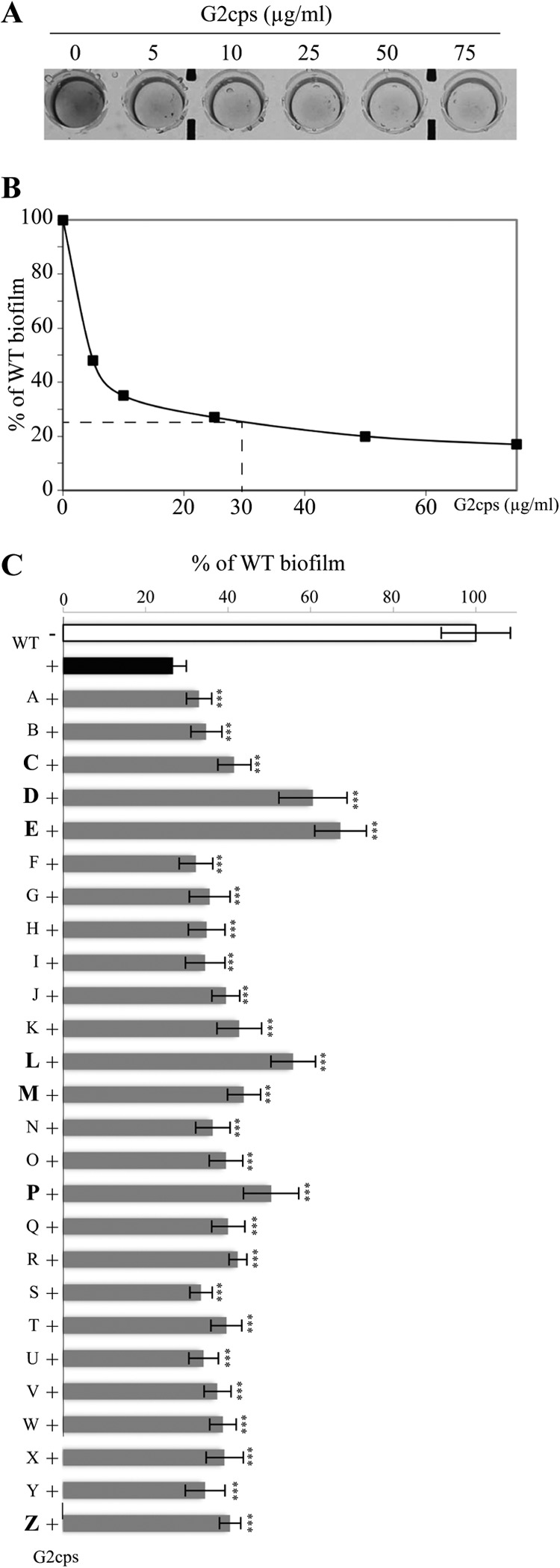
Antibiofilm effect of G2cps and selection of mutants partially resistant to G2cps. (A) Crystal violet staining of biofilm formed by WT strain TG1-c in a PVC microtiter plate in the presence of increasing concentrations of G2cps extract. (B) Quantification of biofilm formed as shown in panel A by the WT in a PVC microtiter plate with increasing concentrations of G2cps. For the rest of the study, we chose to use 30 μg/ml G2cps, which cause a biofilm reduction of about 75%. (C) Quantification of the biofilm formed by the WT and the 26 mutants resistant to G2cps in the presence of 30 μg/ml G2cps. Biofilm quantities were standardized with the quantity of the biofilm formed by WT without G2cps (value, 100; white bar). ***, P ≤ 0.005. P values were determined with respect to the biofilm formed by the WT with G2cps (black bar). The seven mutants chosen for use in the rest of the study are in bold.
Partial resistance to the antibiofilm activity of G2cps results from multiple mutations.
To study the genetic basis of partial resistance to the antibiofilm activity of G2cps, we first determined the localization of transposon insertions in the chromosomes of the C, D, E, L, M, P, and Z mutants, and we observed that they were inserted in seven different loci (Table 1).
Table 1.
Genetic analysis of mutants partially resistant to G2cpsa
| Mutant | Insert | Genome insertionb | Relevant information | Cellular location |
|---|---|---|---|---|
| C | Tn | 4499672, 1,954 bp before yjhB | EcoGene accession no. EG12544 (1,218 bp), uncharacterized member of the major facilitator superfamily of transporters (49) | M |
| Tn | 4499672, 515 bp after yjgZ | EcoGene accession no. G7899 (330 bp), KpLE2 phage-like element; predicted protein | ND | |
| Mu | 1969684, 1,031 bp within tar | EcoGene accession no. EG10988 (1,662 bp), chemoreceptor that senses aspartate and exists as a functional homodimer (50) | M | |
| D | Tn | 2531759, 25 bp before ptsH | EcoGene accession no. EG10788 (258 bp), phosphohistidinoprotein-hexose phosphotransferase, the second of two sugar-nonspecific protein constituents of the phosphotransferase system (51) | C |
| Mu | 1485199, 60 bp before ydcF | EcoGene accession no. EG12110 (801 bp), potential S-adenosylmethionine-dependent enzyme which may play a role in anaerobic respiration (52) | ND | |
| E | Tn | 839158, 1,596 bp within ybiL | EcoGene accession no. G6414 (2,283 bp), putative outer membrane receptor for iron transport (53) | OM |
| Mu | 1984638, 487 bp before araF | EcoGene accession no. EG10057 (990 bp), member of the arabinose ABC transporter (54) | P | |
| Mu | 1984638, 310 bp before yecI | EcoGene accession no. G7033 (504 bp), predicted ferritin-like protein | C | |
| L | Tn | 951258, 1,519 bp within pflB | EcoGene accession no. EG10701 (2,283 bp), pyruvate formate-lyase (inactive) (55) | C/M |
| Mu | 3089457, 304 bp within yggJ | EcoGene accession no. EG12366 (732 bp), methyltransferase responsible for methylation of 16S rRNA at the N-3 position of the U1498 nucleotide (56) | C | |
| M | Tn | 4321916, 14 bp within phnD | EcoGene accession no. EG10714 (1,017 bp), periplasmic binding component of the alkylphosphonate ABC transporter (cryptic in E. coli K-12) (57) | P/OM |
| Mu | 2076442, 313 bp after yeeW | EcoGene accession no. G7086 (168 bp), CP4-44 prophage, predicted protein | ND | |
| Mu | 2076442, 157 bp after yoeF | EcoGene accession no. G0-10456 (357 bp), conserved protein | ND | |
| P | Tn | 4321916, 14 bp within phnD | See mutant M | P/OM |
| Mu | 2159484, 3,076 bp within yegO | EcoGene accession no. G7115 (3,078 bp), member of the MdtABC-TolC multidrug efflux transport system (58) | OM | |
| Z | Tn | 832367, 74 bp within dinG | EcoGene accession no. EG11357 (2,151 bp), ATP-dependent DNA helicase (59) | C |
| Mu | 1068913, 149 bp within ycdG | EcoGene accession no. G6517 (1,329 bp), uncharacterized member of the NCS2 family of nucleobase transporters (60) | M |
Tn, transposon; C, cytoplasmic; M, membrane associated; OM, outer membrane; P, periplasm; ND, not determined. Accession numbers, lengths of potential mutated genes, the function and cellular location of the encoded proteins, and references were obtained from the EcoCyc server (http://www.ecocyc.org/).
Numbers correspond to nucleotide positions obtained from the EcoCyc server.
To verify that the transposon insertions were directly linked to the phenotypes observed in the original mutants, we transduced each transposon-interrupted gene in a fresh WT strain TG1-c background and tested its resistance to G2cps. With the exception of mutants D (transposon in ptsH; Fig. 2A) and L (transposon in pflB; Fig. 2B), neither full nor partial restoration of the phenotype displayed by the original transposon mutants could be obtained upon transduction of the Tn-psc189km transposon, suggesting that most mutants carry additional mutations. We hypothesized that these mutations might be due to delivery of the Mu bacteriophage present in helper strain E. coli S17-1 λpir used to perform our transposon mutagenesis, leading to additional markerless mutagenesis events in all recipient strains (23). Indeed, we established that all seven mutants partially resistant to G2cps (mutants C, D, E, L, M, P, and Z) contained a Mu prophage, the insertion points of which were also mapped (Table 1).
Fig 2.

Determination of genes implicated in partial resistance to G2cps. Biofilm ratio of WT TG1-c, mutants C, D, E, L, and Z partially resistant to G2cps, and transduction mutants with either a single deletion or multiple deletions made according to the different insertion points of both Tn and Mu within the genomes of mutants D (A), L (B), C (C), and Z (D). ***, P ≤ 0.005; N.S., not significant. P values were determined by comparison to both the WT biofilm ratio (black) and the C, D, L, or Z mutant biofilm ratio (gray).
We then constructed mutants with individual and combined deletions of all genes interrupted by the Tn-psc189km transposon and the Mu prophage against a fresh E. coli WT strain TG1-c background, and we analyzed the G2cps resistance of these new mutants (Table 1). We observed that, in mutants D and L, additional deletion of genes inactivated by the Mu prophage insertion (ydcF and yggJ, respectively) did not improve the already restored phenotype of partial resistance to G2cps (Fig. 2A and B, respectively). In contrast, although the mutant C phenotype was not restored upon individual deletion of the yjhB region or tar, combining these two mutations significantly restored the initial phenotype of mutant C (Fig. 2C). Similarly, the phenotype of mutant Z was partially restored only upon deletion of both the dinG and ycdG genes (Fig. 2D). Finally, in the case of mutant E (Tn in ybiL), the Mu prophage was inserted between the divergent genes araF and yecI and was therefore able to either impair or increase expression of these genes due to internal promoter activities. Mutants with deletions in ybiL, araF, and/or yecI consistently remained sensitive to G2cps, but overexpression of either the araF or the yecI gene fully restored G2cps partial resistance (see Fig. S2A in the supplemental material). In contrast, none of these approaches led to significant improvement in resistance to G2cps in mutants M and P (see Fig. S2B and C in the supplemental material).
These results therefore indicate that partial resistance to the G2cps antiadhesion polysaccharide could result from highly diverse mutation events involving at least nine distinct genes or loci (yjhB, yjgZ, tar, ptsH, pflB, dinG, ycdG, araF, and yecI).
Mutants partially resistant to G2cps display modified surface properties.
As G2cps impairs bacterial adhesion by interacting with the bacterial surface (19), we hypothesized that partially resistant mutants could share common surface properties antagonizing G2cps antiadhesion activity. We first compared the surface charges by measuring the motility in an electric field of WT strain E. coli TG1-c and mutants C, D, E, L, M, P, and Z partially resistant to G2cps. We observed that all tested strains displayed reduced electrophoretic motility compared to the WT strain, indicative of a reduced negative net charge of their cell wall at neutral pH (Fig. 3A) (32).
Fig 3.
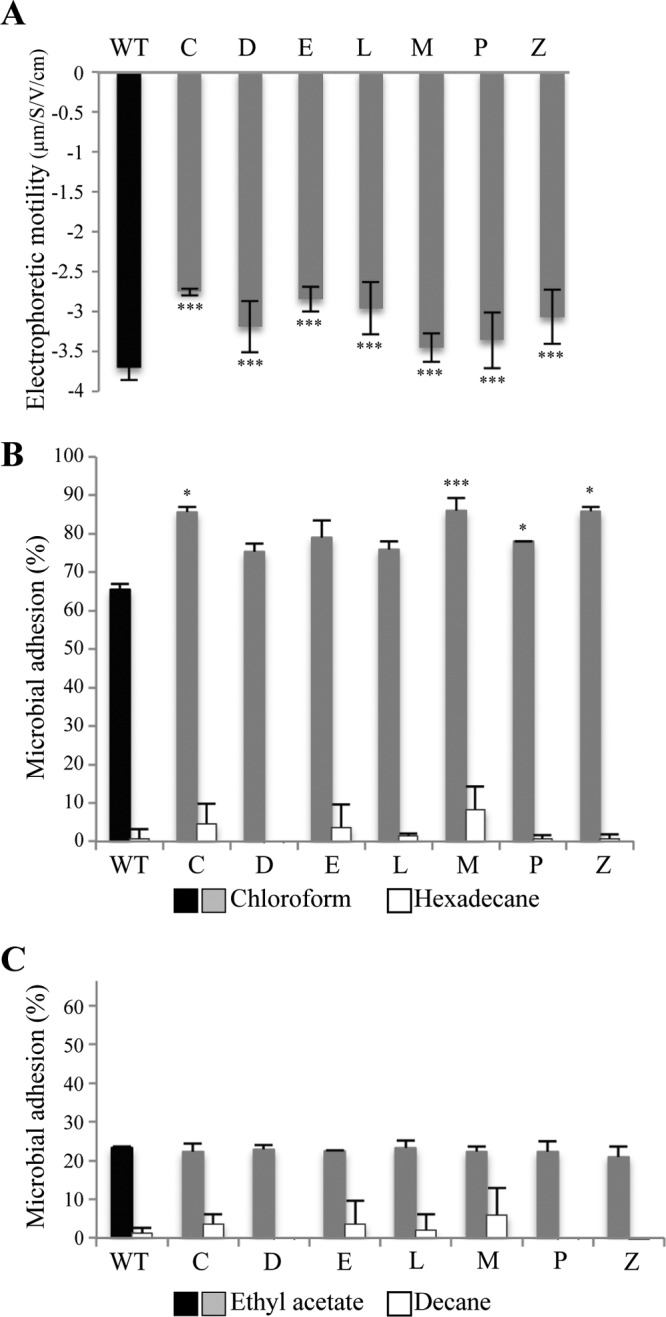
Surface biophysical properties of the seven mutants partially resistant to G2cps. (A) Measurement of electrophoretic motility displayed by WT strain TG1-c and the seven mutants partially resistant to G2cps. ***, P ≤ 0.005. P values were determined by comparison to the WT electrophoretic motility (black bar). (B) Percent microbial adhesion to chloroform and hexadecane. A difference in bacterial affinity between these two solvents is indicative of the bacterial Lewis base properties. P values were determined with respect to the WT microbial adhesion percentage to chloroform (black). (C) Percent microbial adhesion to ethyl acetate and decane. A difference in bacterial affinity between these two solvents is indicative of the bacterial Lewis acid properties. *, P ≤ 0.05; ***, P ≤ 0.005. P values were determined with respect to the percentage of WT microbial adhesion to ethyl acetate (black).
We then analyzed the affinity of mutants partially resistant to G2cps to different solvents, using the microbial adhesion to solvents (MATS) assay. This method enables determination of hydrophobic properties when bacteria display an affinity for hexadecane or decane (nonpolar solvents). MATS analysis is also indicative of Lewis acid properties, determined by comparing the bacterial interaction with decane and with ethyl acetate (electron pair donor solvent), and Lewis base properties, determined by comparing bacterial interaction with hexadecane and with chloroform (electron pair acceptor solvent) (29). WT strain TG1-c and the seven identified partially resistant mutants displayed a marginal affinity for hexadecane and decane (Fig. 3B and C), showing that the WT and the mutants are highly hydrophilic and that the differences in bacterial affinity between the two pairs of solvents, chloroform versus hexadecane (Fig. 3B) and ethyl acetate versus decane (Fig. 3C), were due to Lewis acid-base interactions. We found that 68% of the WT bacteria were associated with chloroform (Fig. 3B) and 23% were associated with ethyl acetate (Fig. 3C), thereby showing strong Lewis base and weak Lewis acid properties, respectively. The C, D, E, L, M, P, and Z mutants showed equivalent affinities to ethyl acetate, indicative of Lewis acid properties equivalent to those of the WT (Fig. 3C). However, all mutants displayed between a 10% and a 20% increased affinity for chloroform compared to that of the WT (Fig. 3B).
Our results demonstrate that most mutants partially resistant to G2cps displayed increased surface Lewis base properties resulting from an increased electron donor nature. This suggests that surface modifications mediate bacterial resistance to G2cps and potentially to other surface-active compounds.
G2cps mainly targets the Lewis base properties of the bacterial surface.
To determine whether the observed modifications in bacterial surface properties displayed by the C, D, E, L, M, P, and Z mutants were implicated in partial resistance to G2cps, MATS assays were performed in the presence of G2cps to compare the changes in surface properties induced by G2cps. We first observed that addition of G2cps reduced by 2 to 3% the affinity of WT strain TG1-c to hexadecane, ethyl acetate, and decane (Fig. 4A), indicating that G2cps only slightly reduces hydrophobic properties and does not modify Lewis acid properties. We also observed that G2cps reduced the WT affinity to chloroform by 35%, thus indicating that contact with G2cps strongly reduces bacterial Lewis base properties (Fig. 4A). We observed that G2cps induced equivalent modifications in mutant surface properties (Fig. 4B and C) but that in the presence of G2cps, the C, D, E, L, M, P, and Z mutants continued to display a 7% to 18% increased affinity to chloroform and therefore increased Lewis base properties compared to WT adhesion (Fig. 4B). Finally, the C, D, E, L, M, P, and Z mutants displayed WT affinity to ethyl acetate in the presence or absence of G2cps (Fig. 4C), confirming that the partial resistance to G2cps was unrelated to the Lewis acid properties of the bacterial surface. These results demonstrate that G2cps strongly reduces bacterial Lewis base properties and that mutants partially resistant to G2cps exposed to G2cps displayed higher Lewis base properties than the G2cps-susceptible WT.
Fig 4.
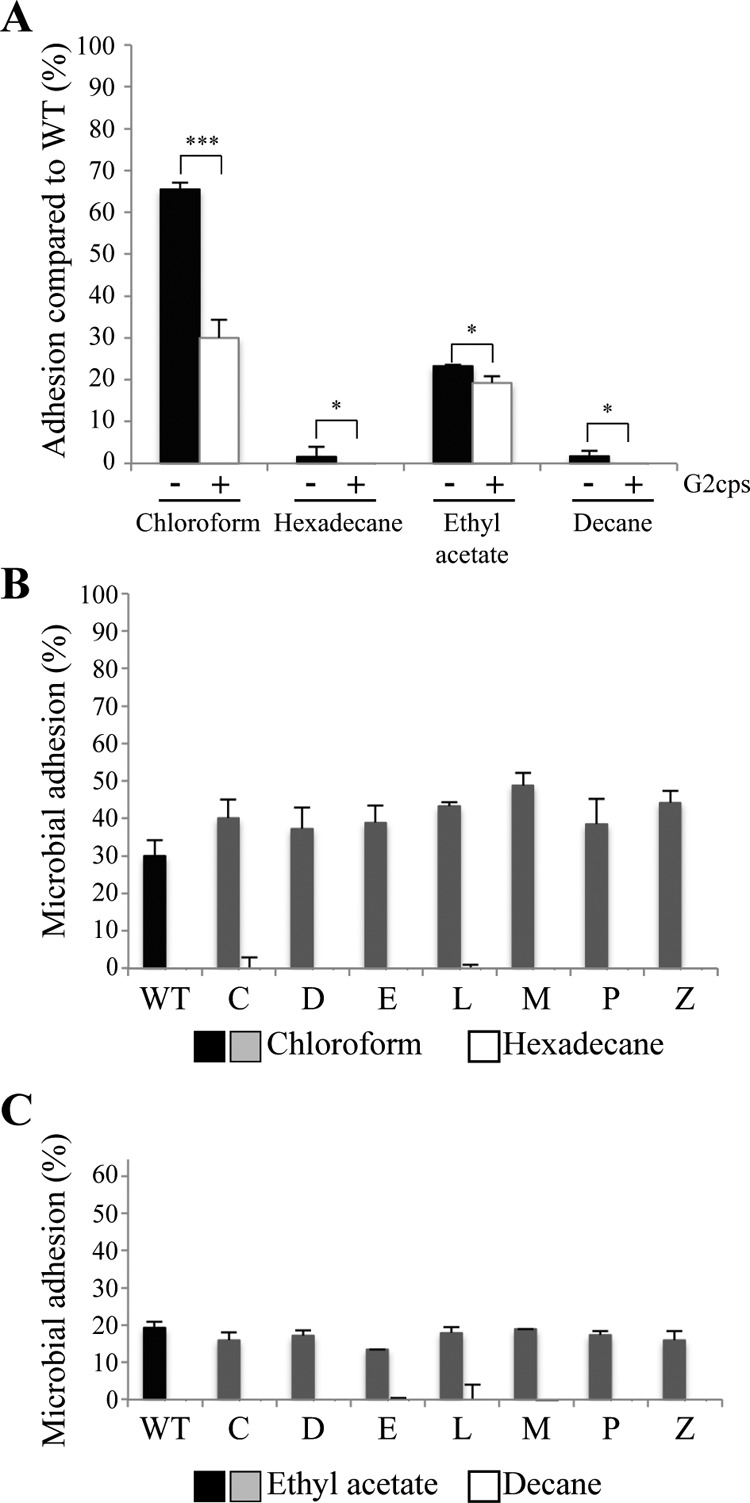
Modifications in surface biophysical properties induced by G2cps. (A) Comparison of percent microbial adhesion to chloroform, hexadecane, decane, and ethyl acetate of WT strain TG1-c with G2cps. *, P ≤ 0.05; ***, P ≤ 0.005. (B) Comparison of the E. coli TG1-c WT and mutant affinity for chloroform and hexadecane solvents in the presence of the G2cps antibiofilm polysaccharide. (C) Comparison of the E. coli TG1-c WT and mutant affinity for ethyl acetate and decane in the presence of the G2cps antibiofilm polysaccharide.
Increased resistance to G2cps does not lead to multiresistance to the antimicrobial polymyxin B or antibiofilm molecules.
To test whether surface modifications of mutants partially resistant to G2cps could modify the efficiency of antimicrobial compounds targeting the bacterial membrane, we tested the susceptibility of the G2cps-resistant mutants to the cationic antimicrobial polymyxin B. We first determined that 2 ng/ml of polymyxin B was sufficient to inhibit WT strain TG1-c growth in liquid culture and observed that the seven identified mutants were as susceptible as the WT under planktonic conditions (data not shown). We then determined that the WT biofilm displayed increased polymyxin B MICs (8 to 15 ng/ml; Fig. 5A). Whereas biofilms formed by mutants C, M, and Z displayed WT susceptibility to polymyxin B, mutant E and L biofilms were more susceptible than the WT. Inversely, mutant D and P biofilms were less susceptible to polymyxin B (Fig. 5A). These results indicated that surface physicochemical property modifications associated with increased resistance to G2cps do not lead to general resistance to the cationic antimicrobial peptide polymyxin B.
Fig 5.

Resistance to the antimicrobial polymyxin B and other antibiofilm molecules. (A) Assay of polymyxin B susceptibility of WT strain TG1-c and mutant strains partially resistant to G2cps biofilms. (B) Biofilm ratio of the WT in the presence of the other antibiofilm molecules produced by strains iai44, Ec094, iai73, and H19, surfactin, and Tween 80. (C to H) Comparison of biofilm ratios of the WT and mutants partially resistant to G2cps in the presence of antibiofilm supernatants of strains Ec094 (C), iai44 (D), iai73 (E), and H19 (F) and in the presence of surfactin (G) and Tween 80 (H). *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.005. P values were determined with respect to the WT biofilm ratio (black bars).
We then asked whether G2cps resistance could also lead to increased resistance in the presence of G2cps-unrelated antiadhesion compounds. We used previously described antibiofilm molecules, including surfactin, Tween 80, and four uncharacterized antibiofilm polysaccharides produced by four natural E. coli isolates (iai44, Ec094, iai73, and H19) (33–35) (see Table S1 in the supplemental material). We first confirmed that these compounds did not affect the growth rate of WT strain TG1-c (data not shown), while its biofilm-forming ability was strongly reduced when grown in the presence of the iai44, Ec094, iai73, or H19 supernatant, surfactin, or Tween 80 (Fig. 5B). We then analyzed the ability of mutants C, D, E, L, M, P, and Z partially resistant to G2cps to form biofilms in the presence of the chosen antibiofilm compounds. We observed that the L mutant (impaired in the pflB and yggJ genes) was also partially resistant to Ec094, iai44, iai73, and H19 supernatants (Fig. 5C to F). Mutants C, D, and E were more resistant to surfactin than the WT (Fig. 5G), while mutant Z was more resistant to Tween 80 (Fig. 5H). In contrast, we observed an increased efficiency of the iai73 antibiofilm supernatant toward the D mutant (Fig. 5E) and of the H19 antibiofilm supernatant toward the D, E, and M mutants (Fig. 5F).
These results indicate that five of the seven mutants partially resistant to G2cps identified in this study also displayed partial resistance to some of the tested antibiofilm compounds. However, all mutants displayed a specific resistance spectrum, indicating that increased resistance to G2cps does not lead to general increased resistance to nonbiocidal surface-active antibiofilm molecules.
DISCUSSION
Use of biosurfactants to prevent biofilm formation constitutes a promising approach to reducing bacterial surface contamination without applying high selection pressure potentially leading to elusive resistance selection (8, 16–18). Here, we tested this hypothesis by screening for E. coli mutants able to form a biofilm in the presence of the G2cps antibiofilm molecule. While we first considered the use of direct identification of mutants able to form a biofilm under dynamic flow conditions (31) in the presence of the G2cps antiadhesion polysaccharide, this strategy gave too many false positives to be practical. We instead used a direct strategy based on the individual screening of the residual adhesion ability of a large number of biofilm-forming E. coli TG1-c transposon mutants. Although we did not identify fully resistant mutants, we characterized several partially resistant mutants forming 10% to 45% more biofilm than WT TG1-c in the presence of G2cps.
Nonbiocidal antimicrobial strategies targeting the onset of virulence factors have recently been proposed, including inhibition of quorum sensing, cyclic di-GMP-dependent biofilm regulation, or adhesin-based adhesion (9, 36). These approaches are based on inhibition of bacterial traits with relatively simple genetic determinants that can be rapidly inactivated in resistant mutants. For instance, single mutational events in the mexR or nalC gene, both of which are involved in efflux pump regulation, were shown to be sufficient to lead to Pseudomonas aeruginosa resistance to the anti-quorum-sensing molecule furanone C-30 (37, 38). Here we showed that the partial resistance in five out of seven mutants identified to be partially resistant to G2cps corresponded to multiple unrelated mutations, thereby indicating that resistance to the G2cps antibiofilm polysaccharide is achieved through potentially rare pleiotropic mutational events. Resistance to G2cps could not be linked to a reduced growth rate or biofilm formation ability of the identified mutants. Moreover, although some of the mutants partially resistant to G2cps also exhibited partial resistance to other tested antibiofilm compounds, G2cps resistance did not favor resistance to other antibiofilm molecules.
The genetic characterization of mutants showed that the tar, yjhB, yjgZ, ptsH, araF, yecI, pflB, dinG, and ycdG genes were implicated in partial resistance to G2cps. In light of the surface-active nature of G2cps, which impairs bacterium-surface interactions via alteration of surface biophysical properties, we hypothesized that partial resistance to this antibiofilm molecule could depend on modified properties of the identified mutants. Indeed, some of the identified mutations in genes encoding membrane proteins (YjhB, Tar, PflB, and YcdG) or a membrane-associated protein (AraF) could alter susceptibility to G2cps through modifications in bacterial surface properties. Using MATS assays, we showed that the reduction in bacterial surface Lewis base properties induced upon exposure to G2cps was not as strong in the mutants as in the WT strain. Indeed, all seven identified mutants still displayed increased Lewis base properties in the presence of G2cps, indicating that partial resistance to G2cps correlates with increased Lewis base properties in the identified mutants. Cell wall composition and properties such as surface net charge play important roles in resistance to biocidal molecules (39–41). For instance, alteration of the bacterial lipopolysaccharide (LPS) negative charge upon LPS mutations or addition of glycine residues mediates resistance to antimicrobial peptides and polymyxin B, respectively (19, 20). However, modifications of the bacterial surface properties of the identified mutants resistant to G2cps do not lead to a general modification of the biofilm susceptibility to the antimicrobial peptide polymyxin B.
Moreover, in contrast to resistance to most biocides, we did not identify single mutations conferring significant resistance against G2cps. This therefore suggests that resistance to G2cps is a pleiotropic mechanism and supports the hypothesis that resistance to this antiadhesion molecule does not occur rapidly in most contexts.
Some models predict that drug resistance often arises from the preexistence of resistant mutants at low frequencies in targeted populations prior to treatment (42, 43). Given the observed pleiotropy of mutants partially resistant to G2cps, resistance to this type of antiadhesion molecule is not likely to rapidly emerge in natural bacterial populations. Consistently, coculture of Staphylococcus aureus for 1 year with Staphylococcus epidermidis-secreting antibiofilm serine protease Esp did not impact the sensitivity of S. aureus to this compound, indicating a striking absence of resistance to this nonbiocidal antibiofilm molecule (44). We therefore propose that physicochemical modifications induced by surface-active compounds such as G2cps target complex cooperative behavior with multifactorial determinants and that strains with such physicochemical modifications are potentially less susceptible to the rapid onset of resistance (45).
Our results therefore indicate that full resistance to the G2cps nonbiocidal antibiofilm polysaccharide could constitute a rare phenotype potentially achieved through pleiotropic mutational events. Anti-infection strategies combining antibiotics with antivirulence and antiadhesion compounds could thus reduce the emergence of multiresistant pathogens (36, 46, 47). Investigation of antibiofilm molecules such as G2cps in relevant in vivo models (48) will allow evaluations of the therapeutic significance of controlling pathogen surface adhesion and colonization, while avoiding selection for new types of resistance in clinical and industrial situations.
Supplementary Material
ACKNOWLEDGMENTS
We thank Marie-Noëlle Bellon-Fontaine for her scientific help during this study. We thank Christophe Beloin, Romain Briandet, Philippe Delepelaire, David Lebeaux, and Bianca Audrain for helpful discussions and critical reading of the manuscript.
L.T. was supported by an ANR fellowship (ERA-NET EuroPathogenomics). O.R. was supported by a fellowship from the Network of Excellence EuroPathogenomics, European Community grant LSHB-CT-2005-512061. This work was supported by a grant from the Région Ile-de-France (DIM Malinf) and the French Government's Investissement d'Avenir program, Laboratoire d'Excellence, Integrative Biology of Emerging Infectious Diseases (grant no. ANR-10-LABX-62-IBEID).
Footnotes
Published ahead of print 3 June 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.02606-12.
REFERENCES
- 1. Barczak AK, Hung DT. 2009. Productive steps toward an antimicrobial targeting virulence. Curr. Opin. Microbiol. 12:490–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Martinez JL, Fajardo A, Garmendia L, Hernandez A, Linares JF, Martínez-Solano L, Sánchez MB. 2009. A global view of antibiotic resistance. FEMS Microbiol. Rev. 33:44–65 [DOI] [PubMed] [Google Scholar]
- 3. Hoiby N, Bjarnsholt T, Givskov M, Molin S, Ciofu O. 2010. Antibiotic resistance of bacterial biofilms. Int. J. Antimicrob. Agents 35:322–332 [DOI] [PubMed] [Google Scholar]
- 4. Lewis K. 2008. Multidrug tolerance of biofilms and persister cells. Curr. Top. Microbiol. Immunol. 322:107–131 [DOI] [PubMed] [Google Scholar]
- 5. Danese PN. 2002. Antibiofilm approaches: prevention of catheter colonization. Chem. Biol. 9:873–880 [DOI] [PubMed] [Google Scholar]
- 6. Vasilev K, Cook J, Griesser HJ. 2009. Antibacterial surfaces for biomedical devices. Expert Rev. Med. Devices 6:553–567 [DOI] [PubMed] [Google Scholar]
- 7. Martins MCL, Costa F, Carvalho IF, Montelaro RC, Gomes P. 2010. Covalent immobilization of antimicrobial peptides (AMPs) onto biomaterial surfaces. Acta Biomater. 7:1431–1440 [DOI] [PubMed] [Google Scholar]
- 8. Rasko DA, Sperandio V. 2010. Anti-virulence strategies to combat bacteria-mediated disease. Nat. Rev. Drug Discov. 9:117–128 [DOI] [PubMed] [Google Scholar]
- 9. Rendueles O, Ghigo JM. 24 January 2012. Multi-species biofilms: how to avoid unfriendly neighbors. FEMS Microbiol. Rev. [Epub ahead of print.] 10.1111/j.1574-6976.2012.00328.x [DOI] [PubMed] [Google Scholar]
- 10. Hentzer M, Riedel K, Rasmussen TB, Heydorn A, Andersen JB, Parsek MR, Rice SA, Eberl L, Molin S, Høiby N, Kjelleberg S, Givskov M. 2002. Inhibition of quorum sensing in Pseudomonas aeruginosa biofilm bacteria by a halogenated furanone compound. Microbiology (Reading, Engl.) 148:87–102 [DOI] [PubMed] [Google Scholar]
- 11. Antoniani D, Bocci P, Maciag A, Raffaelli N, Landini P. 2010. Monitoring of diguanylate cyclase activity and of cyclic-di-GMP biosynthesis by whole-cell assays suitable for high-throughput screening of biofilm inhibitors. Appl. Microbiol. Biotechnol. 85:1095–1104 [DOI] [PubMed] [Google Scholar]
- 12. Eguchi Y, Kubo N, Matsunaga H, Igarashi M, Utsumi R. 2011. Development of an antivirulence drug against Streptococcus mutans: repression of biofilm formation, acid tolerance, and competence by a histidine kinase inhibitor, walkmycin C. Antimicrob. Agents Chemother. 55:1475–1484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Pinkner JS, Remaut H, Buelens F, Miller E, Aberg V, Pemberton N, Hedenström M, Larsson A, Seed P, Waksman G, Hultgren SJ, Almqvist F. 2006. Rationally designed small compounds inhibit pilus biogenesis in uropathogenic bacteria. Proc. Natl. Acad. Sci. U. S. A. 103:17897–17902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Banat IM, Franzetti A, Gandolfi I, Bestetti G, Martinotti MG, Fracchia L, Smyth TJ, Marchant R. 2010. Microbial biosurfactants production, applications and future potential. Appl. Microbiol. Biotechnol. 87:427–444 [DOI] [PubMed] [Google Scholar]
- 15. Ron EZ, Rosenberg E. 2001. Natural roles of biosurfactants. Environ. Microbiol. 3:229–236 [DOI] [PubMed] [Google Scholar]
- 16. Boles B, Thoendel M, Singh P. 2005. Rhamnolipids mediate detachment of Pseudomonas aeruginosa from biofilms. Mol. Microbiol. 57:1210–1223 [DOI] [PubMed] [Google Scholar]
- 17. Rivardo F, Turner RJ, Allegrone G, Ceri H, Martinotti MG. 2009. Anti-adhesion activity of two biosurfactants produced by Bacillus spp. prevents biofilm formation of human bacterial pathogens. Appl. Microbiol. Biotechnol. 83:541–553 [DOI] [PubMed] [Google Scholar]
- 18. Kiran GS, Sabarathnam B, Selvin J. 2010. Biofilm disruption potential of a glycolipid biosurfactant from marine Brevibacterium casei. FEMS Immunol. Med. Microbiol. 59:432–438 [DOI] [PubMed] [Google Scholar]
- 19. Valle J, Da Re S, Henry N, Fontaine T, Balestrino D, Latour-Lambert P, Ghigo JM. 2006. Broad-spectrum biofilm inhibition by a secreted bacterial polysaccharide. Proc. Natl. Acad. Sci. U. S. A. 103:12558–12563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Da Re S, Le Quere B, Ghigo JM, Beloin C. 2007. Tight modulation of Escherichia coli bacterial biofilm formation through controlled expression of adhesion factors. Appl. Environ. Microbiol. 73:3391–3403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dubois M, Gilles K, Hamilton JK, Rebers PA, Smith F. 1951. A colorimetric method for the determination of sugars. Nature 168:167. [DOI] [PubMed] [Google Scholar]
- 22. Hacek DM, Dressel DC, Peterson LR. 1999. Highly reproducible bactericidal activity test results by using a modified National Committee for Clinical Laboratory Standards broth macrodilution technique. J. Clin. Microbiol. 37:1881–1884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ferrières L, Hémery G, Nham T, Guérout A-M, Mazel D, Beloin C, Ghigo J-M. 2010. Silent mischief: bacteriophage Mu insertions contaminate products of Escherichia coli random mutagenesis performed using suicidal transposon delivery plasmids mobilized by broad-host-range RP4 conjugative machinery. J. Bacteriol. 192:6418–6427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Da Re S, Ghigo J-M. 2006. A CsgD-independent pathway for cellulose production and biofilm formation in Escherichia coli. J. Bacteriol. 188:3073–3087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H. 2006. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio Collection. Mol. Syst. Biol. 2:2006.0008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chaveroche MK, Ghigo JM, d'Enfert C. 2000. A rapid method for efficient gene replacement in the filamentous fungus Aspergillus nidulans. Nucleic Acids Res. 28:E97. 10.1093/nar/28.22.e97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lesic B, Bach S, Ghigo JM, Dobrindt U, Hacker J, Carniel E. 2004. Excision of the high-pathogenicity island of Yersinia pseudotuberculosis requires the combined actions of its cognate integrase and Hef, a new recombination directionality factor. Mol. Microbiol. 52:1337–1348 [DOI] [PubMed] [Google Scholar]
- 28. Cherepanov PP, Wackernagel W. 1995. Gene disruption in Escherichia coli: TcR and KmR cassettes with the option of Flp-catalyzed excision of the antibiotic-resistance determinant. Gene 158:9–14 [DOI] [PubMed] [Google Scholar]
- 29. Briandet R, Herry J, Bellon-Fontaine M. 2001. Determination of the van der Waals, electron donor and electron acceptor surface tension components of static Gram-positive microbial biofilms. Colloids Surf. B 21:299–310 [DOI] [PubMed] [Google Scholar]
- 30. Bernier SP, Lebeux D, DeFrancesco AS, Valomon A, Soubigou G, Coppée JY, Ghigo JM, Beloin C. 2012. Starvation together with the SOS response mediates high biofilm-specific tolerance to the fluoroquinolone ofloxacin. PLoS Genet. 9:e1003144. 10.1371/journal.pgen.1003144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ghigo JM. 2001. Natural conjugative plasmids induce bacterial biofilm development. Nature 412:442–445 [DOI] [PubMed] [Google Scholar]
- 32. Palmer J, Flint S, Brooks J. 2007. Bacterial cell attachment, the beginning of a biofilm. J. Ind. Microbiol. Biotechnol. 34:577–588 [DOI] [PubMed] [Google Scholar]
- 33. Eginton PJ, Holah J, Allison DG, Handley PS, Gilbert P. 1998. Changes in the strength of attachment of micro-organisms to surfaces following treatment with disinfectants and cleansing agents. Lett. Appl. Microbiol. 27:101–105 [DOI] [PubMed] [Google Scholar]
- 34. Mireles JR, II, Toguchi A, Harshey RM. 2001. Salmonella enterica serovar Typhimurium swarming mutants with altered biofilm-forming abilities: surfactin inhibits biofilm formation. J. Bacteriol. 183:5848–5854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Rendueles O, Travier L, Latour-Lambert P, Fontaine T, Magnus J, Denamur E, Ghigo J-M. 2011. Screening of Escherichia coli species biodiversity reveals new biofilm-associated antiadhesion polysaccharides. mBio 2(3):e00043–11. 10.1128/mBio.00043-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Cegelski L, Marshall GR, Eldridge GR, Hultgren SJ. 2008. The biology and future prospects of antivirulence therapies. Nat. Rev. Microbiol. 6:17–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kohler T, Perron GG, Buckling A, van Delden C. 2010. Quorum sensing inhibition selects for virulence and cooperation in Pseudomonas aeruginosa. PLoS Pathog. 6:e1000883. 10.1371/journal.ppat.1000883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Maeda T, Garcia-Contreras R, Pu M, Sheng L, Garcia LR, Tomas M, Wood TK. 2012. Quorum quenching quandary: resistance to antivirulence compounds. ISME J. 6:493–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kraus D, Peschel A. 2006. Molecular mechanisms of bacterial resistance to antimicrobial peptides. Curr. Top. Microbiol. Immunol. 306:231–250 [DOI] [PubMed] [Google Scholar]
- 40. Meehl M, Herbert S, Gotz F, Cheung A. 2007. Interaction of the GraRS two-component system with the VraFG ABC transporter to support vancomycin-intermediate resistance in Staphylococcus aureus. Antimicrob. Agents Chemother. 51:2679–2689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hankins JV, Madsen JA, Giles DK, Brodbelt JS, Trent MS. 2012. Amino acid addition to Vibrio cholerae LPS establishes a link between surface remodeling in Gram-positive and Gram-negative bacteria. Proc. Natl. Acad. Sci. U. S. A. 109:8722–8727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ribeiro RM, Bonhoeffer S. 2000. Production of resistant HIV mutants during antiretroviral therapy. Proc. Natl. Acad. Sci. U. S. A. 97:7681–7686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Komarova NL, Wodarz D. 2005. Drug resistance in cancer: principles of emergence and prevention. Proc. Natl. Acad. Sci. U. S. A. 102:9714–9719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Iwase T, Uehara Y, Shinji H, Tajima A, Seo H, Takada K, Agata T, Mizunoe Y. 2010. Staphylococcus epidermidis Esp inhibits Staphylococcus aureus biofilm formation and nasal colonization. Nature 465:346–349 [DOI] [PubMed] [Google Scholar]
- 45. Mellbye B, Schuster M. 2011. The sociomicrobiology of antivirulence drug resistance: a proof of concept. mBio 2(5):e00131–11. 10.1128/mBio.00131-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Paul M, Leibovici L. 2009. Combination antimicrobial treatment versus monotherapy: the contribution of meta-analyses. Infect. Dis. Clin. North Am. 23:277–293 [DOI] [PubMed] [Google Scholar]
- 47. Mitchell G, Lafrance M, Boulanger S, Seguin DL, Guay I, Gattuso M, Marsault E, Bouarab K, Malouin F. 2012. Tomatidine acts in synergy with aminoglycoside antibiotics against multiresistant Staphylococcus aureus and prevents virulence gene expression. J. Antimicrob. Chemother. 67:559–568 [DOI] [PubMed] [Google Scholar]
- 48. Chauhan A, Lebeaux D, Decante B, Kriegel I, Escande MC, Ghigo JM, Beloin C. 2012. A rat model of central venous catheter to study establishment of long-term bacterial biofilm and related acute and chronic infections. PLoS One 7:e37281. 10.1371/journal.pone.0037281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Pao S, Paulsen I, Saier M., Jr 1998. Major facilitator superfamily. Microbiol. Mol. Biol. Rev. 62:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Vaknin A, Berg H. 2008. Direct evidence for coupling between bacterial chemoreceptors. J. Mol. Biol. 382:573–577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Postma PW, Lengeler JW, Jacobson GR. 1993. Phosphoenolpyruvate: carbohydrate phosphotransferase systems of bacteria. Microbiol. Rev. 57:543–594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Chao KL, Lim K, Lehmann C, Doseeva V, Howard AJ, Schwarz FP, Herzberg O. 2008. The Escherichia coli YdcF binds S-adenosyl-l-methionine and adopts an alpha/beta-fold characteristic of nucleotide-utilizing enzymes. Proteins 72:506–509 [DOI] [PubMed] [Google Scholar]
- 53. Molloy M, Herbert B, Slade M, Rabilloud T, Nouwens A, Williams K, Gooley A. 2000. Proteomic analysis of the Escherichia coli outer membrane. Eur. J. Biochem. 267:2871–2881 [DOI] [PubMed] [Google Scholar]
- 54. Horazdovsky BF, Hogg RW. 1989. Genetic reconstitution of the high-affinity l-arabinose transport system. J. Bacteriol. 171:3053–3059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Sawers G, Bock A. 1989. Novel transcriptional control of the pyruvate formate-lyase gene: upstream regulatory sequences and multiple promoters regulate anaerobic expression. J. Bacteriol. 171:2485–2498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Basturea GN, Rudd KE, Deutscher MP. 2006. Identification and characterization of RsmE, the founding member of a new RNA base methyltransferase family. RNA 12:426–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Yakovleva G, Kim S, Wanner B. 1998. Phosphate-independent expression of the carbon-phosphorus lyase activity of Escherichia coli. Appl. Microbiol. Biotechnol. 49:573–578 [DOI] [PubMed] [Google Scholar]
- 58. Nagakubo S, Nishino K, Hirata T, Yamaguchi A. 2002. The putative response regulator BaeR stimulates multidrug resistance of Escherichia coli via a novel multidrug exporter system, MdtABC. J. Bacteriol. 184:4161–4167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Voloshin ON, Vanevski F, Khil PP, Camerini-Otero RD. 2003. Characterization of the DNA damage-inducible helicase DinG from Escherichia coli. J. Biol. Chem. 278:28284–28293 [DOI] [PubMed] [Google Scholar]
- 60. Kim KS, Pelton JG, Inwood WB, Andersen U, Kustu S, Wemmer DE. 2010. The Rut pathway for pyrimidine degradation: novel chemistry and toxicity problems. J. Bacteriol. 192:4089–4102 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.