

Abstract
Alphavirus dogma has long dictated the production of a discrete set of structural proteins during infection of a cell: capsid, pE2, 6K, and E1. However, bioinformatic analyses of alphavirus genomes (A. E. Firth, B. Y. Chung, M. N. Fleeton, and J. F. Atkins, Virol. J. 5:108, 2008) suggested that a ribosomal frameshifting event occurs during translation of the alphavirus structural polyprotein. Specifically, a frameshift event is suggested to occur during translation of the 6K gene, yielding production of a novel protein, termed transframe (TF), comprised of a C-terminal extension of the 6K protein in the −1 open reading frame (ORF). Here, we validate the findings of Firth and colleagues with respect to the production of the TF protein and begin to characterize the function of TF. Using a mass spectrometry-based approach, we identified TF in purified preparations of both Sindbis and Chikungunya virus particles. We next constructed a panel of Sindbis virus mutants with mutations which alter the production, size, or sequence of TF. We demonstrate that TF is not absolutely required in culture, although disrupting TF production leads to a decrease in virus particle release in both mammalian and insect cells. In a mouse neuropathogenesis model, mortality was <15% in animals infected with the TF mutants, whereas mortality was 95% in animals infected with the wild-type virus. Using a variety of additional assays, we demonstrate that TF retains ion-channel activity analogous to that of 6K and that lack of production of TF does not affect genome replication, particle infectivity, or envelope protein transit to the cell surface. The TF protein therefore represents a previously uncharacterized factor important for alphavirus assembly.
INTRODUCTION
The alphaviruses are a group of positive-sense, enveloped, single-stranded RNA viruses belonging to the Togaviridae family. The group contains many medically important species, including eastern, western, and Venezuelan equine encephalitis viruses, Semliki Forest virus (SFV), Ross River virus, Chikungunya virus (CHIKV), and the type member, Sindbis virus (SINV) (1). Recent epidemics of CHIKV, coupled with its spread to a novel mosquito vector, have reemphasized the medical importance of the alphaviruses (2).
The ∼12-kb RNA genome encodes two open reading frames (ORFs). The first ORF, encompassing the 5′ two-thirds of the genome, encodes the nonstructural proteins. These proteins possess the enzymatic functions necessary to replicate the genome of the virus and often antagonize the host innate immune system as well as modulate cellular transcription and translation (3). The second ORF, found at the 3′ end of the genome, encodes the structural proteins. These proteins are initially translated as a polyprotein and are co- and posttranslationally cleaved into their individual, mature proteins by viral and host proteases. Specifically, the capsid protein, found at the N terminus of the structural polyprotein, is an autoprotease and cleaves itself out of the growing polypeptide in cis (4). The capsid protein associates with viral genomic RNA and forms a nucleocapsid core that is later enveloped by the viral envelope protein-enriched plasma membrane (PM) (5). The bulk of the polyprotein contains the unprocessed envelope proteins, synthesized in the order pE2, 6K, and E1. The envelope polyprotein inserts into the endoplasmic reticulum (ER) in a signal sequence-mediated manner (6). Host signal peptidase liberates pE2, 6K, and E1 from each other, and pE2 and E1 likely cotranslationally fold, initially forming heterodimers. As the envelope proteins traffic via the secretory pathway to the PM, the site of virus budding, the pE2/E1 heterodimers assemble into trimers, often referred to as spikes, and pE2 is cleaved by a furin-like protease into the small E3 and the larger E2 proteins (7).
A wealth of knowledge regarding the structure and function of the two major envelope proteins, E2 and E1, has been accumulated. E2 and E1 are type I integral membrane proteins that comprise the shell of the virus particle (8). E2 is primarily responsible for host receptor engagement (9–11), while E1 contains pH-dependent fusogenic properties required for virus entry (12–14). Crystal structures of E1 as well as the spike complex at both neutral and acidic pH have been determined (15, 16), and pseudoatomic structures of alphavirus particles highlighting the location and organization of E2 and E1 in the virus particle are available (8). Details regarding the structure and function of the 6K envelope protein are lacking, however.
6K is a small 55- to 60-amino-acid protein that is predicted to have two transmembrane domains; the first is potentially involved in ion-channel activity, while the second acts as the signal sequence for E1 (17). Despite being synthesized in equal amounts relative to the other envelope proteins, 6K is incorporated into the virus particle in substoichiometric amounts relative to E2 and E1 (18). Mutants in which 6K has been either deleted or mutated have demonstrated various, related phenotypes, including inefficient particle release and the release of multicored virions (19–25). Taken together, it appears that 6K is important for efficient virus budding but is not absolutely required for particle release.
For years, alphavirus dogma implied that pE2, 6K, and E1 were the only envelope proteins produced during infection of a cell (26). However, recent bioinformatics analyses have suggested the production of a novel alphavirus envelope protein. Firth et al. first demonstrated the presence of a slippery codon motif, UUUUUUA, in the 6K gene that is absolutely conserved among the members of the alphavirus genus (27). This work suggested that, at an ∼10 to 15% frequency, the slippery codon mediates ribosomal frameshifting into the −1 ORF coincident with production of a novel protein that they termed transframe (TF). TF shares the N-terminal amino acid sequence, including the first transmembrane domain implicated in ion-channel activity, with 6K. However, it contains a unique, basic C terminus whose length (about 15 residues longer than that of 6K) is also relatively conserved among the alphaviruses due to the presence of a conserved stop codon in the −1 ORF. Inherently, production of TF via the −1 ORF precludes synthesis of E1 after the infrequent ribosomal frameshifting events. Firth and colleagues demonstrated the synthesis of TF during SFV infection but did not explore the frameshift phenomenon in other alphaviruses or characterize the role of TF during infection (27).
We used a mass spectrometry (MS)-based approach to identify TF in highly purified preparations of both SINV and CHIKV particles, validating the findings of Firth and colleagues (27). Using SINV, we probed for the requirement of TF during alphavirus infection in the cell. Specifically, we constructed a panel of mutants with mutations which alter the production, size, or sequence of TF. Using these mutants, we demonstrated that TF is not essential for virus production, although disrupting TF production leads to an ∼1.5-log-unit decrease in virus particle release in both mammalian and insect cell cultures. In a mouse model of SINV neuropathogenesis, most animals infected with the TF mutants survived infection, unlike animals infected with the wild-type virus, concomitant with a decrease in infectious virus release in the TF mutant background. Using a variety of biochemical and cell-based assays, we demonstrate that TF likely retains ion-channel activity similar to that of 6K and that a lack of production of TF does not affect genome synthesis, specific infectivity, or transit of envelope proteins to the cell surface. Therefore, our data suggest that TF may be involved in virus particle assembly or release. Given the overlapping 6K and TF ORFs, the findings described here directly impact decades of published literature with respect to the composition and function of the alphavirus structural proteins.
MATERIALS AND METHODS
Cell culture.
Both C6/36 and BHK-15 (BHK) cells were maintained in minimal essential medium (MEM; Life Technologies) supplemented with 10% fetal bovine serum (FBS) at 30°C and 37°C, respectively. Cycling AP-7 neuronal cells were grown and differentiated as previously described (28).
Generation of mutants with mutations in virus cDNA.
All mutants were generated in pTE12 SINV cDNA plasmids using standard overlap PCR mutagenesis with Platinum Pfx DNA polymerase (Invitrogen). Infectious full-length RNA was produced by linearizing the plasmids with XhoI, followed by SP6-mediated in vitro transcription (Ambion).
Virus purification.
BHK cells were electroporated (1.5 kV, 25 μF, 200 Ω, 0.2-cm-gap cuvette) with 10 μg of in vitro-transcribed RNAs derived from the TE12 cDNA clones. Infection was allowed to proceed for 12 h, at which time the media were harvested and clarified by centrifugation for 30 min at 9,000 × g. Virus particles were pelleted through a 30% sucrose cushion in a Beckman Ti-50.2 rotor at 32,000 rpm for 2 h. Virus pellets were resuspended, pooled, applied to a continuous 0 to 30% iodixanol gradient in TNE (50 mM Tris, pH 7.4, 100 mM NaCl, 1 mM EDTA), and centrifuged at 32,000 rpm in a Beckman SW-41 rotor for 2 h. Virus was manually removed from the gradient tube with a needle and syringe. For mass spectrometry analyses, the gradient step was repeated one time prior to peptide preparation. All steps were carried out at 4°C.
Mass spectrometry.
SINV or CHIKV was purified as described above. Purified virions were delipidated by addition of a 2:1 chloroform-methanol solution, followed by acetone-mediated precipitation of protein. Recovered protein was reduced and alkylated in 8 M urea, using dithiothreitol, 2-iodoethanol, and triethylphosphine. Protein was then digested with trypsin, and peptides were analyzed using a capillary liquid chromatography (LC) system (1100 series LC; Agilent, Santa Clara, CA) by concentrating the peptides on an Agilent 300SB-C18 enrichment column (5 μm, 5 by 0.3 mm), followed by separation on a C18 reversed-phase Zorbax 300SB-C18 analytical column (3.5 μm, 0.75 μm by 150 mm; Agilent) coupled to the electrospray ionization (ESI) source. The peptides were loaded onto the enrichment column, washed with 5% acetonitrile (ACN), 0.01% trifluoroacetic acid (TFA) for 5 min, eluted with a 35-min linear gradient from 5% to 40% buffer B (100% acetonitrile, 0.01% TFA), followed by a 10-min gradient from 40% to 90% buffer B, and, finally, reequilibrated with an isocratic flow (5% buffer B) for 20 min, all at a flow rate of 0.4 μl/min. The LC system was coupled to a hybrid linear ion-trap Orbitrap mass spectrometer (ThermoFisher, San Jose, CA), and the capillary temperature and electrospray voltage were 200°C and +2.2 kV, respectively. LC-MS/MS chromatograms were acquired in positive ion mode, and the Orbitrap mass spectrometer was used as the mass analyzer during MS survey scans over an m/z range from 200 to 2,000 with a duty cycle of 1.2 s. Data-dependent MS/MS was performed in the linear trap quadrupole (LTQ) for the top 5 ions with a normalized collision energy of ∼35%. Dynamic exclusion in the LTQ during data-dependent MS/MS experiments was enabled as follows: repeat count of 2, repeat duration of 30 s, exclusion list size of 250, and exclusion duration of 60 s. LC-MS/MS spectra were analyzed using Spectrum Mill A.03.02.060 software (Agilent Technologies), and searches of the data against the data in the National Center for Biotechnology Information protein database were performed. The data were also searched against those in a custom SINV- or CHIKV-specific database. The parameters were as follows: no more than two tryptic miscleavages were allowed, cysteine was searched as iodoethanol, the peptide mass tolerance was 1.0 Da, and the MS/MS mass tolerance was 0.7 Da. Only peptides with a scored peak intensity (SPI) of >70% and a score of >5 were considered positive hits.
Virus infection of mice.
Two-week-old CD-1 mice (Charles River Laboratories) were infected intracerebrally with 1,000 PFU of either the wild-type or the mutant viruses diluted in 10 μl of phosphate-buffered saline (PBS). Groups of 20 mice per virus were monitored daily for the appearance of clinical symptoms and death for a duration of 12 days. Mice were scored on a 4-point scale: a score of 1 was given for ruffled fur and hunched back, a score of 2 was given for paralysis of one hind limb, a score of 3 was given for paralysis of two hind limbs and/or a moribund state, and a score of 4 was given for death. All animal studies were conducted in accordance with the protocols approved by the Johns Hopkins University Animal Care and Use Committee.
In vivo viral replication.
Two-week-old CD1 mice were infected intracranially with 1,000 PFU of wild-type virus or virus with a TF deletion (ΔTF). Brains were harvested at 3 and 5 days postinfection (dpi), and 10% (wt/vol) homogenates were made in PBS using one hemisphere of the brain and further clarified. The homogenate was titrated on BHK-21 cells to assess viral titers.
Immunofluorescence assays.
6K or TF was subcloned into either the pcDNA4 mammalian expression vector or a custom vector constructed with the pcDNA4 backbone, but with the cytomegalovirus promoter and bovine growth hormone poly(A) signal replaced with those from the baculovirus Orgyia pseudotsugata (29, 30) to allow expression in mosquito cells. Both proteins contained a signal sequence derived from the cytoplasmic domain of SINV E2 followed by an epitope tag consisting of envelope protein G of the vesicular stomatitis virus (VSV-G) at the N termini. The presence of a VSV-G tag at the N terminus of 6K/TF does not attenuate the virus (J. Jose and R. J. Kuhn, unpublished data). The 6K gene was modified such that the slippery codon motif was silently removed, and the TF gene was modified by deletion of a single nucleotide within the slippery codon motif, such that the normal and −1 ORFs were fused appropriately. Cells were transfected with plasmid using the Lipofectamine 2000 reagent (Invitrogen) and fixed and permeabilized with 3.7% formaldehyde in PBS and 0.1% Triton X-100 in PBS, respectively, at 16 h (BHK cells) or 32 h (AP-7 and C6/36 cells). Cells were stained with mouse anti-VSV-G (Sigma), rabbit anticalreticulin (Abcam), or rabbit antigiantin (Abcam) antibodies, followed by fluorescein isothiocyanate (FITC)- or tetramethyl rhodamine isocyanate (TRITC)-labeled secondary antibodies and DAPI (4′,6-diamidino-2-phenylindole). Microscopy was performed on an Olympus IX81 fluorescence microscope.
Protein expression in Escherichia coli.
The 6K or TF genes (modified as described above) were subcloned into the pET11a expression vector. E. coli Rosetta 2(DE3)pLysS was transformed with plasmid, and heterologous protein synthesis was induced with 0.5 mM IPTG (isopropyl-β-d-thiogalactopyranoside).
Luciferase assays.
Mutations were cloned into a SINV background in which firefly luciferase is driven by an internal ribosome entry site (IRES) element inserted 3′ to the E1 gene (for mammalian cells) or in which the Gaussia luciferase gene was driven by a duplicated SINV subgenomic promoter inserted 3′ to the E1 gene (for mosquito cells). Cells were infected at a multiplicity of infection (MOI) of 10, and luciferase levels were measured at the time points indicated per the manufacturer's protocol with substrate from Promega (firefly luciferase) or New England BioLabs (Gaussia luciferase). Measurements were performed on a Molecular Devices LMaxII 384 instrument.
Immunoblotting.
At 8 h postinfection (MOI = 10), BHK cell lysates were separated on a 12% SDS-polyacrylamide gel, electroeluted to 0.2-μm nitrocellulose, and probed with rabbit polyclonal antibodies to actin, SINV capsid, SINV E2, or SINV E1 and a goat anti-rabbit IR680 secondary antibody (LiCor).
Specific infectivity assay and qRT-PCR.
Virus was purified as described above. The amount of infectious virus material was quantified by titration on BHK cells, and the total number of genome copies was determined by quantitative reverse transcription-PCR (qRT-PCR). Specifically, RNA was purified with the TRIzol LS reagent, and the RNA concentration was determined on an ABI 7300 real-time PCR instrument using SYBR green and carboxy-X-rhodamine chemistry with the SINV-specific primers 5′-TTCCCTGTGTGCACGTACAT-3′ and 5′-TGAGCCCAACCAGAAGTTTT-3′. A standard curve was generated using viral RNA isolated from purified virions.
Flow cytometry.
For each experiment, 1 million cells in 100 μl PBS supplemented with 5% calf serum were stained with 1:100 dilutions of a monoclonal anti-E2 primary antibody (SV127) and a goat anti-mouse FITC secondary antibody at the time points postinfection indicated (MOI = 10). Flow analysis was performed on a Beckman Coulter FC500 flow cytometer and with the FlowJo software package.
Polykaryon formation.
BHK cells were infected at an MOI of 1, and at 8 h postinfection, cells were washed with PBS and treated with MEM, pH 5.2, for 5 min at 37°C. Cells were then washed thrice with PBS and incubated at 37°C for 1 h with medium at pH 7.4. Cells were subsequently fixed, permeabilized, and stained as described above using a rabbit anti-SINV capsid antibody, in addition to a goat anti-rabbit FITC secondary antibody and DAPI. A fusion index was calculated as [1 − (number of cells/number of nuclei)]. Twenty-five cells were counted per sample per experiment.
RESULTS
The TF protein is detected in both Sindbis and Chikungunya virus particles.
Firth and colleagues characterized TF synthesis in a single alphavirus, SFV (27). To ascertain whether or not TF production occurred in other alphaviruses, we sought to detect TF synthesis by analyzing a second alphavirus in the same clade as SFV, CHIKV, as well as an alphavirus from a separate clade, SINV. Both viruses were grown in cultured BHK cells, and virus particles were purified to near homogeneity in an ultracentrifuge (see Materials and Methods). Tryptic peptides were generated and subject to LC-MS/MS identification. Peptides corresponding to the unique C-terminal domains of both 6K and TF were detected for SINV (Table 1). Interestingly, while we were able to detect analogous TF peptides for CHIKV, we did not detect any peptides corresponding to 6K in CHIKV particles. Furthermore, despite delipidation prior to trypsin digestion, we were unable to detect any peptides corresponding to the shared N-terminal domain of 6K and TF for either SINV or CHIKV. These data likely do not indicate an absence of 6K in the CHIKV particle, however. Rather, the inability to detect CHIKV 6K is probably reflective of its fewer trypsin cleavage sites relative to the number in SINV 6K, resulting in peptides too large for efficient ionization. Indeed, SDS-PAGE and silver stain analysis of purified CHIKV particles revealed two low-molecular-weight bands that migrated similarly to those identified as 6K and TF in purified SINV preparations (see below). Therefore, our data strongly imply that TF is both synthesized and incorporated into virions in CHIKV and SINV.
Table 1.
Mass spectrometry peptide identification
| Protein | Peptide | Parent m/z | Score | Scored peak intensity (%) |
|---|---|---|---|---|
| 6K | (R)CCSCCLPFLVVAGAYLAK(V) | 1,019.013 | 22.75 | 100 |
| 6K | (R)CCSCCLPFLVVAGAYLAKVDA(−) | 1,161.548 | 25.00 | 100 |
| TF | (R)LRTCDHCSKCATDTV(−) | 892.9551 | 23.50 | 100 |
| TF | (R)TCDHCSKCATDTV(−) | 758.391 | 23.95 | 98.2 |
| TF | (R)LRTCDHCSK(C) | 575.7975 | 21.65 | 97.7 |
TF is essential for efficient infectious virus release.
To determine if the production of TF during alphavirus infection was important, we constructed a panel of mutants using SINV. Four separate mutants (Fig. 1A and B) with mutations that perturbed the production or amino acid sequence of TF yet left the amino acid sequence of 6K and E1 unaltered were generated. In the first mutant, the ΔTF mutant, the slippery codon motif UUUUUUA was mutated such that the motif itself was disrupted but the amino acids encoded by the normal ORF were left unchanged. Thus, ribosomal frameshifting would be ablated and production of TF would not be expected to occur. In the second mutant, the TF-stop mutant, we inserted a premature stop codon in the −1 ORF that was, again, silent for the normal ORF, such that although the frameshift event was expected to occur, a truncated version of TF lacking the C-terminal 19 amino acids would be synthesized. The third mutant, the TF-ext mutant, was constructed such that the normal TF stop codon in the −1 ORF was removed, as was a second stop codon in the −1 ORF three amino acids downstream. Thus, the TF-ext virus would produce a version of TF with a long C-terminal extension. In the fourth mutant, the TF-rdm mutant, we mutated, wherever possible, the third nucleotide in the normal ORF in the region of the 6K/E1 genes that encode TF. These nucleotide changes yielded nonsynonymous mutations in the TF −1 ORF, thus changing the identity of the TF amino acid encoded only by the −1 ORF. Importantly, this mutation drastically altered the amino acid identity of TF and changed many of the cysteines and the basic residues found in the unique TF C terminus (Fig. 1A). Of note, all four mutants were expected to predominantly produce 6K and E1 via the normal ORF, with the ΔTF mutant actually producing excess E1 protein relative to wild-type virus, given the disruption of the frameshift.
Fig 1.

Summary of the mutations constructed to perturb TF synthesis. (A) Amino acid sequences of 6K, TF, and TF of mutant TF-rdm with a mutation in which the unique C-terminal domain of TF contained multiple nonsynonymous mutations in the −1 ORF. The wild-type TF C-terminal domain is underlined. The residues comprising the transmembrane domain implicated in ion-channel activity are italicized. (B) The specific nucleotide changes made in the viral cDNA to construct the ΔTF, TF-stop, or TF-ext mutant are shown. For each mutant, the top row represents the wild-type (WT) nucleotide sequence, the middle row shows the mutant nucleotide sequence, and the bottom row shows the amino acids corresponding to these codons in the normal ORF. The −1 ORF stop codons that were altered in the TF-ext mutant are underlined.
We performed both multistep (MOI = 0.5) and one-step (MOI = 5.0) growth curves in BHK cells, C6/36 cells, and both cycling and differentiated AP-7 neuronal cells for the four mutant viruses. Representative results are shown in Fig. 2 for multistep growth curves, as the trends in one-step growth curve experiments (data not shown) were similar. Specifically, relative to the wild-type virus, cells infected with any of the four mutants demonstrated a ≥1.5-log-unit reduction in infectious particle release during infection. These data suggest that the length and amino acid sequence as well as the production of TF are important for infectious particle release. In the case of the ΔTF mutant, we verified the absence of TF incorporation into virions by analyzing purified virus preparations by denaturing PAGE and silver staining (Fig. 2D). To confirm the presence of both 6K and TF in the wild type and solely 6K in the ΔTF mutant, the bands shown in Fig. 2D were removed and identified by LC-MS/MS. Indeed, the more quickly migrating band was identified as 6K, and the more slowly migrating species, absent in the lane for the ΔTF mutant, was identified as the TF protein (data not shown). Additionally, we estimated, using densitometry, a 50% greater concentration of TF in the purified virions relative to that of 6K (data not shown). Given previous radiodensitometry data from Gaedigk-Nitschko and Schlesinger (18), we therefore estimate ∼16 copies of TF and ∼8 copies of 6K per Sindbis virus virion. This aligns with the findings of Firth and colleagues (27). Furthermore, to interrogate the minimum length of TF required for wild-type-like infectious particle release, we constructed four additional mutants with mutations that modulated the length of TF such that the protein would lack either the C-terminal 17, 14, or 7 amino acids or contain a 4-amino-acid C-terminal extension. Interestingly, in multistep growth curves in BHK cells, all of the C-terminal-truncation mutants grew like the ΔTF virus, while the 4-amino-acid C-terminal-extension mutant grew like the wild type, reinforcing the importance of the length of TF (Fig. 3).
Fig 2.

Multistep growth curve assay analysis of infectious particle release. Cultured cells were inoculated at an MOI of 0.5, and media were collected at the time points indicated. The amount of infectious material in each sample was quantified by plaque assay on BHK cells. (A) BHK cells; (B) C6/36 cells; (C) differentiated AP-7 cells. (D) Wild-type (left) and ΔTF (right) viruses were purified from BHK cells and analyzed by silver staining on a 12% SDS-polyacrylamide gel.
Fig 3.

Multistep growth curve assay analysis of infectious particle release for C-terminal (C-term)-truncation mutants. Cultured BHK cells were inoculated at an MOI of 0.5, and media were collected at the time points indicated. The amount of infectious material in each sample was quantified by plaque assay on BHK cells. The Δ17, Δ14, and Δ7 mutants had a premature stop codon placed in the −1 ORF, such that TF lacked the 17, 14, and 7 C-terminal residues, respectively. The +4 mutant lacks the normal TF stop codon, instead utilizing a stop codon present in the −1 ORF five codons downstream. All mutants were silent for the normal ORF.
TF mutants are attenuated in a mouse model of SINV neuropathogenesis.
To determine the phenotypes of the TF mutants in an animal model, we utilized a mouse model of SINV neuropathogenesis. Mice were challenged intracerebrally with wild-type TE12 or a TF mutant virus, and morbidity and mortality were measured through 12 days postinfection. As outlined in Fig. 4A, wild-type virus caused 95% mortality, while the majority of the mice challenged with ΔTF, TF-stop, TF-ext, or TF-rdm virus survived. The severity of disease experienced by mice infected with the TF mutants was also greatly reduced relative to that experienced by mice infected with the wild-type virus (Fig. 4B). To investigate if the attenuation in pathogenesis might be due to a decrease in the release of infectious virions, we titrated the amount of infectious material in the brain at 3 and 5 days after intracranial inoculation with either the wild-type or the ΔTF virus. As was observed in cultured cells of both mammalian and arthropod origin, fewer infectious virions were found to be released in animals infected with the ΔTF virus than those infected with the wild type (Fig. 4C) when brain homogenates were titrated for infectious material. These data suggest that the TF protein likely has an important role during infection in vivo.
Fig 4.
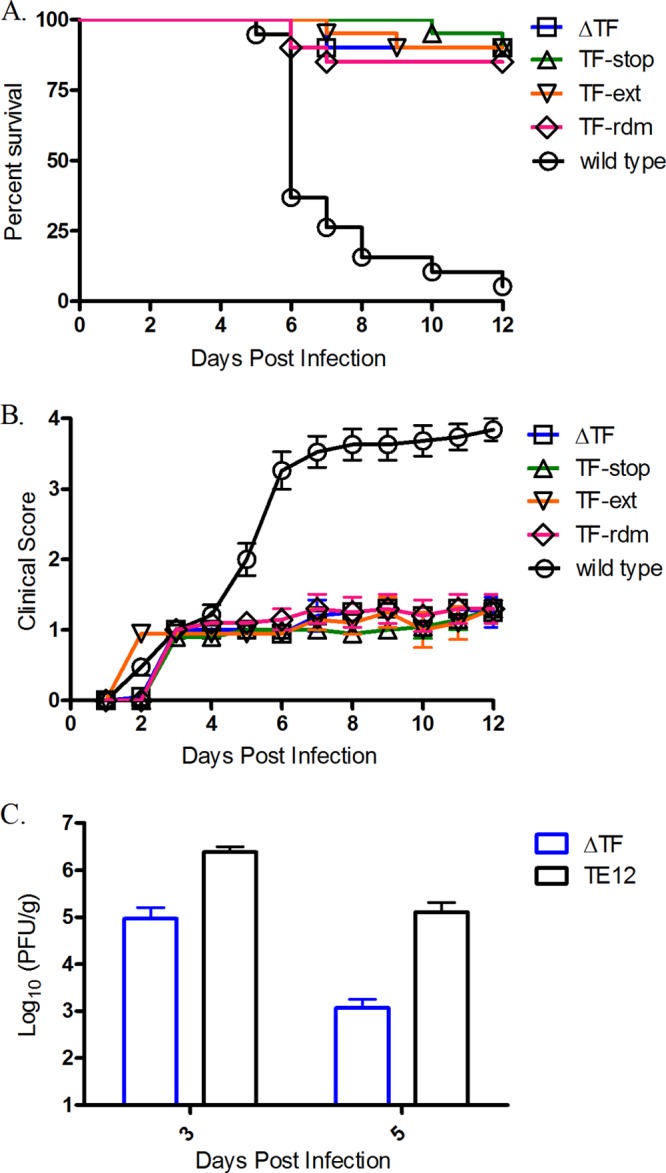
Analysis of TF mutants in a mouse model of SINV neuropathogenesis. Two-week-old CD-1 mice were inoculated intracerebrally with 1,000 PFU of the virus indicated. (A) The survival rates are shown as a function of time. (B) Morbidity was scored on a 4-point scale: a score of 0 was given for no obvious symptoms, a score of 1 was given for kyphoscoliosis and ruffling, a score of 2 was given for one hind limb paralyzed, a score of 3 was given for two hind limbs paralyzed, and a score of 4 was given for death. (C) Two-week-old CD1 mice were infected intracranially with 1,000 PFU of wild-type or ΔTF virus. Brains were harvested at 3 and 5 dpi, and 10% (wt/vol) homogenates were made in PBS using one hemisphere of the brain and further clarified. The homogenate was titrated on BHK-21 cells to assess viral titers.
TF appears to be retained in the ER after synthesis.
Given that virus release kinetics were impaired in mammalian and insect cell culture, as well as in the mouse, when TF synthesis was abrogated, we sought to understand what role the TF protein might play during the course of infection. We first interrogated the subcellular localization of the protein. As we lacked SINV 6K- and TF-specific antibodies, the 6K or TF genes were subcloned into mammalian or insect expression vectors. The proteins were fused to a signal sequence, to ensure translocation of the protein into the ER, and a VSV-G epitope tag at the N terminus. Initial studies in BHK, C6/36, and cycling AP-7 cells demonstrated that while 6K trafficked to intracellular vesicles and the plasma membrane, the TF protein appeared to be retained in a region adjacent to the nucleus (data not shown). To ascertain if TF might be retained in some component of the secretory pathway, we stained transfected BHK cells with antigiantin and anticalreticulin antibodies, which stain the Golgi and ER structures, respectively. As shown in Fig. 5A, much of 6K was concentrated in structures that costained with the Golgi apparatus marker giantin, whereas TF costaining with the Golgi apparatus was much less intense. Conversely, TF appeared to localize to the ER, as colocalization of the TF protein with calreticulin was readily observed (Fig. 5B). Staining of cells infected with a ΔTF virus that produced only 6K and in which the VSV-G epitope tag was retained demonstrated a Golgi apparatus localization pattern similar to that of ectopically expressed 6K, but staining near the ER was absent (data not shown). Additionally, Firth and colleagues raised an SFV TF-specific polyclonal antibody and showed that the bulk of TF staining in BHK cells infected with SFV was around the nucleus, in an ER-like subcellular localization (27). Taken together, these data may indicate retention of TF in the ER, whereas 6K appears to be able to traffic to the Golgi apparatus and eventually the PM. Interestingly, though, TF was detected in purified virus particles, suggesting that some fraction of TF may traffic to the PM because this is the major site of virion budding in mammalian cells.
Fig 5.
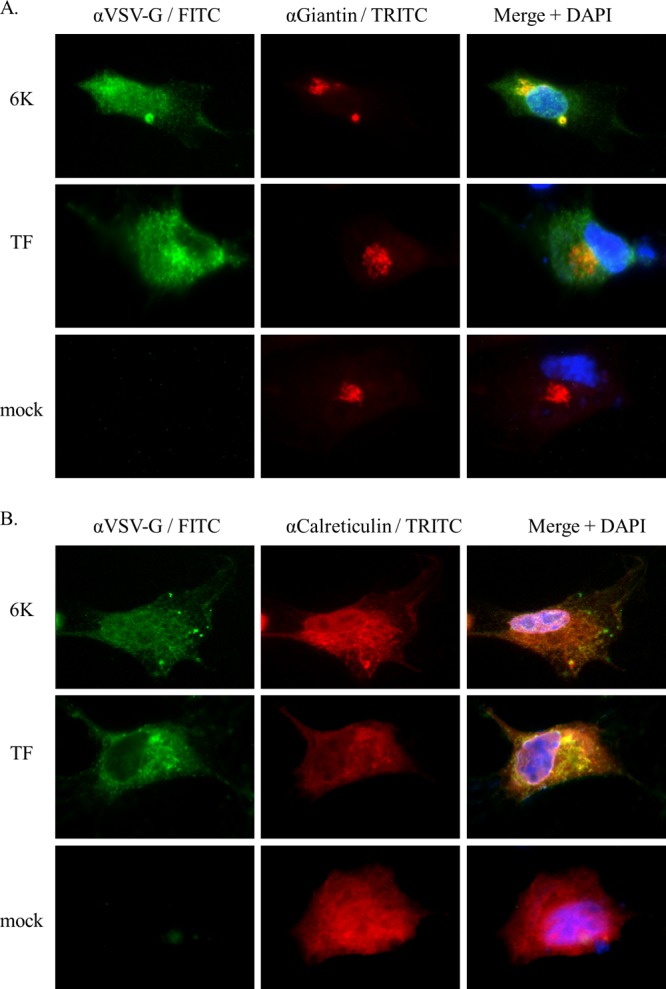
Immunofluorescence analysis of 6K and TF protein subcellular localization. BHK cells were transfected with a plasmid expressing VSV-G epitope-tagged versions of the 6K or TF proteins. At 16 h posttransfection, cells were processed for immunocytochemistry and stained with an anti-VSV-G antibody, DAPI, and either an antigiantin (A) or anticalreticulin (B) antibody.
TF leads to rapid perturbation of E. coli growth after synthesis.
It has long been suspected that 6K contains ion-channel activity. Patch-clamp experiments performed after 6K synthesis in Xenopus oocytes (31), as well as with reconstituted 6K protein in planar lipid bilayers (32), have demonstrated the ability of 6K to preferentially allow the diffusion of cations across membranes. Similarly, induction of expression of 6K in E. coli leads to increased membrane permeability and concomitant perturbation of growth (32, 33). Given that the location of the TF frameshift site is downstream of the region encoding the first transmembrane domain implicated in 6K's ion-channel activity, we suspected that TF would likely retain such activity. As a surrogate for direct ion-channel activity studies, we investigated whether TF has the ability to clear E. coli cultures like 6K does. The 6K or TF gene was subcloned into the pET11a-inducible prokaryotic expression vector. After induction, culture density was measured via determination of the OD600. Figure 6 provides growth data from a single, representative experiment. As outlined in Fig. 6, induction of TF expression rapidly led to a decrease in culture density, as did 6K expression. As a negative control, we expressed the SINV capsid protein, and its synthesis did not affect the growth of the culture. Importantly, as additional controls, we expressed both 6K and TF in the R34A mutant background. The R34 residue of 6K has been implicated in modulating ion-channel activity, with the R34A mutation disrupting such activity (Jose and Kuhn, unpublished). Both the 6K and the TF R34A proteins failed to induce the perturbation of growth like their wild-type counterparts. Therefore, the cytotoxicity of 6K and TF depends on a background in which ion-channel activity is not perturbed and is not because of the inherent cytotoxicity observed upon expression of some membrane proteins in E. coli. Thus, these data strongly suggest that TF retains the ion-channel properties of 6K.
Fig 6.

Analysis of E. coli growth during synthesis of the 6K or TF proteins. LB media were inoculated with media derived from an overnight culture of E. coli harboring a pET11a expression plasmid containing the gene for the proteins indicated below. Inoculated cultures were allowed to grow to an OD600 of between 0.5 and 1.0 and split into two aliquots, and protein synthesis was induced by addition of IPTG to a final concentration of 0.5 mM in one of the aliquots. The OD600 was further monitored after induction. The arrow represents the time at which IPTG was added. (A) 6K; (B) TF; (C) SINV capsid; (D) 6K R34A; (E) TF R34A.
The TF-knockout mutant does not affect genome synthesis.
To determine if the TF protein might somehow be involved in genome synthesis, we used a luciferase reporter system as a surrogate to gauge the relative amount of viral RNA genomes in the infected cell. Specifically, we cloned the ΔTF mutation into a SINV luciferase reporter virus where luciferase protein expression was driven either by an IRES element at the 3′ end of the genome, in the case of mammalian cell studies, or via a duplicated subgenomic promoter at the 3′ end of the genome, in the case of insect cell experiments. The amount of luciferase-derived signal correlates with the concentration of genomic RNA in the cell. Figure 7 shows that, in the ΔTF background, mutant viral RNA levels were not reduced relative to those of wild-type RNA in either mammalian or insect cells and were, in some cases, slightly elevated. In a separate experiment, we validated the data from the luciferase assay as a surrogate assay for genome copy number by also quantifying the genome copy number via qRT-PCR at multiple time points postinoculation of BHK cells. Again, the viral RNA synthesis of the TF mutants was unaffected relative to that of the wild type (data not shown). That genome synthesis was unaffected was not surprising, given that synthesis of the structural polyprotein is inherently dependent on a functional viral replicase; synthesis of the structural polyprotein does not occur until after a functional replicase complex has been established (26). To also ascertain if TF might be involved in structural protein synthesis or maturation, we performed immunoblots with BHK cell lysates infected with wild-type or ΔTF viruses. Synthesis of capsid, E2, E1, and the pE2 precursor was unaffected (Fig. 7D), and nucleocapsid assembly, determined by ultracentrifuge-mediated analysis of infected cell lysates, was not perturbed either (data not shown).
Fig 7.

RNA synthesis analysis using luciferase-expressing virus. Firefly or Gaussia luciferase-expressing viruses containing either the intact TF slippery codon motif or the ΔTF mutation were used as surrogates to analyze viral RNA synthesis. The indicated cells were infected at an MOI of 10, and cell lysates (BHK and AP-7 cells) or cell culture medium (C6/36 cells) was analyzed for luciferase activity at the indicated time point. (A) BHK cells; (B) C6/36 cells; (C) cycling AP-7 cells. RLU, relative light units. (D) BHK cell lysates were also analyzed by Western blotting for the amount and presence of full processed capsid, E2, and E1.
TF does not play a role in particle infectivity.
Plaque assay and growth curve data provide only the number of infectious virus particles released from a cell and not the total number of virus particles. Given that TF was incorporated into virions in the wild-type virus, one possibility is that TF assists with the infectivity of the particle, perhaps by contributing structural rigidity or aiding in virion disassembly during entry. To gauge if there was a difference in the infectivity of the ΔTF virus particle, we determined the specific infectivity of wild-type and ΔTF virus particles released from BHK, C6/36, and cycling AP-7 cells. Specifically, we stringently purified virus particles by ultracentrifugation and, in parallel, determined the number of infectious particles by plaque assay and the total number of virus particles by quantitative RT-PCR using SINV genomic RNA-specific primers. The results are shown in Table 2. While, the particle number/PFU ratio differed among cell types, the results were not significantly different between the wild-type and ΔTF virus particles in any one cell line. Given that a Δ6K virus, in which both 6K and TF production was ablated, showed similar results, our data are not surprising (22). To investigate if TF might play a role in genome-free particle release (34), we analyzed equivalent amounts of purified infectious units by SDS-PAGE and silver staining. Subsequent densitometric analysis of the stained capsid, E2, and E1 bands showed no significant difference in the amount of total virus particles released (data not shown). Thus, TF does not appear to play a role in the infectivity of the alphavirus virion.
Table 2.
Specific infectivity of purified virus particles
| Virus | Ratio for the following cell typea: |
||
|---|---|---|---|
| BHK | cAP7 | C6/36 | |
| Wild type | 450 ± 55 | 1,308 ± 26 | 1,870 ± 163 |
| ΔTF | 499 ± 24 | 1,213 ± 149 | 1,858 ± 90 |
The ratio is the average number of viral RNA molecules per milliliter divided by the number of PFU per milliliter ± 1 standard deviation.
The cell surface concentration and fusogenic ability of the envelope proteins are unaffected in the TF-knockout mutant.
We next interrogated if spike protein transport to the cell surface was impaired in the ΔTF virus background. BHK, C6/36, or cycling AP-7 cells were infected with wild-type or ΔTF virus, and the amount of E2 at the cell surface in fixed, nonpermeabilized cells was determined by flow cytometry at multiple intervals after infection. The concentration of E2 at the cell surface was similar in the ΔTF background and the wild type and in some cases was slightly higher in the ΔTF background than in the wild type (Fig. 8). Because this approach does not provide either information about the conformation of the spike at the plasma membrane or details about the amount of E1 at the cell surface, we performed a polykaryon formation assay (35). E2/E1 spike complexes at the cell surface transition to a fusogenic state if the pH of the cell medium is lower than the pH threshold for fusion, typically ∼pH 5.8. These fusion-active spikes are capable of fusing with the plasma membrane of adjacent cells, leading to polykaryon, or multinucleated, cells. We infected BHK cells with either the wild-type or ΔTF virus and briefly exposed cells to acidic medium. The fusogenic capacity of the infected cells was ascertained by calculating a fusion index. As shown in Fig. 9, spikes at the cell surface of ΔTF mutant-infected cells were capable of initiating cell-cell fusion, as in the wild-type-infected cells at acidic pH. While these data do not provide any kinetic parameters of fusion, they demonstrate the fusogenic nature of the ΔTF spike, the presence of E1 in the ΔTF spike, and, overall, that the data align with the specific infectivity data. Taken together, these data suggest that TF does not play a role in the virus entry process.
Fig 8.

Analysis of E2 cell surface expression. Cells were infected with either wild-type or ΔTF virus at an MOI of 10 and processed for flow cytometry at the indicated time points by fixation and anti-E2 antibody staining. (A) BHK cells; (B) C6/36 cells; (C) cycling AP-7 cells.
Fig 9.
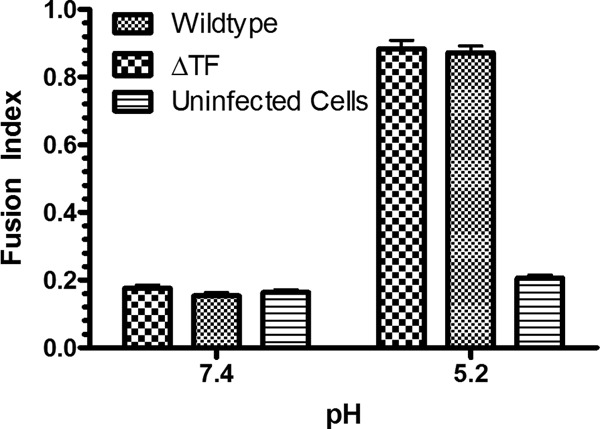
Polykaryon formation in BHK cells. BHK cells were infected with either wild-type or ΔTF virus at an MOI of 1. At 8 h postinoculation, polykaryon formation was initiated by a brief treatment with medium at pH 5.2. Before calculating a fusion index, the cell media were returned to neutral pH and the cells were fixed and immunostained.
TF protein provided in trans cannot rescue a ΔTF virus.
Sanz and Carrasco demonstrated that in SINV Δ6K the 6K protein could not be provided in trans to rescue the attenuation of infectious particles released (23). However, it is now obvious that the Δ6K background ablated the synthesis of both 6K and TF; only 6K, and not TF, was provided in trans in these experiments. We thus sought to understand if, in our SINV ΔTF mutant background, whether or not providing a functional copy of TF might rescue the attenuated phenotype of the ΔTF mutant. We utilized a SINV construct in which the subgenomic promoter was duplicated at the 3′ end of the genome, allowing expression of heterologous protein. In either the wild-type or the ΔTF mutant background, we expressed the wild-type TF protein or the TF-rdm protein, both with N-terminal signal sequences and with VSV-G epitope tags, and the latter was used as a negative control. One-step growth curves were performed in BHK cells, and the results are shown in Fig. 10. Interestingly, providing the wild-type TF protein in trans failed to rescue the attenuated phenotype of the ΔTF mutant. Immunocytochemical analysis of infected cells validated the expression of TF in trans in the ΔTF mutant background and demonstrated its localization to a cellular compartment adjacent to the nucleus (data not shown).
Fig 10.

One-step growth curve analysis of constructs in which TF was provided in trans. Cells were inoculated at an MOI of 5, and virus was harvested at the indicated time points. In either the wild-type or ΔTF mutant background, a duplicated subgenomic promoter drove expression of the TF protein or the TF-rdm protein.
DISCUSSION
The discovery that the alphaviruses employ a frameshifting technique to encode separate proteins in overlapping reading frames is quite intriguing, but not surprising, given that multiple viruses employ this strategy to minimize the size of their genome (36). Historically, electrophoretic analyses of purified alphavirus preparations have consistently revealed two closely migrating bands in the 4- to 8-kDa range, and it was presumed that these bands represented alternative acylation states of 6K (18). The data presented here and from Firth and colleagues (27) show that, instead, one of these bands is a novel structural protein, TF, which shares its N terminus with 6K but has a unique C-terminal amino acid sequence that is very hydrophilic, unlike the hydrophobic C terminus of 6K (27). Given that the slippery codon motif that mediates the frameshift is conserved among the alphaviruses and that TF synthesis has now been demonstrated in SFV, SINV, and CHIKV, it is likely that production of the TF protein occurs universally among the members of the alphavirus genus.
Using SINV as a model system, we have begun to characterize the role of TF in the alphavirus life cycle. We show here that in both mammalian and insect cell cultures, the production of TF is dispensable for virus particle release. However, disruption of TF synthesis or its amino acid sequence perturbs the release of infectious virus particles during infection such that <10% of infectious material is released relative to the amount for wild-type virus. Interestingly, Firth and colleagues reported only an ∼56% reduction in infectious particle release in an SFV ΔTF background, although their report did not provide growth curve analyses (27). Rather, they monitored accumulation of infectious material in the medium after collection at a single time point, which is not an accurate measure of viral release kinetics (27). The observation that mice infected with the TF mutant constructs did not experience severe or fatal disease like mice infected with the wild-type virus did highlights the importance of the TF protein during the virus life cycle in vivo. Additional experimentation revealed that in the animal, as in in vitro studies, the ΔTF virus released fewer infectious particles than the wild type. Given this deficiency in infectious particle release in vivo, it may be that the attenuation of morbidity and mortality seen in the animal was due to the ability of the innate immune system to successfully restrict the infection in the absence of efficient virion release from infected cells. In other words, for successful dissemination throughout the animal, a certain rate or threshold of alphavirus release from the cell may need to occur such that the balance between viral spread and immune-mediated clearance is tipped in favor of the former. The evolution of the TF protein may be an example of gene overprinting (37) selected via pressure for survival in the infected host.
Our data strongly suggest that the TF protein, as opposed to some downstream event in the virus life cycle, is required for efficient virus assembly or release. Specifically, we demonstrated that, in an SINV ΔTF background, genome synthesis and viral structural protein synthesis were not impaired and nucleocapsid assembly was unaffected. Furthermore, it is almost certain that virus entry was not perturbed, as the ΔTF virus was not deficient in infectivity, the ΔTF mutant spike showed fusogenic potential equivalent to that of wild type, and growth kinetics in both one-step and multistep growth curves were similar for the ΔTF mutant. Additionally, the cell surface concentrations of the major envelope proteins were similar between wild type and the ΔTF virus, suggesting that TF is not involved in transport of the envelope proteins to the cell surface. Taken together, these data imply that TF is involved in a late stage in virus assembly and/or budding.
Interestingly, TF could not rescue particle release in trans, suggesting that it requires interaction, soon after synthesis, with an additional structural protein, as TF is produced as part of the structural polyprotein. Given that E1 is not produced during frameshift events and that capsid is soluble in the cytoplasm, TF may require interaction with E2. Indeed, genetic evidence suggests an interaction between a domain of 6K shared with TF and the E2 protein (38, 39). Our data also suggest that the length of the TF protein is important, as even a deletion of the C-terminal 7 residues of TF reduced multiplication to a level similar to that in the ΔTF virus. Indeed, the position of the TF stop codon in the −1 ORF is also conserved among the alphaviruses, suggesting that the length of the TF protein is critical. As TF appears to be retained in the ER, one role of the unique C terminus of TF might be to act as an ER retention signal. Given its very hydrophobic nature, the unique C terminus of TF is unlikely to span a lipid bilayer a second time, like that of 6K, and thus, TF probably contains only one transmembrane domain. However, TF, unlike 6K, may have a cytoplasmic C-terminal tail. Of note, coimmunoprecipitations with a VSV-G-tagged version of TF failed to identify any interacting partners using BHK cell lysates (data not shown).
The similar virus release kinetics among the wild-type and TF mutant viruses early in infection suggests that TF may play a role within a cell only late in infection. Indeed, some virus-induced structures are not apparent in the infected cell early in infection. Virus-induced cytopathic vacuole type II (CPV-II) structures, for example, which likely cotraffic nucleocapsids and envelope protein spikes to the cell surface, accumulate only later in infection (40). Little is known regarding the viral determinants for CPV-II formation, and it is possible that TF might be involved. Furthermore, TF is important in both insect and mammalian cells, suggesting the involvement of TF in a fundamental aspect of virus assembly shared between host and vector. A novel alphavirus, Eilat virus, that replicates only in arthropods was recently described (41). Interestingly, analysis of the nucleotide sequence of Eilat virus shows that both the slippery codon motif in the 6K gene and the conserved stop codon in the −1 ORF are intact, suggesting that TF indeed plays a role in the vector.
Using an E. coli growth assay, our data likely indicate that TF retains ion-channel activity. Furthermore, immunocytochemical analysis suggests that much of TF is retained in the ER. Several other viruses, including members of the picornaviruses and rotavirus, encode ER-resident ion-channel proteins (42). For example, both the 2B protein of coxsackievirus and the nsp4 protein of rotavirus perturb ER calcium stores to facilitate their replication. It will be of interest to explore whether TF might have a similar role, given that the ion-channel domain of 6K, shared with TF, allows passage of calcium ions. Interestingly, treating SINV-infected cells with pharmacological antagonists of cellular calcium release resulted in a phenotype similar to that of the ΔTF virus described here (43).
The discovery of TF confounds the data reported to date regarding the role of 6K. Similar to what we have reported here with respect to TF, an SFV mutation consisting of a deletion of 6K decreases the efficiency of budding without affecting the infectivity of the particle (22). Others have also reported a budding deficiency when 6K chimeras were analyzed in cell culture (25, 39). Additionally, an SINV mutant with a Δ6K mutation which ablates production of both 6K and TF demonstrated a 3- to 4-log-unit reduction in virus particle release (Jose and Kuhn, unpublished), which is more severe than the attenuation of the ΔTF virus reported here, in which 6K synthesis was maintained. Thus, 6K and TF may have distinct but important roles in the alphavirus budding process. Indeed, multiple groups have suggested that 6K subtly influences the organization and stability of the spike. McInerney and colleagues (44) postulated that 6K is involved in the proper folding of the E2-E1 heterodimer, and Loewy et al. (22) demonstrated that an SFV Δ6K mutant particle was significantly more thermolabile that the wild-type particle. Here we show that TF likely does not have a role in spike morphology, and using an approach nearly identical to that of Loewy and colleagues (22), we found that the ΔTF mutant virion, in which 6K was still synthesized and incorporated into virions, was just as thermostable as the wild-type SINV virion (data not shown).
The data reported here validate the production of the alphavirus TF protein and show that TF, while not essential during alphavirus infection, is an important factor in alphavirus budding and/or assembly. Further characterization of the TF protein is required to delineate not only the role of TF but also that of 6K.
ACKNOWLEDGMENTS
We thank Anita Robinson for clerical assistance and Justin Meyers for assistance with flow cytometry experiments. We acknowledge the use of the Flow Cytometry and Cell Separation Facility of the Bindley Bioscience Center.
We acknowledge support from the NIH through NIGMS award GM56279 to R.J.K., biophysics training grant 5T32GM008296 to J.E.S., NINDS award NS038932 to D.E.G., and NCRR grant RR025761 to the Flow Cytometry and Cell Separation Facility.
Footnotes
Published ahead of print 29 May 2013
REFERENCES
- 1. Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE. (ed). 2007. Fields virology, 5th ed Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 2. Thiboutot MM, Kannan S, Kawalekar OU, Shedlock DJ, Khan AS, Sarangan G, Srikanth P, Weiner DB, Muthumani K. 2010. Chikungunya: a potentially emerging epidemic? PLoS Negl. Trop. Dis. 4:e623. 10.1371/journal.pntd.0000623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jose J, Snyder JE, Kuhn RJ. 2009. A structural and functional perspective of alphavirus replication and assembly. Future Microbiol. 4:837–856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Melancon P, Garoff H. 1987. Processing of the Semliki Forest virus structural polyprotein: role of the capsid protease. J. Virol. 61:1301–1309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Soderlund H. 1973. Kinetics of formation of the Semliki Forest virus nucleocapsid. Intervirology 1:354–361 [DOI] [PubMed] [Google Scholar]
- 6. Bonatti S, Migliaccio G, Blobel G, Walter P. 1984. Role of signal recognition particle in the membrane assembly of Sindbis viral glycoproteins. Eur. J. Biochem. 140:499–502 [DOI] [PubMed] [Google Scholar]
- 7. Zhang X, Fugere M, Day R, Kielian M. 2003. Furin processing and proteolytic activation of Semliki Forest virus. J. Virol. 77:2981–2989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tang J, Jose J, Chipman P, Zhang W, Kuhn RJ, Baker TS. 2011. Molecular links between the E2 envelope glycoprotein and nucleocapsid core in Sindbis virus. J. Mol. Biol. 414:442–459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Byrnes AP, Griffin DE. 1998. Binding of Sindbis virus to cell surface heparan sulfate. J. Virol. 72:7349–7356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Klimstra WB, Ryman KD, Johnston RE. 1998. Adaptation of Sindbis virus to BHK cells selects for use of heparan sulfate as an attachment receptor. J. Virol. 72:7357–7366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Smith TJ, Cheng RH, Olson NH, Peterson P, Chase E, Kuhn RJ, Baker TS. 1995. Putative receptor binding sites on alphaviruses as visualized by cryoelectron microscopy. Proc. Natl. Acad. Sci. U. S. A. 92:10648–10652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Garoff H, Frischauf AM, Simons K, Lehrach H, Delius H. 1980. Nucleotide sequence of cDNA coding for Semliki Forest virus membrane glycoproteins. Nature 288:236–241 [DOI] [PubMed] [Google Scholar]
- 13. Omar A, Koblet H. 1988. Semliki Forest virus particles containing only the E1 envelope glycoprotein are infectious and can induce cell-cell fusion. Virology 166:17–23 [DOI] [PubMed] [Google Scholar]
- 14. Wahlberg JM, Bron R, Wilschut J, Garoff H. 1992. Membrane fusion of Semliki Forest virus involves homotrimers of the fusion protein. J. Virol. 66:7309–7318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Li L, Jose J, Xiang Y, Kuhn RJ, Rossmann MG. 2010. Structural changes of envelope proteins during alphavirus fusion. Nature 468:705–708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Voss JE, Vaney MC, Duquerroy S, Vonrhein C, Girard-Blanc C, Crublet E, Thompson A, Bricogne G, Rey FA. 2010. Glycoprotein organization of Chikungunya virus particles revealed by X-ray crystallography. Nature 468:709–712 [DOI] [PubMed] [Google Scholar]
- 17. Liljestrom P, Garoff H. 1991. Internally located cleavable signal sequences direct the formation of Semliki Forest virus membrane proteins from a polyprotein precursor. J. Virol. 65:147–154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gaedigk-Nitschko K, Schlesinger MJ. 1990. The Sindbis virus 6K protein can be detected in virions and is acylated with fatty acids. Virology 175:274–281 [DOI] [PubMed] [Google Scholar]
- 19. Gaedigk-Nitschko K, Ding MX, Levy MA, Schlesinger MJ. 1990. Site-directed mutations in the Sindbis virus 6K protein reveal sites for fatty acylation and the underacylated protein affects virus release and virion structure. Virology 175:282–291 [DOI] [PubMed] [Google Scholar]
- 20. Gaedigk-Nitschko K, Schlesinger MJ. 1991. Site-directed mutations in Sindbis virus E2 glycoprotein's cytoplasmic domain and the 6K protein lead to similar defects in virus assembly and budding. Virology 183:206–214 [DOI] [PubMed] [Google Scholar]
- 21. Ivanova L, Lustig S, Schlesinger MJ. 1995. A pseudo-revertant of a Sindbis virus 6K protein mutant, which corrects for aberrant particle formation, contains two new mutations that map to the ectodomain of the E2 glycoprotein. Virology 206:1027–1034 [DOI] [PubMed] [Google Scholar]
- 22. Loewy A, Smyth J, von Bonsdorff CH, Liljestrom P, Schlesinger MJ. 1995. The 6-kilodalton membrane protein of Semliki Forest virus is involved in the budding process. J. Virol. 69:469–475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sanz MA, Carrasco L. 2001. Sindbis virus variant with a deletion in the 6K gene shows defects in glycoprotein processing and trafficking: lack of complementation by a wild-type 6K gene in trans. J. Virol. 75:7778–7784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Schlesinger MJ, London SD, Ryan C. 1993. An in-frame insertion into the Sindbis virus 6K gene leads to defective proteolytic processing of the virus glycoproteins, a trans-dominant negative inhibition of normal virus formation, and interference in virus shut off of host-cell protein synthesis. Virology 193:424–432 [DOI] [PubMed] [Google Scholar]
- 25. Yao JS, Strauss EG, Strauss JH. 1996. Interactions between PE2, E1, and 6K required for assembly of alphaviruses studied with chimeric viruses. J. Virol. 70:7910–7920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Strauss JH, Strauss EG. 1994. The alphaviruses: gene expression, replication, and evolution. Microbiol. Rev. 58:491–562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Firth AE, Chung BY, Fleeton MN, Atkins JF. 2008. Discovery of frameshifting in alphavirus 6K resolves a 20-year enigma. Virol. J. 5:108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Burdeinick-Kerr R, Govindarajan D, Griffin DE. 2009. Noncytolytic clearance of Sindbis virus infection from neurons by gamma interferon is dependent on Jak/STAT signaling. J. Virol. 83:3429–3435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Theilmann DA, Stewart S. 1992. Tandemly repeated sequence at the 3′ end of the IE-2 gene of the baculovirus Orgyia pseudotsugata multicapsid nuclear polyhedrosis virus is an enhancer element. Virology 187:97–106 [DOI] [PubMed] [Google Scholar]
- 30. Theilmann DA, Stewart S. 1992. Molecular analysis of the trans-activating IE-2 gene of Orgyia pseudotsugata multicapsid nuclear polyhedrosis virus. Virology 187:84–96 [DOI] [PubMed] [Google Scholar]
- 31. Antoine AF, Montpellier C, Cailliau K, Browaeys-Poly E, Vilain JP, Dubuisson J. 2007. The alphavirus 6K protein activates endogenous ionic conductances when expressed in Xenopus oocytes. J. Membr. Biol. 215:37–48 [DOI] [PubMed] [Google Scholar]
- 32. Melton JV, Ewart GD, Weir RC, Board PG, Lee E, Gage PW. 2002. Alphavirus 6K proteins form ion channels. J. Biol. Chem. 277:46923–46931 [DOI] [PubMed] [Google Scholar]
- 33. Sanz MA, Perez L, Carrasco L. 1994. Semliki Forest virus 6K protein modifies membrane permeability after inducible expression in Escherichia coli cells. J. Biol. Chem. 269:12106–12110 [PubMed] [Google Scholar]
- 34. Akahata W, Yang ZY, Andersen H, Sun S, Holdaway HA, Kong WP, Lewis MG, Higgs S, Rossmann MG, Rao S, Nabel GJ. 2010. A virus-like particle vaccine for epidemic Chikungunya virus protects nonhuman primates against infection. Nat. Med. 16:334–338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Levy-Mintz P, Kielian M. 1991. Mutagenesis of the putative fusion domain of the Semliki Forest virus spike protein. J. Virol. 65:4292–4300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chirico N, Vianelli A, Belshaw R. 2010. Why genes overlap in viruses. Proc. Biol. Sci. 277:3809–3817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sabath N, Wagner A, Karlin D. 2012. Evolution of viral proteins originated de novo by overprinting. Mol. Biol. Evol. 29:3767–3780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ivanova L, Le L, Schlesinger MJ. 1995. Characterization of revertants of a Sindbis virus 6K gene mutant that affects proteolytic processing and virus assembly. Virus Res. 39:165–179 [DOI] [PubMed] [Google Scholar]
- 39. Strauss EG, Lenches EM, Strauss JH. 2002. Molecular genetic evidence that the hydrophobic anchors of glycoproteins E2 and E1 interact during assembly of alphaviruses. J. Virol. 76:10188–10194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Soonsawad P, Xing L, Milla E, Espinoza JM, Kawano M, Marko M, Hsieh C, Furukawa H, Kawasaki M, Weerachatyanukul W, Srivastava R, Barnett SW, Srivastava IK, Cheng RH. 2010. Structural evidence of glycoprotein assembly in cellular membrane compartments prior to alphavirus budding. J. Virol. 84:11145–11151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Nasar F, Palacios G, Gorchakov RV, Guzman H, Da Rosa AP, Savji N, Popov VL, Sherman MB, Lipkin WI, Tesh RB, Weaver SC. 2012. Eilat virus, a unique alphavirus with host range restricted to insects by RNA replication. Proc. Natl. Acad. Sci. U. S. A. 109:14622–14627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zhou Y, Frey TK, Yang JJ. 2009. Viral calciomics: interplays between Ca2+ and virus. Cell Calcium 46:1–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Schlesinger MJ, Cahill D. 1989. Verapamil and chlorpromazine inhibit the budding of Sindbis and vesicular stomatitis viruses from infected chicken embryo fibroblasts. Virology 168:187–190 [DOI] [PubMed] [Google Scholar]
- 44. McInerney GM, Smit JM, Liljestrom P, Wilschut J. 2004. Semliki Forest virus produced in the absence of the 6K protein has an altered spike structure as revealed by decreased membrane fusion capacity. Virology 325:200–206 [DOI] [PubMed] [Google Scholar]