

Abstract
Previous studies have demonstrated that type I interferon (IFN-I) restricts West Nile virus (WNV) replication and pathogenesis in peripheral and central nervous system (CNS) tissues. However, the in vivo role of specific antiviral genes that are induced by IFN-I against WNV infection remains less well characterized. Here, using Ifit2−/− mice, we defined the antiviral function of the interferon-stimulated gene (ISG) Ifit2 in limiting infection and disease in vivo by a virulent North American strain of WNV. Compared to congenic wild-type controls, Ifit2−/− mice showed enhanced WNV infection in a tissue-restricted manner, with preferential replication in the CNS of animals lacking Ifit2. Virological analysis of cultured macrophages, dendritic cells, fibroblasts, cerebellar granule cell neurons, and cortical neurons revealed cell type-specific antiviral functions of Ifit2 against WNV. In comparison, small effects of Ifit2 were observed on the induction or magnitude of innate or adaptive immune responses. Our results suggest that Ifit2 restricts WNV infection and pathogenesis in different tissues in a cell type-specific manner.
INTRODUCTION
West Nile virus (WNV) is a mosquito-transmitted, neurotropic, positive-stranded RNA virus in the Flaviviridae family, which includes other pathogens of global health concern, such as dengue, yellow fever, and Japanese encephalitis viruses. WNV is maintained in an enzootic cycle between Culex species mosquitoes and several avian hosts but can cause disease in vertebrate animals, including horses and humans (1). Before 1999, the distribution of WNV was localized largely to Africa, central and southern Asia, the Middle East, southern Europe, and Oceania (2). Since its entry into North America in New York City in 1999, WNV has spread across the continental United States into Canada, Mexico, and parts of South America, likely due to bird migration and the presence of competent mosquito hosts (3, 4). While human transmission usually occurs through mosquito inoculation, other routes (blood transfusion, organ transplantation, and intrauterine transmission) have been reported (1). Although the majority (∼50 to 70%) of human infections are asymptomatic, WNV infection in the elderly or immunocompromised can cause severe neuroinvasive disease, including meningitis, encephalitis, and acute flaccid paralysis (5–7). In the United States alone between 1999 and 2012, ∼36,000 cases and ∼1,500 deaths have been confirmed (http://www.cdc.gov/ncidod/dvbid/westnile/surv&control.htm). Despite this, no approved vaccines or therapeutics are available to treat or prevent human WNV infection.
RNA intermediates of viral replication are recognized by cytosolic and endosomal pattern recognition receptors (PRR), such as RIG-I-like receptors (RLR) or Toll-like receptors (TLR), which signal specific transfection factors (e.g., Irf3 or Irf7) to induce type I interferon (IFN-I) expression and secretion. Autocrine and paracrine binding of IFN-I to its receptor (Ifnar) results in a signaling cascade that includes Stat1, Stat2, and Irf9 and results in the induction of many interferon-stimulated genes (ISGs), some of which inhibit virus infections. Although IFN-I responses control the cell and tissue tropism of several families of RNA and DNA viruses, the specific molecules that mediate this remain poorly characterized. While earlier studies defined Mx1, Pkr, and RNaseL as broad-spectrum antiviral ISGs against several families of RNA viruses, more recent investigations have identified other ISGs (e.g., Ifit1, Ifitm genes, Bst2, Apobec3g, and Adar) that restrict infection of a more limited range of viruses (8–10). Several individual ISGs have been suggested to have antiviral activity against WNV. C6orf150, Hpse, Nampt, Phf15, Ifitm2, Ifitm3, and Isg20 inhibit WNV infection in vitro (10, 11), and viperin (Rsad2), Pkr, and RNaseL restrict WNV pathogenesis in vivo and in vitro in a tissue- and cell type-specific manner (8, 12).
Ifit2 (also known as ISG54) is a member of the IFN-induced proteins with tetratricopeptide repeats (IFIT) family. IFIT proteins contain multiple tetratricopeptide repeats, which are domains implicated in the regulation of cell cycle, transcription, protein transport, and protein folding processes (13). Initial experiments indicated that Ifit2 could restrict infection of several viruses by binding to and inhibiting subunits of eIF3, a key protein involved in initiation of host translation (14). More recent structural and functional studies suggest that IFIT family members also inhibit infection of cytoplasmic RNA and DNA viruses by directly binding nonself RNA, including moieties displaying 5′-ppp RNA (15, 16) and possibly 5′ cap 0 structures (7mGpppN) lacking 2′-O methylation (17, 18). Ifit2, in particular, has been demonstrated to have antiviral activity in cell culture against WNV (19). While in vivo studies have established an antiviral effect of Ifit2 against vesicular stomatitis virus (VSV) infection (20), analogous experiments have not been conducted with WNV or any other flavivirus.
Beyond its antiviral function, Ifit2 has been implicated in the regulation of host cell immune responses. One study showed that human IFIT1 and IFIT2 bind to and inhibit MITA (also known as STING), which functions as a mitochondrial adaptor protein that recruits TANK-binding kinase 1 (TBK1) and IRF-3 to a complex with MAVS (also known as IPS-1, CARDIF, and VISA), resulting in the downstream induction of IFN-β expression in response to viral RNA or DNA (21). However, these data conflict with the results from mouse fibroblasts, macrophages, and dendritic cells in which silencing of IFIT2 expression did not alter IFN-I responses (22). Finally, expression of human IFIT2 also has been suggested to promote apoptosis via a mitochondrial pathway (23).
Here, we examined in detail the function of Ifit2 in vivo against WNV infection and pathogenesis using Ifit2−/− mice. Our data indicate that Ifit2 restricts WNV infection in a cell- and tissue-specific manner yet had small effects on the induction and magnitude of the innate and adaptive antiviral immune responses. Thus, Ifit2 is a key antiviral effector molecule that functions downstream of host pathogen recognition signaling to inhibit infection by WNV and possibly other viruses.
MATERIALS AND METHODS
Virus propagation and titration.
The WNV strain (3000.0259) was isolated in New York in 2000 and passaged once in C6/36 Aedes albopictus cells to generate an insect cell- derived stock virus as described previously (24). Mammalian cell-derived WNV was generated from an infectious cDNA clone of the New York 1999 strain, propagated once in C6/36 Aedes albopictus cells, and passaged a second time in Vero cells (25). WNV-NS5-E218A was propagated in BHK-21-15 cells as described previously (26). BHK-21-15 and Vero cells were used for titration of virus in tissue and cells by plaque or focus-forming assay as described previously (27, 28). Levels of virus in serum and lymph nodes were determined by quantitative reverse transcription (RT)-PCR using WNV-specific primers and probe as described previously (8).
Mouse experiments and tissue preparation.
C57BL/6 wild-type mice were obtained commercially (Jackson Laboratory), and the generation of congenic Ifit2−/− mice was described previously (20). Mice were bred in the animal facility of Washington University School of Medicine, and experiments were performed according to the guidelines and with approval of the Washington University Animal Studies Committee. For infection, 8- to 10-week-old age-matched mice were used. Mice were infected subcutaneously in the footpad (102 PFU in 50 μl) or intracranially (101 PFU in 10 μl) with virus diluted in Hanks balanced salt solution (HBSS) supplemented with 1% heat-inactivated fetal bovine serum (FBS). On specific days after infection, mice were perfused extensively with phosphate-buffered saline (PBS), tissues were harvested and weighed, and virus titers were determined by plaque assay on BHK-21-15 cells (27). For survival studies, 8-week-old mice were infected via footpad inoculation and monitored for 21 days.
IFN bioassay.
Levels of biologically active IFN-I in serum of mice infected with WNV were determined using an encephalomyocarditis virus L929 cell cytopathic effect bioassay as described previously (28, 29).
Cytokine analysis.
Mice were infected subcutaneously with 102 PFU of WNV in the footpad, and serum was collected at 2 days after infection. Cytokine levels in serum were measured using a Bio-Plex Pro 8-plex custom cytokine kit (Bio-Rad) and Bio-Plex 200 (Bio-Rad).
Measurement of WNV-specific antibodies.
The levels of WNV-specific IgM and IgG were measured using an enzyme-linked immunosorbent assay (ELISA) with purified WNV E protein as described previously (30). The titer of neutralizing antibody in serum was quantitated by a focus reduction neutralization assay in Vero cells as described previously (31).
Analysis of splenic CD4+ and CD8+ T cells.
Splenocytes were harvested from wild-type or Ifit2−/− mice on day 8 after WNV infection. Intracellular IFN-γ or tumor necrosis factor alpha (TNF-α) staining of CD8+ T cells was performed after ex vivo restimulation with an immunodominant Db-restricted NS4B peptide as described previously (32). Samples were processed, stained with antibodies against IFN-γ and TNF-α (BD Pharmingen), and analyzed using an LSR II flow cytometer (Becton, Dickinson) and FlowJo software (Treestar).
CNS leukocyte isolation and phenotyping.
Brains were harvested from wild-type and Ifit2−/− mice on day 8 after WNV infection, and CNS leukocytes were isolated after Percoll gradient centrifugation as described previously (33). Cells were stained for CD3, CD4, CD8, CD45, and CD11b with directly conjugated antibodies (BD Pharmingen). Intracellular staining for IFN-γ and TNF-α was performed after NS4B peptide restimulation as described before (32).
Primary cell isolation and infection.
Primary macrophages, dendritic cells, embryonic fibroblasts, cortical neurons, and cerebellar granule cell neurons were prepared from embryonic, neonatal, and adult wild-type and Ifit2−/− mice as detailed in prior studies (34, 35). For multistep growth curves, cells were infected at a low multiplicity of infection (MOI of 0.001) with or without IFN-β pretreatment (12 h, 10 IU/ml; PBL Interferon Source) or IFN-ζ pretreatment (4 h, 0.4 ng/ml; PBL Interferon Source). Virus was harvested from supernatants at specific times and titrated by focus-forming assay on Vero cells.
Statistical analysis.
Kaplan-Meier survival curves were analyzed by the log rank test. A two-tailed Student t test was used to determine statistically significant differences for in vitro experiments. The Mann-Whitney test was used to analyze differences in viral burden. All data were analyzed by using Prism software (GraphPad Software).
RESULTS
Ifit2 is required for control of WNV infection in vivo.
To determine whether a deficiency of Ifit2 resulted in increased susceptibility to lethal WNV infection, we infected wild-type and congenic Ifit2−/− mice via a subcutaneous route with 102 PFU of WNV, and survival was monitored (Fig. 1A). A higher percentage of Ifit2−/− mice succumbed to lethal WNV infection than wild-type mice (92% compared to 38%; P < 0.001). Moreover, we observed a significant decrease in the mean time to death (10.0 compared to 12.2 days; P < 0.01) in Ifit2−/− compared to wild-type mice. As IFIT genes are implicated in the recognition and control of flaviviruses, coronaviruses, and poxviruses lacking 2′-O methylation of the 5′ viral RNA cap structures (17, 18, 36), we also tested whether an absence of Ifit2 affected pathogenicity of WNV-NS5-E218A. Previous studies established that this WNV mutant virus lacked 2′-O methyltransferase activity and was attenuated in wild-type mice (26) yet more virulent in Ifit1−/− mice (17, 18). Unexpectedly, WNV-NS5-E218A remained attenuated in Ifit2−/− adult mice, as no illness or mortality was observed after subcutaneous or intracranial challenge (data not shown). Thus, while Ifit2 had a protective effect against a virulent strain of WNV in vivo, it was not required for restricting viruses lacking 2′-O methylation.
Fig 1.

Survival and viral burden analysis of wild-type and Ifit2−/− mice. (A) Eight-week-old age-matched wild-type (n = 28) and Ifit2−/− (n = 29) mice were infected via the subcutaneous route with 102 PFU of WNV and monitored for mortality for 21 days. Survival differences were judged by the log rank test and were statistically significant (P < 0.0001). (B to G) WNV tissue burden and spread in mice after subcutaneous infection. WNV levels in serum (B), draining lymph node (C), spleen (D), kidney (E), brain (F), and spinal cord (G) of wild-type and Ifit2−/− mice were measured by quantitative RT-PCR (qRT-PCR) (B and C) or by infectious plaque assay (D to G) of samples harvested at the indicated time points. Data are shown as WNV genome equivalents per microgram of 18S rRNA (r18S) or PFU per gram of tissue for 3 to 15 mice per time point. Solid lines represent the median viral titers, and dotted lines denote the limits of detection of the respective assays. Asterisks indicate values that are statistically significant (*, P < 0.05; ***, P < 0.0001).
Ifit2 restricts WNV replication in different tissues.
To begin to understand how Ifit2 restricts WNV infection in vivo, we measured viral burdens in serum, peripheral organs (spleen, kidney, and draining lymph node), and CNS tissues (brain and spinal cord) at different days after subcutaneous infection.
In the serum and draining lymph nodes, we observed a small (3- to 140-fold) yet statistically significant (P < 0.02) increase in WNV replication in Ifit2−/− mice, but only at day 4 after infection (Fig. 1B and C). However, we failed to detect differences in the kinetics or magnitude of WNV infection in the spleen or kidney (Fig. 1D and E) compared to that in mice deficient in IFN-I signaling (34, 37). Overall, compared to mice lacking other innate immune signaling molecules (e.g., Mavs, Irf3, Irf7, or Ifnar) (37–40), a loss of Ifit2 expression had a limited effect on WNV infection in peripheral organs.
We next assessed the effects of an absence of Ifit2 on WNV infection in CNS tissues (Fig. 1F and G). At day 4 after subcutaneous infection, we observed higher levels (P = 0.02) of infection in the brains of Ifit2−/− mice than in wild-type mice. At day 8 after infection, there was a trend toward increased viral burden in the brain and spinal cord of Ifit2−/− mice, although this failed to attain statistical significance.
Given that a deficiency of Ifit2 resulted in a trend toward increased viral burden in the CNS at day 8 after infection, we hypothesized that Ifit2 might restrict infection in some but not all regions, which could obscure small differences. To test this, we infected wild-type and Ifit2−/− mice and at day 9 harvested tissues from different regions of the CNS and analyzed their viral burden. Notably, viral infection in Ifit2−/− mice was higher in the olfactory bulb (105.0 versus 103.9 PFU/g; P = 0.004), brain stem (106.4 versus 104.9 PFU/g; P = 0.004), and cerebellum (105.3 versus 104.6 PFU/g; P = 0.009) than that in wild-type mice (Fig. 2A, C, and D). In comparison, no difference in viral burden was observed in the cerebral cortex or the spinal cord (P > 0.1; Fig. 2B and E). Overall, these data indicate that Ifit2 has a role in restricting WNV infection in specific regions of the brain.
Fig 2.

WNV replication in regions of the CNS of Ifit2−/− mice after subcutaneous infection. Wild-type and Ifit2−/− mice were infected with 102 PFU of WNV via the subcutaneous route. Different regions of the brain were harvested at day 9: olfactory bulb (A), cerebral cortex (B), brain stem (C), cerebellum (D), and spinal cord (E). Tissue homogenates were analyzed for viral burden by plaque assay. Data are shown as PFU per gram of tissue from 11 to 14 mice per time point. Solid lines represent the median viral titers, and dotted lines indicate the limits of detection of the respective assays. Asterisks indicate values that are statistically significant (**, P < 0.001).
Cytokine levels in circulation of Ifit2−/− mice.
Because Ifit2 has been suggested to inhibit STING, a mitochondrial adaptor protein that recruits TBK1 and IRF-3 to a complex with MAVS to induce IFN-β expression (21), we assessed whether a deficiency of Ifit2 affected IFN-I production. Wild-type and Ifit2−/− mice were infected with WNV, and the level of biologically active IFN-I in serum was monitored in a validated encephalomyocarditis virus (EMCV) L929 cell bioassay (29). Notably, in Ifit2−/− mice we observed IFN-I activity levels that were moderately higher (2- to 15-fold; P < 0.05) than those in the wild type at every time point tested (Fig. 3). This may be due to increased viral replication in cells of Ifit2−/− mice or to the proposed loss of negative regulatory effects on cytokine production (41). Because of the elevated IFN-I levels and prior reports suggesting that Ifit2 might regulate inflammatory responses (41), we also measured cytokine levels in serum from wild-type and Ifit2−/− mice at day 2 after infection. No differences in interleukin 1β (IL-1β), IL-6, IL-10, IL-12 (p40 subunit), IFN-γ, TNF-α, CXCL1, and CCL4 cytokines and chemokines were observed (data not shown).
Fig 3.
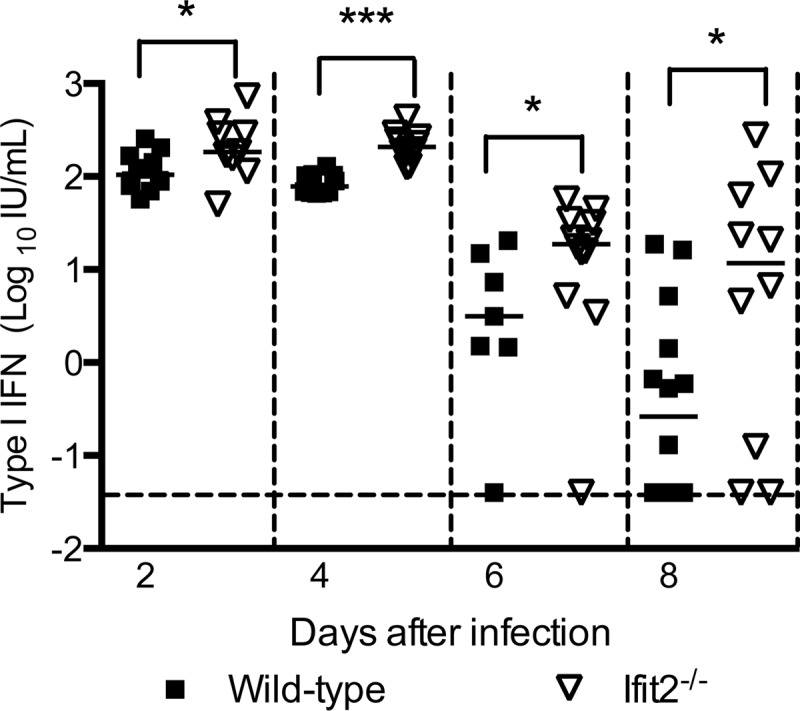
Cytokine levels in serum of wild-type and Ifit2−/− mice after infection with WNV. Wild-type and Ifit2−/− mice were inoculated subcutaneously with 102 PFU of WNV, and serum was collected on days 2 to 8 after infection. IFN-I activity was determined in a bioassay. Solid lines represent the median concentrations, and dotted lines indicate the limits of detection of the respective assays. Asterisks indicate differences that are statistically significant by the Mann-Whitney test (*, P < 0.05; ***, P < 0.0001).
Effect of Ifit2 on adaptive immune responses to WNV infection.
We next investigated whether an absence of Ifit2 influenced the development of an effective adaptive immune response during infection, as prior studies suggest that depressed T and B cell responses can result in enhanced WNV replication in the CNS (27, 42–44). Initially, we evaluated T cell responses in the spleen. Equivalent percentages of CD4+ and CD8+ T cells were isolated from spleens of wild-type and Ifit2−/− mice (Fig. 4A and B). Moreover, we observed no difference in the percentage of WNV-specific CD8+ T cells that expressed IFN-γ, TNF-α, or IFN-γ and TNF-α (double positive) after ex vivo NS4B peptide restimulation in wild-type and Ifit2−/− mice (Fig. 4C to E).
Fig 4.

Peripheral T cell responses after WNV infection in Ifit2−/− mice. Wild-type and Ifit2−/− mice were inoculated subcutaneously with 102 PFU of WNV, and spleens were harvested at day 8 after infection. (A and B) Percentage of CD3+ CD4+ (A) or CD3+ CD8+ (B) cells in the spleen after being gated on live cells; (C and D) data are shown as the percentage of CD3+ CD8+ T cells that expressed intracellular IFN-γ or TNF-α after Db-restricted NS4B peptide restimulation ex vivo. The differences were not statistically significant, and the data were pooled from two independent experiments with a total of 7 or 8 mice. (E) Flow cytometry dot plots showing intracellular IFN-γ and TNF-α staining after NS4B peptide restimulation and gating on CD8+ T cells.
Although peripheral T cell priming was equivalent, we assessed whether leukocyte migration into the CNS might be altered in Ifit2−/− mice, as infiltrating leukocytes into the brain clears WNV infection (43–45). In the brain, at day 8 after infection, we observed similar percentages and numbers of CD8+ T cells (P > 0.6) (Fig. 5A). Antigen specificity was inferred after intracellular staining of IFN-γ or TNF-α in brain CD8+ T cells that were restimulated with the NS4B peptide. No statistical differences were observed in the percentage or number of antigen-specific IFN-γ+ CD8+ or TNF-α+ CD8+ T cells in the brain (P > 0.2) (Fig. 5B and C). We also observed similar numbers of CD11bhigh CD45high macrophages (P = 0.4) and CD11bhigh CD45low microglia (P > 0.7) (Fig. 5D and E).
Fig 5.

Leukocyte accumulation in the CNS of Ifit2−/− mice after WNV infection. Wild-type and Ifit2−/− mice were infected with 102 PFU of WNV by a subcutaneous route, and 8 days later brains were harvested and leukocytes were isolated by Percoll gradient centrifugation. (A) Total percentages and numbers of CD3+ CD8+ T cells; (B) total percentages and numbers of CD3+ CD8+ T cells that expressed intracellular IFN-γ or TNF-α after restimulation with a Db-restricted NS4B peptide; (C) flow cytometry dot plots showing intracellular IFN-γ and TNF-α cells after peptide restimulation and gating on CD8+ T cells; (D) total percentages and numbers of activated microglia (CD11bhigh CD45low) and macrophages (CD11bhigh CD45high). The differences were not statistically significant, and the data were pooled from two independent experiments with a total of 7 or 8 mice. (E) Flow cytometry dot plots indicating gating strategy for defining activated microglia and macrophages.
To assess the effect of Ifit2 on WNV-specific humoral responses, we analyzed serum from Ifit2−/− and wild-type mice for binding to the WNV E protein. At days 6 and 8 after infection, no appreciable differences in levels of anti-WNV E protein IgM were observed between Ifit2−/− and wild-type mice (P > 0.08) (Fig. 6A). Similar levels of WNV-specific IgG also were detected in Ifit2−/− and wild-type mice at day 8 after infection (P > 0.1) (Fig. 6B). Consistent with this, no defect in the levels of neutralizing antibodies was observed in Ifit2−/− mice (P > 0.3) (Fig. 6C). Together, these results suggest that the virological phenotype observed in Ifit2−/− mice was not due to major defects in T or B cell function.
Fig 6.
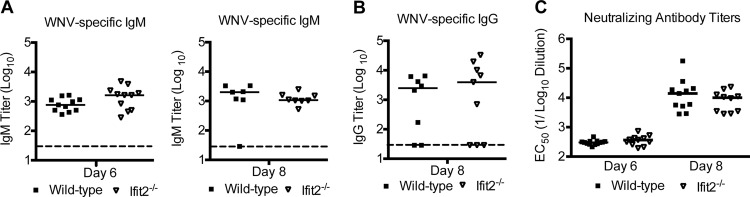
WNV-specific IgM and IgG responses in wild-type and Ifit2−/− mice. Wild-type and Ifit2−/− mice were infected with WNV via the subcutaneous route, and serum was collected at the indicated time points. The development of WNV-specific IgM (A) or IgG (B) was determined by ELISA using purified WNV E protein. (C) Sera were harvested from animals at the indicated times postinfection and tested for neutralization activity using a focus reduction assay (see Materials and Methods). Data represent the effective reciprocal dilution of sera that produced 50% neutralization of WNV infection (EC50). Data are from 7 to 11 mice per group. Differences were not statistically significant (P > 0.05) as judged by the Mann-Whitney test.
Ifit2 controls WNV replication in subsets of primary cells.
Given the variation in tissue-specific virological phenotypes in Ifit2−/− mice, we speculated that there might be cell-specific functions of Ifit2 in restricting WNV replication. To assess this, we compared multistep growth kinetics in several different wild-type and Ifit2−/− primary cells, including macrophages, myeloid dendritic cells, embryonic fibroblasts (MEFs), cortical neurons, and cerebellar granule cell neurons after WNV infection at a low MOI (0.001 to 0.01). In macrophages, cortical neurons, and MEFs, we observed no difference in WNV infection between wild-type and Ifit2−/− cells (Fig. 7A to C). In comparison, small (2- to 3-fold) yet statistically significant (P < 0.05) increases in WNV infection were observed at the 48- and 72-h time points in Ifit2−/− cerebellar granule cells and at the 72-h time point in Ifit2−/− dendritic cells (Fig. 7D and E). The antiviral effect of Ifit2 was magnified when cells were primed with different subtypes of IFN-I. The level of WNV replication was higher in IFN-β-pretreated Ifit2−/− MEFs (2- to 5-fold; P < 0.05) and macrophages (20-fold; P < 0.01) than in wild-type cells (Fig. 7F and G). Similar results were observed after treatment with another mouse IFN-I, IFN-ζ (also known as limitin) (Fig. 7H). Overall, our results establish a cell type-specific effect of Ifit2 on WNV replication, which can be magnified in the context of an IFN-I response.
Fig 7.

WNV replication in wild-type and Ifit2−/− primary MEFs, myeloid cells, and neurons. Primary cells from wild-type and Ifit2−/− mice were infected with WNV, and viral replication from 6 to 72 h was measured by focus-forming assay. Shown are data from bone marrow-derived macrophages (MOI, 0.01) (A), cortical neurons (MOI, 0.001) (B), MEFs (MOI, 0.01) (C), cerebellar granule cell neurons (MOI, 0.001) (D), and bone marrow-derived dendritic cells (MOI, 0.001) (E). (F to H) MEFs (F) and bone marrow-derived macrophages (G and H) were pretreated with 10 IU/ml of IFN-β (F and G) or 0.4 ng/ml of IFN-ζ (H) for 4 h and infected with WNV, and viral replication was measured. Asterisks indicate differences that are statistically significant by the Mann-Whitney test (*, P < 0.05; **, P < 0.001; ***, P < 0.0001).
Ifit2 has a direct role in restricting WNV replication in the CNS.
Previous studies suggested a direct role of Ifit2 in controlling virus replication in the CNS (20, 46). Because our survival and virological experiments through a peripheral route suggested that Ifit2 contributed to the control of WNV infection in selected CNS tissues, we directly introduced virus into the brain after intracranial injection and monitored spread to the contralateral brain regions (olfactory bulb, cerebral cortex, brain stem, spinal cord, cerebellum) on days 2, 4, and 6 after inoculation (Fig. 8A to E). Threshold levels of infection were detected at day 2 after infection in wild-type and Ifit2−/− mice. By day 4, viral titers of Ifit2−/− mice were substantially higher in the olfactory bulb (105.0 versus 103.1 PFU/g; P = 0.002), cerebral cortex (106.9 versus 103.5 PFU/g; P = 0.0001), brain stem (105.6 versus 102.9 PFU/g; P < 0.0001), cerebellum (105.5 versus 102.8 PFU/g; P = 0.0002), and spinal cord (105.1 versus 102.6 PFU/g; P < 0.0001) than in wild-type mice. Nonetheless, by day 6, viral titers of wild-type mice became equivalent to those observed in Ifit2−/− mice. These results suggest that the antiviral action of Ifit2 restricts WNV spread in the CNS, especially during the early stages of virus spread.
Fig 8.

WNV replication in regions of the CNS of Ifit2−/− mice after intracranial infection. Wild-type and Ifit2−/− mice were infected with 101 PFU of WNV in 10 μl of HBSS supplemented with 1% FBS via the intracranial route. Different regions of the contralateral brain were harvested at the indicated time points: olfactory bulb (A), cerebral cortex (B), brain stem (C), cerebellum (D), and spinal cord (E). Tissue homogenates were analyzed for viral burden by plaque assay. Data are shown as PFU per gram of tissue for 7 to 18 mice per time point. Solid lines represent the median viral titers, and dotted lines indicate the limits of detection of the respective assays. Asterisks indicate values that are statistically significant (**, P < 0.001; ***, P < 0.0001).
DISCUSSION
Subsets of ISGs are responsible for the antiviral effector functions of IFN-I. While several hundred ISGs have been identified by transcriptional profiling and RNAseq studies (47), relatively few have been shown directly to inhibit viral infections (48). IFIT genes encode a family of proteins that are induced after IFN treatment, viral infection, or pathogen-associated molecular pattern (PAMP) recognition (49, 50). Four family members have been characterized in humans (IFIT1 [ISG56], IFIT2 [ISG54], IFIT3 [ISG60], and IFIT5 [ISG58]), and three members are expressed in mice (Ifit1 [Isg56], Ifit2 [Isg54], and Ifit3 [Isg49]). We initiated these pathogenesis studies because preliminary data suggested that Ifit2 had antiviral activity against attenuated WNV strains in cell culture (17, 19). Indeed, our analysis here shows a key role for Ifit2-dependent restriction of WNV infection in vivo. Ifit2−/− mice showed enhanced susceptibility to virulent WNV infection, and this was associated with elevated levels of infection in subsets of CNS tissues. A direct effect of Ifit2 on neuronal cell infection and spread in vivo also was suggested, as deficient mice exhibited a higher viral burden in CNS tissues following intracranial inoculation. Finally, a multistep viral growth analysis of primary cells confirmed a cell type-specific antiviral function of Ifit2, as Ifit2−/− myeloid cells, embryonic fibroblasts, and cerebellar granule cell neurons but not cortical neurons showed enhanced infectivity. This phenotype was magnified in the context of priming with IFN-I, which is consistent with the hypothesis that IFN-induced Ifit2 expression in some cell types restricts WNV replication.
A previous study also suggested an antiviral function of Ifit2 in a subset of CNS tissues against VSV in vivo (20). In that report, Ifit2 inhibited VSV replication in the cortex, brain stem, midbrain, and cerebellum after intranasal infection. Analogously, in our study, Ifit2 limited WNV infection in multiple regions of the brain after intracranial infection. These results suggest that Ifit2 contributes to protection against viral pathogenesis in the brain, which is consistent with the observation that Ifit2 is induced in many regions of the brain upon viral infection (51). Nonetheless, in our subcutaneous infection model, some regions of the CNS (cerebral cortex and spinal cord) failed to show enhanced infection in Ifit2−/− mice, although this could reflect the time point that was assessed in these experiments. Moreover, Ifit2−/− mice did not show increased viral infection in visceral organs such as the spleen and kidney, which could reflect the restricted expression pattern of Ifit2 in those tissues (46). The antiviral role of Ifit2 has been described in the context of other virus infections. Our data showing an antiviral role of Ifit2 are consistent with recent data with influenza A virus, although the mechanisms of action may differ. Ifit2 was suggested to inhibit influenza A virus by forming a complex with Ifit1 and Ifit3 to recognize the 5′-ppp moiety on genomic viral RNA (22). In comparison, Ifit2 expression failed to show any antiviral effects on alphavirus infection in vitro (52).
The identification of Ifit2 as an ISG that restricts WNV infection in vivo adds to a small number of studies suggesting that the deletion of individual ISGs can impact WNV pathogenesis. While a redundancy of antiviral ISGs against a given virus might preclude phenotypes when a single antiviral ISG is targeted for deletion, recent studies with WNV suggest otherwise. Using genetically deficient mice, several ISGs, including the PKR (protein kinase R, also known as Eif2ak2), RNase L, and viperin (Rsad2) genes, were shown to inhibit WNV in vivo. Activated PKR phosphorylates the α subunit of eukaryotic translation initiation factor 2 (eIF2-α), resulting in a block of protein synthesis (26). RNase L is an endoribonuclease, and, once activated, it cleaves viral RNA and mRNA, leading to a decrease in protein synthesis and viral replication (23). Pkr−/− × RnaseL−/− mice were vulnerable to subcutaneous WNV infection, with increased mortality and viral replication. Viperin (Rsad2) is an endoplasmic reticulum (ER)-associated protein that has inhibitory activity against several viruses, possibly because it inhibits bulk protein secretion, lipid raft formation, and virus budding and localizes to ER-derived lipid droplets, which are required for efficient replication by some RNA viruses (53). viperin−/− mice infected with WNV also showed increased lethality and/or enhanced viral replication in CNS tissues (12).
How does Ifit2 inhibit WNV infection? Although more detailed mechanism-of-action studies are required, Ifit2 has been proposed to inhibit viral translation through its binding to and inhibition of subunits of eIF3 (14). eIF3 is a multisubunit complex that functions during the translation initiation step, including assembly of the eIF2-GTP-Met-tRNA ternary complex, formation of the 43S preinitiation complex, and mRNA recruitment to the 43S preinitiation complex (54). Mouse Ifit2 and human IFIT2 can block the formation of the 48S complex by binding to eIF3c, and human IFIT2 can block eIF3 binding to the ternary complex by interacting with eIF3e (14). Another IFIT family member, human IFIT1, has been suggested to inhibit influenza and Rift Valley fever virus replication by binding to the 5′-ppp moiety and sequestering RNA from replication (22), and this phenotype reportedly required a complex with IFIT2 and IFIT3. While capped WNV genomic RNA lacks a 5′-ppp end, the negative-strand intermediate may have this moiety exposed (55). Thus, it is possible that Ifit2 inhibits WNV replication by recognizing a 5′-ppp motif on the negative-strand RNA in a complex with Ifit1. Against the need for a complex of Ifit proteins for inhibiting wild-type WNV infection, no increased replication of lethality was observed in Ifit1−/− mice (18). In comparison, recent studies showed that flavivirus mutants lacking 2′-O methyltransferase activity (e.g., WNV-NS5-E218A) were attenuated in wild-type mice (26, 56, 57) yet more virulent in Ifit1−/− mice (17, 18), suggesting that Ifit1 recognizes RNA cap structures lacking 2′-O methylation to inhibit viral replication. Although our prior studies suggested that ectopic expression of Ifit2 might inhibit such viruses, WNV-NS5-E218A remained attenuated in Ifit2−/− mice; this demonstrates that Ifit2 is not absolutely required for inhibition of WNV strains lacking 2′-O methylation.
Our experiments also revealed no appreciable difference in B and T cell responses between Ifit2−/− and wild-type mice during WNV infection. This suggests that the absence of Ifit2 does not influence the development of effective adaptive immunity during viral infection.
In summary, our results show that Ifit2 contributes to the antiviral response against WNV in vivo, as its targeted deletion was associated with increased lethality and selectively enhanced replication in specific tissues, without an appreciable negative effect of the innate or adaptive T cell or antibody responses. Based on the tissue-specific antiviral effects of Ifit2, we speculate that different cell types might differentially require Ifit2 to restrict WNV replication. The importance of Ifit2 as an antiviral molecule in a given cell type may reflect the qualitative and quantitative ISG signature that is induced (10, 58). A more detailed biochemical and cellular analysis is planned to reveal mechanistically how Ifit2 restricts infectivity of WNV in different subsets of cells.
ACKNOWLEDGMENTS
The authors would like to acknowledge the National Institutes of Health for support of this work: grants U19 AI083019, R01 AI104972, R01 AI104002 (M.S.D.), and CA068782 (G.C.S.).
Footnotes
Published ahead of print 5 June 2013
REFERENCES
- 1. Hayes EB, Komar N, Nasci RS, Montgomery SP, O'Leary DR, Campbell GL. 2005. Epidemiology and transmission dynamics of West Nile virus disease. Emerg. Infect. Dis. 11:1167–1173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kleiboeker SB. 2011. West Nile virus, p 761–768 InNriagu JO. (ed), Encyclopedia of environmental health. Elsevier, Burlington, MA [Google Scholar]
- 3. Artsob H, Gubler DJ, Enria DA, Morales MA, Pupo M, Bunning ML, Dudley JP. 2009. West Nile virus in the New World: trends in the spread and proliferation of West Nile virus in the Western Hemisphere. Zoonoses Public Health 56:357–369 [DOI] [PubMed] [Google Scholar]
- 4. Guerrero-Sanchez S, Cuevas-Romero S, Nemeth NM, Trujillo-Olivera MTJ, Worwa G, Dupuis A, Brault AC, Kramer LD, Komar N, Estrada-Franco JG. 2011. West Nile virus infection of birds, Mexico. Emerg. Infect. Dis. 17:2245–2252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Omalu BI, Shakir AA, Wang G, Lipkin WI, Wiley CA. 2003. Fatal fulminant pan-meningo-polioencephalitis due to West Nile virus. Brain Pathol. 13:465–472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Armah HB, Wang G, Omalu BI, Tesh RB, Gyure KA, Chute DJ, Smith RD, Dulai P, Vinters HV, Kleinschmidt-DeMasters BK, Wiley CA. 2007. Systemic distribution of West Nile virus infection: postmortem immunohistochemical study of six cases. Brain Pathol. 17:354–362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Busch MP, Wright DJ, Custer B, Tobler LH, Stramer SL, Kleinman SH, Prince HE, Bianco C, Foster G, Petersen LR, Nemo G, Glynn SA. 2006. West Nile virus infections projected from blood donor screening data, United States, 2003. Emerg. Infect. Dis. 12:395–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Samuel MA, Whitby K, Keller BC, Marri A, Barchet W, Williams BRG, Silverman RH, Gale M, Jr, Diamond MS. 2006. PKR and RNase L contribute to protection against lethal West Nile virus infection by controlling early viral spread in the periphery and replication in neurons. J. Virol. 80:7009–7019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Scherbik SV, Paranjape JM, Stockman BM, Silverman RH, Brinton MA. 2006. RNase L plays a role in the antiviral response to West Nile virus. J. Virol. 80:2987–2999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Schoggins JW, Wilson SJ, Panis M, Murphy MY, Jones CT, Bieniasz P, Rice CM. 2011. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 472:481–485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jiang D, Weidner JM, Qing M, Pan Guo X-BH, Xu C, Zhang X, Birk A, Chang J, Shi P-Y, Block TM, Guo J-T. 2010. Identification of five interferon-induced cellular proteins that inhibit West Nile virus and dengue virus infections. J. Virol. 84:8332–8341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Szretter KJ, Brien JD, Thackray LB, Virgin HW, Cresswell P, Diamond MS. 2011. The interferon-inducible gene viperin restricts West Nile virus pathogenesis. J. Virol. 85:11557–11566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. D'Andrea LD, Regan L. 2003. TPR proteins: the versatile helix. Trends Biochem. Sci. 28:655–662 [DOI] [PubMed] [Google Scholar]
- 14. Terenzi F, Hui DJ, Merrick WC, Sen GC. 2006. Distinct induction patterns and functions of two closely related interferon-inducible human genes, ISG54 and ISG56. J. Biol. Chem. 281:34064–34071 [DOI] [PubMed] [Google Scholar]
- 15. Abbas YM, Pichlmair A, Górna MW, Superti-Furga G, Nagar B. 2013. Structural basis for viral 5′-PPP-RNA recognition by human IFIT proteins. Nature 494:60–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Katibah GE, Lee HJ, Huizar JP, Vogan JM, Alber T, Collins K. 2013. tRNA binding, structure, and localization of the human interferon-induced protein IFIT5. Mol. Cell 49:743–750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Daffis S, Szretter KJ, Schriewer J, Li J, Youn S, Errett J, Lin Schneller T-YS, Zust R, Dong H, Thiel V, Sen GC, Fensterl V, Klimstra WB, Pierson TC, Buller RM, Gale M, Jr, Shi P-Y, Diamond MS. 2010. 2′-O methylation of the viral mRNA cap evades host restriction by IFIT family members. Nature 468:452–456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Szretter KJ, Daniels BP, Cho H, Gainey MD, Yokoyama WM, Gale M, Jr, Virgin HW, Klein RS, Sen GC, Diamond MS. 2012. 2′-O methylation of the viral mRNA cap by West Nile virus evades ifit1-dependent and -independent mechanisms of host restriction in vivo. PLoS Pathog. 8:e1002698. 10.1371/journal.ppat.1002698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Perwitasari O, Cho H, Diamond MS, Gale M., Jr 2011. Inhibitor of κB kinase epsilon (IKK(epsilon)), STAT1, and IFIT2 proteins define novel innate immune effector pathway against West Nile virus infection. J. Biol. Chem. 286:44412–44423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fensterl V, Wetzel JL, Ramachandran S, Ogino T, Stohlman SA, Bergmann CC, Diamond MS, Virgin HW, Sen GC. 2012. Interferon-induced Ifit2/ISG54 protects mice from lethal VSV neuropathogenesis. PLoS Pathog. 8:e1002712. 10.1371/journal.ppat.1002712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Li Y, Li C, Xue P, Zhong B, Mao AP, Ran Y, Chen H, Wang Y-Y, Yang F, Shu H-B. 2009. ISG56 is a negative-feedback regulator of virus-triggered signaling and cellular antiviral response. Proc. Natl. Acad. Sci. U. S. A. 106:7945–7950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Pichlmair A, Lassnig C, Eberle C-A, Górna MW, Baumann CL, Burkard TR, Bürckstümmer T, Stefanovic A, Krieger S, Bennett KL, Rülicke T, Weber F, Colinge J, Müller M, Superti-Furga G. 2011. IFIT1 is an antiviral protein that recognizes 5′-triphosphate RNA. Nat. Immunol. 12:624–630 [DOI] [PubMed] [Google Scholar]
- 23. Stawowczyk M, Van Scoy S, Kumar KP, Reich NC. 2011. The interferon stimulated gene 54 promotes apoptosis. J. Biol. Chem. 286:7257–7266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ebel GD, Carricaburu J, Young D, Bernard KA, Kramer LD. 2004. Genetic and phenotypic variation of West Nile virus in New York, 2000–2003. Am. J. Trop. Med. Hyg. 71:493–500 [PubMed] [Google Scholar]
- 25. Vogt MR, Moesker B, Goudsmit J, Jongeneelen M, Austin SK, Oliphant T, Nelson S, Pierson TC, Wilschut J, Throsby M, Diamond MS. 2009. Human monoclonal antibodies against West Nile virus induced by natural infection neutralize at a postattachment step. J. Virol. 83:6494–6507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhou Y, Ray D, Zhao Y, Dong H, Ren S, Li Z, Guo Y, Bernard KA, Shi P-Y, Li H. 2007. Structure and function of flavivirus NS5 methyltransferase. J. Virol. 81:3891–3903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Diamond MS, Shrestha B, Marri A, Mahan D, Engle M. 2003. B cells and antibody play critical roles in the immediate defense of disseminated infection by West Nile encephalitis virus. J. Virol. 77:2578–2586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lazear HM, Lancaster A, Wilkins C, Suthar MS, Huang A, Vick SC, Clepper L, Thackray L, Brassil MM, Virgin HW, Nikolich-Zugich J, Moses AV, Gale M, Jr, Früh K, Diamond MS. 2013. IRF-3, IRF-5, and IRF-7 coordinately regulate the type I IFN response in myeloid dendritic cells downstream of MAVS signaling. PLoS Pathog. 9:e1003118. 10.1371/journal.ppat.1003118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Austin BA, James C, Silverman RH, Carr DJJ. 2005. Critical role for the oligoadenylate synthetase/RNase L pathway in response to IFN-beta during acute ocular herpes simplex virus type 1 infection. J. Immunol. 175:1100–1106 [DOI] [PubMed] [Google Scholar]
- 30. Mehlhop E, Diamond MS. 2006. Protective immune responses against West Nile virus are primed by distinct complement activation pathways. J. Exp. Med. 203:1371–1381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Fuchs A, Pinto AK, Schwaeble WJ, Diamond MS. 2011. The lectin pathway of complement activation contributes to protection from West Nile virus infection. Virology 412:101–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Purtha WE, Myers N, Mitaksov V, Sitati E, Connolly J, Fremont DH, Hansen TH, Diamond MS. 2007. Antigen-specific cytotoxic T lymphocytes protect against lethal West Nile virus encephalitis. Eur. J. Immunol. 37:1845–1854 [DOI] [PubMed] [Google Scholar]
- 33. Szretter KJ, Samuel MA, Gilfillan S, Fuchs A, Colonna M, Diamond MS. 2009. The immune adaptor molecule SARM modulates tumor necrosis factor alpha production and microglia activation in the brainstem and restricts West Nile virus pathogenesis. J. Virol. 83:9329–9338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lazear HM, Pinto AK, Vogt MR, Gale M, Jr, Diamond MS. 2011. Beta interferon controls West Nile virus infection and pathogenesis in mice. J. Virol. 85:7186–7194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Klein RS, Lin E, Zhang B, Luster AD, Tollett J, Samuel MA, Engle M, Diamond MS. 2005. Neuronal CXCL10 directs CD8+ T-cell recruitment and control of West Nile virus encephalitis. J. Virol. 79:11457–11466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Züst R, Cervantes-Barragan L, Habjan M, Maier R, Neuman BW, Ziebuhr J, Szretter KJ, Baker SC, Barchet W, Diamond MS, Siddell SG, Ludewig B, Thiel V. 2011. Ribose 2′-O-methylation provides a molecular signature for the distinction of self and non-self mRNA dependent on the RNA sensor Mda5. Nat. Immunol. 12:137–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Samuel MA, Diamond MS. 2005. Alpha/beta interferon protects against lethal West Nile virus infection by restricting cellular tropism and enhancing neuronal survival. J. Virol. 79:13350–13361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Daffis S, Samuel MA, Keller BC, Gale M, Jr, Diamond MS. 2007. Cell-specific IRF-3 responses protect against West Nile virus infection by interferon-dependent and -independent mechanisms. PLoS Pathog. 3:e106. 10.1371/journal.ppat.0030106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Daffis S, Samuel MA, Suthar MS, Keller BC, Gale M, Jr, Diamond MS. 2008. Interferon regulatory factor IRF-7 induces the antiviral alpha interferon response and protects against lethal West Nile virus infection. J. Virol. 82:8465–8475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Suthar MS, Ma DY, Thomas S, Lund JM, Zhang N, Daffis S, Rudensky AY, Bevan MJ, Clark EA, Kaja M-K, Diamond MS, Gale M., Jr 2010. IPS-1 is essential for the control of West Nile virus infection and immunity. PLoS Pathog. 6:e1000757. 10.1371/journal.ppat.1000757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Berchtold S, Manncke B, Klenk J, Geisel J, Autenrieth IB, Bohn E. 2008. Forced IFIT-2 expression represses LPS induced TNF-alpha expression at posttranscriptional levels. BMC Immunol. 9:75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Brien JD, Uhrlaub JL, Nikolich-Zugich J. 2007. Protective capacity and epitope specificity of CD8(+) T cells responding to lethal West Nile virus infection. Eur. J. Immunol. 37:1855–1863 [DOI] [PubMed] [Google Scholar]
- 43. Shrestha B, Diamond MS. 2004. Role of CD8+ T cells in control of West Nile virus infection. J. Virol. 78:8312–8321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wang Y, Lobigs M, Lee E, Müllbacher A. 2003. CD8+ T cells mediate recovery and immunopathology in West Nile virus encephalitis. J. Virol. 77:13323–13334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Shrestha B, Samuel MA, Diamond MS. 2006. CD8+ T cells require perforin to clear West Nile virus from infected neurons. J. Virol. 80:119–129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Terenzi F, White C, Pal S, Williams BRG, Sen GC. 2007. Tissue-specific and inducer-specific differential induction of ISG56 and ISG54 in mice. J. Virol. 81:8656–8665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. de Veer MJ, Holko M, Frevel M, Walker E, Der S, Paranjape JM, Silverman RH, Williams BRG. 2001. Functional classification of interferon-stimulated genes identified using microarrays. J. Leukoc. Biol. 69:912–920 [PubMed] [Google Scholar]
- 48. Schoggins JW, Rice CM. 2011. Interferon-stimulated genes and their antiviral effector functions. Curr. Opin. Virol. 1:519–525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Diamond MS, Farzan M. 2013. The broad-spectrum antiviral functions of IFIT and IFITM proteins. Nat. Rev. Immunol. 13:46–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sen GC, Sarkar SN. 2007. The interferon-stimulated genes: targets of direct signaling by interferons, double-stranded RNA, and viruses. Curr. Top. Microbiol. Immunol. 316:233–250 [DOI] [PubMed] [Google Scholar]
- 51. Wacher C, Müller M, Hofer MJ, Getts DR, Zabaras R, Ousman SS, Terenzi F, Sen GC, King NJC, Campbell IL. 2007. Coordinated regulation and widespread cellular expression of interferon-stimulated genes (ISG) ISG-49, ISG-54, and ISG-56 in the central nervous system after infection with distinct viruses. J. Virol. 81:860–871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zhang Y, Burke CW, Ryman KD, Klimstra WB. 2007. Identification and characterization of interferon-induced proteins that inhibit alphavirus replication. J. Virol. 81:11246–11255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Fitzgerald KA. 2011. The interferon inducible gene: viperin. J. Interferon Cytokine Res. 31:131–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hinnebusch AG. 2006. eIF3: a versatile scaffold for translation initiation complexes. Trends Biochem. Sci. 31:553–562 [DOI] [PubMed] [Google Scholar]
- 55. Brinton MA. 2002. The molecular biology of West Nile virus: a new invader of the Western Hemisphere. Annu. Rev. Microbiol. 56:371–402 [DOI] [PubMed] [Google Scholar]
- 56. Dong H, Chang DC, Xie X, Toh YX, Chung KY, Zou G, Lescar J, Lim SP, Shi P-Y. 2010. Biochemical and genetic characterization of dengue virus methyltransferase. Virology 405:568–578 [DOI] [PubMed] [Google Scholar]
- 57. Li S-H, Dong H, Li X-F, Xie X, Zhao H, Deng Y-Q, Wang X-Y, Ye Q, Zhu S-Y, Wang H-J, Zhang B, Leng Q-B, Zuest R, Qin E-D, Qin C-F, Shi P-Y. 2013. Rational design of a flavivirus vaccine through abolishing viral RNA 2′-O methylation. J. Virol. 87:5812–5819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Cho H, Proll SC, Szretter KJ, Katze MG, Gale M, Jr, Diamond MS. 2013. Differential innate immune response programs in neuronal subtypes determine susceptibility to infection in the brain by positive-stranded RNA viruses. Nat. Med. 19:458–464 [DOI] [PMC free article] [PubMed] [Google Scholar]