

Abstract
Mitochondria are important for the viral life cycle, mainly by providing the energy required for viral replication and assembly. A highly complex interaction with mitochondria is exerted by rubella virus (RV), which includes an increase in the mitochondrial membrane potential as a general marker for mitochondrial activity. We aimed in this study to provide a more comprehensive picture of the activity of mitochondrial respiratory chain complexes I to IV. Their activities were compared among three different cell lines. A strong and significant increase in the activity of mitochondrial respiratory enzyme succinate:ubiquinone oxidoreductase (complex II) and a moderate increase of ubiquinol:cytochrome c oxidoreductase (complex III) were detected in all cell lines. In contrast, the activity of mitochondrial respiratory enzyme cytochrome c oxidase (complex IV) was significantly decreased. The effects on mitochondrial functions appear to be RV specific, as they were absent in control infections with measles virus. Additionally, these alterations of the respiratory chain activity were not associated with an elevated transcription of oxidative stress proteins, and reactive oxygen species (ROS) were induced only marginally. Moreover, protein and/or mRNA levels of markers for mitochondrial biogenesis and structure were elevated, such as nuclear respiratory factors (NRFs) and mitofusin 2 (Mfn2). Together, these results establish a novel view on the regulation of mitochondrial functions by viruses.
INTRODUCTION
Mitochondria are required for the maintenance of cell function and integrity. Their most important role lies in energy production, but they are also at the intersection of regulatory pathways that coordinate metabolic processes (e.g., calcium homeostasis and cellular proliferation), cellular fate (apoptosis and necrosis), and antiviral defense (1, 2). Even a participation of mitochondria in the innate immune response was identified (2). There are a number of viruses that interfere with the important role of mitochondria in cellular antiviral response pathways, mainly with the regulation of apoptosis (1). Additionally, as the powerhouses of the cell, mitochondria provide most of the energy for viral replication and assembly. Up to 90% of the cellular ATP is produced in the inner mitochondrial membrane (IMM) by oxidative phosphorylation (OXPHOS), (3). OXPHOS comprises a series of redox reactions carried out by four multisubunit enzyme complexes (complexes I to IV) of the electron transport chain (ETC). Electrons are transferred in a stepwise manner through this series of electron carriers from NADH (and FADH2) as reducing equivalents to the final acceptor molecular oxygen. A small percentage of electrons that are transported through the respiratory complexes leaks out, which results in generation of reactive oxygen species (ROS). The main ROS species is hydrogen peroxide, which is converted to water by enzymes such as catalase, peroxiredoxin, or glutathione peroxidase as components of the cellular antioxidant system. Respiratory complexes I and III are the main generators of mitochondrial ROS (4). The energy that is released during the flow of electrons is stored as an electrochemical proton gradient across the IMM, which is finally used by the ATP synthase (complex V) to generate ATP (3). A voltage potential, the mitochondrial membrane potential (Δψm), and a pH gradient are part of this proton motive force. Therefore, Δψm serves as a general indicator for mitochondrial activity.
Mitochondria also participate in the assembly of membrane-associated viral replication complexes or might even function as the replication organelle itself. They also provide host replication factors (5). A prominent example for these replication factors is the mitochondrial matrix protein p32 (gC1q-R), (5, 6). Among its viral interaction partners is rubella virus (RV), an efficient teratogen and the only member of the Rubivirus genus within the family Togaviridae. The molecular mechanisms involved in the teratogenic outcome of RV infection of seronegative women especially during the first trimester of pregnancy are not yet clear. RV itself exerts a complex interaction with mitochondria. The RV capsid protein localizes to mitochondria and associates with p32 (7), which is required for RV replication (8). Additionally, the perinuclear accumulation of mitochondria in the proximity of RV replication complexes requires p32 (9). Moreover, the RV capsid protein is so far the only known RNA virus-encoded protein that interferes with protein import into mitochondria (10). While many viral infections affect the mitochondrial energy and oxidative balance, RV appears to exert an overall positive influence on mitochondrial and bioenergetic function. This is reflected by an increase in respiratory chain (RC) complex III activity, in the Δψm, and by a high level of intracellular ATP in RV-infected cells in comparison to the mock-infected control even at late stages of infection (9).
In this study, the modulation of mitochondrial respiratory chain complexes by RV was addressed systematically. The results shown indicate that in the course of RV infection, the activities of all four RC complexes are significantly altered in three different cell lines irrespective of their metabolic (tumorogenic and nontumorogenic) background. Especially the increase in respiratory complex II activity was profound, revealing an increase by up to 141% compared to the control. These alterations appear to be RV specific, as they were absent after application of measles virus (MV) as a control virus. Additionally, the increase of complex II activity was only marginally associated with oxidative stress induction. A mechanism for the positive influence of RV on the ETC is discussed. The results presented reveal a complex interaction of RV with mitochondria, which ensures ongoing supply of energy during its long replication cycle. A deeper understanding of the modulation of mitochondrial respiratory complexes by viruses such as RV could contribute to our understanding of mitochondrial functions and the treatment of these viruses with metabolic antagonists (11).
MATERIALS AND METHODS
Reagents.
Digitonin was purchased from Fluka Biochemica (Ulm, Germany). All other reagents and general laboratory chemicals were from Sigma-Aldrich (Hannover, Germany). Media, supplements for cell culture, and fetal calf serum (FCS) were purchased from Life Technologies-Invitrogen (Darmstadt, Germany). A549 and Vero cells were from the American Type Culture Collection (ATCC; Manassas, VA). MCF-7 cells were obtained from IAZ (Munich, Germany).
Cell culture and virus infection.
The epithelial cell line MCF-7 was maintained in Eagle's minimum essential medium (MEM) with 2 mM l-glutamine, 1 mM sodium pyruvate, 1% nonessential amino acids, 10% FCS, and antibiotics. Vero cells and A549 cells were propagated in Dulbecco's modified Eagle's medium (DMEM) with antibiotics. Medium for A549 cells was supplemented with 1 mM HEPES. All cell lines were cultivated at 37°C under 5% CO2 in a humidified incubator. All cell lines were either mock infected (control) with medium excluding virus or infected with RV wild-type strain Therien at a multiplicity of infection (MOI) of 5 PFU per cell. MV laboratory strain Edmonston was applied to A549 and MCF-7 cells at an MOI of 5 and to Vero cells at an MOI of 0.01. Fresh medium was added after 2 h of incubation at 37°C. Assays were performed at 12, 24, 48, and 72 h postinfection (hpi). Viral titers were determined by standard plaque assay as described previously (12).
Isolation of a mitochondrion-enriched cellular fraction.
To obtain a mitochondrion-enriched cellular fraction, intact mitochondria were isolated from 60-mm dishes in a medium of 0.2 mM EDTA, 0.25 M sucrose, and 10 mM Tris-HCl (pH 7.8) (mito buffer) by differential centrifugation of cell homogenates obtained after mechanical disruption of cells (MagNA Lyser; Roche, Mannheim, Germany). Differential centrifugation was done according to published protocols (www.Mitosciences.com/PDF/mitos.pdf [MitoSciences]and technical bulletin for mitochondrion isolation kit [Sigma-Aldrich]). Briefly, 10 mM triethanolamine and 0.1 mg/ml of digitonin were added before homogenization. Undisrupted cells and nuclei were removed by centrifugation at 1,000 × g for 10 min at 4°C. Mitochondria were pelleted from the supernatant by centrifugation at 3,500 × g for 15 min at 4°C and subsequently solubilized in 100 μl of mito buffer supplemented with 0.5 mM phenylmethylsulfonyl fluoride (PMSF) and 0.05 mM pepstatin A. The protein concentration was determined by the bicinochinic acid (BCA) test. The yield was typically 80 to 150 μg (in a total volume of 100 μl) per 60-mm dish of cultured cells.
Assays for the activities of respiratory complexes I to IV.
Freshly prepared mitochondria (10 μl of a 0.4-μg/μl mitochondrial fraction for complexes I, III, and IV and 10 μl of an undiluted mitochondrial preparation for complex II) were used for the spectrophotometric determination of the activity of RC complexes I to IV by biochemical assays with a total volume of 200 μl. Protocols were adapted from previous publications (references 13 and 14 for complexes I and II, reference 15 for complex III, and reference 13 for complex IV). Activities of respiratory chain complexes were calculated as units per 1 μg of isolated mitochondrial fraction and then normalized to citrate synthase activity. Complex I (NADH:ubiquinone oxidoreductase) activity was determined in assay buffer composed of 25 mM potassium phosphate, 3.5 mg/ml of BSA, 0.06 mM 2,6-dichlorophenolindophenol, 0.0125 mM antimycin A, and 0.7 mM decylubiquinone. The reaction was started by adding 0.2 mM NADH. Complex I activity was measured spectrophotometrically by the oxidation of NADH with decylubiquinone at 600 nm. Complex II (succinate:ubiquinone oxidoreductase) activity was determined in assay buffer containing 25 mM potassium phosphate, 2 mg/ml of BSA, 20 mM succinate, 0.05 mM DCPIP, 20 mM NaN3, and 0.1 mM ATP. The reaction was started through the addition of 0.1 mM decylubiquionone. Complex II activity was assessed spectrophotometrically by the oxidation of succinate with ubiquinone at 600 nm. Complex III (ubiquinol:ferrocytochrome c oxidoreductase) activity was assessed in assay buffer containing 50 mM Tris-HCl, 4 mM NaN3, 0.1 mg/ml of BSA, and 0.05% (vol/vol) Tween 20. The reaction was started through addition of 0.25 mM decylubiquinol and 0.0625 mM cytochrome c. Reduction of cytochrome c with decylubiquinol was determined at 550 nm. Complex IV (cytochrome c oxidase) activity was determined through addition of 150 μl of reduced cytochrome c (50 μM) to 10 μl of diluted mitochondrial fraction. Complex IV activity was measured by the oxidation of reduced cytochrome c at 550 nm.
Measurement of ROS.
Intracellular ROS generation was detected using the probe 2′,7′-dichlorofluorescein diacetate (DCF-DA) as a nonfluorescent cell-permeative compound. Intracellular esterases cleave the acetate groups such that the nonfluorescent dye 2′,7′-dichlorofluorescein (DCF) is retained intracellularly, which, in turn, is oxidized by intracellular ROS (mainly by hydrogen peroxides) and thus becomes fluorescent. The fluorescent product DCF was detected with an excitation wavelength of 485 nm and an emission wavelength of 530 nm on a fluorescence microscope (DM IRB; Leica, Wetzlar, Germany) and a FACSCalibur flow cytometer (Becton, Dickinson, San Jose, CA) using CellQuest software. Vero cells were plated at a density of 1 × 106 per 60-mm dish and after 24 h either mock or RV infected. At 72 hpi, Vero cells were incubated with DMEM containing 1% FCS and 70 μM DCF-DA for 30 min at 37°C in the dark. After being washed with phosphate-buffered saline (PBS), cells were trypsinized, pelleted, and resuspended in DMEM with 1% FCS. After an incubation period of 60 min under shaking conditions, cells were pelleted, fixed with 2% (wt/vol) paraformaldehyde (PFA), and resuspended in PBS. Thereafter, intracellular ROS generation was measured by flow cytometry. Three independent samples of 10,000 cells were analyzed for each experimental condition, and the percentage of RV-infected cells with high DCF fluorescence was calculated in relation to the corresponding mock sample as described below. As negative controls, both mock-infected, unstained and mock-infected, dye-loaded Vero cells were employed. As a positive control, oxidative activity was increased through preincubation of mock-infected Vero cells with 30 μM oligomycin and 0.03% H2O2 for 15 h before loading with the DCF-DA dye. For fluorescence microscopy analysis, Vero cells were loaded with DCF-DA as described above. During an additional incubation period of 1 h at 37°C, cells were incubated with 5 μg/ml of Hoechst bisbenzamide 33285 (Invitrogen). Thereafter, cells were directly subjected to microscopic analysis. Postacquisition analysis was performed with Corel Photo-Paint 11 software with minimal alterations to background and contrast, which were applied equally to samples and controls.
Western blot analysis.
Equal amounts (30 to 70 μg) of mitochondrion-enriched fractions were separated on a 10% bisacrylamide gel electrophoresis gel under denaturing conditions and transferred to a polyvinylidene difluoride (PVDF) membrane. After blocking of the membrane with 5% dry milk, probing was performed with a 1:200 dilution of rabbit polyclonal antibody against Mfn2 and rabbit polyclonal antibody against VDAC1 (Santa Cruz Biotechnology, Inc., Heidelberg, Germany), a 1:200 dilution of polyclonal antibody to VDAC, and a 1:500 dilution of rabbit SDHA (see below) polyclonal antibody (Acris Antibodies, Herford, Germany) overnight at 4°C. Incubation with secondary antibody and detection by chemiluminescence were done as described previously (9).
Gene expression analysis by RT-qPCR.
RNA was prepared and samples were processed as described previously (16). Briefly, total RNA was isolated with TriFast reagent (PeqLab, Erlangen, Germany), and 1.25 μg of RNA was reverse transcribed. Quantitative real-time PCR (RT-qPCR) was performed with the SYBR green dye technique on a Light Cycler system (Roche) to determine the mRNA expression level of heme oxygenase 1 (HMOX-1), NF-κB, nucleus-encoded subunit NDUFB1 of complex I, subunits A and B (SDHA and SDHB) of complex II (succinate dehydrogenase [SDH]), subunit UQCRC1 of complex III, COX4I1 of complex IV, and nuclear factor E2-related factor 1 (NRF1) and NRF2. Gene expression was calculated with geNorm, version 3.5, and normalized to the housekeeping genes for hypoxanthine phosphoribosyltransferase 1 (HPRT1) and HUEL (solute carrier family 30 member 9). For quantification of viral subgenomic and genomic RNAs, 1 μg of RNA was reverse transcribed and the primer pair TBR2 and TBR7 was employed to amplify a 177-bp region from the capsid protein. A plasmid standard curve was generated in the range of 1 × 109 to 1 × 102 based on RV infectious cDNA clone Robo503 (a kind gift of T. Frey, Georgia State University, Atlanta, GA). The standard curve was used to determine the copy number of viral RNA in each respective sample. Primer sequences are given in Table 1. Primer specificity for the selected cellular genes was confirmed by sequence analysis.
Table 1.
Sequences of oligonucleotide primers and annealing temperatures used for RT-qPCR
| Gene | Sequence (5′–3′)a | Temp (°C) | Reference or sourceb |
|---|---|---|---|
| HPRT1 | TGACACTGGCAAAACAATGCA | 62 | 16 |
| GGTCCTTTTCACCAGCAAGCT | |||
| HUEL | TCAGACGACGAAGTCCCCATGAAG | 65 | 16 |
| TCCTTACGCATTTTTTCCTCTGGC | |||
| NRF1 | ATCAGCAAACGCAAACACAG | 59 | 49 |
| TTTGTTCCACCTCTCCATCAG | |||
| NRF2 | CAGACTGGCTGGGAACTTCT | 59 | 49 |
| AGCACCATGCTTAAGTAAAACCTC | |||
| NDUFB1 | GGACCACTGGGTTCATGTTC | 59 | This study |
| GGTAACTTCTTCACTGGGTTGC | |||
| SDHA | TGGGAACAAAGGGCATCTG | 62 | 50 |
| CCACCACTGCATCAAATTCATG | |||
| SDHB | AGTTGACTCTACTTTGACCTTCCGAAG | 63 | 51 |
| GACCTTATTGAGGTTGGTGTCAATCCT | |||
| UQCRC1 | TTACAAGGCCCCTCGAATG | 59 | This study |
| GGCGAGGTCTAACAGTTGCT | |||
| COX4I1 | TCCACCTCTGTGTGTGTACGA | 59 | This study |
| CAGACAGGTGCTTGACATGG | |||
| HMOX-1 | GGGTGATAGAAGAGGCCAAGA | 59 | This study |
| AGCTCCTGCAACTCCTCAAA | |||
| NF-κB/p65 | ATCTGCCGAGTGAACCGAAACT | 63 | 52 |
| CCAGCCTGGTCCCGTGAA A | |||
| Mfn2 | TGCCTCAGAGCCCGAGTA | 60 | This study |
| CTGGTACAACGCTCCATGTG | |||
| TBR2 | CAACACGCCGCACGGACAAC | 69 | |
| TBR7 | CCACAAGCCGCGAGCAGT CA | 69 |
For each set of primers, the primer in the sense orientation is listed first and the primer in the antisense orientation is listed second.
All primers designed in this study were designed using the ProbeFinder software (version 2.45. Roche).
Statistical analyses.
All data in the diagrams are expressed as means ± standard errors (SE) of at least three individual experiments. Level of significance from the control population is indicated by asterisks (*, P < 0.05; **, P < 0.01; and ***, P < 0.001). Standard two-tailed paired Student's t tests were used for evaluating the significance of the difference between the means of RV- and mock-infected cultures.
RESULTS
Effects of RV infection on the activities of respiratory chain complexes I to IV.
Three RV-permissive cell lines with different metabolic backgrounds (Vero as a nontumorogenic cell line, the human breast adenocarcinoma cell line MCF-7, and the human lung carcinoma cell line A549) were used for a comprehensive analysis of the activities of RC complexes under RV infection. An MOI of 5 was chosen to achieve a high initial infection rate. All three cell lines are susceptible to high replication of RV; however, the replication kinetics differ among them (Fig. 1A). A549 cells are characterized by a high initial RNA load already observable at 12 h postinfection (hpi). In contrast to A549 cells, in Vero and MCF-7 cells viral RNA reached a plateau and was maintained at a high level until 72 hpi. All three cell lines were mock infected, and the activities of RC complexes I to IV were determined after incubation periods of 24, 48, and 72 h. Figure 1B indicates that the activity of RC complex I exceeded the activity of the remaining complexes. MCF-7 cells possessed a slightly higher activity of complex II. However, the activity rates of all four RC complexes were similar between the three cell lines tested. Thereafter, Vero, MCF-7, and A549 cells were infected with RV and samples were collected at 12, 24, 48, and 72 hpi in order to determine RC complex activity as infection proceeds (corresponding mock-infected controls were set to 100%). Figure 2 shows the alterations in the activities of mitochondrial RC complexes, which are further summarized in Table 2. Peak activities of respiratory chain complexes I to IV and the time point of RV-induced alterations were determined for the indicated cell lines. RV-induced alterations in the activities of complexes I to IV were similar between the three cell lines tested, especially between Vero (Fig. 2A) and MCF-7 (Fig. 2B) cells. However, Vero (decrease to 64% ± 10%) and MCF-7 (increase to 145% ± 14%) cells differ in the activity of RC complex IV at 48 hpi. In all three cell lines tested, alterations were noticeable only after 12 hpi. Complexes I and IV are apparently least altered in RV-infected cells. However, complex I activity is strongly reduced in RV-infected A549 cells, to 53% ± 5%, at 72 hpi (Fig. 2C). In all three cell lines tested, complex II represents the main target of RV-induced alterations within the ETC. In comparison to that in the mock-infected population, its activity was significantly increased by RV infection, by 141% (to 241% ± 15%) in A549 cells, by 131% (to 231% ± 31%) in MCF-7 cells, and by 99 (to 199% ± 31%) in Vero cells. Hence, the extents of upregulation of complex II activity were very similar between the three cell lines. The only noticeable difference in complex II activity between RV-infected Vero and MCF-7 cells (72 hpi) versus A549 cells (24 hpi) lies in the time point of complex II peak activity. Complex IV activity, except at 48 hpi for MCF-7 cells, is significantly downregulated in all three cell lines. The highest level of downregulation of complex IV, by 46% (to 54% ± 15%), was observed for A549 cells. The activity of complex III was moderately increased, by 60% (to 160% ± 27%) in A549 cells, by 42% (to 142% ± 23%) in MCF-7 cells, and by 44% (to 144% ± 17%) in Vero cells. The same applies for complex I, with a slight increase, of 22% (increase to 122% ± 26%), in Vero cells and an increase of 53% (increase to 153% ± 26%) in A549 cells. MCF-7 cells, however, showed no increase in complex I activity. The next attempt was to determine the specificity of these alterations. As a control virus, MV strain Edmonston was used, as all three cell lines applied in this study are susceptible to MV. MV, like RV, replicates in the cytoplasm, but in contrast to RV, it is not known to be associated with mitochondria. As with RV, MCF-7 and A549 cells could be infected with MV at an MOI of 5. Due to extensive syncytium formation, Vero cells require a lower MOI, 0.01. Samples were collected at 24 and 48 hpi, as MV induces a strong cytopathic effect after 48 h of infection. With just a few exceptions, the activities of most RC complexes remained almost unchanged after MV infection (Fig. 3). Among the three cell lines tested, MCF-7 showed the strongest alterations, which are reflected by increases in complex I, II, and IV activities at 48 hpi by 32% (to 132% ± 1%), by 34% (to 134% ± 15%), and by 34% (to 134% ± 12%), respectively. In Vero cells, only complex IV activity was increased, by 69% (to 169% ± 15%) at 48 hpi. The increase in complex IV activity in Vero cells could be due to extensive syncytium formation, which was not observable in A549 or MCF-7 cells. Syncytium formation was reduced after application of MV at an MOI of 0.001, which also lacked the induction of complex IV activity (data not shown).
Fig 1.

(A) Analysis of the replication kinetics of RV in Vero, MCF-7, and A549 cells and (B) activities of mitochondrial respiratory chain complexes I, II, III, and IV. (A) Mean RV genomic and subgenomic RNA copies were determined for 12, 24, 48, and 72 hpi by RT-qPCR. (B) Enzyme activities were determined by spectrophotometric assays and normalized to citrate synthase activity. Vero, MCF-7, and A549 cells were mock infected after 24 h of cultivation. Samples were collected at further incubation periods of 24, 48, and 72 h.
Fig 2.
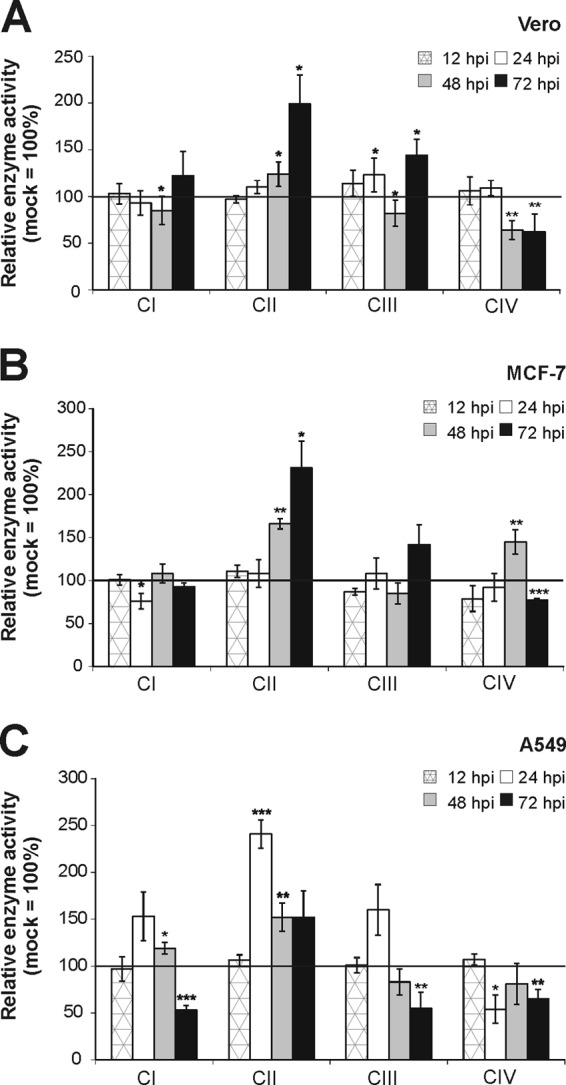
Activities of mitochondrial respiratory chain complexes I, II, III, and IV were measured in mitochondria isolated from RV-infected Vero (A), MCF-7 (B), and A549 (C) cells. Enzyme activities were determined by spectrophotometric assays and normalized to citrate synthase activity. Data are shown as percentages of the corresponding mock-infected controls, which were set as 100%. Level of significance from the control population is indicated by asterisks (*, P < 0.05; **, P < 0.01; and ***, P < 0.001).
Table 2.
Summary of mitochondrial ETC enzyme activities in three different cell lines after RV infection
| No. of days postinfection | % relative enzyme activity in cell linea |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Complex I |
Complex II |
Complex III |
Complex IV |
|||||||||
| A549 | MCF-7 | Vero | A549 | MCF-7 | Vero | A549 | MCF-7 | Vero | A549 | MCF-7 | Vero | |
| 1 | 153 ± 26 (n = 3) | 76 ± 9 (n = 4) | 93 ± 13 (n = 5) | 241 ± 15 (n = 4) | 108 ± 16 (n = 3) | 110 ± 7 (n = 3) | 160 ± 27 (n = 3) | 108 ± 18 (n = 3) | 123 ± 18 (n = 6) | 54 ± 15 (n = 3) | 92 ± 16 (n = 3) | 109 ± 8 (n = 6) |
| 2 | 119 ± 6 (n = 3) | 108 ± 11 (n = 3) | 85 ± 15 (n = 8) | 152 ± 15 (n = 4) | 166 ± 6 (n = 3) | 124 ± 13 (n = 5) | 83 ± 14 (n = 5) | 85 ± 12 (n = 4) | 82 ± 14 (n = 6) | 81 ± 22 (n = 3) | 145 ± 14 (n = 4) | 64 ± 10 (n = 5) |
| 3 | 53 ± 5 (n = 5) | 93 ± 4 (n = 3) | 122 ± 26 (n = 3) | 152 ± 28 (n = 3) | 231 ± 31 (n = 3) | 199 ± 31 (n = 3) | 55 ± 17 (n = 5) | 142 ± 23 (n = 3) | 144 ± 17 (n = 4) | 65 ± 10 (n = 4) | 77 ± 2 (n = 4) | 62 ± 19 (n = 5) |
Values are group means ± SE in comparison with mock-infected cells, set at 100%. n, sample size.
Fig 3.
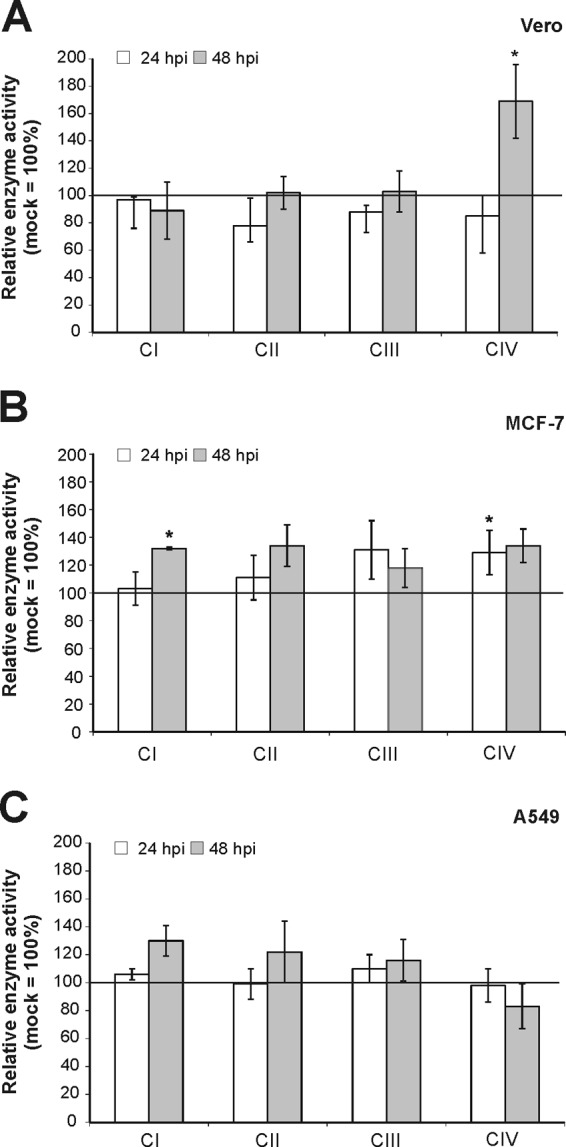
Activities of mitochondrial respiratory chain complexes I, II, III, and IV were measured in mitochondria isolated from MV-infected Vero (A), MCF-7 (B), and A549 (C) cells. Enzyme activities were determined by spectrophotometric assays and normalized to citrate synthase activity. Data are shown as percentages of the corresponding mock-infected controls, which were set as 100%. *, P < 0.05 compared with the control population.
In summary, the RV-induced alteration of the activities of RC enzymes is virus specific, is complex, and occurred in the three different cell lines in similar manners and to similar extents. Especially for complex II, a strong increase, to up to 231% (± 31%), in activity was observed.
Gene expression analysis of oxidative stress markers.
RV infection results in a higher activity of respiratory complexes, especially of complex II (Fig. 2). Based on these results, we hypothesized that the high mitochondrial activity in RV-infected cells could be accompanied by oxidative stress induction. Hence, the effect of RV infection on the mRNA expression of two representative markers for oxidative stress induction was measured by RT-qPCR in Vero and A549 cells, as they are the most frequently used cell lines for RV infection studies. NF-κB regulates as a transcription factor various cellular processes, including immune and stress response and proliferation (17). The predominant form within the mammalian NF-κB family is a heterodimer of family members p65 and p50. Heme oxygenase is a cellular stress protein required for the cleavage of heme to generate biliverdin, a potent antioxidant. Heme oxygenase 1 (HMOX-1) represents its inducible isozyme, which is activated by various stimuli, including oxidative stress, heat shock, and inflammatory agents (18). Figure 4 shows no significant increase in the mRNA expression of NF-κB/p65 in Vero cells or of HMOX-1 in Vero and A549 cells. Only A549 cells displayed an alteration in the mRNA expression of NF-κB/p65 in comparison to the mock-infected controls (set to 100%). The increase in the NF-κB/p65 mRNA level was already significant on the first day of infection (to 212% ± 27%) and increased further until the third day of infection (to 304% ± 111%). To confirm the suitability of NF-κB and HMOX-1 as oxidative stress markers, the respiratory complex V (ATP synthase) inhibitor oligomycin (30 μM) as an inducer of oxidative stress was applied to mock-infected Vero and A549 cells at 48 hpi. Oligomycin treatment resulted in an upregulation of the mRNA expression of NF-κB and HMOX-1, with the exception of HMOX-1 in A549 cells (Fig. 4). However, an increase in HMOX-1 mRNA expression was detected after treatment of A549 cells with 0.03% hydrogen peroxide (data not shown). Hydrogen peroxide was not included as an inducer of oxidative stress in Fig. 4, as there was a broad variation in HMOX-1 and NF-κB mRNA expression. These data indicate that RV-infected cells lack an upregulation of oxidative stress markers. As a next step, these data were to be complemented by the determination of reactive oxygen species (ROS) generation as a second and important parameter for oxidative stress induction.
Fig 4.
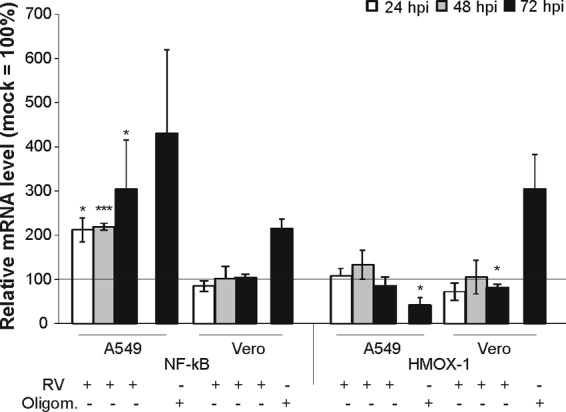
Gene expression of oxidative stress markers in RV-infected Vero and A549 cells. RT-qPCR was applied for analysis of gene expression of the oxidative stress markers HMOX-1 and NF-κB/p65. As a positive control, oligomycin (30 μM) was applied to mock-infected controls at 48 hpi. Samples were collected at 72 hpi for RNA extraction. Data are shown as percentages of the corresponding mock-infected controls and, in the case of the positive control oligomycin, as a percentage of the untreated control. Gene expression was normalized against HUEL and HPRT-1.*, P < 0.05, and ***, P < 0.001, compared with the control population.
Evaluation of ROS generation.
An enhanced electron flow rate could lead to increased electron leakage and, finally, oxidative stress through generation of ROS. Mainly respiratory complexes I and III contribute to generation of ROS (4). To determine, therefore, whether RV infection is accompanied by production of ROS, mock- and RV-infected Vero and A549 cells were incubated at 72 hpi with the dye DCF-DA. This time frame was selected because it is the time point of the highest increase of complex I, II, and III activities in RV-infected Vero cells, which lacked an increase in mRNA expression level of selected markers for oxidative stress induction (Fig. 4). Thus, if ROS is generated, it should be detectable at this time point. Additionally, at 72 hpi, a cytopathic effect (indicative of cytopathogenicity) was clearly visible in RV-infected Vero and A549 cells (data not shown).
The ROS-dependent generation of the fluorescent compound DCF was measured by both flow cytometric (Fig. 5A and C) and microscopic (Fig. 5B and D) analyses. As a positive control, Vero and A549 cells were pretreated with either of the oxidative stress inducers oligomycin (30 μM) and 0.03% H2O2 (0.03%) for 15 h. In comparison to the untreated control, both inducers of ROS resulted in a strong increase in intracellular DCF fluorescence (Fig. 5). In comparison to the mock-infected controls, the flow cytometric analysis revealed for RV-infected Vero and A549 cells (72 hpi) only a minimal increase in ROS levels (Fig. 5A and C, respectively). The extents of ROS generation were comparable between Vero and A549 cells. There was only a slight shift in DCF fluorescence detectable. To be able to calculate the shift in DCF fluorescence in flow cytometry histograms and to compare the results of three independent experiments, the marker M1 was set such that in the right portion of the histogram, 50% of the total DCF-DA-stained and untreated Vero and A549 (mock) cell population was encompassed. The marker was copied to histograms of oligomycin (30 μM)-pretreated or 0.03% H2O2-pretreated or RV-infected Vero and A549 cells. Figure 5A and C also include the mean (±SE) for the marker M1 obtained for the indicated samples in three independent experiments. For Vero cells, the M1 marker included 60% (±2%) of the total cell population for an infection with RV (MOI = 5). For A549 cells, the M1 marker included 66% (±7%) of the total cell population for an infection with RV (MOI = 5).This indicates that RV infection results in only a slight increase of DCF fluorescence. Pretreatment with 30 μM oligomycin or 0.03% H2O2 resulted in Vero cells in an increase in the 50% population included by the marker M1 of 36% (to 86% ± 10%) or of 24% (to 74% ± 6%) and in A549 cells in an increase of 39% (to 89% ± 7%) or of 26% (to 76% ± 6%) (Fig. 5A and C, respectively). The generation of ROS was also analyzed by fluorescence microscopy. Figure 5B and D illustrate the slight increase in ROS generation in RV-infected Vero (Fig. 5Bb) and A549 (Fig. 5Db) cells in comparison to the mock-infected population (Fig. 5Ba and Da), respectively. In contrast to RV, pretreatment of Vero and A549 cells with 30 μM oligomycin resulted in a strong increase in intracellular ROS levels (Fig. 5Bc and Dc). Hence, despite a significant increase in the activities of selected mitochondrial respiratory complexes, RV infection induces only a moderate increase in intracellular ROS compared to that in the mock-infected cell population.
Fig 5.

Flow cytometric (A and C) and microscopic (B and D) analyses of ROS production in RV-infected Vero (A and B) and A549 (C and D) cells using the hydrogen peroxide-sensitive probe DCF-DA. ROSlevels were measured by flow cytometric analysis. Representative flow cytometry profiles of DCF fluorescence are shown for RV- and mock-infected Vero (A) and A549 (C) cells obtained at 72 hpi. Oligomycin (30 μM)-pretreated or 0.03% H2O2-pretreated, mock-infected cells were included as controls. The marker M1 was set to include in the right portion of the flow cytometry histogram 50% of the total DCF-DA-stained and untreated (mock) cell population. This marker was copied to histograms of oligomycin (30 μM)-pretreated or 0.03% H2O2-pretreated or RV-infected cells. Additionally, values are given as the means (±SE) of three independent experiments. a. U., arbitrary unit; n, sample size. Fluorescence microscopy images are shown for intracellular ROS in mock-infected and RV-infected (MOI = 5) Vero (B) and A549 (D) cells at 72 hpi. As positive controls, cells were pretreated with 30 μM oligomycin for 15 h. Cells were stained with DCF-DA (a to c), and nuclei were counterstained with Hoechst 33285 (d to f).
Analysis of the expression of transcription factors regulating mitochondrial respiratory function and of subunits of respiratory complexes.
In an attempt to identify factors that could contribute to the observed changes in mitochondrial activity, the mRNA expression of master regulators of mitochondrial biogenesis and function and of subunits of RC complexes was determined. As representative master regulators, nuclear respiratory factor 1 (NRF1) and NRF2 were chosen. They are key nuclear transcription factors, which regulate the expression of nuclear genes required for mitochondrial respiratory function and mitochondrial DNA transcription and replication. Their gene targets also include subunits of the five respiratory complexes. While RC complexes I, III, and IV are encoded by mitochondrial and nuclear DNA, complex II is entirely nucleus encoded (19). The effect of RV infection on the mRNA expression of selected nucleus-encoded subunits of RC complexes I, II, III, and IV was examined. For this purpose, the NDUFB1 subunit of complex I, subunits A and B (SDHA and SDHB) of complex II (succinate dehydrogenase [SDH]), subunit UQCRC1 of complex III, and the COX4I1 subunit of complex IV were selected. The mRNA expression profiles of these genes were investigated in A549 and Vero cells at 24, 48, and 78 h postinfection. RV-infected samples were compared to the corresponding mock-infected control (set to 100%) (Fig. 6A and B). The mRNA expression profiles of NRF1 and -2 showed similar increases in both cell lines (Fig. 6A). The NRF2 gene was the most significantly upregulated gene. In comparison to that in the mock-infected control, NRF2 mRNA expression increased by 115% (to 215% ± 16% in A549 cells) and by 151% (to 251% ± 21% in Vero cells) over the time of infection. The mRNA of the NDUFB1 subunit was downregulated at all selected time points, by up to 43% (to 57% ± 2%) in Vero cells at 48 hpi and by up to 29% (to 71% ± 7%) in A549 cells at 48 hpi (Fig. 6B). At 24 hpi, a significant increase over the mock-infected population was detected for the mRNA expression of the SDHA subunit of complex II in both Vero (to 165% ± 19%) and A549 (to 200% ± 4%) cells. However, at 72 hpi, a significant decrease, by 39% (to 61% ± 17%), was detected for SDHA in Vero cells. Significant increases (to 169% ± 14% in A549 cells at 24 hpi and to 161% ± 14% in Vero cells at 72 hpi) were seen in both cell lines for the expression of the SDHB gene (Fig. 6B). Hence, only the time point of the highest mRNA expression level of SDHB varied between the two cell lines. The mRNA level of the UQCRC1 subunit of complex III was increased only in A549 cells at 24 hpi by 46% (to 146% ± 9%). The mRNA expression level of the COX4I1 subunit of complex IV was increased at 24 hpi in both Vero cells (to 137% ± 21%) and A549 cells (to 129% ± 23%). The data presented show that RV induces alterations of the mRNA expression of NRF1 and -2 as key regulators of mitochondrial biogenesis and of selected RC subunits as their subsequent targets. Among the examined RC subunits, the mRNA expression changes were most profound for the SDHA and SDHB subunits of complex II. Hence, the next attempt was to determine the protein expression level of SDHA in mitochondrion-enriched fractions of Vero (Fig. 6C) and A549 (Fig. 6D) cells by Western blot analysis. In A549 cells, no changes in the protein expression of SDHA were detected after RV infection in comparison to the mock-infected control. However, the picture was different for Vero cells. At 48 and 72 h postinfection, an additional band of higher molecular weight was seen only in RV-infected samples (Fig. 6C). In summary, RV infection alters the mRNA expression level of the transcription factors NRF1 and NRF2 and of selected subunits of RC complexes, most notably of complex II. This is also reflected by an altered protein expression state, possibly corresponding to heterodimer formation.
Fig 6.

mRNA expression analysis of mitochondrial biomarkers (A and B) and protein expression analysis of a complex II subunit (C and D) in RV-infected Vero and A549 cells. RT-qPCR was applied for the analysis of the gene expression of the nuclear transcription factors NRF1 and -2 (A) and of nucleus-encoded subunit NDUFB1 of complex I, SDHA and SDHB of complex II (succinate dehydrogenase), subunit UQCRC1 of complex III, and COX4I1 of complex IV (B). Data are shown as percentages of the corresponding mock-infected controls. Gene expression was normalized against HUEL and HPRT-1. The mitochondrial protein expression level of SDHA was resolved by a 10% SDS-PAGE using 70 μg of mitochondria isolated from RV- and mock-infected Vero (C) and A549 (D) cells. The blot was probed with SDHA and VDAC (loading control) antibodies. Level of significance from the control population is indicated by asterisks (*, P < 0.05; **, P < 0.01; and ***, P < 0.001).
Expression of Mfn2 as a marker for mitochondrial morphology and metabolism.
The peroxisome proliferator-activated receptor-γ coactivator 1α (PGC-1α) belongs to a family of transcription coactivators and therefore regulates several key metabolic processes. PGC-1α induces the transcription of the aforementioned transcription factors NRF1 and NRF2 (19) but also of mitofusin 2 (Mfn2) as an important regulatory protein for mitochondrial structure and metabolic activity (20). Therefore, the next attempt was to determine the mRNA and protein expression levels of Mfn2 in RV-infected Vero and A549 cells. Figure 7A shows that the mRNA expression of Mfn2 was significantly elevated at 72 hpi in A549 (to 773% ± 168%) and Vero (to 151% ± 51%) cells. As Mfn2 is a mitochondrial protein, a mitochondrion-enriched cellular fraction was used for analysis of its protein expression level. Figure 7B and C suggest differences in the mitochondrial Mfn2 protein expression level over the time of infection. Western blot analysis of isolated mitochondria shows that at 3 days postinfection, mitochondria in RV-infected Vero and A549 cells had a higher portion of Mfn2 than did the mock-infected control. Additionally, the effect of heat stress (as an inducer of oxidative stress) on Mfn2 expression was examined in Vero cells. Vero cells were kept under 42°C for 3 h and then incubated for 24 h at 37°C and subjected to isolation of a mitochondrion-enriched cellular fraction. Western blot analysis of this fraction from heat stress-induced Vero cells revealed a higher expression level of Mfn2 than in the control population (Fig. 7C). These results indicate that RV infection induces an upregulation of Mfn2 at its mRNA and protein levels in two representative RV-permissive cell lines.
Fig 7.
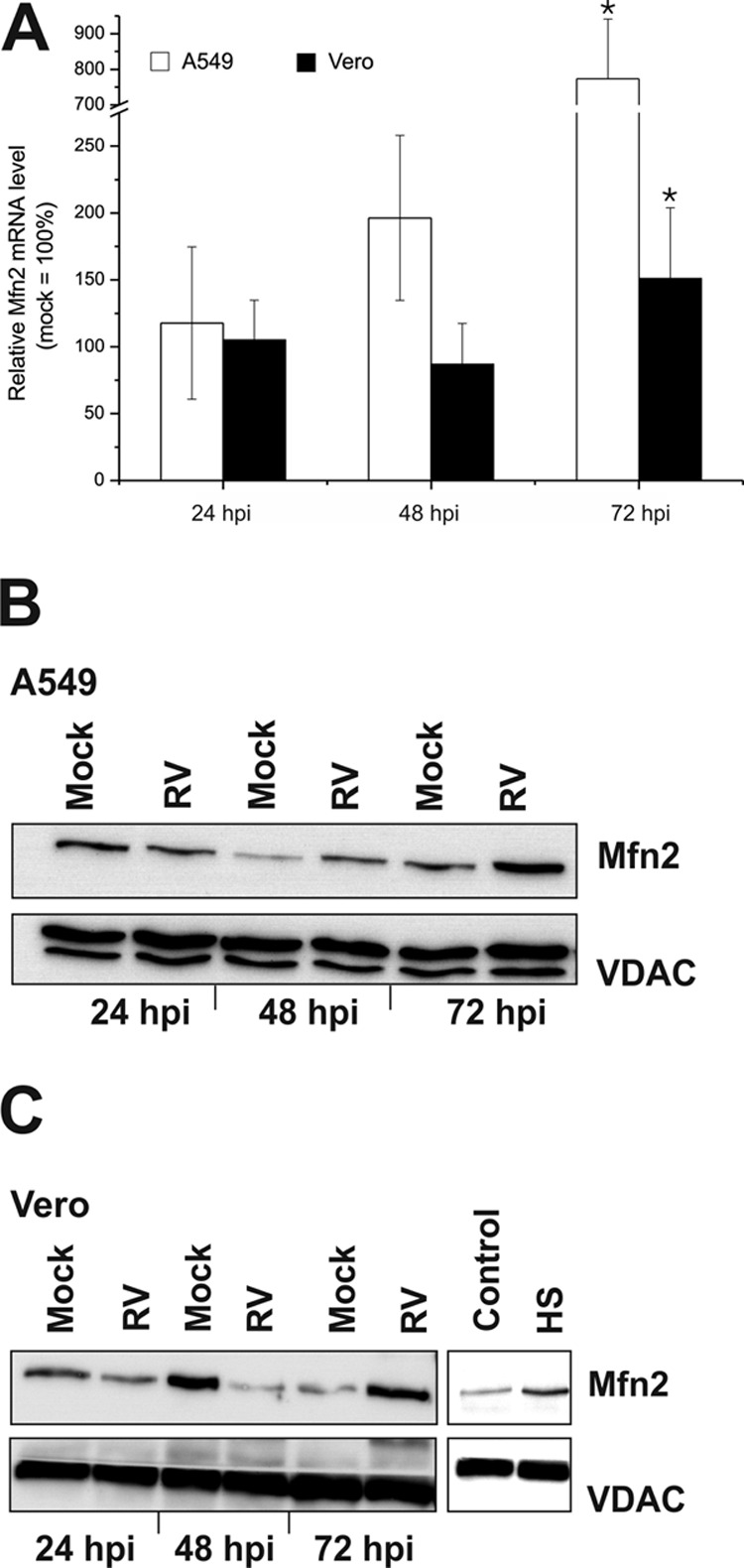
Effect of RV infection on the mRNA and protein expression level of Mfn2 in A549 and Vero cells. (A) The transcript level of Mfn2 was assessed by RT-qPCR. Data are shown as percentages of the corresponding mock-infected controls. Gene expression was normalized against HUEL and HPRT-1. The mitochondrial protein expression level of Mfn2 was resolved by a 10% SDS-PAGE using 30 μg of mitochondria isolated from RV- and mock-infected A549 (B) and Vero (C) cells. The blot was probed with Mfn2 and VDAC3 (loading control) antibodies. HS, heat shock treatment; control, untreated Vero cells. *, P < 0.05 compared with the control.
DISCUSSION
As intracellular parasites, viruses rely on their cellular host for providing the frame, the building blocks, and the energy required for their replication machinery. Hence, viruses impose a burden on the infected cell, especially through exhaustion of its metabolism. Data presented here indicate that RV as a slow-replicating virus has evolved means to ensure ongoing energy supply for its replication while circumventing the induction of oxidative stress. This clearly distinguishes RV from other viruses. Evidence is provided in this paper that RV infection strongly upregulates the activity of RC complex II and leads to an increase of protein and/or mRNA expression level of markers for mitochondrial biogenesis and function. This is in line with the presumption in our previous publication in which it was shown that the influence of RV on cellular metabolism is rather positive. In comparison to mock-infected controls, the mitochondrial membrane potential and the intracellular ATP content in RV-infected cells were increased (9).
Despite a renewed focus on metabolic engineering by viruses (21), data on the underlying mechanism remain to be obtained. Some viruses, such as influenza or measles virus, do not rely on cellular metabolism (22) or mitochondrial respiration (23), respectively. However, in most cases, virus infection leads to mitochondrial dysfunction and subsequently to induction of oxidative stress. This has been shown for rabies virus-infected mouse dorsal root ganglion neurons, for which induction of oxidative stress contributes to the reduced axonal outgrowth (24). Moreover, in dengue virus-infected hepatocytes, Δψm and intracellular ATP content are decreased, indicating mitochondrial dysfunction (25). Also, for hepatitis C virus (HCV)-infected cells, massive generation of ROS and a decreased Δψm were described (2). Mitochondrial dysfunction and ROS generation were observed 24 h after infection of a mouse neuroblastoma cell line by Sindbis virus, the prototype member of the alphavirus genus within the family Togaviridae (26). In contrast to these observations, we found that the metabolic activities of mitochondria are maintained through the entire RV replication cycle. Even at the clearly visible onset of the cytopathic effect at 72 hpi, the activity of distinct RC complexes is upregulated.
Our investigations show comparable results on the activity of the respiratory chain in three different RV-infected cell lines, highlighting complex II as the main target of the metabolic modulation by RV. This is especially noteworthy, as breast cancer cells such as the MCF-7 cell line are characterized by higher OXPHOS rates and a higher activity of RC complex II (27). There are only a few investigations on the modulation of the activity of RC complexes by chemical or natural compounds or by viral infections. One such example is melatonin, which has antioxidant functions and increases the activities of RC complexes I and IV, probably through enhancement of the electron flow along the ETC (28). Streptozotocin-induced diabetic rats are characterized by an increase in the activities of respiratory complexes I and II and a decrease in complex III and IV activities (29). HCV-infected cells show an impairment of OXPHOS, which appears to be mainly due to a reduction in the activity of RC complex I of up to 50% (30). The respiratory capacities of complexes I and II decrease when Sindbis virus infection of a mouse neuroblastoma cell line proceeds (26). An increase in the activities of both the respiratory complex IV and the ATP synthase has been described to occur after HIV-1 infection (31). Mouse models for coxsackievirus B3-induced myocarditis suggest an involvement of mitochondria in virus elimination and viral pathology (32). In this regard, the elevated activities of RC complexes I (by 21.7% ± 6.1%) and III (by 44% ± 10.3%) appear to contribute to generation of ROS and subsequently to mitochondrion-related apoptosis and virus elimination (32). However, the observed alterations of respiratory chain complex activities in response to RV infection appear to be much more profound and, more interestingly, show only marginal induction of markers for oxidative stress such as ROS. Respiratory complexes I and III are the main generators of mitochondrial ROS (4). Hence, the upregulation of RC complex II by RV should be favorable in the context of ROS generation. The increase in the mRNA expression level of NF-κB (as a marker for oxidative stress) in RV-infected A549 cells is probably due to an antiviral interferon response. Vero cells lack the type I interferon response (33), which particularly activates the NF-κB pathway (17).
In the three cell lines tested, a downregulation of complex IV activity was detected at some time point of infection with RV. There are two possibilities for a compensatory modulation of RC complex activities. First, the downregulation of complex IV could be an adaptive response to the increase in complex II activity to avoid an overfunctioning of the respiratory chain. Second, the downregulation of complex IV might be compensated by an increase in complex II activity to ensure ongoing electron flow through the electron transport chain. Similar compensatory mechanisms were also reported for cell lines derived from patients with an ATP synthase deficiency (complex V) which was balanced by an upregulation of RC complex III and IV activity (34). Similar to RV infection (but at a different scale), ionizing radiation of A549 cells induces an upregulation of complex II activity (by about 23%) and a downregulation of complex IV activity (by about 12%) (35).
There are several possibilities to explain how RV modulates the activity of the respiratory chain. RV infection results in slight alterations of the mRNA expression level of subunits of complexes I, III, and IV, whereas the mRNA expression level of SDHA and SDHB shows the highest rate of induction among the RC subunits, and especially SDHB is kept at an almost constantly increased level in Vero cells. This supports the important role played by respiratory complex II during the course of RV infection. Complex II is the only enzyme that participates in both the tricarboxylic acid (TCA) cycle and the respiratory chain. This may consequently lead to an enhancement of the TCA cycle, because all enzymes of the TCA cycle react as a group of enzymes and form a metabolon. The TCA cycle fuels the respiratory chain with reducing equivalents. Higher mRNA levels of the SDHA subunit of complex II in RV-infected cells seems to result in an altered SDHA protein expression state. A higher-molecular-weight species of SDHA appears on Western blots (Fig. 6C) together with the increased complex II activity at 48 hpi. This species is absent from mock-infected Vero cells and in RV- and mock-infected A549 cells. A549 cells show the strongest upregulation of complex II activity already at 24 hpi. Hence, a higher-molecular-weight species of SDHA could possibly not have been formed. The mechanism of RV-induced upregulation of complex II activity in Vero cells appears to differ from the one found in A549 cells. The higher-molecular-weight species of SDHA might correspond to heterodimers formed with SDHB. SDHA and SDHB both represent the catalytic moiety of complex II, and thus, RV infection could support heterodimer formation, which could result in higher complex II activity. In A549 cells, the viral RNA level is at 12 hpi higher than the one found in Vero or MCF-7 cells, which could contribute to the observed early increase in respiratory complex II activity in A549 cells at 24 hpi. We could not find differences in the inherent activities of respiratory complexes I to IV in mock-infected A549, MCF-7, or Vero cells (Fig. 1B) that could help to explain the slight variations in respiratory complex activity between Vero and A549 cells after RV infection (Fig. 2). The activity rates of each complex of the RC are comparable between Vero, A549, and MCF-7 cells. Important contributors to RV-induced alterations of RC activities appear to be the transcription factors NRF1 and NRF2. The mitochondrial respiratory machinery is regulated by both nuclear and mitochondrial genes. Their coordinated action is ensured by the PGC-1 family of coactivators, which, in turn, activate transcription factors such as NRF1 and NRF2 (36). NRF1 and NRF2 have overlapping but also distinct functions. NRF1 mainly coordinates the expression of genes that are necessary for both mitochondrial respiration (including genes encoding RC subunits) and the DNA replication apparatus (19). An elevation of NRF-1 mRNA is already sufficient to stimulate mitochondrial activity (19). Especially NRF2 is involved in the stress response pathway and regulates the expression of antioxidant enzymes (37). Both RV-infected Vero and A549 cells show elevated mRNA expression levels for NRF1 and NRF2. Hence, both transcription factors probably contribute to the positive effect of RV on mitochondrial activity, and especially NRF2 to the low level of oxidative stress induction.
How an altered activity rate of complex II could be beneficial for RV and could possibly contribute to the previously observed increase in mitochondrial membrane potential and the intracellular ATP content in RV-infected cells (9) remains to be resolved. The data presented here add to the complexity of known and unknown functions of complex II. The respiratory chain complexes are not a linear arrangement of enzymatic supercomplexes. They show different activity rates among each other (Fig. 1B) and also tissue-specific activity rates and expression levels (38). Each subunit of the RC complexes differs in its ability to relate its protein expression level to its enzymatic activity (39). For example, one-third of the regular protein content of the 8-kDa subunit of complex I is sufficient to maintain 70% of regular complex I activity. More importantly, complex IV activity has to decrease to a critical value until mitochondrial respiration in general is affected (39). This so-called biochemical threshold effect is specific for each complex. During the initial stages of physiological stress, an activation of SDH activity was noticed in rat blood lymphocytes (40). Hence, an increased activity of complex II could be beneficial under physiological or metabolic stress, which is also posed on the infected cell by RV. Defects in complex II functions are relatively rare in humans, and complex II is the only component of the RC that is capable of maintaining its activity under reduction of the RC and in the presence of ATP (41). Moreover, complex II can also induce reverse electron flow through complex I, which, in turn, supports NADH production (41). This emphasizes the importance and unique properties of complex II.
Another important point to consider is how the reported interference of RV with mitochondrial protein import fits into this picture (10). This phenomenon has so far been demonstrated only in yeast cells and certainly needs further investigation. The protein import and assembly process of mitochondrial subunits is complex and requires several cofactors, which to some extent differ among yeast and mammalian cells (42). Moreover, protein import into mitochondria requires the translocase of the outer membrane (TOM). This also includes the import of most of the subunits of the RC complexes (43). However, surface-exposed TOM receptors are not required for the interference of RV with mitochondrial protein import (10).
Besides the regulation of NRF1 and NRF2 transcriptions factors, PGC-1-dependent pathways positively influence mitochondrial activity and biogenesis and are thus involved in the adaptation of mitochondrial metabolism to the energy needs of the cell. Mfn2 represents one of the targets of the nuclear coactivator PGC-1α (20). Mfn2 is a dynamin-related GTPase protein that fulfills several important functions within the cell in addition to directing mitochondrial fusion. Mfn2 expression is increased at mRNA and protein expression levels after RV infection (Fig. 7). The slight discrepancy between the constant increases of Mfn2 at its mRNA level in A549 cells to its decreased protein expression rate at 48 hpi might not be significant. This discrepancy could be due to differences in Mfn2 expression rate between Vero and A549 cells or an impaired rate of import of Mfn2 into mitochondria at 48 hpi. Expression of Mfn2 is induced in skeletal muscle by exercise (20) and by mild heat stress (44). Mfn2 was also reported to protect against cold stress, which is associated with induction of apoptosis and reactive oxygen (45). The Vpr protein of HIV-1 reduces the expression level of Mfn2, which results in loss of mitochondrial membrane potential and apoptotic cell death (46). Moreover, Mfn2 gain of function increases Δψm and stimulates mitochondrial activity (20), which was also seen in RV-infected Vero and MCF-7 cells (9).
The low level of oxidative stress induction in RV-infected cells is consistent with the recent observation that in response to stress, including oxidative stress and virus infection, cells generate Ras-GAP SH3 domain-binding protein (G3BP)-containing stress granules. However, these granules were detectable only in less than half of the infected cells and only at late stages of RV infection (47).
Taken together, the data presented here indicate that RV has evolved a mechanism through which mitochondrial functions are positively influenced. This occurs without the induction of oxidative stress which is usually inevitable during a virus infection. The study of viral modulation of mitochondrial functions could reveal important aspects of signaling and metabolic pathways that are involved in the regulation of mitochondrial activity. The alteration of mitochondrial respiratory and redox functions by viruses such as RV could contribute to therapeutic strategies comprising metabolic antagonists against viral infections. Even metabolic agonists for compensation of mitochondrial defects without oxidative stress induction could be designed (11, 48).
ACKNOWLEDGMENTS
We thank Grit Szczepankiewicz and Viktoria Bothe for technical assistance in performing measles virus infections of cell lines and in performing SDHA Western blotting, respectively.
Footnotes
Published ahead of print 29 May 2013
REFERENCES
- 1. Ohta A, Nishiyama Y. 2011. Mitochondria and viruses. Mitochondrion 11:1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Quarato G, Scrima R, Agriesti F, Moradpour D, Capitanio N, Piccoli C. 2013. Targeting mitochondria in the infection strategy of the hepatitis C virus. Int. J. Biochem. Cell Biol. 45:156–166 [DOI] [PubMed] [Google Scholar]
- 3. Saraste M. 1999. Oxidative phosphorylation at the fin de siècle. Science 283:1488–1493 [DOI] [PubMed] [Google Scholar]
- 4. Dröse S, Brandt U. 2012. Molecular mechanisms of superoxide production by the mitochondrial respiratory chain. Adv. Exp. Med. Biol. 748:145–169 [DOI] [PubMed] [Google Scholar]
- 5. Fernandez de Castro I, Volonte L, Risco C. 2013. Virus factories: biogenesis and structural design. Cell. Microbiol. 15:24–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lagaudriere-Gesbert C, Purvina M, Assrir N, Rossignol JM. 2012. Role of the cellular protein gC1qR in the virus life cycles. Virologie 16:85–94 [DOI] [PubMed] [Google Scholar]
- 7. Beatch MD, Hobman TC. 2000. Rubella virus capsid associates with host cell protein p32 and localizes to mitochondria. J. Virol. 74:5569–5576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Beatch MD, Everitt JC, Law LJ, Hobman TC. 2005. Interactions between rubella virus capsid and host protein p32 are important for virus replication. J. Virol. 79:10807–10820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Claus C, Chey S, Heinrich S, Reins M, Richardt B, Pinkert S, Fechner H, Gaunitz F, Schafer I, Seibel P, Liebert UG. 2011. Involvement of p32 and microtubules in alteration of mitochondrial functions by rubella virus. J. Virol. 85:3881–3892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ilkow CS, Weckbecker D, Cho WJ, Meier S, Beatch MD, Goping IS, Herrmann JM, Hobman TC. 2010. The rubella virus capsid protein inhibits mitochondrial import. J. Virol. 84:119–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. El-Bacha T, Da Poian AT. 2013. Virus-induced changes in mitochondrial bioenergetics as potential targets for therapy. Int. J. Biochem. Cell Biol. 45:41–46 [DOI] [PubMed] [Google Scholar]
- 12. Claus C, Hofmann J, Uberla K, Liebert UG. 2006. Rubella virus pseudotypes and a cell-cell fusion assay as tools for functional analysis of the rubella virus E2 and E1 envelope glycoproteins. J. Gen. Virol. 87:3029–3037 [DOI] [PubMed] [Google Scholar]
- 13. Medja F, Allouche S, Frachon P, Jardel C, Malgat M, Mousson de Camaret B, Slama A, Lunardi J, Mazat JP, Lombes A. 2009. Development and implementation of standardized respiratory chain spectrophotometric assays for clinical diagnosis. Mitochondrion 9:331–339 [DOI] [PubMed] [Google Scholar]
- 14. Janssen AJ, Trijbels FJ, Sengers RC, Smeitink JA, van den Heuvel LP, Wintjes LT, Stoltenborg-Hogenkamp BJ, Rodenburg RJ. 2007. Spectrophotometric assay for complex I of the respiratory chain in tissue samples and cultured fibroblasts. Clin. Chem. 53:729–734 [DOI] [PubMed] [Google Scholar]
- 15. Luo C, Long J, Liu J. 2008. An improved spectrophotometric method for a more specific and accurate assay of mitochondrial complex III activity. Clin. Chim. Acta 395:38–41 [DOI] [PubMed] [Google Scholar]
- 16. Chey S, Claus C, Liebert UG. 2010. Validation and application of normalization factors for gene expression studies in rubella virus-infected cell lines with quantitative real-time PCR. J. Cell. Biochem. 110:118–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pfeffer LM, Kim JG, Pfeffer SR, Carrigan DJ, Baker DP, Wei L, Homayouni R. 2004. Role of nuclear factor-kappaB in the antiviral action of interferon and interferon-regulated gene expression. J. Biol. Chem. 279:31304–31311 [DOI] [PubMed] [Google Scholar]
- 18. Le WD, Xie WJ, Appel SH. 1999. Protective role of heme oxygenase-1 in oxidative stress-induced neuronal injury. J. Neurosci. Res. 56:652–658 [DOI] [PubMed] [Google Scholar]
- 19. Scarpulla RC. 2008. Transcriptional paradigms in mammalian mitochondrial biogenesis and function. Physiol. Rev. 88:611–638 [DOI] [PubMed] [Google Scholar]
- 20. Zorzano A. 2009. Regulation of mitofusin-2 expression in skeletal muscle. Appl. Physiol. Nutr. Metab. 34:433–439 [DOI] [PubMed] [Google Scholar]
- 21. Maynard ND, Gutschow MV, Birch EW, Covert MW. 2010. The virus as metabolic engineer. Biotechnol. J. 5:686–694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ritter JB, Wahl AS, Freund S, Genzel Y, Reichl U. 2010. Metabolic effects of influenza virus infection in cultured animal cells: intra- and extracellular metabolite profiling. BMC Syst. Biol. 4:61. 10.1186/1752-0509-4-61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Derakhshan M, Willcocks MM, Salako MA, Kass GE, Carter MJ. 2006. Human herpesvirus 1 protein US3 induces an inhibition of mitochondrial electron transport. J. Gen. Virol. 87:2155–2159 [DOI] [PubMed] [Google Scholar]
- 24. Jackson AC, Kammouni W, Fernyhough P. 2011. Role of oxidative stress in rabies virus infection. Adv. Virus Res. 79:127–138 [DOI] [PubMed] [Google Scholar]
- 25. El-Bacha T, Midlej V, Pereira da Silva AP, Silva da Costa L, Benchimol M, Galina A, Da Poian AT. 2007. Mitochondrial and bioenergetic dysfunction in human hepatic cells infected with dengue 2 virus. Biochim. Biophys. Acta 1772:1158–1166 [DOI] [PubMed] [Google Scholar]
- 26. Silva da Costa L, Pereira da Silva AP, Da Poian AT, El-Bacha T. 2012. Mitochondrial bioenergetic alterations in mouse neuroblastoma cells infected with Sindbis virus: implications to viral replication and neuronal death. PLoS One 7:e33871. 10.1371/journal.pone.0033871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Whitaker-Menezes D, Martinez-Outschoorn UE, Flomenberg N, Birbe RC, Witkiewicz AK, Howell A, Pavlides S, Tsirigos A, Ertel A, Pestell RG, Broda P, Minetti C, Lisanti MP, Sotgia F. 2011. Hyperactivation of oxidative mitochondrial metabolism in epithelial cancer cells in situ: visualizing the therapeutic effects of metformin in tumor tissue. Cell Cycle 10:4047–4064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Paradies G, Petrosillo G, Paradies V, Reiter RJ, Ruggiero FM. 2010. Melatonin, cardiolipin and mitochondrial bioenergetics in health and disease. J. Pineal Res. 48:297–310 [DOI] [PubMed] [Google Scholar]
- 29. Raza H, Prabu SK, John A, Avadhani NG. 2011. Impaired mitochondrial respiratory functions and oxidative stress in streptozotocin-induced diabetic rats. Int. J. Mol. Sci. 12:3133–3147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ripoli M, D'Aprile A, Quarato G, Sarasin-Filipowicz M, Gouttenoire J, Scrima R, Cela O, Boffoli D, Heim MH, Moradpour D, Capitanio N, Piccoli C. 2010. Hepatitis C virus-linked mitochondrial dysfunction promotes hypoxia-inducible factor 1 alpha-mediated glycolytic adaptation. J. Virol. 84:647–660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tripathy MK, Mitra D. 2010. Differential modulation of mitochondrial OXPHOS system during HIV-1 induced T-cell apoptosis: up regulation of complex-IV subunit COX-II and its possible implications. Apoptosis 15:28–40 [DOI] [PubMed] [Google Scholar]
- 32. Ebermann L, Wika S, Klumpe I, Hammer E, Klingel K, Lassner D, Volker U, Erben U, Zeichhardt H, Schultheiss HP, Dorner A. 2012. The mitochondrial respiratory chain has a critical role in the antiviral process in coxsackievirus B3-induced myocarditis. Lab. Invest. 92:125–134 [DOI] [PubMed] [Google Scholar]
- 33. Diaz MO, Ziemin S, Le Beau MM, Pitha P, Smith SD, Chilcote RR, Rowley JD. 1988. Homozygous deletion of the alpha- and beta 1-interferon genes in human leukemia and derived cell lines. Proc. Natl. Acad. Sci. U. S. A. 85:5259–5263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Havlíčková Karbanová V, Cizkova Vrbacka A, Hejzlarova K, Nuskova H, Stranecky V, Potocka A, Kmoch S, Houstek J. 2012. Compensatory upregulation of respiratory chain complexes III and IV in isolated deficiency of ATP synthase due to TMEM70 mutation. Biochim. Biophys. Acta 1817:1037–1043 [DOI] [PubMed] [Google Scholar]
- 35. Yamamori T, Yasui H, Yamazumi M, Wada Y, Nakamura Y, Nakamura H, Inanami O. 2012. Ionizing radiation induces mitochondrial reactive oxygen species production accompanied by upregulation of mitochondrial electron transport chain function and mitochondrial content under control of the cell cycle checkpoint. Free Radic. Biol. Med. 53:260–270 [DOI] [PubMed] [Google Scholar]
- 36. Scarpulla RC. 2012. Nucleus-encoded regulators of mitochondrial function: Integration of respiratory chain expression, nutrient sensing and metabolic stress. Biochim. Biophys. Acta 1819:1088–1097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nguyen T, Nioi P, Pickett CB. 2009. The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress. J. Biol. Chem. 284:13291–13295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Duborjal H, Beugnot R, Mousson de Camaret B, Issartel JP. 2002. Large functional range of steady-state levels of nuclear and mitochondrial transcripts coding for the subunits of the human mitochondrial OXPHOS system. Genome Res. 12:1901–1909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rossignol R, Faustin B, Rocher C, Malgat M, Mazat JP, Letellier T. 2003. Mitochondrial threshold effects. Biochem. J. 370:751–762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zakharchenko MV, Zakharchenko AV, Khunderyakova NV, Tutukina MN, Simonova MA, Vasilieva AA, Romanova OI, Fedotcheva NI, Litvinova EG, Maevsky EI, Zinchenko VP, Berezhnov AV, Morgunov IG, Gulayev AA, Kondrashova MN. 2013. Burst of succinate dehydrogenase and alpha-ketoglutarate dehydrogenase activity in concert with the expression of genes coding for respiratory chain proteins underlies short-term beneficial physiological stress in mitochondria. Int. J. Biochem. Cell Biol. 45:190–200 [DOI] [PubMed] [Google Scholar]
- 41. Rustin P, Munnich A, Rotig A. 2002. Succinate dehydrogenase and human diseases: new insights into a well-known enzyme. Eur. J. Hum. Genet. 10:289–291 [DOI] [PubMed] [Google Scholar]
- 42. Mick DU, Dennerlein S, Wiese H, Reinhold R, Pacheu-Grau D, Lorenzi I, Sasarman F, Weraarpachai W, Shoubridge EA, Warscheid B, Rehling P. 2012. MITRAC links mitochondrial protein translocation to respiratory-chain assembly and translational regulation. Cell 151:1528–1541 [DOI] [PubMed] [Google Scholar]
- 43. Kulawiak B, Hopker J, Gebert M, Guiard B, Wiedemann N, Gebert N. 2013. The mitochondrial protein import machinery has multiple connections to the respiratory chain. Biochim. Biophys. Acta 1827:612–626 [DOI] [PubMed] [Google Scholar]
- 44. Liu CT, Brooks GA. 2012. Mild heat stress induces mitochondrial biogenesis in C2C12 myotubes. J. Appl. Physiol. 112:354–361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zhang W, Chen Y, Yang Q, Che H, Chen X, Yao T, Zhao F, Liu M, Ke T, Chen J, Luo W. 2010. Mitofusin-2 protects against cold stress-induced cell injury in HEK293 cells. Biochem. Biophys. Res. Commun. 397:270–276 [DOI] [PubMed] [Google Scholar]
- 46. Huang CY, Chiang SF, Lin TY, Chiou SH, Chow KC. 2012. HIV-1 Vpr triggers mitochondrial destruction by impairing Mfn2-mediated ER-mitochondria interaction. PLoS One 7:e33657. 10.1371/journal.pone.0033657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Matthews JD, Frey TK. 2012. Analysis of subcellular G3BP redistribution during rubella virus infection. J. Gen. Virol. 93:267–274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Vafai SB, Mootha VK. 2012. Mitochondrial disorders as windows into an ancient organelle. Nature 491:374–383 [DOI] [PubMed] [Google Scholar]
- 49. Mueller EE, Mayr JA, Zimmermann FA, Feichtinger RG, Stanger O, Sperl W, Kofler B. 2012. Reduction of nuclear encoded enzymes of mitochondrial energy metabolism in cells devoid of mitochondrial DNA. Biochem. Biophys. Res. Commun. 417:1052–1057 [DOI] [PubMed] [Google Scholar]
- 50. Fischer M, Skowron M, Berthold F. 2005. Reliable transcript quantification by real-time reverse transcriptase-polymerase chain reaction in primary neuroblastoma using normalization to averaged expression levels of the control genes HPRT1 and SDHA. J. Mol. Diagn. 7:89–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Schildgen V, Wulfert M, Gattermann N. 2011. Impaired mitochondrial gene transcription in myelodysplastic syndromes and acute myeloid leukemia with myelodysplasia-related changes. Exp. Hematol. 39:666–675.e661. 10.1016/j.exphem.2011.03.007 [DOI] [PubMed] [Google Scholar]
- 52. Sun W, Guo MM, Han P, Lin JZ, Liang FY, Tan GM, Li HB, Zeng M, Huang XM. 2012. Id-1 and the p65 subunit of NF-kappaB promote migration of nasopharyngeal carcinoma cells and are correlated with poor prognosis. Carcinogenesis 33:810–817 [DOI] [PubMed] [Google Scholar]