

Abstract
Plant pararetroviruses integrate serendipitously into their host genomes. The banana genome harbors integrated copies of banana streak virus (BSV) named endogenous BSV (eBSV) that are able to release infectious pararetrovirus. In this investigation, we characterized integrants of three BSV species—Goldfinger (eBSGFV), Imove (eBSImV), and Obino l'Ewai (eBSOLV)—in the seedy Musa balbisiana Pisang klutuk wulung (PKW) by studying their molecular structure, genomic organization, genomic landscape, and infectious capacity. All eBSVs exhibit extensive viral genome duplications and rearrangements. eBSV segregation analysis on an F1 population of PKW combined with fluorescent in situ hybridization analysis showed that eBSImV, eBSOLV, and eBSGFV are each present at a single locus. eBSOLV and eBSGFV contain two distinct alleles, whereas eBSImV has two structurally identical alleles. Genotyping of both eBSV and viral particles expressed in the progeny demonstrated that only one allele for each species is infectious. The infectious allele of eBSImV could not be identified since the two alleles are identical. Finally, we demonstrate that eBSGFV and eBSOLV are located on chromosome 1 and eBSImV is located on chromosome 2 of the reference Musa genome published recently. The structure and evolution of eBSVs suggest sequential integration into the plant genome, and haplotype divergence analysis confirms that the three loci display differential evolution. Based on our data, we propose a model for BSV integration and eBSV evolution in the Musa balbisiana genome. The mutual benefits of this unique host-pathogen association are also discussed.
INTRODUCTION
Endogenous retroviruses in animal genomes were discovered initially in the late 1960s and early 1970s (1). Integration into the host chromosome of a provirus mediated by a virus-encoded integrase is obligatory to the replication of retroviruses. Proviruses integrated into the DNA of germ line cells as endogenous provirus are indeed inherited by the host as Mendelian traits, and these viruses are named endogenous retroviruses (ERVs) to distinguish them from horizontally transmitted exogenous retroviruses (2). Until recently, retroviruses were the only endogenous viruses known. This feature has now been extended to many other viruses: from RNA viruses that do not require integration to replicate, such as bornaviruses and filoviruses (3, 4), to circoviruses and parvoviruses with single-stranded DNA genomes (5) and to hepadnaviruses with partially double-stranded DNA genomes (6). Such integration events play no role in viral replication, cannot give rise to infectious virus even if their endogenous retrovirus is infectious, and constitute a fossil record useful to determine the age of viruses (7, 8). Whether such sequences confer any biological advantage on the host remains an interesting question.
In contrast to animal viruses, no known plant virus encodes an integrase or requires integration into the host genome to replicate. Nowadays, access to plant genome sequencing data reveals a significant proportion of foreign sequences, including viral sequences. All the viral sequences found so far belong to the Caulimoviridae family. The members of this family are double-stranded DNA viruses exploiting a reverse transcription process for replication. Interestingly, some of these integrated viral sequences are infectious, leading frequently to plant infection. Such sequences have undergone extensive viral genome rearrangements and contain more than one copy of the viral genome (9). Surprisingly, these sequences appear to be inherited by their host plant as Mendelian traits and to be transmitted vertically like ERVs. However, and unlike members of the animal kingdom, plants infected in this manner are always hybrids resulting from interspecific crosses. To date, three plant viruses have been described as endogenous and infectious viruses in hybrids: (i) Tobacco vein clearing virus (TVCV) in Nicotiana edwardsonii, which is an allohexaploid derived from a cross between Nicotiana glutinosa (n = 24) and Nicotiana clevelandii (n = 12) (10); (ii) Petunia vein clearing virus (PVCV) in petunia hybrid Petunia hybrida resulting from a wild cross between Petunia integrifolia subsp. inflata and Petunia axillaris subsp. Axillaris (11); and (iii) Banana streak virus (BSV) in several interspecific banana hybrids obtained by crosses between Musa balbisiana (B genome) and Musa acuminata (A genome) (12, 13). Mechanisms that lead to integration, activation, and subsequent episomal infection are complex and still largely unknown. Over the last decade, some data relating to integrated viral sequences, the locus of integration, and activation processes occurring following different stresses such as wounding and tissue culture have been accumulating (10, 11, 14, 15), revealing several differences between the three different models described above. Indeed, the PVCV genome is integrated in tandem arrays in a complete, continuous form as a “retro provirus”; its activation resembles that of retroviruses and involves the production of a greater-than-full-length transcript that could be reverse transcribed into DNA (16, 17). On the other hand, BSV Goldfinger species (BSGFV) is present in the diploid M. balbisiana (BB) Pisang klutuk wulung (PKW) plant as a diallelic integration much longer than a single viral genome, exhibiting a succession of partial viral sequences that are sometimes inverted and partially duplicated, representing at least one total viral genome (13). Based on the endogenous BSGFV sequence (eBSGFV), an activation process based on a direct transcription event seems unlikely to occur alone. Indeed, in the theoretical model proposed in reference 18, two initial homologous recombination (HR) steps could be required to reconstitute a full-length circular BSGFV genome.
Of the three plant species affected by viral disease outbreaks from these endogenous pararetrovirus sequences, banana remains the most critical in terms of economic impact. Indeed, banana (Musa spp.) ranks as the world's fourth most important food crop in terms of gross value of production after rice, wheat, and maize. In the past 20 years, the emergence of BSV in all banana-producing countries resulted from the awakening of eBSVs and correlates with the massive use of newly created interspecific banana hybrids. Consequently, all reported interspecific banana hybrids are considered at risk for the wide and rapid dissemination of BSV, and within a very short period, BSV has become the major constraint to banana breeding programs worldwide. Recently, we established that these problematic infectious eBSVs are present in the B genome of banana only (12, 15, 19, 20). The B genome forms part of the genotype of many important banana cultivars, such as the famous plantain subgroup that is a staple food for millions of people in Africa and Latin America. Moreover, it is often associated with desirable traits of agronomic interest such as vegetative vigor, biotic and abiotic stress tolerance, and resistance to pathogens such as the fungus responsible for the severe black Sigatoka disease. Consequently, a better knowledge of eBSV structure, genomic organization, and chromosomal localization in the B genome as well as infection capacity to produce a functional viral genome will provide molecular tools that can be used to widely screen banana hybrids, genitors, and germplasm. This step becomes a prerequisite not only for future crop-oriented breeding programs aimed at producing safe interspecific banana hybrids but also to estimate and limit the risk of BSV outbreak on natural hybrids spread intensively in developing countries as a food source. Previous studies (12, 18) have revealed the presence of three species of BSV—Banana streak Obino l'Ewai virus (BSOLV), Banana streak Goldfinger virus (BSGFV), and Banana streak Imove virus (BSImV)—in an F1 triploid (AAB) population obtained from an interspecific genetic cross between PKW, seedy diploid Musa balbisiana (BB), and IDN 110 tetraploid Musa acuminata (AAAA). Both are virus free, and PKW is the only one to harbor the eBSV counterpart of each BSV species. PKW is therefore solely responsible for virus transmission and expression among the progeny.
The aim of the present study was to fully characterize the three eBSVs present within the B genome that contribute to BSV epidemics worldwide. This in-depth characterization was performed on the diploid Musa balbisiana PKW genome by combining molecular, genomic, genetic, and cytogenetic approaches. Based on our data, we propose a model representing the most probable scenario to have occurred from the initial eBSV integration to the picture observed nowadays in PKW, and we discuss the mutual benefits of this particular plant-pathogen interaction.
MATERIALS AND METHODS
Screening of BAC library.
Bacterial artificial chromosome (BAC) libraries were obtained from diploid M. balbisiana PKW (21) and two M. acuminata banana plants: the diploid Calcutta 4 (AA) (22) and the triploid “Cavendish” subgroup cv. Grande Naine (AAA) (23). Clones of BAC libraries were spotted onto high-density Hybond N+ filters (AP Biotech, Little Chalfont, United Kingdom) using a Flexys robot. The filters were hybridized with BSV probes covering the entire genome of four BSV species—Obino l'ewai (BSOLV) (NC_003381.1) (24), Imove (BSImV) (HQ659760/) (25), Mysore (BSMyV) (AY805074) (26), and Vietnam (BSVNV) (AY750155) (27)—as described in reference 13 for Goldfinger (BSGFV). BSV-positive BAC DNA was digested with four different enzymes (HindIII, BamHI, PstI, and XhoI) and separated on a 0.8% agarose gel in 1× Tris-acetate-EDTA at 60 V, run for 20 h. The separated fragments were denatured and transferred to a nylon membrane (Hybond-N+; Amersham Pharmacia Biotech). Filters were hybridized with probes corresponding to the full-length genome of each BSV species. Restriction profiles were scored manually. One BAC clone was selected by fingerprint profiles for sequencing.
BAC sequencing.
BACs containing eBSGFV were obtained from GenBank (accession numbers AP009325 and AP009326, corresponding to MBP_71C19 and MBP_94I16, respectively [13]). BAC MBP_31007 containing eBSOLV was obtained from GenBank (accession number AP009334).
Selected BAC clones containing eBSOLV (BAC_73B22 and BAC_17D14) as well as eBSImV (BAC_68C24) were sequenced at Genoscope (http://www.genoscope.cns.fr/spip/). Libraries from the three BAC clones were obtained after mechanical shearing of BAC DNA and cloning of 5-kbp and 10-kbp fragments into pcdna 2.1 (Invitrogen) and pCNS (pSU18-derived) plasmids, respectively. Vector DNAs were purified and end sequenced using dye terminator chemistry on ABI 3730 sequencers (Applied Biosystems, France) until 12× coverage was reached. BACs 73B22 and 17D14 were assembled using the Phred/Phrap/Consed software package (CodonCode Corporation), whereas Arachne assembler was used for BAC 68C24. Primer walks and PCR were needed to complete the final phases.
For BACs harboring eBSImV (MBP_68C24) or eBSOLV (MBP_17D14c) sequences, the GenBank accession numbers are HE983625 and HE983609, respectively.
Gene model prediction, sequence alignment, and synteny analysis.
Plant and virus gene structures were predicted using the EuGène combiner release 3.2 (28) with rice-specific parameters that integrate several lines of evidence. Gene models were predicted with the ab initio gene finders, EuGéneIMM and Fgenesh (29). Translation start sites and splice sites were predicted by SpliceMachine (30). Available monocotyledon expressed sequence tags (ESTs) from EMBL were aligned on the genome using Sim4 (31). Similarities to protein sequences were identified using BLASTX (NCBI-BLASTALL) (32) as described in reference 33.
Polypeptide functions were also predicted by integrating several lines of evidence. Protein similarities were searched for using tBLASTn on translated monocotyledon ESTs and BLASTp on UniProt. Protein domains were predicted with InterproScan (34). Clusters of orthologous genes between the predicted polypeptides and the proteomes of Oryza sativa (TIGR release 5.0) and Sorghum bicolor (JGI release 1.0) were also identified using the pipeline GreenPhyl (35). Predicted genes were annotated manually using Artemis (36). A gene is considered complete when its coding sequence (CDS) is canonical and significantly matches a known sequence in the public data banks with coverage parameters Qcov and Scov greater or equal to 0.8. Under these parameters, CDS is also predicted to be functional. When a gene contains mutations that could prevent correct expression (i.e., a missing start or stop codon, noncanonical splice site, frameshift, or in-frame stop codon), it is considered a pseudogene. A polypeptide was annotated as a fragment when its coverage (Qcov) was less than 0.8 as determined by comparing its length to the length of the match with the best significant hit. We annotated a gene as a remnant when it was composed of a small fragment (Qcov < 0.3) or more than three fragments (Qcov < 0.5) and/or when it had more than two mutations preventing correct CDS expression. BAC sequence annotation is available on a genome browser at http://gnpannot.musagenomics.org/cgi-bin/gbrowse/musa/. Sequence comparison between haplotypes was performed using dot plot analysis (37). Local alignments were performed using BLASTN (32) and visualized with the Artemis Comparison Tool (ACT) (36). Synteny analysis with Musa acuminata reference genome (38) was performed using BLAST analysis. BAC sequences were compared to pseudomolecules using a BLASTN search with an E value of 10−10 and visualized with ACT. The synteny between BAC proteomes and the reference Musa genome was performed using BLASTp analysis against translated CDS at http://banana-genome.cirad.fr/blast.html.
Fluorescent in situ hybridization (FISH) of plant material and chromosome preparations.
Roots tips of M. balbisiana diploid PKW grown in a tropical greenhouse were collected at different times from 8.45 a.m. to 11.30 a.m. The young growing roots were treated with 8-hydroxyquinoleine (0.04%) for 4 h, fixed in a solution of ethanol-glacial acetic acid (3:1, vol/vol) for 24 h (12 h at room temperature and 12 h at 4°C), and then kept in 70% ethanol. Chromosome preparations were prepared as described in reference 39, with slight modifications: the enzymatic mixture was composed of 1% cellulase (Sigma), 1% cytohelicase (Sigma), and 1% pectolyase (Sigma). Before being squashed, the roots were kept in water overnight at 4°C.
The hybridizations were performed with full-length genome probes for each BSV species and the 45S ribosomal DNA (rDNA) sequence as a control. The virus probes were labeled by random priming with biotin-14-dUTP (Invitrogen Life Technology) and digoxigenin-11-dUTP (High Prime DNA labeling kit; Roche) and with biotin for 45S. The hybridization protocol was done as described in reference 40, with slight modifications: selected slides were pretreated with 1 μg/ml of RNase in 2× SSC (1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate) for 45 min at 37°C and then rinsed twice in 2× SSC at room temperature. Slides were then denatured in a solution of 70% (vol/vol) formamide in 2× SSC for 2 min at 70°C in a bath, immersed for 3 min in 2× SSC in a bath previously put on ice, and, finally, dehydrated through an alcohol series (5 min each in ethanol baths of 70%, 90%, and 100% at −20°C). The hybridization mixture contained 50% (vol/vol) deionized formamide, 10% (wt/vol) sodium dextran sulfate, 2× SSC, 0.5 μg/μl of sonicated salmon sperm DNA, 0.66% (wt/vol) SDS, and 5 μl of virus probes. This mixture was denatured in a boiling bath for 10 min and kept on ice for at least 15 min; 50 μl of hybridization mixture was applied to each slide and covered with a plastic coverslip. Hybridization was carried out overnight at 37°C in a moist chamber. The next day, the slides were washed in 2× SSC, 0.5× SSC, and 0.1× SSC for 10 min each at 42°C and in 2× SSC at room temperature. Slides were pretreated with 5% (wt/vol) bovine serum albumin (BSA) in 4× SSC with Tween 20 for 10 min at 37°C. The detection solution contained 6 ng/μl of avidin-rhodamine and 10 ng/μl of sheep antidigoxigenin (anti-DIG)-fluorescein isothiocyanate (FITC) diluted in the pretreatment BSA solution. Fifty microliters of this mixture was loaded on each slide, and detection was carried out for 45 min at 37°C in a moist chamber. Slides were rinsed 3 times in 4× SSC with Tween 20 for 5 min at 42°C. Then 50 μl of a solution of 7.5 ng/μl of rabbit anti-sheep-FITC antibodies and, if amplification was required for probes labeled with biotin (in the case of virus biotin probes), 5 ng/μl of biotinylated antiavidin antibodies in goat serum diluted in 4× SSC with Tween 20 were loaded on each slide and left for 45 min in a moist chamber at 37°C. Slides were rinsed 3 times in 4× SSC with Tween 20 for 5 min at 42°C. Then, chromosomes were counterstained with Vectashield mounting medium with 4′,6-diamidino-2-phenylindole (DAPI) (Vector Laboratories). In the case of virus biotin probes, before this step, a final detection step with a solution of 6 ng/μl of avidine-rhodamine diluted in 5% (wt/vol) BSA and 4× SSC with Tween 20 was applied for 45 min in a moist chamber at 37°C, followed by 3 rinses in 4× SSC with Tween 20 for 5 min at 42°C.
Observations were carried out on an epifluorescence microscope (Leica DMRXA 2); images were captured using a cooled Hamamatsu Orca AG camera and processed using Velocity software (PerkinElmer).
Genetic analysis. (i) Interspecific genetic cross of PKW with cv. IDN 110 4x.
The plant population used in the present study consisted of 165 F1 allotriploid hybrids (AAB). This population derives from an interspecific genetic cross between the virus-free diploid (BB) Musa balbisiana female parent PKW and the virus-free autotetraploid (AAAA) Musa acuminata male parent cv. IDN 110 4x. The absence of viruses in both parents was confirmed by immunosorbent electron microscopy and by immunocapture PCR (IC-PCR) (12). This genetic cross is fully described and characterized in reference 12. Leaf samples were stored at −80°C.
(ii) DNA extraction.
Total DNA was extracted by the method described in reference 41 from leaf tissue of AAB progeny stored at −80°C. The quality and amount of DNA were estimated visually after separation of 5 μl of DNA extraction in a 0.8% agarose gel, staining with ethidium bromide, and visualizing on a UV transilluminator.
(iii) PCR.
All PCRs were performed on 5 to 20 ng of template DNA using a common mix composed of 20 mM Tris-HCl (pH 8.4), 50 mM KCl, 0.1 mM each deoxynucleoside triphosphate, 1.5 mM MgCl2, 400 nM forward and reverse primers, and 1 U of Taq DNA polymerase (Eurogentec, Seraing, Belgium) in a final volume of 25 μl. DNA was amplified using the following program: 1 cycle at 94°C for 4 min; 35 cycles at 94°C for 30 s, primer annealing temperature for 30 s, and 72°C for 1 min per kb; and a final extension at 72°C for 10 min. Amplicons were visualized after migration of 8 μl of PCR products on an agarose gel (1 to 3% according to the expected size of the amplicon) in 0.5× TBE (45 mM Tris-borate, 1 mM EDTA [pH 8]). The gel was stained with ethidium bromide, and amplified bands were visualized under UV light.
(iv) eBSV genotyping.
To perform genetic analysis of eBSV loci, segregation analysis was performed on the triploid progeny (AAB) described above. Allelic segregation was estimated in the eBSV region using two types of molecular markers: (i) markers derived from the eBSV structure itself when the specific structure of integration allows for specific amplification of the integration locus (see below) and (ii) simple sequence repeat (SSR) markers defined from the BAC sequence with the dedicated pipeline SAT (http://sat.cirad.fr/sat (42) of the South Green bioinformatic platform (http://southgreen.cirad.fr/), using default parameters.
The targeted eBSV, the type of marker developed, the name and the sequence of the primer, and the size of the PCR product are given in the table provided in File S1 in the supplemental material. For allelic markers, discrimination between alleles is also described in detail in File S1.
BSV genotyping.
BSV was genotyped by immunocapture PCR. The immunocapture step consisted of coating sterile polypropylene thin-walled 0.2-ml tubes (Axygen, Union City, CA) for 4 h at 37°C with 25 μl of immunoglobulin G purified from polyclonal antiserum raised against BSV species and Sugarcane bacilliform virus species (a kind gift from B. E. L. Lockhart), diluted at 2 μg/ml in carbonate coating buffer (15 mM sodium carbonate, 34 mM sodium bicarbonate [pH 9.6]). The tubes were then washed three times with 100 μl of PBT washing buffer (136 mM NaCl, 1.4 mM KH2PO4, 2.6 mM KCl, 8 mM Na2HPO4, 0.05% Tween 20 [pH 7.4]). Plant extracts were prepared by grinding 0.5-g leaf samples in 5 ml of grinding buffer (2% polyvinylpyrrolidone 40, 0.2% sodium sulfite, and 0.2% bovine serum albumin prepared in PBT) using a manual bead grinder and plastic grinding bags (Bio-Rad Phytodiagnostics, Marnes-la-Coquette, France). Portions (1 ml) of plant extracts were transferred to microcentrifuge tubes and clarified by centrifugation at room temperature for 5 min at 7,000 rpm. Then, 25 μl of the supernatant was loaded into coated tubes, followed by incubation for 1 h 30 min at room temperature. The tubes were washed five times with 100 μl of PBT and three times with 100 μl of sterile water and then dried briefly.
BSV was genotyped by PCR directly in tubes using specific BSV species primers. The following primers were used: for BSOLV, OL-R (5′-GCT CAC TCC GCA TCT TAT CAG TC-3′) and OL-F (5′-ATC TGA AGG TGT GTT GAT CAA TGC-3′); for BSImV, Im-R (5′-CAC CCA GAC TTT TCT TTC TAG C-3′) and Im-F (5′-TGC CAA CGA ATA CTA CAT CAA C-3′); and for BSGFV, GF-R (5′-TCG GTG GAA TAG TCC TGA GTC TTC-3′) and GF-F (5′-ACG AAC TAT CAC GAC TTG TTC AAG C-3′). PCRs were performed as described above at an annealing temperature of 58°C for 25 cycles (BSImV) or 30 cycles (BSOLV and BSGFV). Genomic DNA contamination was controlled using Musa sequence-tagged microsatellite site primers AGMI025 (5′-TTA AAG GTG GGT TAG CAT TAG G-3) and AGMI026 (5′-TTT GAT GTC ACA ATG GTG TTC C-3′) (43). These primers were used in multiplex PCR with the specific BSV primers described above.
Haplotype divergence of eBSV in PKW.
The divergence time between allelic Musa regions corresponding to overlapping BAC sequences was calculated based on genic, intergenic, and repetitive sequence divergence according to the formula T = K/(2r), where T is the time of divergence, K is the number of base substitutions per site, and r is the substitution rate (44). Nucleotide substitutions were calculated using MEGA4 (45) with the Kimura 2-parameter substitution model. A rate 2-fold higher than that determined for coding sequences in banana (46) was used based on the assumption that noncoding sequences evolve more rapidly (47).
In order to estimate if sequential insertions of BSV occurred in eBSV, we calculated the p-distance (proportion of nucleotide differences between two sequences) within each eBSV allele for their duplicated open reading frames (ORFs) and intergenic sequence (IG). The same sequence was used to calculate the p-distance between eBSV alleles. Due to the high degree of reorganization of each eBSV, we compared only parts of the viral genome, calculated pairwise distances, and, finally, used the average p-distances to compare their evolution. For eBSGFV, we compared ORF1 and ORF2 included in fragments III, Va, Vb, and Vc; 0.6 kb of ORF3 included in fragment II, IV, Va, and Vc; and 0.24 kb of IG in fragments III, Va, Vb, and Vc. For eBSOLV, we compared ORF1 and ORF2 in fragments 1-I, 1-VI, 1-VII, 2-I, 2-VII, and 2-VIII; 3 kb of ORF3 included in fragments 1-I, 1-V, 2-III, 2-VII, and 2-VIII; 1 kb of ORF3 included in fragments 1-I, 1-V, 1-VIII, 2-III, 2-VII, 2-VIII, and 2-IX; and 0.66 kb of IG included in fragments 1-I, 1-VI, 1-VII, 2-I, 2-VII, and 2-VIII. For BSImV, we compared 1 kb of ORF3 included twice in fragments Im-II and once in Im-III and 1 kb of IG included twice in fragment Im-II.
The complete distance matrix is available in File S2 in the supplemental material.
Nucleotide sequence accession numbers.
GenBank accession numbers for BAC harboring eBSImV sequence (MBP_68C24) and eBSOLV sequences (MBP_17D14c) are HE983625 and HE983609, respectively.
RESULTS
Presence of eBSV in the genome of PKW.
We screened high-density filters of the 9× Musa balbisiana Pisang klutuk wulung (PKW) BAC library and two additional Musa acuminata BAC libraries (the seedy diploid Calcutta 4 and the triploid “Cavendish” cv. Grande Naine, with coverages of 9× and 4.5×, respectively) with probes covering the full-length genomes of four distinct BSV species—Obino l'ewai (BSOLV), Imove (BSImV), Mysore (BSMyV), and Vietnam (BSVNV)—as described in reference 13 for BSGFV. The M. acuminata BAC libraries did not produce any signal with either BSV species, while the M. balbisiana BAC library had signals with BSOLV, BSImV, and BSMyV (Table 1). We analyzed the fingerprints of all BAC clones positive for each BSV species after hybridization with the corresponding BSV probes. BSOLV, BSGFV, and BSMyV had two distinct restriction patterns, while BSImV exhibited a single pattern despite the use of four different enzymes (data not shown). All BSV species showed a number of hits that never exceeded three times the coverage of the BAC library. Furthermore, each eBSV showed only one or two distinct patterns. For this work, we focused on BSV species that are present, expressed, and consequently infectious in the banana F1 population described in reference 12, i.e., BSOLV, BSGFV, and BSImV.
Table 1.
Overview of BSV integrations in the PKW BAC library
| Probe | No. of hits | Fingerprint eBSV pattern | BAC(s) selected |
|---|---|---|---|
| BSGFV | 9 | 2 | MBP_071C19, MBP_094I16 (13) |
| BSOLV | 10 | 2 | MBP_031O07, MBP_073B22, MBP_017D14 |
| BSImV | 24 | 1 | MBP_068C24 |
| BSMyV | 15 | 2 | None |
| BSVNV | 0 | None |
We set up a fluorescent in situ hybridization (FISH) experiment on metaphase chromosome preparations of PKW using probes corresponding to the full-length viral genome. eBSV hybridization was carried out independently for each of the three BSV species (Fig. 1). We observed signals on two distinct chromosomes for each BSV species; double dots on chromosomes correspond to the hybridization of eBSV on both sister chromatids. eBSImV and eBSGFV are located in chromosomes distinct from those carrying rDNA. Taken together, our data clearly indicated low-copy-number integration of all three BSV species.
Fig 1.
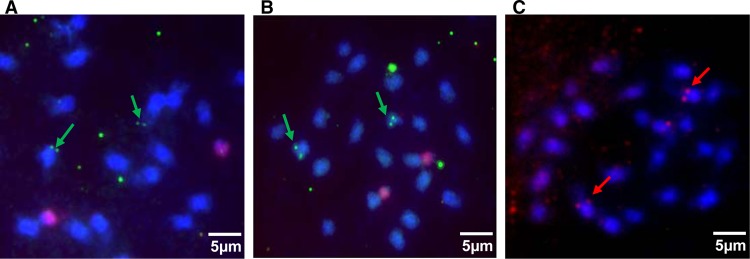
Localization of the three BSV integrations by fluorescent in situ hybridization in root cells of PKW banana chromosomes. Hybridizations were performed with full-length genome probes for each BSV species and detected with FITC (green for BSGFV in panel A and BSImV in panel B) or rhodamine (red for BSOLV in panel C). Pink diffuse signals in panels A and B correspond to the 45S rDNA sequence labeled with Alexa 555. Chromosomes are counterstained with DAPI (blue).
Structure of eBSV in the genome of PKW.
Of the three BSV species detected within the Musa balbisiana BAC library, we have already extensively described integrations of BSGFV termed eBSGFV (endogenous BSGFV) in reference 13. eBSGFV is located in a single locus of the genome and presents two different alleles with distinct structures and infectious properties.
Here, following sequencing of three BAC clones, we determined the structure of eBSOLV. The sequences of BAC clones MBP_73B22 and MBP_17D14, which exhibited identical restriction patterns, were grouped into a contig based on an identical sequence overlap of 69,938 bp to obtain a sequence of 185,044 bp named MBP_017D14c (HE983609). We observed eBSV in both BACs MBP_31O07 and MBP017D14c as a continuous stretch of sequences highly similar to that of BSOLV. The viral integrants in BACs MBP_31O07 and MBP017D14c are hereafter referred to as eBSOLV-1 and eBSOLV-2, respectively. The integrants are much longer than a single BSV genome: 22,900 bp for eBSOLV-1 and 23,200 bp for eBSOLV-2, compared to 7,839 bp for BSOLV. These integrants are composed only of viral sequences, with no Musa embedded genome sequences (Fig. 2). Both exhibit a complex rearrangement of viral sequences, with most viral regions being duplicated and therefore present in several copies within each eBSV, either in the same or in the opposite orientation with respect to the organization of the episomal BSV genome.
Fig 2.
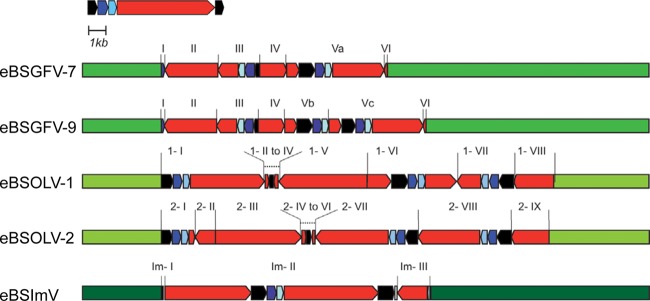
Overview of eBSV structures in PKW. Banana genomic sequences are in green. The BSV genome is represented in linear view with dark blue, light blue, and red boxes indicating ORF 1, ORF2, and ORF3 of the virus, respectively. The intergenic region is in black. eBSV fragments are indicated.
eBSOLV-1 is composed of 8 fragments, numbered 1-I to 1-VIII, that are structurally identical to BSOLV (Fig. 3A). Fragments 1-I, 1-VI, and 1-VII contain intergenic sequence (IG) followed by a complete ORF1 and ORF2 and part of ORF3. In fragment 1-VI, this is preceded by part of ORF3. IG is complete in fragment 1-VI and truncated by 315 bp in 1-I, 668 bp in 1-III, and 91 bp in 1-VII. All IGs are similar and differ from the BSOLV intergenic region by a 9-bp insertion (5′-ATAGCTGTA-3′) at position 90 (except in fragment 1-VI) and a 12-bp insertion (5′-GACTGGCTAGGT-3′) at position 434 of the virus. Fragments 1-II and 1-IV have only a small part of ORF3 (200 to 300 bp), while fragments 1-V and 1-VIII harbor larger parts (>1 kb). No full-length ORF3 was present in any of the 7 fragments that contain it. However, considering all fragments, the entire ORF3 can be reconstituted (Fig. 3A).
Fig 3.

Positions of eBSV fragments in the counterpart BSV genome. BSOLV (A) and BSImV (B) genomes are each represented in linear view, with a kilobase pair scale bar at the top. IG, ORF1, ORF2, and ORF3 are labeled. Boxes below the BSV genome represent fragments of eBSV in the same orientation (white boxes) or in reverse orientation (black boxes). Coordinates indicate boundaries of eBSV fragments in reference to the viral genome. The names of fragments are indicated on the left side. The gray box below the scale bar indicates the missing fragment of ORF3 in eBSOLV-2. Black lines below the scale bar represent the zone of ORF3 used for dating analysis of both BSOLV and BSImV.
eBSOLV-2 is composed of 9 fragments (Fig. 3A). Fragments 2-I, 2-VII, and 2-VIII contain IG followed by complete ORF1 and ORF2 and part of ORF3. All IGs are truncated, by 315 bp in 2-I, 668 bp in 2-V, 234 bp in 2-VII, and 91 bp in 2-VIII, and were similar to those found in eBSOLV-1, including both 9- and 12-bp insertions. Unlike eBSOLV-1, none of them was complete. Indeed, a fragment of 91 bp at the 5′ end of IG was always missing. Fragments 2-IV and 2-VI corresponded only to a small part of ORF3, while fragments 2-II, 2-III, and 2-IX corresponded to larger parts. No full-length ORF3 was present in any of the 8 fragments that contained it. Moreover, and in contrast to eBSOLV-1, a fragment of 217 bp (corresponding to nucleotides 6732 to 6949 on the BSOLV genome) at the 3′ end of ORF3 was always missing.
The 5′ and 3′ flanking regions of eBSOLV-1 and eBSOLV-2 were similar over 6,063 bp and 2,426 bp, respectively, despite the structural reorganization of eBSOLVs. This indicated a common locus of insertion in the Musa genome.
We described eBSImV based on sequencing of BAC MBP_068C24 (HE983625). This 121,693-bp sequence contained one eBSV as a unique stretch of sequence similar to BSImV (Fig. 2). The integrant was again much longer than a single BSV genome: 7,827 bp for BSImV and 15,800 bp for eBSImV. eBSImV is composed of 3 fragments: Im-I, Im-II, and Im-III. Im-I is composed of 35 bp of IG. Im-II is a long stretch of sequences containing more than 1 copy of the complete circular viral genome (1.76 viral genomes). Starting in the middle of ORF3, this fragment contained the 3′ part of ORF3, followed by the complete sequences of IG, ORF1, ORF2, and ORF3 and finishing with a truncated IG sequence missing about 240 bp at the 3′ end. Im-III contained only the 3′ end of ORF2 and 1.9 kbp of ORF3 in reverse orientation (Fig. 3B).
Genomic organization of eBSV in the genome of PKW.
BSGFV is integrated at a single locus as two alleles (13). We characterized the genomic organization of eBSOLV and eBSImV to see whether they followed a similar organization.
The two eBSOLVs containing BAC clones exhibited very high sequence identity (99.998%) on the 108.6 kbp of overlapping Musa regions. This high gene and transposable element (TE) structure conservation was consistent with the hypothesis of allelic insertion for BSOLV as described for BSGFV, where eBSGFV-7 and eBSGFV-9 are located on homologous chromosomes.
To further analyze allelic insertion in PKW, we first monitored the segregation of eBSOLV-1, eBSOLV-2, and eBSImV among the triploid (AAB) F1 progeny of a genetic cross between PKW (BB) (female parent) and M. acuminata cv. IDN 110 4x (AAAA) (male parent). The parents were confirmed as virus free by multiplex immunocapture PCR (48). As a high degree of sequence conservation between the integrated and episomal forms of BSV existed, we developed molecular markers (PCR and derived cleaved amplified polymorphic sequences [dCAPs]) to genotype each eBSV, discriminating them from their viral counterparts (see Materials and Methods and supplemental material). These PCR markers were specific to either Musa junctions or internal structure and, where possible, specific to each allele. The genotyping of parents confirmed the absence of eBSOLV-1, eBSOLV-2, and eBSImV within the M. acuminata parent (data not shown).
We developed a dCAP (Dif-OL-HaeIII) marker to distinguish between eBSOLV-1 and eBSOLV-2 (Fig. 4). A PCR amplification test using a set of primers specific to eBSOLV-2 and located on another part of eBSV allowed confirmation of the allelic profile with presence/absence scoring. eBSOLV-1 and eBSOLV-2 segregate in 53.5% and 46.5% among the F1 progeny, respectively, which was compatible with the 50/50 ratio expected for single locus segregation (df = 2; χ2 = 0.40; P = 0.70). As for eBSGFV, this indicated a monogenic segregation of eBSOLV.
Fig 4.
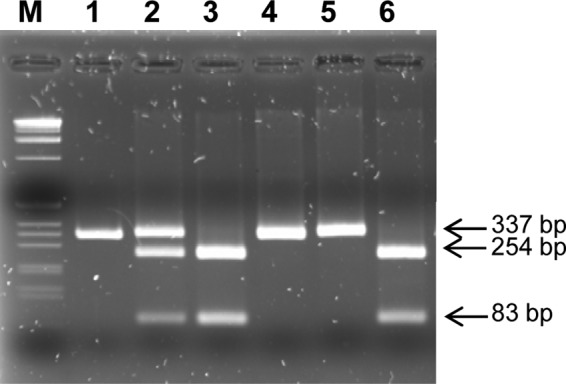
Genotyping of eBSOLV-1 and eBSOLV-2 in the F1 triploid (AAB) population using a dCAP marker. The PCR product obtained from eBSOLV-1 is cut into two bands of 83 and 254 bp by the endonuclease HaeIII (New England BioLabs), whereas the one from eBSOLV-2 is not digested. Lane M, 1-kb DNA ladder (Invitrogen); lane 1, undigested PCR product of the diploid M. balbisiana PKW; lanes 2 to 6, digested PCR product from PKW carrying both eBSOLV alleles, BAC MBP_31007 carrying eBSOLV-1, BAC MBP_73B22 carrying eBSOLV-2, and two triploid (AAB) F1 progeny of a genetic cross between PKW and M. acuminata cv. IDN 110 4x (AAAA) carrying eBSOLV-2 and eBSOLV-1, respectively. Digested DNA was loaded onto a 2.5% agarose gel stained with ethidium bromide, and bands were visualized under UV light.
A unique eBSV is described for eBSImV, and its genotyping by PCR revealed its presence in the entire progeny. Next, we developed various molecular markers and strategies to detect a potential second eBSImV allele: sequencing of the eBSImV ends for the 24 BAC clones positive with BSImV probe and development of two dCAP markers based on point mutations detected on eBSImV (see “Which allele is infectious?” below) and 20 SSR derived from Musa sequences flanking eBSImV. Unfortunately, all SSR were monomorphic in PKW (data not shown), and point mutations were detected in all BAC clones (data not shown). Finally, no single nucleotide polymorphism (SNP) was observed between BAC end sequences and the MBP_068C24 sequence (data not shown).
Based on these genetic results, we demonstrated that the two chromosomes labeled with each BSV species using the FISH technique (Fig. 1) were homologous chromosomes. Integration of the three BSV species is confirmed as allelic, all resulting from a monolocus integration event within the genome of PKW. The integration of BSGFV and BSOLV is diallelic, whereas that of BSImV is monoallelic.
Which allele is infectious?
In the case of BSGFV, we previously demonstrated that only eBSGFV-7 is infectious by in silico sequence analysis and by monitoring the segregation of BSGFV infection among the triploid (AAB) F1 progeny described in “Genomic Organization of eBSV in the Genome of PKW” above (13). We first searched in silico for the presence of a full-length viral genome in all eBSOLV and eBSImV sequences and further analyzed the type of mutations accumulated in each eBSV allele (Table 2). We noticed a deletion of 309 bp at the junction of ORF3 and IG in eBSOLV-2 (Fig. 3A). This deletion corresponded to nucleotides 6732 to 7041 of the BSOLV genome. Conversely, eBSOLV-1 contained the full-length BSV genome at least once (Fig. 3A). We compared the homologous region of each eBSOLV-1 fragment to the corresponding BSV genome. Despite the very close similarity (>99% identity on average) between sequences, some differences observed in eBSV sequences may affect viral gene functions. There were 15 synonymous and 26 nonsynonymous mutations (Table 2), but none of these led to either premature stop codons or frameshifts. Based on these in silico analyses, eBSOLV-1 was the only possible infectious allele.
Table 2.
Number of mutations accumulated within each eBSV fragment compared to the reference genome of BSOLV (A) and BSImV (B)a
| eBSV | Total no. of mutations | No. of Syn | No. of nSyn | Mutation rate |
|---|---|---|---|---|
| eBSOLV-1 | 48 | 15 | 26 | 0.0021 |
| eBSOLV-2 | 65 | 19 | 37 | 0.0028 |
| eBSImV | 23 | 10 | 9 | 0.00145 |
Total number of mutations includes mutations in intergenic region. Syn, synonymous mutations; nSyn, nonsynonymous mutations.
Regarding eBSImV, no comparison of the two alleles was possible, as they are so far identical. We identified at least a full-length viral genome within eBSImV as described in “Structure of eBSV in the Genome of PKW” above. As with eBSOLV and eBSGFV, our in silico analysis showed a very high degree of sequence conservation between integrated and episomal forms of the virus present in the database (25). Similarly, identity was higher than 99%, with only 23 mutations found among the 15.8 kbp of eBSImV (Table 2). Synonymous and nonsynonymous mutations are represented equally, with 10 and 9 mutations, respectively. In addition, we found two deleterious mutations in the two ORF3s in fragment II: (i) a deletion of an adenosine at position 95192 on BAC MBP_068C24 leading to a frameshift and premature stop codon (61 bp downstream of the deletion) and (ii) a substitution of guanine to adenosine at position 102946 leading to a premature stop codon (Fig. 2 and 3). The presence of these two mutations, which was confirmed in the 24 BAC clones isolated during PKW library screening, precludes reconstitution of an episomal virus through simple transcription, as the point mutations are 7,754 bp apart, whereas a full-length BSImV genome is 7,768 bp long. In addition, mutations at both positions would seem to interrupt production of a complete protein.
We then monitored the segregation of either BSOLV or BSImV infection among the triploid (AAB) F1 progeny using immunocapture PCR (IC-PCR) and BSV species-specific primers. In parallel, we genotyped the same population with the molecular markers developed to discriminate plants harboring the eBSOLV-1 or eBSOLV-2 allele. Allelic genotyping is not possible for eBSImV. We found that 99% of the diseased hybrids were infected by BSOLV and that 100% of BSOLV-infected plants harbored the eBSOLV-1 allele. In addition, we sequenced the IG of viral particles infecting five independent triploid offspring to search for the 12-bp and 9-bp insertions present in the IGs of eBSOLV. No insertion was observed at position 90, whereas partial insertions at position 434 were seen in all released virions. These insertions were all shorter than 12 bp and differed from one virion to another, but all had 100% sequence identity to the reference BSOLV genome (Fig. 5). All together, our data demonstrated unambiguously that BSOLV infecting the progeny originated from the eBSOLV-1 allele and that fragment 1-VI is involved in release of the functional episomal genome, since it was the only allele to display an IG without the 9-bp insertion at position 90. One interesting possibility was that the region of the 12-bp insertion could be a hot spot of recombination involved in release of the BSOLV genome.
Fig 5.

Comparison of IGs of BSOLV infecting triploid offspring and eBSOLV. Primers (BSV2, 5′-GTA TCA GAG CAA GGT TCG TTT TT-3′, and BSV525, 5′-ATC CCA AGT TTT CTC GAC CAT AA-3′) surrounding the 9- and 12-bp deletions in the IG of BSOLV were designed and used in IC-PCR on 5 independent infected interspecific AAB hybrids (plants 55, 139, 196, 199, and 207) using the following PCR program: denaturation stage at 94°C for 5 min followed by 30 cycles of 30 s at 94°C, 30 s at 60°C, and 1 min at 72°C and a final extension at 72°C for 10 min. PCR products were purified and sequenced. BSOLV, IG of episomal virus (accession number AJ002234); eBSOLV1 (1-VI), IG present in fragment VI of eBSOLV allele 1; eBSOLV, IG present in both alleles of the wild diploid M. balbisiana PKW (except in fragment VI of eBSOLV1); plants 55 to 207, IG of episomal virus produced from eBSOLV in the different hybrids.
The detection of BSImV particles expressed in the AAB progeny shows that 88% of the diseased hybrids were infected with BSImV (49) (data not shown). Although the BSImV particles present in the hybrids came from eBSV, we cannot say which allele was infectious, as they were structurally identical. We then performed PCR and sequencing of the part of ORF3 originally containing the two point mutations in BSImV particles obtained from five triploid offspring. None of the BSImV sequences obtained harbored either of the two point mutations. This led us to think that either the two alleles recombined during gamete formation or reverse transcriptase of the virus may change one of the point mutations very early after the first transcription event.
Genomic landscape and eBSV chromosome localization.
At first glance, gene and TE content appeared to be highly variable between the different eBSVs (Fig. 6). Indeed, eBSGFV and eBSImV are inserted within two gene-rich regions with an average of 0.17 and 0.15 gene per kbp, while eBSOLV is inserted within a TE-rich region with only one predicted gene among the 190 kbp of total BAC sequence (0.005 gene per kb).
Fig 6.
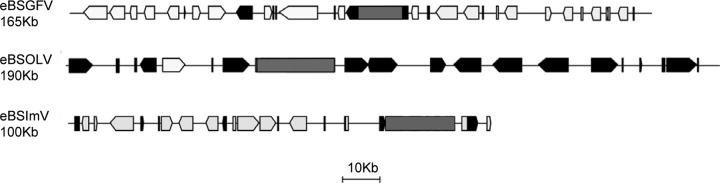
Gene and TE contents surrounding eBSVs in the Musa genome present in BAC clones. The black boxes correspond to TE, the gray boxes to eBSV, and the white boxes to genes in BAC clones containing eBSGFV, eBSImV, and eBSOLV.
We investigated the 2 kb upstream and downstream of each eBSV more precisely by using multiple alignment tools (MAFFT and CLUSTAL) in order to search for a common structure at the insertion locus. We previously established (13) that eBSGFV is inserted into a Ty3/gypsy-like retrotransposon, which is itself inserted into the fifth intron of the mom gene. No similarities or notable structures existed that suggested a common target insertion site for the three BSV species. We performed additional analyses of the 20 first bases upstream and downstream of each eBSV to search for target site duplication (TSD). No similarity was recorded.
eBSOLV is inserted in a TE-rich region between two recently inserted retrotransposons based on long terminal repeat (LTR) similarities (data not shown). Unlike eBSGFV, neither gene fragments nor retrotransposon-interrupted structures are detected flanking eBSVs, indicating that insertion occurred in intergenic sequences. We identified gypsy-like retrotransposons and a copia-like retrotransposon (RE06) close to the 5′ and 3′ flanking regions of eBSOLVs, respectively; however, the gypsy-like retrotransposon was not similar to the one containing eBSGFV.
For eBSImV, the 5′ flanking region is composed of a truncated RE02 LARD element, while the 3′ region is composed of intergenic sequence. We observed an environment similar to that of eBSOLV, although the BAC was rich in genes.
We then performed FISH analysis using the three BSV species as probes to hybridize to the same chromosome preparation. eBSGFV and eBSOLV colocalized on the same chromosome (as demonstrated by the presence of a pair of double green dots on the same chromosomes), whereas eBSImV was on a different chromosome (Fig. 7).
Fig 7.

Localization of the three eBSV on the chromosomes of PKW. Two independent metaphases are shown (A and B). Hybridizations were performed with full-length genome probes for each BSV species and detected with FITC (green) for BSGFV and BSOLV and with Alexa 594 for BSImV. Chromosomes are counterstained with DAPI (blue).
BLAST alignment of all BAC clones to the recent Musa acuminata reference genome sequence (38) allowed synteny analysis to determine potential chromosome localization (Fig. 8). We noted strong similarity for BACs containing eBSGFV with chromosome 1 and for BACs containing eBSImV with chromosome 2. Regarding the two regions surveyed, breaks in synteny are located precisely in the neighborhood of eBSVs and in the vicinity of some TEs. This confirmed the distinct localization of both integrations. No match was found for BAC containing eBSOLV. This was probably due to the high TE content of the BAC clone. Unfortunately, the only gene present on the BAC was not yet anchored in the Musa acuminata reference genome sequence.
Fig 8.

eBSV locus synteny with the Musa acuminata reference genome. “MBP094I16” and “MBP068C24” refer to BACs containing eBSGFV and eBSImV, respectively. The black boxes correspond to TEs and the white boxes to genes. Thick black lines represent the part of chromosomes 1 and 2 of the reference M. acuminata genome (38) matching with BAC containing eBSGFV and eBSImV, respectively. Positions on chromosome are indicated with coordinates.
Divergence and evolution of eBSV since their integration.
Faced with the genomic and genetic organization of eBSV within PKW described in this article, we concluded that the genetic organization was complex and could have resulted from multiple and/or sequential insertions of BSV at the same locus. In all cases, the initial hemizygous integration has been duplicated onto the homologous chromosome, but depending on the scenario, the timing of this duplication was more or less recent. By comparing the nucleotide diversities of similar sequences, we then calculated the p-distance and Ks coefficient (synonymous mutations/synonymous sites) between BAC haplotypes (Tables 3 and 4) and the p-distance within eBSV alleles (Table 5).
Table 3.
Musa haplotype gene divergence of eBSV BAC clone datinga
| Haplotype | Size (bp) | nSyn | Syn | Ks | Time of divergence (My) |
|---|---|---|---|---|---|
| BSGFV haplotypes | |||||
| PAL | 2,139 | 0 | 0 | 0 | 0.00 |
| Zonadhesin | 3,846 | 1 | 1 | 0.0011 | 0.12 |
| HP1 | 1,239 | 0 | 0 | 0 | 0.00 |
| HP2 | 1,419 | 0 | 0 | 0 | 0.00 |
| Auxin-responsive protein | 420 | 0 | 0 | 0 | 0.00 |
| Actin depolymerizing factor | 432 | 1 | 0 | 0 | 0.00 |
| BSOLV haplotype | |||||
| Histidine kinase | 3,001 | 0 | 0 | 0 | 0 |
Syn, synonymous mutation; nSyn, nonsynonymous mutation; Ks:, synonymous mutations/synonymous sites; MY, million years.
Table 4.
Musa haplotype nucleic acid divergence of eBSV BAC clones
| Element | Size (bp) | No. of nucleic acid differences between haplotypes | p-distance |
|---|---|---|---|
| Colinear region except eBSGFV | 82,579 | 210 | 0.002554 |
| Colinear region except eBSOLV | 101,400 | 130 | 0.00129 |
Table 5.
Divergence (p-distance) within eBSVs
| eBSV allele | p-distance |
|||
|---|---|---|---|---|
| ORF1 | ORF2 | ORF3 | IG | |
| eBSGFV-7 | 0.0000 | 0.0000 | 0.0042 | 0.0000 |
| eBSGFV-9 | 0.0025 | 0.0040 | 0.0074 | 0.0056 |
| eBSOLV-1 | 0.0012 | 0.0000 | 0.0011 | 0.0010 |
| eBSOLV-2 | 0.0000 | 0.0016 | 0.0014 | 0.0031 |
| eBSImV | NAa | NA | 0.0018 | 0.0010 |
NA, not applicable.
The haplotype divergence of gene sequences surrounding eBSV sites is presented in Table 3. No divergence was recorded for the only gene present in eBSOLV BACs, indicating recent duplication or a high selection constraint. Similarly, we observed weak divergence among the six genes present in eBSGFV BACs, with only two genes containing three mutations (2 nonsynonymous and 1 synonymous). The divergences of nucleic acid sequences flanking eBSVs are 0.0026 for the eBSGFV locus and 0.0013 for the eBSOLV locus. These values were similar to those of eBSVs themselves, in which divergence between eBSGFV-7 and eBSGFV-9 is 0.0037 and divergence between eBSOLV-1 and eBSOLV-2 is 0.0013 (see File S1 in the supplemental material). Consequently, the data presented here do not allow calculation of the time of divergence based on synonymous mutations accumulated into gene sequences. However, the nucleotide divergence data obtained on almost 100 kb of sequence may indicate that divergence of the eBSGFV locus may be twice as old as that of the eBSOLV locus.
Within eBSV, the divergence recorded varies from gene to gene (Table 5). The divergence recorded within eBSGFV-7 and eBSGFV-9 in ORF3 was higher than that of other genes (ORF1 and ORF2) or IG. The divergence observed within eBSGFV-9 was systematically higher than that observed within eBSGFV-7. This certainly indicated a differential evolution of each allele.
The values obtained for both genes and IG within eBSOLV-1 and eBSOLV-2 were similar, with the exception of IG in eBSOLV-2, which accumulated more mutations. The divergence observed within eBSOLV-2 was systematically higher than that observed within eBSOLV-1 except for ORF1, indicating a differential evolution of each allele here also, as observed for eBSGFV. Values were globally lower than those recorded for eBSGFV.
The eBSImV divergence of duplicated ORF3 and IG within the available allele was similar to that recorded for ORF3 of eBSOLV.
DISCUSSION
Similar integration patterns for each BSV species in PKW.
In this study, we fully characterized for the first time the integration of three widespread BSV species present within the genome of PKW and restricted to Musa balbisiana. We found that these eBSVs are absent not only from the two other common Musa acuminata accessions screened during this study but also from the Musa acuminata subsp. malaccensis DH Pahang reference sequence (38) and more widely from the other Musa species (20; P.-O. Duroy et al., unpublished data). Both genetic studies and FISH analysis confirmed that integration of BSOLV, BSGFV, and BSImV each resulted from independent events at a single locus. The PKW genome harbors at least three different BSV species integrated once each, which is rare compared to integration events reported for other plant pararetroviruses. More than a hundred clustered copies of viral integrants exist in tobacco (N. edwardsonii) (10), and ca. 100 to 200 copies of PVCV are integrated at five loci in petunia (11). No other viral integrants or molecular fossils of BSOLV, BSImV, or BSGFV, such as those reported for endogenous retroviruses (50), were found in the PKW genome other than those described here. These examples illustrate the diversity of infectious integrant patterns known nowadays in plants, ranging from a few copies to several hundred copies.
Unlike the patterns described for PKW, noninfectious BSV-like sequences ranging from 700 bp to 18,000 bp have been discovered recently within the Musa acuminata genome of the newly sequenced DH-Pahang (38). These correspond to 24 loci spread over the entire banana genome, on 10 out of 11 chromosomes. Similar structures are reported for rice, in which Kunii et al. (51) found 29 endogenous noninfectious rice tungro bacilliform virus (RTBV)-like sequences, and in tobacco (N. tabacum), in which thousands of noninfectious TVCV-like insertions have been uncovered (52). In contrast to infectious integrants, noninfectious integrants seem to exhibit similar patterns, with numerous integrations all over the plant genome.
We noted that the 5′ flanking region of eBSOLV in PKW is identical (100% identity) to the musa6 clone previously sequenced (accession number AF106946) from the polyploid cultivated AAB banana clone Obino l'Ewai (53). In addition, based on the use of specific PCR markers for each integrant, we have previously reported that eBSGFV and eBSImV conserve the same locus of integration among all the Musa balbisiana diploids available worldwide (20). Using the same approach, our group (Duroy et al., unpublished) confirmed that the eBSV/Musa balbisiana genome junctions are extremely well conserved in all three BSV species in all B genomes available among 77 accessions of diploids and triploid banana plants representing the entire Musa balbisiana diversity available. All together, our data indicate that BSV integration occurred in natural Musa balbisiana diploids before domestication and highlight the fact that current diploids and A/B interspecific cultivars originated from the same M. balbisiana ancestor carrying eBSVs.
The vicinity of endogenous pararetroviruses (EPRVs) with retrotransposons, mainly Ty3-gypsy elements of the family Metaviridae, has been reported frequently for several plants (11, 54, 55). In PKW, this is supported by the systematic presence of TEs closely surrounding eBSV. All eBSVs are inserted into, or very close to, repeated elements. This suggests that the process of BSV integration may be opportunistic. Quasi-LTR (QTR) regions similar to those flanking ePVCV in the petunia genome do not exist in banana. No specific sequence signatures required for retroelement integration (e.g., target site duplication or inverted repeats) is observed flanking eBSVs. We found that the three BSV integrations occur in different types of Musa regions (gene rich for eBSGFV and eBSImV, and a TE-rich region for eBSOLV) and on different chromosomes (chromosome 1 for eBSOLV and eBSGFV and chromosome 2 for eBSImV). Clearly, little is known about the molecular mechanisms of integration of pararetroviruses into plant genomes, and additional data are required to elucidate this process. However, as viral DNA gets into the plant nucleus during infection, it has the chance of becoming integrated during DNA break repairs, as suggested for eBSGFV inserted into the Ty3/gypsy retrotransposon (13) or by illegitimate recombination following the mechanisms of nonhomologous end joining (NHEJ) as reported for several endogenous viral elements (EVE) (56, 57). In this case, integration is thought to occur during the minichromosome viral phase due either to the two gaps existing within the open circular viral DNA, allowing access to single-stranded DNA, or to single-stranded overhanging sequences (flaps) constituting the end of the open circular viral DNA being readily available to initiate the recombination process (9, 11, 51, 52). This could explain the structure of eBSImV, i.e., 1.76 linear genomes. Taken together, these data support the hypothesis of stochastic insertion of each BSV into a single locus of the Musa genome.
Sequential integration of BSV species into the genome of PKW.
It is difficult to estimate the time of BSV integration because BSV probably evolves with a higher substitution rate than that of its host and, consequently, of the eBSV counterpart, which is subject to the host rate. Consequently, as described for other EVE (56), eBSV should reflect an ancestral state of the virus genome. We recorded strong identity at the nucleic acid level between eBSV and BSV for the three species (>99%).
The nucleotide divergence recorded between BSV and eBSV is between 1.5 × 10−3 and 2.8 × 10−3 mutations per site among the three BSV species (Table 2). This divergence is due to the differential rate of mutation between viruses and the plant genome. With a rate for retroviruses assumed to be between 10−6 and 10−4 mutations per site per cell infection (58), we can estimate the divergence time between BSV and eBSV at 15 to 2,800 cell infection cycles (59). This could indicate a fairly recent integration, whereas our data suggest an integration event after the divergence of A/B but before M. balbisiana diversification. This is in agreement with our earlier work (13), which estimated BSGFV integration at or after 0.640 million years ago. Therefore, the low divergence observed between eBSV and BSV more probably reflects the massive and almost exclusive contribution of eBSVs to the current viral population, since no epidemic of BSOLV, BSImV, and BSGFV is reported worldwide.
The nucleotide divergences recorded within eBSVs (Table 5), between eBSVs (Table 5), and between BSV and eBSV (Table 2), as well as the structural organization of integration, suggest sequential integration of BSV species into the PKW genome. It is difficult to distinguish which arrived first between BSOLV and BSGFV, since the p-distance seems to indicate an older insertion for BSGFV but the structure of eBSOLV, which is more rearranged, may suggest the opposite. However, eBSImV contains fewer mutations that other eBSVs, shows a relative linear viral genome, and is monoallelic, all of which clearly suggest that eBSImV is the most recent.
Integration and rapid evolution of eBSV loci.
The different patterns of integration in PKW for the three BSV species range from structures similar to the concatenated linear viral genome for eBSImV to the more complex organization of eBSGFV and eBSOLV. Integrant size is always over a full-length viral genome, with no embedded Musa sequences, and each eBSV locus contains one functional genome. All three eBSVs have two allelic copies.
Integration of tandem copies of a virus genome has been documented for plants (11). In eBSImV, we found a continuous structure of a 1.76-length viral genome, suggesting that a tandem integration process occurred also in banana. This duplicated structure is inserted in the neighborhood of repetitive elements that may promote rapid evolution of eBSV, as has been documented for resistance gene cluster evolution (60, 61). Indeed, the current structure of the eBSV locus may be compared to that described extensively for the same plant in the case of the RGA08 locus (62). Unlike the RGA08 locus, where intergenic sequences and TEs are clearly implicated in the evolution of the locus, we found that only viral sequences are involved in evolution of eBSV locus structure. We observed that the variation in eBSV structure concerns only viral sequences, whereas flanking sequences are remarkably conserved in all contexts, especially at the eBSOLV integration locus, which is flanked by dozens of TEs. Indeed, we estimated nucleotide diversity at 0.25% (0.03% for coding sequences) for eBSGFV haplotypes and 0.13% (no variation for coding sequences) for eBSOLV haplotypes (Tables 3 and 4).
To arrive at the current picture of integration in PKW, we assume that the most probable scenario is based on the unequal recombinations that follow duplication of the initial integration on the homologous chromosome. However, we cannot totally rule out other scenarios. Indeed, sequential targeted insertion of virus at the same locus might occur. We observed that point mutations accumulate in eBSVs at the same rate as in flanking Musa sequences, indicating that novel viral sequences have not integrated since haplotype divergence. Moreover, we searched for accumulation of mutations within duplicated viral fragments of eBSV. The absence of any difference allows us to discard the scenario of sequential waves of virus integration at the same locus, contrary to what is observed in the Musa acuminata HD genome (38; M. Chabannes and F. C. Baurens, unpublished data). A further possibility concerns recombination between eBSV and virions; however, this is very unlikely because it requires at least two crossovers within a very short distance (virus genome length) to produce viable gametes.
Current integration structures are driven by both virus and host.
eBSGFV, eBSOLV, and eBSImV are present in the genomes of all M. balbisiana isolates surveyed (20). We established in this study by genomic, genetic, and cytogenetic analyses that each eBSV is present on a homologous chromosome in the PKW genome. In a wild population with N random-mating diploids, if the new DNA does not confer any selective advantage on the host plant, the probability that the mutation becomes fixed (i.e., that the entire population contains homozygous new DNA) is 1/2N. The fixation process takes, on average, 4N generations (63). Fixation is unlikely to occur without selective pressure in the M. balbisiana context due to pollen dispersion over a large geographical area, which implies an extensive banana population (64). In addition, the flower morphology of Musa does not usually allow self-pollination in a single bunch because male and female flowers bloom not at the same time but sequentially. However, vegetative multiplication of bananas produces clumps of plants composed of the same genotype that can produce multiple flowers at the same time, thus allowing self-fertilization. Our data support the early statement that wild bananas are outbred but tolerate occasional generations of inbreeding (65).
PKW eBSV loci are widely conserved in almost all banana cultivars with a B genome (Duroy et al., unpublished). This is explained easily if a small number of plants form the origin of these cultivars. Indeed, new multidisciplinary findings concerning domestication of banana (66) suggest that the species M. balbisiana has been selected by humankind and transferred out of its geographical area of origin before contributing to some important groups of interspecific cultivars. However, the finding of similar eBSV structures also in seedy M. balbisiana populations (20) suggests that a strong bottleneck might have occurred as previously suggested in reference 64 to explain the relatively low diversity observed in M. balbisiana nowadays.
Based on the data collected and elements discussed in this article, we propose a model explaining the integration and evolution process of BSV from virions to the currently observed eBSV (Fig. 9). Our model uses the property of BSV to concatenate its DNA during replication as a template for the initial integration in germ cells. This was followed by a duplication process (self-pollination) of eBSV on the homologous chromosome. Alleles then evolved by both unequal recombination and point mutation accumulation. This process took place independently and sequentially for the three eBSVs described here.
Fig 9.
Proposed scenario for BSGFV, BSOLV, and BSImV integrations into the M. balbisiana nuclear genome. The life cycle of the virus produces concatenated viral genomes in the nucleus of the cell. This concatenated genome is integrated into the nuclear genome of PKW by illegitimate recombination. The three BSV species integrate sequentially into the PKW genome. After each BSV integration, the homozygous state is obtained by self-pollination and selected. Between each new integration and self-pollination, the previously integrated BSV evolves continuously by unequal recombination and point mutation insertions, leading to the present-day structure. The BSV genome is represented in linear view, with dark blue, light blue, and red boxes indicating the three ORFs of the virus. The IG is in black.
Why do eBSVs persist?
It is difficult to imagine maintenance of a functional viral sequence in the host genome without any selective advantage. The initial context of BSV integration favors a harmonious coexistence between the two partners. This proximity allowed passive and stochastic movement of DNA. It is therefore likely that the initial integration conferred a better fitness on the infected plant, leading to fixation of eBSVs in the banana population. Once fixed, the endogenous viral genome duplications and inversions observed may indicate a plant strategy to disarm any viral activity with potentially lethal effect. The presence of the viral genome in the plant allows the establishment of a resistance mechanism based on sequence homology between eBSV and BSV to prevent further external infection. Gene silencing-based resistance has been reported to occur in tobacco and petunia for TVCV and PVCV, respectively (11, 67, 68).
However, we observed that a differential selection between eBSV alleles exists (Table 5). This differential selection seems to favor the infectious allele, as more mutations accumulate in the noninfectious allele in both eBSOLV and eBSGFV. Moreover, the infectious allele (eBSGFV-7) is more prevalent in the seedy BB plant diversity than the noninfectious allele (20). This suggests that selection tends to retain functional alleles. We assume that after the initial integration of viral DNA into the plant genome, two antagonist forces act on the eBSV locus: one aiming to keep functional integrations in the population and the other aiming to disrupt the viral genome to prevent “self-infection.”
In PKW, eBSV structures imply a minimum of two steps of recombination to release a functional viral genome. These recombination steps have been described extensively by us previously (18) for eBSGFV, and our data suggest that recombination also occurs in the expression process for both eBSOLV and eBSImV. The structure of eBSOLV does not permit direct transcription, and virions obtained in the Musa population always contain fragment 1-VI and a hot spot of recombination existing in the IG that is probably involved in viral genome recircularization (Fig. 5). Functional BSImV most likely arises from recombination because of the presence of deleterious mutations in eBSV (frameshift or a stop codon) that prevent direct transcription of functional viral genomes. Thus, we assume that a first step of structural evolution, leading to viral genome shuffling in eBSV, is necessary to prevent the large-scale production of viral genomes and viruses from eBSVs, which might threaten plant populations.
In the case of PKW, this process must have occurred with the three BSV species at different times, and probably in response to different epidemic or abiotic constraints.
Supplementary Material
ACKNOWLEDGMENTS
We thank Gaëtan Droc for the setup of the Gnpannot annotation platform, Nathalie Laboureau for technical help in population segregation analysis, and Sophie Mangenot for technical help with BAC sequencing data. We are very grateful to P. Capy and A. D'Hont for critical readings of the manuscript.
P.-O. Duroy is supported by a CIRAD Ph.D. grant and P. Gayral by a CIRAD/Région Languedoc Roussillon Ph.D. grant.
Footnotes
Published ahead of print 29 May 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.00899-13.
REFERENCES
- 1. Weiss RA. 2006. The discovery of endogenous retroviruses. Retrovirology 3:67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Vogt PK. 1997. Historical introduction to the general properties of retroviruses, p 1–26 In Coffin JM, Hughes SH, Varmus HE. (ed), Retroviruses. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY: [PubMed] [Google Scholar]
- 3. Belyi VA, Levine AJ, Skalka AM. 2010. Unexpected inheritance: multiple integrations of ancient bornavirus and ebolavirus/marburgvirus sequences in vertebrate genomes. PLoS Pathog. 6:e1001030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Taylor DJ, Leach RW, Bruenn J. 2010. Filoviruses are ancient and integrated into mammalian genomes. BMC Evol. Biol. 10:193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Belyi VA, Levine AJ, Skalka AM. 2010. Sequences from ancestral single-stranded DNA viruses in vertebrate genomes: the Parvoviridae and Circoviridae are more than 40 to 50 million years old. J. Virol. 84:12458–12462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gilbert C, Feschotte C. 2010. Genomic fossils calibrate the long-term evolution of hepadnaviruses. PLoS Biol. 8:e1000495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Horie M, Honda T, Suzuki Y, Kobayashi Y, Daito T, Oshida T, Ikuta K, Jern P, Gojobori T, Coffin JM, Tomonaga K. 2010. Endogenous non-retroviral RNA virus elements in mammalian genomes. Nature 463:84–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Katzourakis A, Gifford RJ. 2010. Endogenous viral elements in animal genomes. PLoS Genet. 6:e1001191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hohn T, Richert-Poeggeler KR, Staginnus C, Harper G, Schwarzacher T, Chee How T, Teycheney PY, Iskra Caruana ML, Hull R. 2008. Evolution of integrated plant viruses, p 53–81 In Roossinck MJ. (ed), Plant virus evolution. Springer, Heidelberg, Germany [Google Scholar]
- 10. Lockhart BE, Menke J, Dahal G, Olszewski NE. 2000. Characterization and genomic analysis of tobacco vein clearing virus, a plant pararetrovirus that is transmitted vertically and related to sequences integrated in the host genome. J. Gen. Virol. 81:1579–1585 [DOI] [PubMed] [Google Scholar]
- 11. Richert-Pöggeler KR, Noreen F, Schwarzacher T, Harper G, Hohn T. 2003. Induction of infectious petunia vein clearing (pararetro) virus from endogenous provirus in petunia. EMBO J. 22:4836–4845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lheureux F, Carreel F, Jenny C, Lockhart BE, Iskra-Caruana ML. 2003. Identification of genetic markers linked to banana streak disease expression in inter-specific Musa hybrids. Theor. Appl. Genet. 106:594–598 [DOI] [PubMed] [Google Scholar]
- 13. Gayral P, Noa-Carrazana JC, Lescot M, Lheureux F, Lockhart BE, Matsumoto T, Piffanelli P, Iskra-Caruana ML. 2008. A single Banana streak virus integration event in the banana genome as the origin of infectious endogenous pararetrovirus. J. Virol. 82:6697–6710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dallot S, Acuna P, Rivera C, Ramirez P, Cote F, Lockhart BE, Caruana ML. 2001. Evidence that the proliferation stage of micropropagation procedure is determinant in the expression of banana streak virus integrated into the genome of the FHIA 21 hybrid (Musa AAAB). Arch. Virol. 146:2179–2190 [DOI] [PubMed] [Google Scholar]
- 15. Côte FX, Galzi S, Folliot M, Lamagnere Y, Teycheney PY, Iskra-Caruana ML. 2010. Micropropagation by tissue culture triggers differential expression of infectious endogenous Banana streak virus sequences (eBSV) present in the B genome of natural and synthetic interspecific banana plantains. Mol. Plant Pathol. 11:137–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Harper G, Hull R, Lockhart B, Olszewski N. 2002. Viral sequences integrated into plant genomes. Annu. Rev. Phytopathol. 40:119–136 [DOI] [PubMed] [Google Scholar]
- 17. Staginnus C, Richert-Poggeler KR. 2006. Endogenous pararetroviruses: two-faced travelers in the plant genome. Trends Plant Sci. 11:485–491 [DOI] [PubMed] [Google Scholar]
- 18. Iskra-Caruana ML, Baurens FC, Gayral P, Chabannes M. 2010. A four-partner plant-virus interaction: enemies can also come from within. Mol. Plant Microbe Interact. 23:1394–1402 [DOI] [PubMed] [Google Scholar]
- 19. Gayral P, Iskra-Caruana ML. 2009. Phylogeny of Banana Streak Virus reveals recent and repetitive endogenization in the genome of its banana host (Musa sp.). J. Mol. Evol. 69:65–80 [DOI] [PubMed] [Google Scholar]
- 20. Gayral P, Blondin L, Guidolin O, Carreel F, Hippolyte I, Perrier X, Iskra-Caruana ML. 2010. Evolution of endogenous sequences of banana streak virus: what can we learn from banana (Musa sp.) evolution? J. Virol. 84:7346–7359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Safár J, Noa-Carrazana JC, Vrana J, Bartos J, Alkhimova O, Sabau X, Simkova H, Lheureux F, Caruana ML, Dolezel J, Piffanelli P. 2004. Creation of a BAC resource to study the structure and evolution of the banana (Musa balbisiana) genome. Genome 47:1182–1191 [DOI] [PubMed] [Google Scholar]
- 22. Vilarinhos AD, Piffanelli P, Lagoda P, Thibivilliers S, Sabau X, Carreel F, D'Hont A. 2003. Construction and characterization of a bacterial artificial chromosome library of banana (Musa acuminata Colla). Theor. Appl. Genet. 106:1102–1106 [DOI] [PubMed] [Google Scholar]
- 23. Piffanelli P, Vilarinhos A, Safar J, Sabau X, Dolezel J. 2008. Construction of bacterial artificial chromosome (BAC) libaries of banana (Musa acuminata and Musa balbisiana). Fruits 63:375–379 [Google Scholar]
- 24. Harper G, Hull R. 1998. Cloning and sequence analysis of banana streak virus DNA. Virus Genes 17:271–278 [DOI] [PubMed] [Google Scholar]
- 25. Geering AD, Parry JN, Thomas JE. 2011. Complete genome sequence of a novel badnavirus, banana streak IM virus. Arch. Virol. 156:733–737 [DOI] [PubMed] [Google Scholar]
- 26. Geering AD, Pooggin MM, Olszewski NE, Lockhart BE, Thomas JE. 2005. Characterisation of Banana streak Mysore virus and evidence that its DNA is integrated in the B genome of cultivated Musa. Arch. Virol. 150:787–796 [DOI] [PubMed] [Google Scholar]
- 27. Lheureux F, Laboureau N, Muller E, Lockhart BE, Iskra-Caruana ML. 2007. Molecular characterization of banana streak acuminata Vietnam virus isolated from Musa acuminata siamea (banana cultivar). Arch. Virol. 152:1409–1416 [DOI] [PubMed] [Google Scholar]
- 28. Foissac S, Gouzy J, Rombauts S, Mathe C, Amselem J, Sterck L, Van de Peer Y, Rouze P, Schiex T. 2008. Genome annotation in plants and fungi: EuGene as a model platform. Curr. Bioinform. 3:87–97 [Google Scholar]
- 29. Salamov AA, Solovyev VV. 2000. Ab initio gene finding in Drosophila genomic DNA. Genome Res. 10:516–522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Degroeve S, Saeys Y, De Baets B, Rouze P, Van de Peer Y. 2005. SpliceMachine: predicting splice sites from high-dimensional local context representations. Bioinformatics 21:1332–1338 [DOI] [PubMed] [Google Scholar]
- 31. Florea L, Hartzell G, Zhang Z, Rubin GM, Miller W. 1998. A computer program for aligning a cDNA sequence with a genomic DNA sequence. Genome Res. 8:967–974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Altschul SF, Madden TL, Schaffer AA, Zhang JH, Zhang Z, Miller W, Lipman DJ. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25:3389–3402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. UniProt Consortium 2009. The Universal Protein Resource (UniProt) 2009. Nucleic Acids Res. 37:D169–D174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Quevillon E, Silventoinen V, Pillai S, Harte N, Mulder N, Apweiler R, Lopez R. 2005. InterProScan: protein domains identifier. Nucleic Acids Res. 33:W116–W120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Conte MG, Gaillard S, Lanau N, Rouard M, Perin C. 2008. GreenPhylDB: a database for plant comparative genomics. Nucleic Acids Res. 36:D991–D998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Carver T, Berriman M, Tivey A, Patel C, Bohme U, Barrell BG, Parkhill J, Rajandream MA. 2008. Artemis and ACT: viewing, annotating and comparing sequences stored in a relational database. Bioinformatics 24:2672–2676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Krumsiek J, Arnold R, Rattei T. 2007. Gepard: a rapid and sensitive tool for creating dotplots on genome scale. Bioinformatics 23:1026–1028 [DOI] [PubMed] [Google Scholar]
- 38. D'Hont A, Denoeud F, Aury JM, Baurens FC, Carreel F, Garsmeur O, Noel B, Bocs S, Droc G, Rouard M, Da Silva C, Jabbari K, Cardi C, Poulain J, Souquet M, Labadie K, Jourda C, Lengelle J, Rodier-Goud M, Alberti A, Bernard M, Correa M, Ayyampalayam S, McKain MR, Leebens-Mack J, Burgess D, Freeling M, Mbeguie AMD, Chabannes M, Wicker T, Panaud O, Barbosa J, Hribova E, Heslop-Harrison P, Habas R, Rivallan R, Francois P, Poiron C, Kilian A, Burthia D, Jenny C, Bakry F, Brown S, Guignon V, Kema G, Dita M, Waalwijk C, Joseph S, Dievart A, Jaillon O, Leclercq J, Argout X, Lyons E, Almeida A, Jeridi M, Dolezel J, Roux N, Risterucci AM, Weissenbach J, Ruiz M, Glaszmann JC, Quetier F, Yahiaoui N, Wincker P. 2012. The banana (Musa acuminata) genome and the evolution of monocotyledonous plants. Nature 488:213–217 [DOI] [PubMed] [Google Scholar]
- 39. D'Hont A, Grivet L, Feldmann P, Rao S, Berding N, Glaszmann JC. 1996. Characterisation of the double genome structure of modern sugarcane cultivars (Saccharum spp.) by molecular cytogenetics. Mol. Gen. Genet. 250:405–413 [DOI] [PubMed] [Google Scholar]
- 40. D'Hont A, Paget-Goy A, Escoute J, Carreel F. 2000. The interspecific genome structure of cultivated banana, Musa spp. revealed by genomic DNA in situ hybridization. Theor. Appl. Genet. 100:177–183 [Google Scholar]
- 41. Gawel NJ, Jarret RL. 1991. A modified CTAB DNA extraction procedure for Musa and Ipomoea. Plant Mol. Biol. Rep. 9:262–266 [Google Scholar]
- 42. Dereeper A, Argout X, Billot C, Rami JF, Ruiz M. 2007. SAT, a flexible and optimized Web application for SSR marker development. BMC Bioinformatics 8:465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lagoda PJ, Noyer JL, Dambier D, Baurens FC, Grapin A, Lanaud C. 1998. Sequence tagged microsatellite site (STMS) markers in the Musaceae. Mol. Ecol. 7:659–663 [PubMed] [Google Scholar]
- 44. SanMiguel P, Gaut BS, Tikhonov A, Nakajima Y, Bennetzen JL. 1998. The paleontology of intergene retrotransposons of maize. Nat. Genet. 20:43–45 [DOI] [PubMed] [Google Scholar]
- 45. Tamura K, Dudley J, Nei M, Kumar S. 2007. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol. Biol. Evol. 24:1596–1599 [DOI] [PubMed] [Google Scholar]
- 46. Lescot M, Piffanelli P, Ciampi AY, Ruiz M, Blanc G, Leebens-Mack J, da Silva FR, Santos CM, D'Hont A, Garsmeur O, Vilarinhos AD, Kanamori H, Matsumoto T, Ronning CM, Cheung F, Haas BJ, Althoff R, Arbogast T, Hine E, Pappas GJ, Jr, Sasaki T, Souza MT, Jr, Miller RN, Glaszmann JC, Town CD. 2008. Insights into the Musa genome: syntenic relationships to rice and between Musa species. BMC Genomics 9:58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ma J, Bennetzen JL. 2004. Rapid recent growth and divergence of rice nuclear genomes. Proc. Natl. Acad. Sci. U. S. A. 101:12404–12410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Le Provost G, Iskra-Caruana ML, Acina I, Teycheney PY. 2006. Improved detection of episomal Banana streak viruses by multiplex immunocapture PCR. J. Virol. Methods 137:7–13 [DOI] [PubMed] [Google Scholar]
- 49. Lheureux F. 2002. Etude des mécanismes génétiques impliqués dans l'expression des séquences EPRVs pathogènes des Bananiers au cours de croisements génétiques interspécifiques. Ph.D. thesis Université Sciences et Techniques du Languedoc USTL, Montpellier, France [Google Scholar]
- 50. Lovisolo O, Hull R, Rosler O. 2003. Coevolution of viruses with hosts and vectors and possible paleontology. Adv. Virus Res. 62:325–379 [DOI] [PubMed] [Google Scholar]
- 51. Kunii M, Kanda M, Nagano H, Uyeda I, Kishima Y, Sano Y. 2004. Reconstruction of putative DNA virus from endogenous rice tungro bacilliform virus-like sequences in the rice genome: implications for integration and evolution. BMC Genomics 5:80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Jakowitsch J, Mette MF, van Der Winden J, Matzke MA, Matzke AJ. 1999. Integrated pararetroviral sequences define a unique class of dispersed repetitive DNA in plants. Proc. Natl. Acad. Sci. U. S. A. 96:13241–13246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ndowora T, Dahal G, LaFleur D, Harper G, Hull R, Olszewski NE, Lockhart B. 1999. Evidence that badnavirus infection in Musa can originate from integrated pararetroviral sequences. Virology 255:214–220 [DOI] [PubMed] [Google Scholar]
- 54. Gregor W, Mette MF, Staginnus C, Matzke MA, Matzke AJ. 2004. A distinct endogenous pararetrovirus family in Nicotiana tomentosiformis, a diploid progenitor of polyploid tobacco. Plant Physiol. 134:1191–1199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Staginnus C, Gregor W, Mette MF, Teo CH, Borroto-Fernandez EG, Machado ML, Matzke M, Schwarzacher T. 2007. Endogenous pararetroviral sequences in tomato (Solanum lycopersicum) and related species. BMC Plant Biol. 7:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Feschotte C, Gilbert C. 2012. Endogenous viruses: insights into viral evolution and impact on host biology. Nat. Rev. Genet. 13:283–296 [DOI] [PubMed] [Google Scholar]
- 57. Holmes EC. 2011. The evolution of endogenous viral elements. Cell Host Microbe 10:368–377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Menéndez-Arias L. 2009. Mutation rates and intrinsic fidelity of retroviral reverse transcriptases. Viruses 1:1137–1165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Sanjuán R, Nebot MR, Chirico N, Mansky LM, Belshaw R. 2010. Viral mutation rates. J. Virol. 84:9733–9748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Mazourek M, Cirulli ET, Collier SM, Landry LG, Kang BC, Quirin EA, Bradeen JM, Moffett P, Jahn MM. 2009. The fractionated orthology of Bs2 and Rx/Gpa2 supports shared synteny of disease resistance in the Solanaceae. Genetics 182:1351–1364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. David P, Chen NW, Pedrosa-Harand A, Thareau V, Sevignac M, Cannon SB, Debouck D, Langin T, Geffroy V. 2009. A nomadic subtelomeric disease resistance gene cluster in common bean. Plant Physiol. 151:1048–1065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Baurens FC, Bocs S, Rouard M, Matsumoto T, Miller RN, Rodier-Goud M, MBéguié-A-MBéguié D, Yahiaoui N. 2010. Mechanisms of haplotype divergence at the RGA08 nucleotide-binding leucine-rich repeat gene locus in wild banana (Musa balbisiana). BMC Plant Biol. 10:149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Innan H, Kondrashov F. 2010. The evolution of gene duplications: classifying and distinguishing between models. Nat. Rev. Genet. 11:97–108 [DOI] [PubMed] [Google Scholar]
- 64. Ge XJ, Liu MH, Wang WK, Schaal BA, Chiang TY. 2005. Population structure of wild bananas, Musa balbisiana, in China determined by SSR fingerprinting and cpDNA PCR-RFLP. Mol. Ecol. 14:933–944 [DOI] [PubMed] [Google Scholar]
- 65. Simmonds NW. (ed). 1962. The evolution of the bananas. Longmans Green, London, England [Google Scholar]
- 66. Perrier X, De Langhe E, Donohue M, Lentfer C, Vrydaghs L, Bakry F, Carreel F, Hippolyte I, Horry JP, Jenny C, Lebot V, Risterucci AM, Tomekpe K, Doutrelepont H, Ball T, Manwaring J, de Maret P, Denham T. 2011. Multidisciplinary perspectives on banana (Musa spp.) domestication. Proc. Natl. Acad. Sci. U. S. A. 108:11311–11318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Mette MF, Kanno T, Aufsatz W, Jakowitsch J, van der Winden J, Matzke MA, Matzke AJ. 2002. Endogenous viral sequences and their potential contribution to heritable virus resistance in plants. EMBO J. 21:461–469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Noreen F, Akbergenov R, Hohn T, Richert-Poggeler KR. 2007. Distinct expression of endogenous Petunia vein clearing virus and the DNA transposon dTph1 in two Petunia hybrida lines is correlated with differences in histone modification and siRNA production. Plant J. 50:219–229 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.