

Abstract
Pyrrolidine dithiocarbamate (PDTC) is widely used as an antioxidant or an NF-κB inhibitor. It has been reported to inhibit the replication of human rhinoviruses, poliovirus, coxsackievirus, and influenza virus. In this paper, we report that PDTC could inhibit the replication of herpes simplex virus 1 and 2 (HSV-1 and HSV-2). PDTC suppressed the expression of HSV-1 and HSV-2 viral immediate early (IE) and late (membrane protein gD) genes and the production of viral progeny. This antiviral property was mediated by the dithiocarbamate moiety of PDTC and required the presence of Zn2+. Although PDTC could potently block reactive oxygen species (ROS) generation, it was found that this property did not contribute to its anti-HSV activity. PDTC showed no activity in disrupting the mitogen-activated protein kinase (MAPK) pathway activation induced by viral infection that was vital for the virus's propagation. We found that PDTC modulated cellular ubiquitination and, furthermore, influenced HSV-2-induced IκB-α degradation to inhibit NF-κB activation and enhanced PML stability in the nucleus, resulting in the inhibition of viral gene expression. These results suggested that the antiviral activity of PDTC might be mediated by its dysregulation of the cellular ubiquitin-proteasome system (UPS).
INTRODUCTION
Herpes simplex virus 1 and 2 (HSV-1 and HSV-2) are among the most prevalent human pathogens in both industrialized and developing countries. HSV-1 is transmitted predominantly through the oral route, establishing latency in the trigeminal ganglion, while HSV-2 is transmitted sexually and establishes latency in the sacral ganglion (1). HSV-2 is identified as a risk factor for human immunodeficiency virus type 1 (HIV-1) transmission and disease progression (2, 3), while HSV-1 is suspected to be a cause of Alzheimer's disease (4, 5). The ability of the virus to successfully avoid clearance by the immune system by entering a nonreplicating state leads to lifelong infection, accompanied by periodic reactivation that results in viral shedding from the site of the initial infection. The current herpes medication works by mechanisms similar to those of nucleoside and nucleotide analogues by blocking the reproduction of herpesviruses. However, these drugs can prevent/lessen outbreaks but do not stop the virus from spreading, while a prophylactic vaccine still falls short. Therefore, studies of new anti-HSV drugs are of great importance and may help to identify novel targets for therapeutic drug development.
Pyrrolidine dithiocarbamate (PDTC), a stable pyrrolidine derivative of dithiocarbamates, is well suited for modifying the intracellular redox status of a given gene under defined tissue culture conditions. Not only can PDTC block the transcription of endogenous cellular NF-κB-responsive genes, such as the MCP-1 gene (6), but it also induces the expression of certain viral genes (7, 8). PDTC has been used for a variety of biochemical applications, including the inhibition of reactive oxygen species (ROS) (9) and NF-κB activation (10, 11), induction of apoptosis (12, 13), and maintenance of cell viability (14). It was also reported that PDTC showed an inhibitory effect on proteasome-dependent proteolysis (15), implying that PDTC was a potential inhibitor for the ubiquitin-proteasome system (UPS). It was also reported that PDTC could act as a specific inhibitor of the E3 ubiquitin ligase, targeting IκB for ubiquitination to influence proteasome-mediated IκB degradation (9).
Proteasomes, located in the nucleus and cytoplasm, are large protein complexes which mediate intracellular protein proteolysis and maintain cell homeostasis (16). The proteasome pathway, degrading both functional and aberrantly folded proteins through an intracellular proteolytic system, regulates cell cycle, apoptosis, inflammatory responses, and antigen presentation (17, 18). Previous studies demonstrated that several viruses, including HSV, depended on cellular proteasome activity to facilitate their postentry events, such as viral gene expression, replication, and immune evasion (19). Infected-cell polypeptide 0 (ICP0), an HSV immediate early (IE) protein, contains an intact domain known to have an E3-ubiquitin ligase activity (20, 21). ICP0 appears to stimulate the degradation of a number of cellular proteins via the proteasome pathway, such as PML (22–24), Sp100 (24), CENP-A/B/C (25–27), E2FBP1 (28), etc. Recently, Delboy et al. reported that the proteasome pathway played a significant role in HSV capsid postpenetration steps (29), and this effect was ICP0 dependent (30). These findings implied that cellular proteasome activity was important for HSV's life cycle and infection.
Previous studies have shown that PDTC acts as an antiviral agent against several RNA viruses (7, 8, 31–33). In this study, we report that PDTC inhibited HSV gene expression and replication and that this inhibitory activity was not associated with its antioxidant activity and the cellular mitogen-activated protein kinase (MAPK) pathways. We show that PDTC acted as an inhibitor of the UPS, preventing not only proteasome-mediated proteolysis of ubiquitin conjugates and IκB-α but also HSV-induced PML delocalization and degradation. Also, other evidence showed that a functional UPS was critical for HSV replication and viral gene expression. We concluded that PDTC blocked HSV replication and gene expression through dysregulation of the cellular UPS.
MATERIALS AND METHODS
Reagents, cell lines, plasmids, and viruses.
PDTC, sodium diethyldithiocarbamate (DDTC), pyrrolidine, N-acetyl cysteine (NAC), apocynin (APO), ATP, ZnCl2, EDTA, EDTA-Ca, EDTA-Mg, EDTA-Zn, EDTA-Cu, N,N,N′,N′-tetrakis (2-pyridylmethyl) ethylenediamine (TPEN), and pyrazone-41 (PYR-41) were purchased from Sigma-Aldrich (St. Louis, MO). WP1130 (Degrasyn) was obtained from Selleckchem (Houston, TX). 2-Dichlorodihydrofluorescein diacetate (DCFH-DA), MG132, SB203580, SP600125, antibodies for IκB-α, anti-p38 antibody, and anti-phosphorylated p38 (p-p38) antibody were from Beyotime Institute of Biotechnology (Haimen, Jiangsu, China). Goat anti-mouse Alexa Fluor 488-IgG, 4′,6′-diamidino-2-phenylindole (DAPI), and FluoZin-3 AM were from Life Technologies (Carlsbad, CA). IRDye 680 and IRDye 800 secondary antibodies were obtained from LI-COR (Lincoln, NE). Antibodies for the late protein gD-1/2 (gD of HSV-1 and -2), ICP0-1, ICP4-1, p53, p65, phosphorylated Jun N-terminal protein kinase 1 and 2 (p-JNK1/2), JNK2, PML, glyceraldehyde-3-phosphate dehydrogenase (GAPDH), β-catenin, and radioimmunoprecipitation assay (RIPA) lysis buffer were purchased from Santa Cruz (Santa Cruz, CA). Antiubiquitin antibody was obtained from eBioscience (San Diego, CA). Anti-HSV VP5 antibody was from Abcam (Cambridge, MA). Bz-VGR-AMC, Suc-LLVY-AMC, and Z-LLE-AMC (where Bz is benzoyl, Suc is N-succinyl, Z is benzyloxycarbonyl, AMC is 7-amido-4-methylcoumarin, and V, G, R, L, V, Y, and E are amino acids) were synthesized by GL Biochem Ltd. (Shanghai, China).
HEC-1-A, HeLa, and Vero cells were obtained from American Type Culture Collection (ATCC, Manassas, VA). Vero-ICP10P, an HSV-2 infection indicator cell line, was generated from Vero cells stably transfected with an HSV-2 ICP10 promoter-driven luciferase reporter plasmid. NF-κB-luc and AP-1-luc reporter plasmids were from Clontech (Palo Alto, CA). pRK-Ub-WT (plasmid number 17608; wild-type [WT] ubiquitin expression plasmid) and pRK-Ub-KO (plasmid number 17603; dominant negative [DN] ubiquitin expression plasmid) were from Addgene (Cambridge, MA). HSV-1(HF) and HSV-2(G) were propagated and titrated on Vero cells as described previously (34). The HSV-2 virus titration assay could also be performed on Vero-ICP10P cells by measuring luciferase activity. The viral titration was calculated from the standard curve.
Western blot.
Cells were lysed and centrifuged at 12,000 × g for 10 min at 4°C. Total protein in supernatant was quantified by using a bicinchoninic acid (BCA) protein assay kit (Pierce, Rockford, IL). After SDS-PAGE separation, the proteins were transferred onto polyvinylidene difluoride (PVDF) membranes (Millipore, Billerica, MA). The membranes were blocked, incubated in primary antibodies for 2 h at room temperature (RT), washed, and then incubated in IRDye IgG (1:10,000) for 1 h at RT. The bands were visualized with a LI-COR Odyssey infrared imager (LI-COR), and the intensities were analyzed using Odyssey version 3.0 software.
In-Cell Western assay.
Cell monolayers in a 96-well plate were fixed with 4% paraformaldehyde for 20 min at RT and permeabilized by 5 washes in 0.1% Triton X-100 with 5 min for each wash. The wells were blocked for 90 min and then incubated with primary antibodies diluted in blocking buffer (1:200) for 2 h. After washing with phosphate-buffered saline with Tween 20 (PBS-T; pH 7.4) buffer, the cell monolayers were inoculated in IRDye-IgG (1:1,500) for 1 h, rinsed, and scanned in the LI-COR Odyssey infrared imager. The fluorescence intensity of each well was measured by using Odyssey version 3.0 software, and the protein expression level was normalized to that of β-catenin.
Immunofluorescence staining.
HEC-1-A cells cultured on 10-mm glass coverslips were rinsed with PBS and fixed with 4% paraformaldehyde for 15 min at RT before being permeabilized using 0.2% Triton X-100 for 15 min. The cells were blocked for 1 h, and cellular proteins were immunolabeled using the respective primary antibodies followed by Alexa Fluor 488-IgG. Nuclei were visualized by staining with DAPI. Images were acquired using an Olympus FluoView FV10i confocal microscope (Tokyo, Japan).
Cell transfection and luciferase assay.
Cells cultured in a 96-well plate were transiently transfected with 100 ng/well luciferase reporter plasmid or cotransfected with 50 ng/well luciferase reporter plasmid and 150 ng/well ubiquitin expression plasmids using Lipofectamine 2000 transfection reagent (Life Technologies). Cells were cultured for a further 24 h and then infected with virus in the absence or presence of drugs. The relative luminescence units (RLU) were determined by using the Bright-Glo luciferase assay system (Promega, Madison, WI).
RNA extraction and real-time PCR.
Total RNA was extracted using TRIzol reagent (Life Technologies) and reverse transcribed to cDNA using the ReverTra Ace quantitative reverse transcription-PCR kit (Toyobo, Osaka, Japan). Real-time PCR was performed in triplicate on the ABI Prism 7300 sequence detection system using SYBR green PCR master mix (Toyobo) according to the manufacturer's protocol. The levels of HSV-1 ICP0 (forward, TACGTGAACAAGACTATCACGGG, and reverse, TCCATGTCCAGGATGGGC), HSV-1 ICP4 (forward, GGCCTGCTTCCGGATCTC, and reverse, GGTGATGAAGGAGCTGCTGTT), HSV-1 gD (forward, AGCAGGGGTTAGGGAGTTG, and reverse, CCATCTTGAGAGAGGCATC), HSV-2 ICP0 (forward, GTGCATGAAGACCTGGATTCC, and reverse, GGTCACGCCCACTATCAGGTA), HSV-2 ICP4 (forward, GCGAGCTGCGGTTCGT, and reverse, GCCACGCGCAGGTC), and HSV-2 gD (forward, CCAAATACGCCTTAGCAGACC, and reverse, CACAGTGATCGGGATGCTGG) mRNA transcription were standardized against that of the GAPDH housekeeping gene (forward, TGCACCACCAACTGCTTAGC, and reverse, GGCATGGACTGTGGTCATGAG).
ROS detection.
Cells cultured in black opaque 96-well plates were treated with the antioxidants for 30 min prior to HSV-2 infection (multiplicity of infection [MOI] of 1) for 24 h. The cells were washed with PBS and then exposed to DCFH-DA diluted in Dulbecco's modified Eagle's medium (DMEM) medium (10 μM). After 20 min of incubation, the cells were rinsed 3 times, and the plates were read using the Hitachi F7000 spectrofluorometer (Tokyo, Japan) (excitation, 488 nm, and emission, 525 nm).
Cellular 26S proteasome activity assay.
HEC-1-A cells were lysed, and cytoplasmic extracts were obtained by centrifugation at 12,000 × g for 10 min at 4°C. The effects of drugs on cellular 26S proteasome activity were determined as described previously (35). Briefly, 20 μg total protein diluted in assay buffer (20 mM Tris, pH 8.0, 1 mM ATP, and 2 mM MgCl2) was mixed with the compounds indicated below and with 75 μM substrates to a final volume of 100 μl. The substrates, Bz-VGR-AMC, Suc-LLVY-AMC, and Z-LLE-AMC, were used to determine the proteasome trypsinlike, chymotrypsinlike, and peptidylglutamyl-peptide hydrolase (PDGH) activities, respectively. The mixtures were incubated in the absence or presence of drugs for 1 h at 30°C. 26S proteasome cleavage activity was measured in the Hitachi F7000 spectrofluorometer (excitation, 380 nm, and emission, 460 nm).
Intracellular Zn2+ detection.
HEC-1-A cells grown in black opaque 96-well plates were loaded with 5 μM FluoZin-3 AM in PBS for 30 min at 37°C. The cells were then washed with Zn2+-free medium to remove nonspecific dye staining on the cell surface and subsequently incubated for 30 min in complete medium to allow deesterification. PDTC was added to the medium for 30 min, and the plate was read in the spectrofluorometer (excitation, 494 nm, and emission, 516 nm).
Statistics.
Statistical analysis was performed using the two-tailed Student t test. Statistical significance was determined at P values of <0.05 and <0.01.
RESULTS
PDTC inhibited HSV late gene expression and replication.
To investigate PDTC's inhibitory effect on HSV-2 replication, HEC-1-A cells infected by HSV-2 in the presence of serial concentrations of PDTC were subjected to freeze-thawing cycles, and the progeny virus released was titrated in Vero-ICP10P cells. As shown in Figure 1A, PDTC inhibited HSV-2 replication in a dose-dependent manner. We evaluated the cytotoxicity of PDTC by using a cell-counting kit (CCK-8 assay kit; Dojindo, Kumamoto, Japan), and the results showed that more than 90% of the cells were viable at 100 μg/ml PDTC. We also investigated the effect of PDTC on the expression of HSV membrane protein gD, which was used to represent late gene expression in the viral life cycle, via the In-Cell Western assay. PDTC treatment resulted in the reduction of both HSV-1 and HSV-2 gD expression in HEC-1-A cells (Fig. 1B and C), which corresponded to the reduction of viral replication. ICP5, the major HSV capsid protein, was also downregulated by PDTC (data not shown). To verify this observation, we also evaluated PDTC's inhibitory effect on viral gD-1/2 expression at the mRNA transcriptional level via real-time PCR analysis. It was shown that the reduction of gD mRNA transcription was parallel to the reduction of the expression of the protein (Fig. 1D and E). Similar antiviral activity and inhibition of gD expression by PDTC were also observed in Vero and HeLa cells (data not shown), suggesting that these were not cell-specific phenomena.
Fig 1.

PDTC inhibited HSV-gD gene expression and replication. (A) PDTC showed an inhibitory effect on HSV-2 replication. HEC-1-A cells were treated with serial concentrations of PDTC prior to HSV-2 infection (MOI = 1) and subjected to three freeze-thawing cycles 24 h p.i. The released virus was titrated on Vero-ICP10P cells as described in the text. (B, C) PDTC inhibited HSV gD expression. HEC-1-A cells were treated with PDTC and infected with virus at an MOI of 1, and the gD-1 (B) and gD-2 (C) protein expression levels were determined 24 h p.i. via the In-Cell Western assay and normalized to the β-catenin level. Insets show results of representative experiments. (D, E) gD-1 (D) and gD-2 (E) mRNA transcriptional levels were quantified by real-time PCR analysis at 12 h p.i. All experiments were performed three times. Error bars show standard deviations.
The PDTC molecule consists of a dithiocarbamate and a pyrrolidine moiety. To illustrate their roles in viral inhibition, either DDTC or pyrrolidine was investigated in the HSV infection assay. As shown in Figure 2A, DDTC reduced HSV-2 gD expression. In contrast, pyrrolidine showed no effect on viral replication, suggesting that the anti-HSV activity of PDTC was dependent on the dithiocarbamate moiety. We also showed that PDTC's antiviral activity was reversible upon the removal of the agent. As shown in Figure 2B, the removal of PDTC partially restored gD expression within 3 h.
Fig 2.
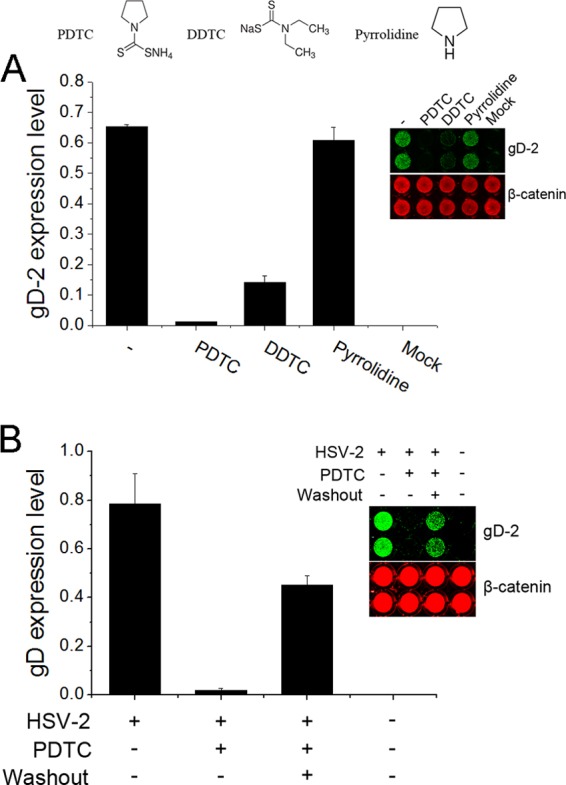
PDTC's antiviral property was mediated by its dithiocarbamate moiety, and this antiviral effect was reversible after removal of PDTC. (A) The dithiocarbamate moiety of the PDTC molecule was essential for its antiviral property. HEC-1-A cells were exposed to 300 μM PDTC, DDTC, or pyrrolidine prior to HSV-2 infection (MOI = 1). (B) PDTC's antiviral effect was reversible. HEC-1-A cells were exposed to PDTC (25 μg/ml) prior to infection. After 3 h of incubation, PDTC was removed by washing and complete medium was supplied. The gD expression levels were determined 24 h p.i. via the In-Cell Western assay and normalized to the β-catenin level. Error bars show standard deviations.
Zn2+ was essential for the anti-HSV activity of PDTC.
We previously found that PDTC failed to inhibit HSV-2 infection in serum-free medium (data not shown). Divalent metal ions, including copper and zinc, were reported to mediate PDTC bioactivities such as inhibition of NF-κB activation and induction of apoptosis (34, 35). To explore the roles of divalent metal ions in the antiviral activity of PDTC, serial concentrations of EDTA were added to fetal bovine serum (FBS)-containing medium to chelate metal ions, and the effects on PDTC viral inhibitory activity were analyzed. As shown in Figure 3A, PDTC antiviral activity was attenuated by the addition of EDTA, implying that the presence of certain divalent ions was required for its antiviral activity.
Fig 3.

Zn2+ was essential for PDTC's antiviral activity. (A, B) HEC-1-A cells cultured in complete medium were exposed to PDTC (25 μg/ml) in the presence of serial concentrations of EDTA (A) or 10 μM EDTA-Ca, -Mg, -Zn, or -Cu (B) and infected with HSV-2 (MOI = 1). (C) HEC-1-A cells were cultured in FBS-free medium containing 0.5 μM Zn2+ or serial concentrations of Cu2+ and PDTC (25 μg/ml) prior to HSV-2 infection (MOI = 1). (D) HEC-1-A cells were exposed to PDTC (25 μg/ml) in the presence of serial concentrations of TPEN and then infected with HSV-2. Each TPEN concentration was in duplicate. Mock-treated cells were exposed to HSV-2 alone. The gD expression levels were determined 24 h p.i. Error bars show standard deviations.
To determine the roles of individual ions, we investigated the effects of EDTA-Ca, -Mg, -Zn, and -Cu on PDTC's antiviral activity, using a method previously described by Lanke et al. (33). The affinities of these ions to EDTA are in the order of Cu2+>Zn2+>Mg2+>Ca2+, and the differential affinities allow the depletion of the ion with higher affinity. The results showed that EDTA-Ca and -Mg abrogated PDTC inhibition of HSV-2 replication, while EDTA-Zn and -Cu did not. To confirm the role of zinc, cells cultured in serum-free medium supplied with 0.5 μM Cu2+ or Zn2+ were infected with HSV-2 in the presence of PDTC. As shown in Figure 3C, only Zn2+ exhibited complete inhibition of HSV-2 gD expression, and Cu2+ showed no significant effect even when it was used at 5 μM, which was 10 times higher than in complete culture medium (∼0.5 μM). TPEN, a cell membrane-permeable specific zinc chelator, was also tested in the PDTC inhibitory assay and shown to abrogate the anti-HSV activity of PDTC.
The effect of PDTC on the expression of HSV IE proteins.
Whether PDTC modulated HSV immediate early (IE) gene expression was investigated by using the In-Cell Western assay and real-time PCR. ICP0 and ICP4 are two important IE regulatory proteins of HSV that play vital roles in viral replication, cell growth, and apoptosis (36–39). ICP0 acts synergistically with ICP4 and is shown to be essential for the activation of early genes and viral replication (36, 38, 40, 41). PDTC exhibited strong inhibitory activity on ICP0 and ICP4 expression in HSV-1-infected cells 24 h postinfection (p.i.) (Fig. 4A and B).
Fig 4.

Effect of PDTC on the expression of HSV IE proteins. (A, B) PDTC inhibited the expression of HSV-1 ICP0 (A) and ICP4 (B). HEC-1-A cells exposed to PDTC were infected with HSV-1 (MOI = 1). ICP0 and ICP4 expression levels were determined 24 h p.i. and normalized to the β-catenin level. (C) PDTC effects on HSV-1 ICP0 and ICP4 expression at different time point were determined via Western blot assay. (D) PDTC effects on HSV-1 ICP0 and ICP4 mRNA transcription at 6 h or 12 h p.i. ICP0 and ICP4 mRNA expression levels were quantified via real-time PCR. (E) PDTC interfered with nuclear-cytoplasmic translocation of ICP0. HEC-1-A cells were infected with HSV-1 (MOI = 1) for 2 h and then incubated with PDTC (25 μg/ml) or MG132 (6 μg/ml). ICP0 localization was observed via immunofluorescence staining 12 h p.i. All experiments were performed 2 to 3 times, and a representative result is shown. Error bars show standard deviations.
The effect of PDTC on HSV-1 IE gene expression at the early stage of the viral life cycle was also investigated. ICP0 was affected weakly by PDTC 12 h p.i., but ICP4 expression was significantly reduced after 8 h (Fig. 4C). ICP0 and ICP4 transcription levels were also downregulated by PDTC 12 h p.i. (Fig. 4D). However, the downregulation at 6 h p.i. was considerably less effective (Fig. 4D), which was also observed in HSV-2 infected cells (data not shown).
We further investigated the HSV-1 ICP0 localization after PDTC treatment. PDTC inhibited the nuclear-cytoplasmic translocation of ICP0 (Fig. 4E) similarly to MG132; the translocation of ICP0 has been reported as an essential step for effective HSV replication (42). Taking these results together, we concluded that PDTC exerted significant inhibition of HSV ICP4 expression at the early stage of the virus life cycle. However, it appeared that PDTC inhibited ICP0 localization instead of reducing its expression level.
PDTC inhibition of HSV replication was neither through its antioxidant activity nor by blocking the MAPK pathway.
PDTC has been widely used as a potent antioxidant. We show that PDTC completely attenuated ROS production in HSV-2-infected cells (Fig. 5A), even at low concentrations. NAC and apocynin, both strong antioxidant agents, completely inhibited HSV-2-induced ROS production. However, neither NAC nor APO could inhibit HSV-2 replication (Fig. 5B), suggesting that the antioxidant activity of PDTC was probably not directly related to its antiviral action.
Fig 5.

PDTC inhibited HSV replication neither through its antioxidant activity nor by blocking the MAPK pathway. (A) PDTC attenuated HSV-2-induced ROS production. HEC-1-A cells were infected with HSV-2 (MOI = 1) in the presence or absence of NAC (1 mM), APO (50 μg/ml), or PDTC. ROS levels were determined 24 h p.i. as described in the text. a.u., arbitrary units. (B) Both NAC and APO showed no anti-HSV-2 activity. Cells were infected with HSV-2 (MOI = 1) in the presence or absence of PDTC (12.5 μg/ml), NAC (1 mM), or APO (50 μg/ml), and the gD expression levels were determined 24 h p.i. (C) Cells transfected with AP-1-luc reporter plasmid were treated with serial concentrations of PDTC, SB203580 (20 μM), or SP600125 (20 μM) before being mock infected or infected with HSV-2 (MOI = 1). The RLU were determined 24 h p.i. and are expressed as the percentage of the amount in the mock-infected cells. (D) Cells were either mock infected or infected with HSV-2 (MOI = 1) in the presence or absence of PDTC. JNK/p38 MAPK activation was determined 12 h p.i. via Western blot assay. Error bars show standard deviations.
It was reported that the activation of c-Jun N-terminal kinase (JNK) and p38 MAPK play an important role in HSV-1 replication (43–45). We observed that the HSV-2 life cycle was also affected by these two pathways in HEC-1-A cells (data not shown). We therefore investigated the effects of PDTC on JNK and p38 MAPK pathways and the activation of transcription factor AP-1. PDTC did not inhibit HSV-2-induced AP-1 activation, as demonstrated by an AP-1 luciferase reporter plasmid (Fig. 5C), while two positive controls, SB203580 (p38 inhibitor) and SP600125 (JNK inhibitor), completely inhibited the AP-1 activation. In addition, the phosphorylation of JNK1/2 and p38 was not inhibited by PDTC (Fig. 5D), suggesting that PDTC's inhibition of HSV-2 replication was probably not mediated by inhibition of the MAPK pathway.
PDTC inhibited HSV replication through dysregulation of the UPS and inhibition of NF-κB activation.
Ubiquitin-mediated protein degradation regulates a variety of cellular processes and is strongly associated with viral pathogenesis (24, 46–56). Evidence showed that PDTC treatment resulted in the accumulation of several short-half-life proteins and inhibited proteasome-dependent proteolysis (15). We also showed that PDTC treatment led to the accumulation of ubiquitinated proteins both in HEC-1-A and Vero cells (Fig. 6A). Further experiments showed that PDTC treatment enhanced the accumulation of ubiquitinated p53 (Fig. 6B), a short-lived intracellular protein. Therefore, we investigated whether the inhibition of the UPS was related to the antiviral activity of PDTC.
Fig 6.

PDTC may inhibit HSV replication through dysregulation of the UPS and inhibition of NF-κB activation. (A) HSV-2 infection changed the cellular profile of ubiquitin conjugates. Cells were incubated with 50 μg/ml PDTC and collected at the indicated time points. The levels of ubiquitin conjugates were determined via Western blot assay. (B) PDTC induced the accumulation of ubiquitinated p53. HEC-1-A cells were cultured in the presence of PDTC (50 μg/ml) or MG132 (6 μg/ml) for 2 h. p53 and its ubiquitinated forms were visualized using p53-specific antibody via Western blot assay. (C) HSV-2 infection changed the cellular profile of ubiquitin conjugates. HEC-1-A cells infected with HSV-2 (MOI = 1) were collected at the indicated times, and the levels of ubiquitin conjugates were determined. (D) PDTC inhibited HSV replication through dysregulation of the UPS and inhibition of HSV-induced IκB-α degradation. Cells treated with PDTC or MG132 were either mock infected or infected with HSV-2 (MOI = 1). The levels of ubiquitin conjugates, gD, and IκB-α were determined 24 h p.i. (E) PDTC-induced dysregulation of the UPS could be mitigated by EDTA. Cells exposed to PDTC were either mock treated or treated with EDTA (10 μM). The levels of ubiquitin conjugates, gD, and IκB-α were determined 24 h p.i. (F) PDTC treatment inhibited HSV-2-induced NF-κB activation. NF-κB activation was determined using NF-κB-luc reporter plasmid as described in the text. The RLU were determined 24 h p.i. and are expressed as the percentage of the amount in the mock-infected cells. (G) PDTC disrupted HSV-2-induced p65 nuclear translocation. p65 translocation in uninfected or infected cells treated with dimethyl sulfoxide (DMSO) or PDTC (50 μg/ml) was determined by immunofluorescence staining 24 h p.i. as described in the text. All experiments were performed 2 or 3 times, and the results of a representative experiment are shown. Error bars show standard deviations.
We observed that HSV-2 infection induced alteration of ubiquitin conjugates in HEC-1-A cells. The viral infection disrupted the normal profiles of ubiquitin conjugates and accelerated the cellular protein degradation, especially that of the high-molecular-weight proteins (Fig. 6C). We showed that PDTC treatment reversed the changes in the ubiquitin conjugate profiles induced by HSV-2 infection and inhibited the loss of ubiquitinated proteins and viral gD protein (Fig. 6D). In addition, HSV-2 infection caused progressive degradation of IκB-α, an endogenous NF-κB inhibitor (data not shown). It was also found that PDTC inhibition of proteasome-dependent proteolysis was associated with the level of IκB-α (Fig. 6D). It was possible that PDTC inhibition of cellular proteasome activity prevented the IκB-α degradation induced by HSV-2 infection. Consistent with our early observations, the addition of EDTA mitigated the PDTC-mediated accumulation of polyubiquitinated proteins and the degradation of IκB-α in HSV-2-infected cells, thus attenuating the antiviral activity of PDTC (Fig. 6E).
Earlier studies reported that inhibition of NF-κB activation reduced HSV-1 replication (57, 58). Using a NF-κB luciferase reporter plasmid, we showed that HSV-2-induced NF-κB activation was down modulated by PDTC in a dose-dependent manner (Fig. 6F). PDTC was also found to interfere with HSV-2-induced p65 nuclear translocation (Fig. 6G). Therefore, we postulated that PDTC's inhibition of HSV-2 replication may be mediated by its dysregulation of the cellular UPS and inhibition of HSV-2-mediated NF-κB activation.
The increased intracellular Zn2+ concentration facilitated by PDTC might contribute to its inhibitory effect on 26S proteasome activity and antiviral activity.
26S proteasome is one of the most common forms of the proteasomal complex. Therefore, we investigated whether PDTC directly affected 26S proteasome function in vitro. Specific synthetic fluorogenic substrates of the 26S proteasome were used to determine the effect of PDTC on cellular 26S proteasome activity. As shown in Figure 7A, MG132 blocked 26S proteasome's trypsinlike, chymotrypsinlike, and PDGH activities, but PDTC showed no effect on its cleavage functions, suggesting that PDTC's inhibition of viral replication was not through direct inhibition of 26S proteasome function.
Fig 7.

The increased intracellular Zn2+ concentration mediated by PDTC might contribute to its inhibitory effect on 26S proteasome activity and its anti-HSV property. (A) PDTC did not inhibit 26S proteasome activity in a cell-free system. 26S proteasome activity was measured in the presence or absence of PDTC as described in the text. MG132 (2 μg/ml) was used as the positive control. (B) PDTC increased the intracellular Zn2+ level. Cells were incubated with PDTC or 200 μM Zn2+ for 30 min. The intracellular Zn2+ level was determined via FluoZin-3 AM labeling. (C) Zn2+ attenuated HSV-2-induced loss of ubiquitin conjugates and IκB-α and showed antiviral activity. HEC-1-A cells were either infected or not infected with HSV-2 (MOI = 1) in the presence or absence of Zn2+ (200 μM). Levels of ubiquitin conjugates, IκB-α, and gD were determined via Western blot assay 24 h p.i. Error bars show standard deviations.
Previous reports have shown that PDTC is a zinc ionophore and able to transport zinc ions into mammalian cells (32, 33). Using FluoZin-3 AM probe, a Zn2+-selective indicator, we demonstrated that PDTC or free Zn2+ treatment increased the intracellular Zn2+ concentration after 30 min (Fig. 7B). Kim et al. found that free Zn2+ could inhibit 26S proteasome's cleavage activity (15). We observed that free Zn2+ blocked the loss of ubiquitin-conjugated proteins and degradation of IκB-α induced by HSV-2 and, thus, inhibited gD-2 expression and HSV-2 replication (Fig. 7C). The evidence showed that the increased intracellular Zn2+ concentration contributed to PDTC-mediated dysregulation of the UPS. Therefore, we postulated that PDTC facilitated Zn2+ influx, leading to dysregulation of the UPS and proteasome-dependent proteolysis.
PDTC stabilized the nuclear localization of PML during HSV infection.
PML nuclear bodies (PML-NBs) have been shown to play an important role against viral infection (59, 60). The IE proteins of many viruses target and colocalize with PML-NBs for initial viral replication (60, 61). Reports have shown that HSV infection leads to proteasome-dependent PML degradation and delocalization (24). Therefore, we investigated whether PDTC treatment would lead to stabilized nuclear localization of PML-NBs.
As shown in Figure 8A, PDTC-treated cells exhibited enlarged NB size and increased NB numbers whether the cells were infected with HSV-2 or not, suggesting that PDTC stabilized the PML-NB complexes localized to the nucleus. The enlarged size and the increased NB numbers in the cellular compartment might compensate for PML degradation caused by HSV infection, resulting in the inhibition of viral replication. EDTA could reverse the PML-NB stabilization induced by PDTC (Fig. 8B), which indicated that Zn2+-mediated inhibition of proteasome activity might be of great importance for PML nuclear stabilization and localization.
Fig 8.
PDTC stabilized nuclear localization of PML during HSV infection. (A) HEC-1-A cells were treated with either PDTC or MG132 (10 μg/ml) and then infected with HSV-2 (MOI = 1). (B) Cells exposed to PDTC (50 μg/ml) were either treated or not treated with EDTA (10 μM) and then infected with HSV-2 (MOI = 1). PML-NBs were visualized by immunofluorescence staining 16 h p.i.
The UPS was critical for HSV replication.
To better understand the importance of ubiquitination for HSV gene expression and replication, a dominant-negative (DN) mutant of a ubiquitin expression plasmid was utilized. We showed that the expression of DN-mutant ubiquitin inhibited the expression of both HSV-1 and HSV-2 gD in HeLa cells (Fig. 9A and B) significantly in comparison to the levels of gD expression in the vehicle control and with wild-type (WT) ubiquitin. Further analysis showed that ubiquitin mutant expression also blocked HSV-1 ICP4 expression at the early stage of viral infection (Fig. 9C). We also investigated whether DN mutation of ubiquitin would inhibit HSV-2-induced NF-κB activation. As shown in Figure 9D and E, this mutant also attenuated NF-κB activation in HEC-1-A and HeLa cells whether the cells were infected or not. Based on these observations, we concluded that functional ubiquitination was essential for effective replication of the virus.
Fig 9.

Ubiquitination was vital for HSV replication. (A, B) HeLa cells were transfected with vehicle, HA-Ub-WT, or HA-Ub-KO and infected with HSV-1 (A) or HSV-2 (B) (MOI = 1). The gD expression levels were determined 24 h p.i. and normalized to the β-catenin level. (C) The ICP4-1 level was determined via Western blot assay 6 h p.i. (D, E) DN mutant of ubiquitin destroyed HSV-2-induced NF-κB activation. HEC-1-A (D) or HeLa (E) cells cotransfected with vehicle, HA-Ub-WT, or HA-Ub-KO and NF-κB-Luc reporter plasmid were either mock infected or infected with HSV-2 (MOI = 1). NF-κB activation was measured 24 h p.i. Each panel shows the means and standard deviations from three independent experiments. *, P < 0.05, and **, P < 0.01, for comparison with vehicle. (F) The inhibitors of DUBs and UBE1 inhibited HSV-2 replication. HEC-1-A cells were either infected or not infected in the presence of WP1130 (2 μg/ml) or PYR-41 (20 μg/ml). Levels of ubiquitin conjugates, IκB-α, and gD were determined 24 h p.i.
We also analyzed whether other components in the UPS were essential for the HSV life cycle. We employed WP1130, a selective deubiquitinase (DUB) inhibitor, and PYR-41, an irreversible inhibitor of ubiquitin-activating enzyme E1 (UBE1), to investigate their effects on the accumulation of ubiquitin conjugates, IκB-α degradation, and antiviral activity. As shown in Figure 9F, treatment with WP1130 and PYR-41 led to the accumulation of ubiquitin conjugates, consistent with a previous report (62). Both inhibitors also had dramatic effects in inhibiting the reduction of ubiquitin conjugates caused by HSV-2, blocked HSV-2-induced IκB-α degradation, and inhibited viral gD-2 expression. These observations suggested that DUBs and UBE1 are vital for the viral replication.
DISCUSSION
PDTC has been previously reported to inhibit the infection of viruses, including influenza virus (31), rhinovirus (8), poliovirus (32), coxsackievirus (7, 33), and mengovirus (33). In this study, we report inhibitory activity of PDTC against HSV-1 and HSV-2, and our mechanistic studies suggested that PDTC might inhibit HSV gene expression and replication through dysregulation of the UPS, leading to inhibition of NF-κB activation.
PDTC is known to be a multifunctional agent, exhibiting antioxidant activity, inhibition of NF-κB, induction of apoptosis, and maintenance of cell viability (9–14, 63). PDTC's bioactivity is dependent on the presence of divalent metal ions, including copper and zinc (12–14, 35). Si et al. reported that the presence of copper or zinc ions is essential for the anticoxsackievirus activity of PDTC (7, 33). FBS contains a much higher level of Zn2+ (50 μM) than of Cu2+ (5 μM), and thus, DMEM supplemented with 10% FBS contains only 5 μM Zn2+ and 0.5 μM Cu2+ (Life Technologies). The competitive chelating experiments using EDTA-metal ions ruled out major roles of both Ca2+ and Mg2+ in PDTC's inhibition of HSV. The results of the Cu2+/Zn2+ chasing assay suggested that Zn2+ but not Cu2+ was a dominant factor for PDTC's antiviral property (Fig. 3C).
The intracellular redox status is essential for the replication and infection of several viruses, including influenza virus (64), HIV-1 (65, 66), etc. Schachtele et al. reported that HSV-1 infection elevated intracellular ROS in wild-type microglial cell cultures (67). Gonzalez-Dosal et al. also found that HSV infection of macrophages could induce ROS generation (68). We also found that HSV-2 infection of human genital epithelial cells elevated the intracellular ROS level dramatically (Fig. 5A). However, although PDTC and two other ROS inhibitors, NAC and APO, inhibited ROS production in infected cells, neither NAC nor APO had any viral inhibitory activity (Fig. 5A and B), suggesting that HSV replication was independent of cellular redox status and that the antiviral activity of PDTC could not be attributed to its anti-ROS activity.
It was reported that HSV-1 infection activated the MAPK pathway (43). Similarly, our results showed that HSV-2 infection of HEC-1-A cells stimulated the JNK/p38 MAPK pathway (data not shown). We found that blocking these two pathways using pharmacological inhibitors inhibited viral replication (data not shown). Further analysis showed that PDTC did not affect the phosphorylation of JNK1/2 and p38 (Fig. 5D). Downstream AP-1 activation was not downregulated in the presence of PDTC, although gD expression was reduced (Fig. 5C).
A previous study demonstrated that PDTC inhibited proteasome-dependent proteolysis (15). In the current study, we observed that PDTC increased the levels of ubiquitin conjugates in both HEC-1-A and Vero cells (Fig. 6A), as well as the accumulation of ubiquitinated p53 (Fig. 6B). HSV-2 infection of human genital epithelial cells accelerated the cellular protein degradation and reduced the accumulation of cellular ubiquitin conjugates (Fig. 6C), and this effect could be reversed by PDTC treatment (Fig. 6D and E). We found that proteasome-dependent IκB-α degradation was also inhibited by PDTC treatment (Fig. 6D), consistent with the observation that PDTC was an NF-κB inhibitor (11, 69). Studies have shown that HSV-1 infection results in persistent activation of NF-κB, which is essential for HSV-1 replication (57, 58). We also observed that p65 nuclear translocation and IκB-α degradation in human genital epithelial cells were enhanced by HSV-2 infection (data not shown). Therefore, we speculated that PDTC might maintain the cytosolic IκB-α level by blocking cellular proteasome function, since the proteasome is the major component mediating IκB-α degradation. It is worth noting that, in contrast to the strong inhibition of proteasome-mediated cleavage by MG132 (Fig. 7A), PDTC did not inhibit cellular 26S proteasome activity directly. Instead, PDTC induced intracellular Zn2+ influx (Fig. 7C), and the free Zn2+ might inhibit 26S proteasome cleavage activity.
PML-NBs have been reported as an important cellular component interacting with HSV-1 regulatory protein ICP0, which controls viral lytic infection (70, 71). PML was also reported to be a repressor for HSV-1 infection, and this PML repression could be mitigated by the expression of ICP0 (23). We showed that PDTC treatment led to enlarged PML-NB complexes and increased NB numbers in HEC-1-A cells (Fig. 8A). The PDTC inhibition of HSV-2-induced, proteasome-mediated PML-NB degradation and delocalization suggested that PML stabilized by PDTC may repress HSV gene expression and replication.
Proteasome activity is an important part of the replication of several viruses (53, 55, 56, 72–78). Previous studies have demonstrated that the effective replication, gene expression, and reactivation of HSV-1 are dependent on proteasome activity, and treatment with proteasome inhibitors potently inhibited viral replication (79, 80). Recently, Delboy et al. reported that cellular proteasome activity facilitated the postpenetration steps of HSV and that the inhibition of the proteasome pathway using specific inhibitors, such as MG-132 or lactacystin, would reduce the transportation of HSV capsids to the nucleus (29). They also showed that this effect was dependent on the presence of ICP0 (30). In our study, we observed that PDTC treatment disturbed cellular ubiquitin-proteasome function, inhibited IκB-α degradation, and led to the disruption of viral gene expression. In addition, our observations that a DUB inhibitor (WP1130) and a UBE1 inhibitor (PYR-41) inhibited HSV-2 replication through interfering with ubiquitin conjugation/deconjugation and the increasing levels of ubiquitinated proteins in HEC-1-A cells (Fig. 9) were further evidence for the roles of polyubiquitination in HSV replication. Also, the proteasome-dependent IκB-α degradation induced by HSV-2 infection was disrupted by WP1130 and PYR-41 treatment. The current study demonstrates that homeostasis of the ubiquitin cycle and a functional UPS are prerequisites for viral gene expression and HSV replication and that the UPS is a potential drug target for HSV infection.
ACKNOWLEDGMENTS
This study was supported by the National Science Foundation (grant number 30870124) and the Major Research and Development Project from the Ministry of Health (grants number 2012ZX10001-007-009-001 and 2013ZX10001005-003).
We thank Qihan Li at Institute of Medical Biology, Chinese Academy of Medical Sciences, for HSV-1(HF) and Erguang Li at the School of Medicine, Nanjing University, China, for HSV-2(G) and for his critical reading of the manuscript.
Footnotes
Published ahead of print 5 June 2013
REFERENCES
- 1. Ahmad N, Ray CG, Drew WL. 2011. Chapter 14. Herpesviruses, p 247–270 In Ryan KJ Ray CG (ed), Sherris medical microbiology, 5th ed McGraw Hill Medical, New York, NY [Google Scholar]
- 2. Kapiga SH, Sam NE, Bang H, Ni Q, Ao TTH, Kiwelu I, Chiduo S, Ndibe U, Seage G, Coplan P. 2007. The role of herpes simplex virus type 2 and other genital infections in the acquisition of HIV-1 among high-risk women in northern Tanzania. J. Infect. Dis. 195:1260–1269 [DOI] [PubMed] [Google Scholar]
- 3. Lama JR, Lucchetti A, Suarez L, Laguna-Torres VA, Guanira JV, Pun M, Montano SM, Celum CL, Carr JK, Sanchez J. 2006. Association of herpes simplex virus type 2 infection and syphilis with human immunodeficiency virus infection among men who have sex with men in Peru. J. Infect. Dis. 194:1459–1466 [DOI] [PubMed] [Google Scholar]
- 4. Itzhaki RF, Wozniak MA. 2008. Herpes simplex virus type 1 in Alzheimer's disease: the enemy within. J. Alzheimers Dis. 13:393–405 [DOI] [PubMed] [Google Scholar]
- 5. Holmes C, Cotterell D. 2009. Role of infection in the pathogenesis of Alzheimers disease: implications for treatment. CNS Drugs 23:993–1002 [DOI] [PubMed] [Google Scholar]
- 6. Lee YW, Hennig B, Toborek M. 2003. Redox-regulated mechanisms of IL-4-induced MCP-1 expression in human vascular endothelial cells. Am. J. Physiol. Heart Circ. Physiol. 284:H185–H192 [DOI] [PubMed] [Google Scholar]
- 7. Si X, McManus BM, Zhang J, Yuan J, Cheung C, Esfandiarei M, Suarez A, Morgan A, Luo H. 2005. Pyrrolidine dithiocarbamate reduces coxsackievirus B3 replication through inhibition of the ubiquitin-proteasome pathway. J. Virol. 79:8014–8023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gaudernak E, Seipelt J, Triendl A, Grassauer A, Kuechler E. 2002. Antiviral effects of pyrrolidine dithiocarbamate on human rhinoviruses. J. Virol. 76:6004–6015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hayakawa M, Miyashita H, Sakamoto I, Kitagawa M, Tanaka H, Yasuda H, Karin M, Kikugawa K. 2003. Evidence that reactive oxygen species do not mediate NF-κB activation. EMBO J. 22:3356–3366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Schreck R, Meier B, Männel DN, Dröge W, Baeuerle PA. 1992. Dithiocarbamates as potent inhibitors of nuclear factor kappa B activation in intact cells. J. Exp. Med. 175:1181–1194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ziegler-Heitbrock H, Sternsdorf T, Liese J, Belohradsky B, Weber C, Wedel A, Schreck R, Bauerle P, Strobel M. 1993. Pyrrolidine dithiocarbamate inhibits NF-kappa B mobilization and TNF production in human monocytes. J. Immunol. 151:6986–6993 [PubMed] [Google Scholar]
- 12. Chen SH, Liu SH, Liang YC, Lin JK, Lin-Shiau SY. 2000. Death signaling pathway induced by pyrrolidine dithiocarbamate-Cu2+ complex in the cultured rat cortical astrocytes. Glia 31:249–261 [DOI] [PubMed] [Google Scholar]
- 13. Erl W, Weber C, Hansson GK. 2000. Pyrrolidine dithiocarbamate-induced apoptosis depends on cell type, density, and the presence of Cu2+ and Zn2+. Am. J. Physiol. Cell Physiol. 278:C1116–C1125 [DOI] [PubMed] [Google Scholar]
- 14. Chung KC, Park JH, Kim CH, Lee HW, Sato N, Uchiyama Y, Ahn YS. 2000. Novel biphasic effect of pyrrolidine dithiocarbamate on neuronal cell viability is mediated by the differential regulation of intracellular zinc and copper ion levels, NF-κb, and MAP kinases. J. Neurosci. Res. 59:117–125 [PubMed] [Google Scholar]
- 15. Kim I, Kim CH, Kim JH, Lee J, Choi JJ, Chen ZA, Lee MG, Chung KC, Hsu CY, Ahn YS. 2004. Pyrrolidine dithiocarbamate and zinc inhibit proteasome-dependent proteolysis. Exp. Cell Res. 298:229–238 [DOI] [PubMed] [Google Scholar]
- 16. Peters J, Franke W, Kleinschmidt J. 1994. Distinct 19 S and 20 S subcomplexes of the 26 S proteasome and their distribution in the nucleus and the cytoplasm. J. Biol. Chem. 269:7709–7718 [PubMed] [Google Scholar]
- 17. Goldberg A. 2007. Functions of the proteasome: from protein degradation and immune surveillance to cancer therapy. Biochem. Soc. Trans. 35:12–17 [DOI] [PubMed] [Google Scholar]
- 18. Pickart CM. 2004. Back to the future with ubiquitin. Cell 116:181–190 [DOI] [PubMed] [Google Scholar]
- 19. Banks L, Pim D, Thomas M. 2003. Viruses and the 26S proteasome: hacking into destruction. Trends Biochem. Sci. 28:452–459 [DOI] [PubMed] [Google Scholar]
- 20. Boutell C, Sadis S, Everett RD. 2002. Herpes simplex virus type 1 immediate-early protein ICP0 and its isolated RING finger domain act as ubiquitin E3 ligases in vitro. J. Virol. 76:841–850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Van Sant C, Hagglund R, Lopez P, Roizman B. 2001. The infected cell protein 0 of herpes simplex virus 1 dynamically interacts with proteasomes, binds and activates the cdc34 E2 ubiquitin-conjugating enzyme, and possesses in vitro E3 ubiquitin ligase activity. Proc. Natl. Acad. Sci. U. S. A. 98:8815–8820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Boutell C, Orr A, Everett RD. 2003. PML residue lysine 160 is required for the degradation of PML induced by herpes simplex virus type 1 regulatory protein ICP0. J. Virol. 77:8686–8694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Everett RD, Rechter S, Papior P, Tavalai N, Stamminger T, Orr A. 2006. PML contributes to a cellular mechanism of repression of herpes simplex virus type 1 infection that is inactivated by ICP0. J. Virol. 80:7995–8005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chelbi-Alix MK. 1999. Herpes virus induced proteasome-dependent degradation of the nuclear bodies-associated PML and Sp100 proteins. Oncogene 18:935–941 [DOI] [PubMed] [Google Scholar]
- 25. Lomonte P, Morency E. 2007. Centromeric protein CENP-B proteasomal degradation induced by the viral protein ICP0. FEBS Lett. 581:658–662 [DOI] [PubMed] [Google Scholar]
- 26. Everett RD, Earnshaw WC, Findlay J, Lomonte P. 1999. Specific destruction of kinetochore protein CENP-C and disruption of cell division by herpes simplex virus immediate-early protein Vmw110. EMBO J. 18:1526–1538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lomonte P, Sullivan KF, Everett RD. 2001. Degradation of nucleosome-associated centromeric histone H3-like protein CENP-A induced by herpes simplex virus type 1 protein ICP0. J. Biol. Chem. 276:5829–5835 [DOI] [PubMed] [Google Scholar]
- 28. Fukuyo Y, Horikoshi N, Ishov AM, Silverstein SJ, Nakajima T. 2011. The herpes simplex virus immediate-early ubiquitin ligase ICP0 induces degradation of the ICP0 repressor protein E2FBP1. J. Virol. 85:3356–3366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Delboy MG, Roller DG, Nicola AV. 2008. Cellular proteasome activity facilitates herpes simplex virus entry at a postpenetration step. J. Virol. 82:3381–3390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Delboy MG, Nicola AV. 2011. A pre-immediate-early role for tegument ICP0 in the proteasome-dependent entry of herpes simplex virus. J. Virol. 85:5910–5918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Uchide N, Ohyama K, Bessho T, Yuan B, Yamakawa T. 2002. Effect of antioxidants on apoptosis induced by influenza virus infection: inhibition of viral gene replication and transcription with pyrrolidine dithiocarbamate. Antiviral Res. 56:207–217 [DOI] [PubMed] [Google Scholar]
- 32. Krenn B, Holzer B, Gaudernak E, Triendl A, Van Kuppeveld F, Seipelt J. 2005. Inhibition of polyprotein processing and RNA replication of human rhinovirus by pyrrolidine dithiocarbamate involves metal ions. J. Virol. 79:13892–13899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lanke K, Krenn B, Melchers W, Seipelt J, van Kuppeveld F. 2007. PDTC inhibits picornavirus polyprotein processing and RNA replication by transporting zinc ions into cells. J. Gen. Virol. 88:1206–1217 [DOI] [PubMed] [Google Scholar]
- 34. Furuta S, Ortiz F, Sun XZ, Wu HH, Mason A, Momand J. 2002. Copper uptake is required for pyrrolidine dithiocarbamate-mediated oxidation and protein level increase of p53 in cells. Biochem. J. 365:639–648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kim CH, Kim JH, Hsu CY, Ahn YS. 1999. Zinc is required in pyrrolidine dithiocarbamate inhibition of NF-κB activation. FEBS Lett. 449:28–32 [DOI] [PubMed] [Google Scholar]
- 36. Everett R. 1984. Trans activation of transcription by herpes virus products: requirement for two HSV-1 immediate-early polypeptides for maximum activity. EMBO J. 3:3135–3141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Everett R. 1984. A detailed analysis of an HSV-1 early promoter: sequences involved in trans-activation by viral immediate-early gene products are not early-gene specific. Nucleic Acids Res. 12:3037–3056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. O'Hare P, Hayward GS. 1985. Evidence for a direct role for both the 175,000-and 110,000-molecular-weight immediate-early proteins of herpes simplex virus in the transactivation of delayed-early promoters. J. Virol. 53:751–760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. O'Hare P, Hayward GS. 1985. Three trans-acting regulatory proteins of herpes simplex virus modulate immediate-early gene expression in a pathway involving positive and negative feedback regulation. J. Virol. 56:723–733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Everett R. 1987. A detailed mutational analysis of Vmw110, a trans-acting transcriptional activator encoded by herpes simplex virus type 1. EMBO J. 6:2069–2076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gelman IH, Silverstein S. 1985. Identification of immediate early genes from herpes simplex virus that transactivate the virus thymidine kinase gene. Proc. Natl. Acad. Sci. U. S. A. 82:5265–5269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lopez P, Van Sant C, Roizman B. 2001. Requirements for the nuclear-cytoplasmic translocation of infected-cell protein 0 of herpes simplex virus 1. J. Virol. 75:3832–3840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. McLean T, Bachenheimer S. 1999. Activation of cJUN N-terminal kinase by herpes simplex virus type 1 enhances viral replication. J. Virol. 73:8415–8426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zachos G, Clements B, Conner J. 1999. Herpes simplex virus type 1 infection stimulates p38/c-Jun N-terminal mitogen-activated protein kinase pathways and activates transcription factor AP-1. J. Biol. Chem. 274:5097–5103 [DOI] [PubMed] [Google Scholar]
- 45. Karaca G, Hargett D, McLean TI, Aguilar J, Ghazal P, Wagner EK, Bachenheimer SL. 2004. Inhibition of the stress-activated kinase, p38, does not affect the virus transcriptional program of herpes simplex virus type 1. Virology 329:142–156 [DOI] [PubMed] [Google Scholar]
- 46. Mehle A, Strack B, Ancuta P, Zhang C, McPike M, Gabuzda D. 2004. Vif overcomes the innate antiviral activity of APOBEC3G by promoting its degradation in the ubiquitin-proteasome pathway. J. Biol. Chem. 279:7792–7798 [DOI] [PubMed] [Google Scholar]
- 47. Shamu CE, Flierman D, Ploegh HL, Rapoport TA, Chau V. 2001. Polyubiquitination is required for US11-dependent movement of MHC class I heavy chain from endoplasmic reticulum into cytosol. Mol. Biol. Cell 12:2546–2555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Schubert U, Antón LC, Bačík I, Cox JH, Bour S, Bennink JR, Orlowski M, Strebel K, Yewdell JW. 1998. CD4 glycoprotein degradation induced by human immunodeficiency virus type 1 Vpu protein requires the function of proteasomes and the ubiquitin-conjugating pathway. J. Virol. 72:2280–2288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Luo H, Zhang J, Dastvan F, Yanagawa B, Reidy MA, Zhang HM, Yang D, Wilson JE, McManus BM. 2003. Ubiquitin-dependent proteolysis of cyclin D1 is associated with coxsackievirus-induced cell growth arrest. J. Virol. 77:1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Luo H, Zhang J, Cheung C, Suarez A, McManus BM, Yang D. 2003. Proteasome inhibition reduces coxsackievirus B3 replication in murine cardiomyocytes. Am. J. Pathol. 163:381–385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Everett RD, Freemont P, Saitoh H, Dasso M, Orr A, Kathoria M, Parkinson J. 1998. The disruption of ND10 during herpes simplex virus infection correlates with the Vmw110- and proteasome-dependent loss of several PML isoforms. J. Virol. 72:6581–6591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Pokrovskaja K, Mattsson K, Kashuba E, Klein G, Szekely L. 2001. Proteasome inhibitor induces nucleolar translocation of Epstein-Barr virus-encoded EBNA-5. J. Gen. Virol. 82:345–358 [DOI] [PubMed] [Google Scholar]
- 53. Satheshkumar P, Anton LC, Sanz P, Moss B. 2009. Inhibition of the ubiquitin-proteasome system prevents vaccinia virus DNA replication and expression of intermediate and late genes. J. Virol. 83:2469–2479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Greene W, Zhang W, He M, Witt C, Ye F, Gao SJ. 2012. The ubiquitin/proteasome system mediates entry and endosomal trafficking of Kaposi's sarcoma-associated herpesvirus in endothelial cells. PLoS Pathog. 8:e1002703. 10.1371/journal.ppat.1002703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Karpe YA, Meng XJ. 2012. Hepatitis E virus replication requires an active ubiquitin-proteasome system. J. Virol. 86:5948–5952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Teale A, Campbell S, Van Buuren N, Magee WC, Watmough K, Couturier B, Shipclark R, Barry M. 2009. Orthopoxviruses require a functional ubiquitin-proteasome system for productive replication. J. Virol. 83:2099–2108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Patel A, Hanson J, McLean TI, Olgiate J, Hilton M, Miller WE, Bachenheimer SL. 1998. Herpes simplex virus type 1 induction of persistent NF-κB nuclear translocation increases the efficiency of virus replication. Virology 247:212–222 [DOI] [PubMed] [Google Scholar]
- 58. Gregory D, Hargett D, Holmes D, Money E, Bachenheimer S. 2004. Efficient replication by herpes simplex virus type 1 involves activation of the IκB kinase-IκB-p65 pathway. J. Virol. 78:13582–13590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Regad T, Chelbi-Alix MK. 2001. Role and fate of PML nuclear bodies in response to interferon and viral infections. Oncogene 20:7274–7286 [DOI] [PubMed] [Google Scholar]
- 60. Everett RD, Chelbi-Alix MK. 2007. PML and PML nuclear bodies: implications in antiviral defence. Biochimie 89:819–830 [DOI] [PubMed] [Google Scholar]
- 61. Saffert RT, Kalejta RF. 2008. Promyelocytic leukemia-nuclear body proteins: herpesvirus enemies, accomplices, or both? Future Virol. 3:265–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Kapuria V, Peterson LF, Showalter H, Kirchhoff PD, Talpaz M, Donato NJ. 2011. Protein cross-linking as a novel mechanism of action of a ubiquitin-activating enzyme inhibitor with anti-tumor activity. Biochem. Pharmacol. 82:341–349 [DOI] [PubMed] [Google Scholar]
- 63. Park SH, Choi WS, Yoon SY, Ahn YS, Oh YJ. 2004. Activation of NF-κB is involved in 6-hydroxydopamine- but not MPP+-induced dopaminergic neuronal cell death: its potential role as a survival determinant. Biochem. Biophy. Res. Comm. 322:727–733 [DOI] [PubMed] [Google Scholar]
- 64. Bureau C, Bernad J, Chaouche N, Orfila C, Béraud M, Gonindard C, Alric L, Vinel JP, Pipy B. 2001. Nonstructural 3 protein of hepatitis C virus triggers an oxidative burst in human monocytes via activation of NADPH oxidase. J. Biol. Chem. 276:23077–23083 [DOI] [PubMed] [Google Scholar]
- 65. Israel N, Gougerot-Pocidalo MA. 1997. Oxidative stress in human immunodeficiency virus infection. Cell. Mol. Life Sci. 53:864–870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Pace GW, Leaf CD. 1995. The role of oxidative stress in HIV disease. Free Radic. Biol. Med. 19:523–528 [DOI] [PubMed] [Google Scholar]
- 67. Schachtele SJ, Hu S, Little MR, Lokensgard JR. 2010. Herpes simplex virus induces neural oxidative damage via microglial cell Toll-like receptor-2. J. Neuroinflamm. 7:35. 10.1186/1742-2094-7-35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Gonzalez-Dosal R, Horan KA, Rahbek SH, Ichijo H, Chen ZJ, Mieyal JJ, Hartmann R, Paludan SR. 2011. HSV infection induces production of ROS, which potentiate signaling from pattern recognition receptors: role for S-glutathionylation of TRAF3 and 6. PLoS Pathog. 7:e1002250. 10.1371/journal.ppat.1002250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Schacker T, Zeh J, Hu H, Shaughnessy M, Corey L. 2002. Changes in plasma human immunodeficiency virus type 1 RNA associated with herpes simplex virus reactivation and suppression. J. Infect. Dis. 186:1718–1725 [DOI] [PubMed] [Google Scholar]
- 70. Everett RD. 2000. ICP 0, a regulator of herpes simplex virus during lytic and latent infection. Bioessays 22:761–770 [DOI] [PubMed] [Google Scholar]
- 71. Everett RD, Boutell C, Orr A. 2004. Phenotype of a herpes simplex virus type 1 mutant that fails to express immediate-early regulatory protein ICP0. J. Virol. 78:1763–1774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Thomas M, Pim D, Banks L. 1999. The role of the E6-p53 interaction in the molecular pathogenesis of HPV. Oncogene 18:7690–7700 [DOI] [PubMed] [Google Scholar]
- 73. Klinger PP, Schubert U. 2005. The ubiquitin-proteasome system in HIV replication: potential targets for antiretroviral therapy. Expert Rev. Anti Infect. Ther. 3:61–79 [DOI] [PubMed] [Google Scholar]
- 74. Widjaja I, de Vries E, Tscherne DM, García-Sastre A, Rottier PJM, de Haan CAM. 2010. Inhibition of the ubiquitin-proteasome system affects influenza A virus infection at a postfusion step. J. Virol. 84:9625–9631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Si X, Gao G, Wong J, Wang Y, Zhang J, Luo H. 2008. Ubiquitination is required for effective replication of coxsackievirus B3. PLoS One 3:e2585. 10.1371/journal.pone.0002585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Raaben M, Posthuma CC, Verheije MH, te Lintelo EG, Kikkert M, Drijfhout JW, Snijder EJ, Rottier PJM, de Haan CAM. 2010. The ubiquitin-proteasome system plays an important role during various stages of the coronavirus infection cycle. J. Virol. 84:7869–7879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Kanlaya R, Pattanakitsakul S, Sinchaikul S, Chen ST, Thongboonkerd V. 2010. The ubiquitin-proteasome pathway is important for dengue virus infection in primary human endothelial cells. J. Proteome Res. 9:4960–4971 [DOI] [PubMed] [Google Scholar]
- 78. Neznanov N, Dragunsky EM, Chumakov KM, Neznanova L, Wek RC, Gudkov AV, Banerjee AK. 2008. Different effect of proteasome inhibition on vesicular stomatitis virus and poliovirus replication. PLoS One 3:e1887. 10.1371/journal.pone.0001887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Everett RD, Orr A, Preston CM. 1998. A viral activator of gene expression functions via the ubiquitin-proteasome pathway. EMBO J. 17:7161–7169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. La Frazia S, Amici C, Santoro MG. 2006. Antiviral activity of proteasome inhibitors in herpes simplex virus-1 infection: role of nuclear factor-kappaB. Antiviral Ther. 11:995–1004 [PubMed] [Google Scholar]