

Abstract
Previous studies have demonstrated that the murine coronavirus mouse hepatitis virus (MHV) nonstructural protein 2 (ns2) is a 2′,5′-phosphodiesterase that inhibits activation of the interferon-induced oligoadenylate synthetase (OAS)-RNase L pathway. Enzymatically active ns2 is required for efficient MHV replication in macrophages, as well as for the induction of hepatitis in C57BL/6 mice. In contrast, following intranasal or intracranial inoculation, efficient replication of MHV in the brain is not dependent on an enzymatically active ns2. The replication of wild-type MHV strain A59 (A59) and a mutant with an inactive phosphodiesterase (ns2-H126R) was assessed in primary hepatocytes and primary central nervous system (CNS) cell types—neurons, astrocytes, and oligodendrocytes. A59 and ns2-H126R replicated with similar kinetics in all cell types tested, except macrophages and microglia. RNase L activity, as assessed by rRNA cleavage, was induced by ns2-H126R, but not by A59, and only in macrophages and microglia. Activation of RNase L correlated with the induction of type I interferon and the consequent high levels of OAS mRNA induced in these cell types. Pretreatment of nonmyeloid cells with interferon restricted A59 and ns2-H126R to the same extent and failed to activate RNase L following infection, despite induction of OAS expression. However, rRNA degradation was induced by treatment of astrocytes or oligodendrocytes with poly(I·C). Thus, RNase L activation during MHV infection is cell type specific and correlates with relatively high levels of expression of OAS genes, which are necessary but not sufficient for induction of an effective RNase L antiviral response.
INTRODUCTION
The murine coronavirus mouse hepatitis virus (MHV) is an enveloped, positive-strand RNA virus of the coronavirus family within the nidovirus order. MHV is a collection of strains with tropisms for different organs, including the liver and central nervous system (CNS), and thus provides models for the study of acute encephalitis and hepatitis, as well as chronic demyelinating disease. The MHV-A59 strain (A59) used in this study induces mild encephalitis and moderate hepatitis. Studies of the pathogenesis of MHV strains and recombinant chimeric MHVs have shown that postentry virus-host interactions have significant impact on organ tropism and virulence in MHV-infected mice (1, 2).
The type I interferon (IFN) response is an early innate response that is crucial to survival of mice following infection with many viruses, including MHV (3–5). During infection, viral double-stranded RNA (dsRNA) is recognized by pattern recognition receptors, such as MDA5 in the case of MHV in most cell types (3–5); this leads to the synthesis of type I IFN (Fig. 1). Alpha/beta IFN (IFN-α/β) induces expression of interferon-stimulated genes (ISGs) encoding pattern recognition receptors, transcription factors, and antiviral effectors, including multiple oligoadenylate synthetase (OAS) proteins. Viral dsRNA directly binds to and activates OAS to synthesize 2′,5′-linked oligoadenylates (2-5A), which induce RNase L dimerization and activity (6–9). RNase L is a particularly potent antiviral effector in that it both directly cleaves viral and host RNA, thereby reducing the amount of viral RNA available for replication and packaging of the genome into progeny virus, and inhibits host and viral protein synthesis. In addition, RNase L-mediated cleavage generates additional small RNAs that can be recognized by cytoplasmic pattern recognition receptors, further amplifying IFN synthesis (7). Importantly, and unlike most other IFN-induced activities, activation of the OAS-RNase L pathway requires both viral infection (production of dsRNA) and type I IFN exposure (upregulation of OAS) in the same cell in order to produce sufficient 2-5A to activate RNase L.
Fig 1.
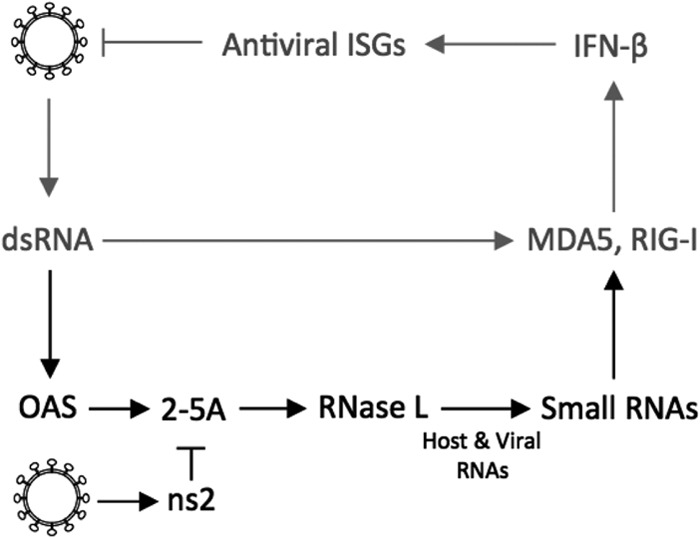
Canonical type I IFN induction and signaling pathway. (Gray) MHV RNA is recognized by MDA5, leading to expression of IFN-β. IFN-β mediates the transcription of antiviral ISGs. (Black) OAS-RNase L pathway. dsRNA activates OAS, which synthesizes 2-5A. The oligonucleotide activates RNase L to degrade viral and cellular RNA. MHV ns2 can degrade 2-5A.
Many viruses, including MHV, have evolved mechanisms to avoid and/or antagonize host type I IFN responses (10), including the OAS-RNase L pathway (7). We have shown previously that the A59 accessory protein nonstructural protein 2 (ns2) is a type I IFN antagonist. ns2 has 2′,5′-phosphodiesterase activity that cleaves 2-5A, thereby antagonizing the activation of the OAS-RNase L pathway (11). An ns2 mutant of A59 (ns2-H126R), expressing an inactive phosphodiesterase, was unable to replicate efficiently in macrophages derived from C57BL/6 (B6) mice, indicating that an active ns2 is required for robust replication of A59 in that cell type. However, ns2-H126R was able to replicate efficiently in macrophages derived from mice deficient in either type I interferon receptor expression (IFNAR−/−) or RNase L expression (RNase L−/−) (2, 11). These and other published data (2, 11) led us to conclude that ns2 antagonizes IFN signaling by downregulating RNase L activation. Furthermore, in vivo, ns2 acts as an organ-specific virulence factor that is required for efficient hepatic virus replication and pathology in B6 mice (2, 12). Macrophage depletion promoted the replication of ns2-H126R in the liver to a level close to that of wild-type virus. This observation emphasizes the crucial role of the type I IFN response in macrophages in protecting the host from MHV-induced hepatitis (2).
In contrast to its infection of the liver, ns2-H126R can replicate efficiently, to an extent similar to that of A59, in the CNS of B6 mice, as well as in cell lines and some primary cells (2). Here, we have further investigated the mechanisms underlying the organ- and cell-type-specific requirements for ns2-mediated IFN antagonism in the efficient replication of MHV. We have compared the OAS mRNA expression levels, as well as the replication of A59 and ns2-H126, between the liver and the brain, and also among macrophages and brain and liver parenchymal cell types. Our data suggest that activation of the OAS-RNase L pathway depends in part on the levels of expression of OAS genes. Furthermore, activation of this pathway is most robust in myeloid lineage cells, and successful replication in myeloid cells is a prerequisite for replication in the liver, but not in the CNS.
MATERIALS AND METHODS
Cell lines, viruses, and mice.
Murine L2 fibroblasts were maintained in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS), HEPES (10 mM), and 1% penicillin-streptomycin. Plaque assays were performed on L2 cells as described previously (13). The recombinant coronaviruses inf-MHV-A59 (referred to as A59 here) and inf-ns2-H126R (referred to as ns2-H126R here) were obtained from Stuart Siddell (University of Bristol, Bristol, United Kingdom) and have been described previously (2, 11, 12). C57BL/6 mice were purchased from the National Cancer Institute (Frederick, MD). RNase L−/− mice (bred for 10 generations to obtain a B6 background) were described previously (9) and were further bred in the facility at the University of Pennsylvania. Sendai virus (SeV) strain Cantell (14) was obtained from Carolina Lopez, University of Pennsylvania; LaCrosse virus (15) was obtained from Samantha Soldan, University of Pennsylvania; encephalomyocarditis virus (EMCV) (16) was obtained from Ian M. Kerr (Cancer Research United Kingdom, London, United Kingdom), and Newcastle disease virus expressing green fluorescent protein (NDV-GFP) (17) was obtained from Adolfo Garcıa-Sastre (Mount Sinai School of Medicine, New York, NY).
Infection of mice.
Four- to 5-week-old B6 mice or RNase L−/− mice were anesthetized with isoflurane (IsoFlo; Abbott Laboratories) and inoculated intracranially in the left cerebral hemisphere with 200 PFU in 25 μl or intranasally in each nostril with 105 PFU of A59 or ns2-H126R in 20 μl of PBS containing 0.75% BSA. Organs were harvested at day 5 postinfection, and viral titers were determined by plaque assay on L2 cells. The University of Pennsylvania IACUC approved all experimental procedures.
qRT-PCR.
RNA was isolated with an RNeasy minikit (Qiagen, Valencia, CA). Quantitative real-time reverse transcriptase PCR (qRT-PCR) was performed as described previously (2). Briefly, 200 ng (cells) or 350 ng (tissue) of total RNA was transcribed into cDNA using reverse transcriptase (Superscript III; Invitrogen) in a total volume of 20 μl. Then, 2 μl of cDNA was combined with 12.5 μl of iQ5 SYBR green mix (Bio-Rad, Hercules, CA), 6.5 μl diethyl pyrocarbonate (DEPC)-treated water, and 4 μl primer mix (5 μM each). DNA was amplified using an iQ5 iCycler (Bio-Rad), and cycle threshold (CT) values were recorded. mRNA levels were quantified as ΔCT values relative to actin mRNA [ΔCT = CT(gene of interest) − CT(β-actin)]. Relative mRNA expression levels were expressed using the formula 2−ΔCT.
Primary cell cultures. (i) Bone marrow-derived macrophages.
Primary bone marrow-derived macrophages (BMM) were generated from the hind limbs of B6 or RNase L−/− mice as described previously (11, 18) and cultured in DMEM supplemented with 10% FBS and 20% L929 cell-conditioned medium for 6 days before infection. Cultures were routinely ≥99% pure as assessed by positive staining for expression of CD11b and negative staining for expression of CD11c.
(ii) Neurons.
Hippocampal neurons were prepared from embryonic day 15 (E15) to E16 mouse embryos, seeded onto poly-l-lysine-coated tissue culture plates, and cultured in neurobasal medium containing B-27 supplement (Invitrogen), 1% penicillin-streptomycin, 2 mM l-glutamine, and 4 μg/ml glutamate for 4 days in the absence of an astrocyte feeder layer, as previously described (19). After 4 days in vitro, the neuron cultures were infected and/or treated with IFN-α as described above. Neuron cultures were routinely 95 to 98% pure, as determined by positive immunostaining for MAP2 and negative immunostaining for CD11b (microglia-specific marker), glial fibrillary acidic protein (GFAP) (astrocyte-specific marker), and OLIG2 (oligodendrocyte-specific marker) (19).
(iii) Astrocytes.
Primary astrocyte cultures were generated from the brains of 1- to 3-day-old neonatal B6 mice. Tissue was dissociated by mechanical disruption through nylon mesh and plated in complete medium consisting of minimal essential medium (MEM) supplemented with 10% FBS, 1% nonessential amino acid solution, 2 mM l-glutamine, 1% penicillin-streptomycin, and 10 mM HEPES. After 9 to 11 days in culture, the flasks were shaken to remove nonadherent cells, and the remaining adherent cells were ≥95% pure astrocytes, as determined by positive immunostaining for GFAP (2).
(iv) Microglia.
Primary microglia cultures were generated from neonatal mice similarly to astrocyte cultures. However, the cells were plated in complete medium consisting of DMEM supplemented with 10% FBS, 2 mM l-glutamine, 50 U/ml penicillin, and 50 μg/ml streptomycin. After 12 days in culture, the flasks were shaken at 200 rpm for 45 min to remove nonadherent cells, including microglia. These cells were plated, and after 30 min, the medium was replaced to remove any floating cells. Microglia cultures were ≥95% pure, as determined by positive immunostaining for CD11b and negative staining for GFAP (2).
(v) Oligodendrocytes.
To establish cultures of mouse oligodendrocytes, a mixed population of cells was isolated from forebrains of C57BL/6 mice and seeded on 100-mm petri dishes in a serum-free growth medium with platelet-derived growth factor (PDGF), fibroblast growth factor 2 (FGF2), and NT-3, as previously described (20). Once confluent, oligodendrocyte lineage cells were purified using a modified wash-down procedure to remove astrocytes and other contaminating cells (20). The oligodendrocyte precursors were plated on polylysine-coated 12-mm coverslips in 24-well dishes. The growth medium was removed from the cultures, and the cells were fed with “differentiation medium” (DM) (21). Immature oligodendrocytes appeared within 2 days, at which time cells were infected or treated with IFN as described below and in Results.
(vi) Hepatocytes.
Mouse primary hepatocytes were isolated using a two-step collagenase perfusion technique involving sequential perfusion of the liver with EDTA and collagenase (Invitrogen). Hepatocytes were plated on collagen-coated plates (BD Biosciences) in William E medium (Sigma), a modified MEM used for long-term culture of adult liver epithelial cells, supplemented with 10% fetal bovine serum and antibiotics. The cells were allowed to attach for 2 h before switching to William E medium supplemented with 1% (vol/vol) nonessential amino acids (3).
Infections of cell cultures.
Virus was added to cells at a multiplicity of infection (MOI) of 1 PFU/cell and allowed to adsorb for 1 h at 37°C. Cultures were washed with PBS (3 times) and then fed with medium as described for each cell type. The culture supernatants were harvested at the times indicated for the specific experiments, and the titers were determined by plaque assay on L2 cells.
Bioassay for interferon.
Supernatants recovered from cells that were infected with A59 at an MOI of 1 PFU/cell were exposed to 6,000 J/m2 UV light in a Stratalinker 1800 (Stratagene) to inactivate the virus. L2 mouse fibroblasts were treated with the UV-inactivated supernatants for 24 h and then infected with NDV-GFP at an MOI of 1 PFU/cell. Control cells were treated with 100 U/ml universal IFN-α for 24 h and then infected with 1 PFU/cell of NDV-GFP. At 24 h postinfection (p.i.), cells were examined for enhanced-GFP (EGFP) expression under a Nikon Eclipse 2000E-Ufluorescence microscope, and images were acquired using SPOT imaging software (Diagnostic Instruments).
Interferon sensitivity assay.
Neurons, astrocytes, oligodendrocytes, and microglia were pretreated with 100 U/ml of universal human IFN-α. After treatment for 16 to 24 h, the medium was removed, and the cells were infected with MHV at an MOI of 1. At the indicated time points, supernatants were harvested and stored at −80°C. The titers of released virus were determined by plaque assay on L2 cells.
RNase L-mediated rRNA cleavage.
For quantification of rRNA cleavage, total RNA from virus-infected cells was isolated using an RNeasy kit (Qiagen) and quantified using a Nanodrop analyzer. Equal amounts of RNA were separated on RNA chips and analyzed with an Agilent Bioanalyzer 2100 (Agilent Technologies) as described previously (11, 22).
Immunofluorescent staining.
Hepatocytes were fixed in PBS containing 4% paraformaldehyde (Electron Microscopy Sciences), blocked with 1.5% normal goat serum, and immunolabeled with a mouse monoclonal antibody (1.16.1) directed against the MHV nucleocapsid protein (a gift from Julian Leibowitz, Texas A&M University). Primary antibody was detected with goat anti-mouse Alexa Fluor 488 (Invitrogen). Fluorescence was visualized with a Nikon Eclipse 2000E-Ufluorescence microscope, and images were acquired using SPOT imaging software (Diagnostic Instruments).
Poly(I·C) transfection.
Confluent astrocyte and neuron cultures in 12-well plates were transfected with 10 μg/ml of poly(rI)·poly(rC) [poly(I·C)] (Sigma) using Lipofectamine 2000 (Life Technologies) according to the manufacturer's protocol. The cells were harvested 6 h after transfection and lysed with TRIzol to isolate RNA for Bioanalyzer analysis, also referred to as RNA chip analysis.
Statistical analysis.
An unpaired two-tailed t test was used to determine statistical significance. All data were analyzed with GraphPad Prism software (GraphPad Software, Inc., CA).
RESULTS
MHV replication depends on ns2-phosphodiesterase activity in an organ-specific manner.
MHV strain A59 infects predominantly the CNS and the liver, resulting in mild encephalitis and moderate hepatitis (23). We have shown previously that amino acid substitution at either of the predicted catalytic phosphodiesterase residues, H46A or H126R, of the A59 ns2 protein confers attenuation of viral replication and pathogenicity in the liver, but not in the brain, in B6 mice (2). We have now compared replication of wild-type A59 and mutant ns2-H126R in the brain and liver between RNase L−/− mice and B6 mice by quantifying the infectious viral titers in each organ 5 days following intracranial inoculation with 200 PFU/mouse of each virus, which corresponds to the peak of viral infection (Fig. 2A). The infectious virus titers did not differ significantly between the brains of B6 and RNase L−/− mice infected with either A59 or mutant ns2-H126R. In contrast, titers of ns2-H126R were more than 100-fold lower than those of A59 (P = 0.0052) in the livers of infected B6 mice at 5 days p.i., consistent with previously observed results of intrahepatic inoculation (2, 11). The titers of ns2-H126R and A59 in the livers of RNase L−/− mice were not significantly different, further illustrating the role of ns2 as an antagonist of RNase L activation.
Fig 2.
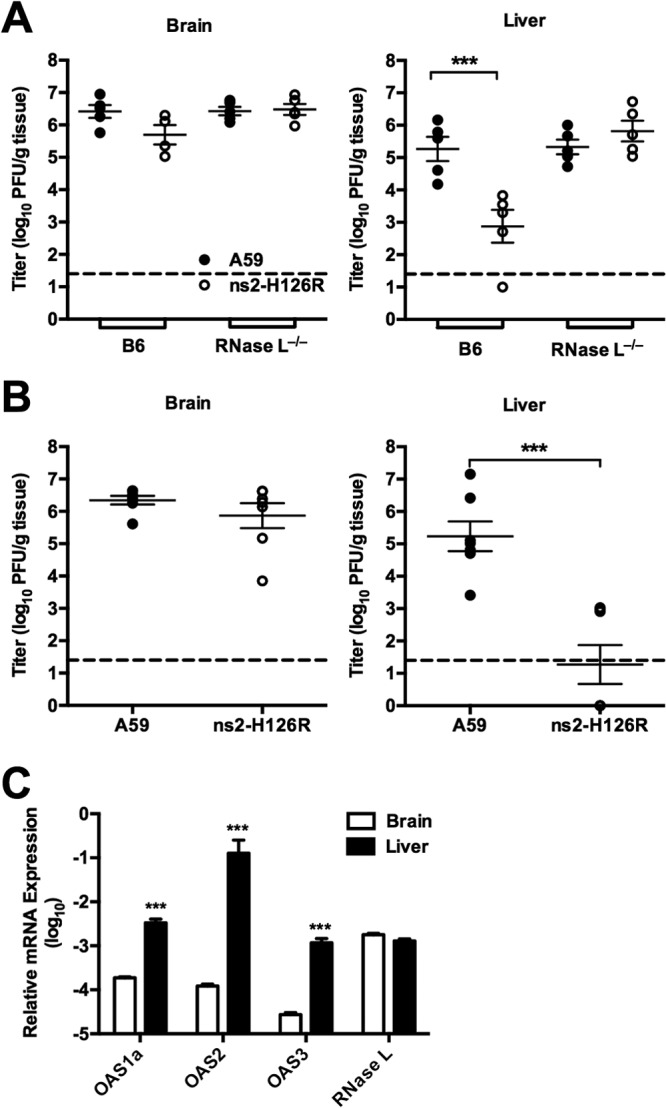
Replication of A59 and ns2-H126R in the liver and the brain in B6 and RNase L−/− mice. (A) B6 and RNase L−/− mice were inoculated intracranially with 200 PFU/mouse of A59 or ns2-H126R (n = 5). The mice were sacrificed at 5 days p.i., the brains and livers were harvested, and the virus titers were determined by plaque assay. (B) B6 and RNase L−/− mice were inoculated intranasally with 2 × 105 PFU/mouse of A59 or ns2-H126R (n = 5). The mice were sacrificed at 5 days p.i., the brains and livers were harvested, and the virus titers were determined by plaque assay. Dashed lines designate the lower limit of detection. (C) RNA was extracted from the livers and brains of uninfected mice (n = 5); expression levels of OAS1a, OAS2, OAS3, and RNase L mRNAs were assessed by qRT-PCR. mRNA expression levels relative to that of β-actin mRNA were expressed as 2−ΔCT [ΔCT = CT(gene of interest) − CT(β-actin)]. The data are from one representative of two experiments performed and are shown as the means ± standard errors of the mean (SEM). ***, P < 0.001.
We reasoned that host defenses might be higher when virus enters the brain by a more natural route of infection (24). Thus, to further investigate the requirement for ns2 expression for MHV replication in the CNS, B6 mice were infected intranasally with 2 × 105 PFU A59 or ns2-H126R and the virus titers in the brain and liver 5 days p.i. were quantified (Fig. 2B). Again, the titers in the brain did not differ significantly between A59- and ns2-H126R-infected mice 5 days p.i., while the titers of A59 in the liver were significantly greater than those of ns2-H126R (P = 0.0006). The A59 titers in the liver were approximately 1,000-fold higher than the minimal titers of ns2-H126R. Thus, dispensability of ns2 activity for replication in the brain was independent of the route of inoculation.
RNase L activation depends on the level of 2-5A, which is produced by several OAS proteins upon activation by viral dsRNA. We used qRT-PCR to compare the basal levels of expression of three active OAS genes, OAS1a, OAS2, and OAS3 genes, between the brains and livers of uninfected B6 mice (11) (Fig. 2C). We found that the basal mRNA expression levels of OAS1a, OAS2, and OAS3 genes in naive mice were significantly higher in the liver than in the brain, as we reported previously for other IFN-induced genes (2). This suggests that the basal level of OAS might influence the host's ability to activate RNase L and restrict MHV infection in an organ-specific manner. The basal levels of RNase L mRNA expression were similar between the brain and liver (Fig. 2C) and do not likely contribute to the differences in the effects of viral phosphodiesterase activity.
Efficient replication of MHV in hepatocytes does not require antagonism of the OAS-RNase L pathway.
We previously observed that macrophages, unlike transformed fibroblast lines and other primary cell types tested, restrict replication of ns2-H126R more effectively than that of A59 and that in vivo depletion of macrophages allowed ns2-H126R to cause hepatitis in B6 mice (2). Based on these data, we hypothesized that liver parenchymal cells were less well prepared than macrophages to restrict MHV. To test this hypothesis, we compared replication of A59 and ns2-H126R in hepatocytes, the most abundant cell type in the liver parenchyma, and in macrophages. Primary hepatocyte cultures (Fig. 3A) and BMM (Fig. 3B) were infected with A59 or ns2-H126R, and the replication kinetics were compared between the isolates for each cell type. While BMM severely restricted ns2-H126R replication relative to A59, the viruses replicated with similar kinetics and to similar final titers in primary hepatocytes. In addition, both viruses produced syncytia in hepatocytes, as observed by staining the cultures with an antibody directed against the MHV nucleocapsid protein (Fig. 3C). We next compared the effects of IFN pretreatment on replication of A59 and ns2-H126R in hepatocytes. Cultures were treated with 100 U/ml of IFN-α for 24 h before infection. Virus was quantified from the supernatant at several times postinfection. IFN treatment of hepatocytes decreased viral titers 100-fold relative to control wells by 16 h p.i. (Fig. 3D). However, ns2-H126R and A59 replicated to similar levels in IFN-treated cells, indicating that IFN treatment of hepatocytes induces a robust anti-MHV response that is not dependent on RNase L activation. Consistent with this observation, RNase L-mediated degradation of rRNA was not detected in hepatocytes infected with A59 or ns2-H126R, as assessed by Bioanalyzer analysis (see Materials and Methods) (Fig. 3E).
Fig 3.
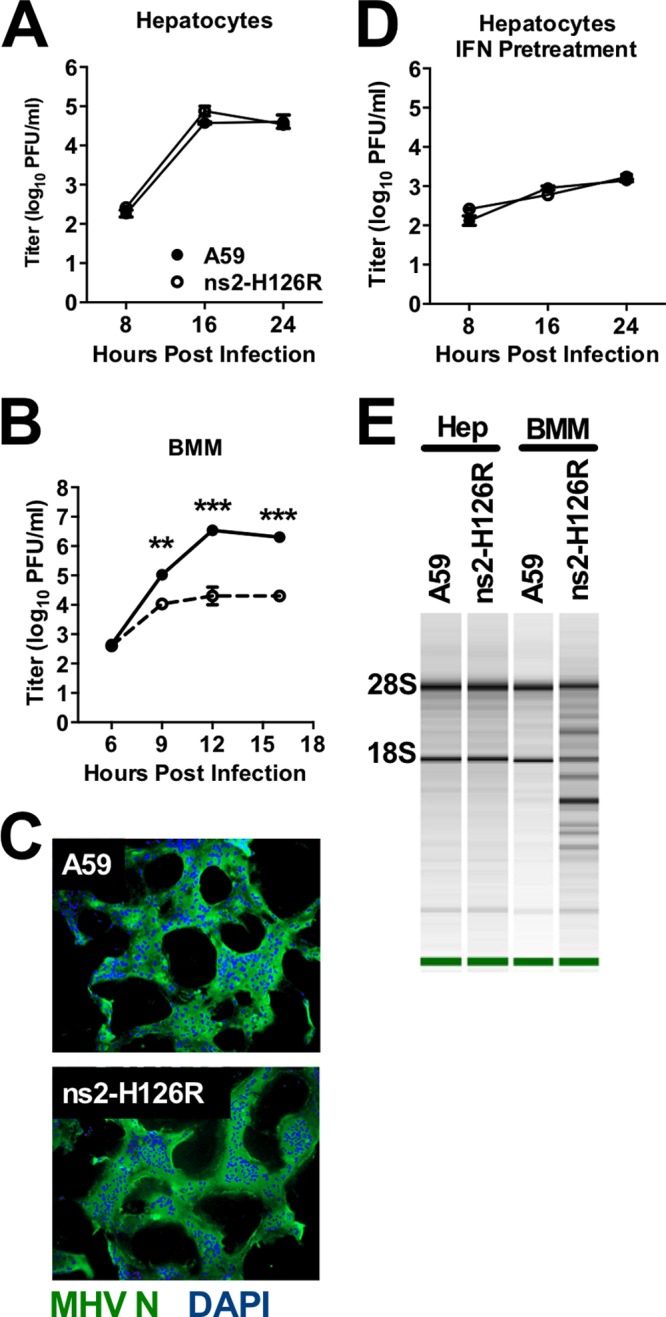
Replication of A59 and ns2-H126R in primary hepatocytes. (A and B) Primary hepatocyte (A) or BMM (B) cultures were infected with A59 or ns2-H126R (1 PFU/cell); at the times indicated, the virus titer was determined from the supernatant. (C) Primary hepatocyte cultures were infected with A59 or ns2-H126R (1 PFU/cell); at 16 h p.i., the cells were stained with DAPI (4′,6-diamidino-2-phenylindole) to locate cellular nuclei (blue) and an antibody directed against MHV nucleocapsid protein to visualize virus (green). The cultures were examined under a Nikon Eclipse 2000E-U-fluorescence microscope. (D) Primary hepatocyte cultures were treated with 100 U/ml IFN for 24 h and then infected with A59 or ns2-H126R and treated as for panel A. The hepatocytes used in panels A and D were prepared from the same mouse. (E) RNA was extracted from infected hepatocytes and BMM (16 h postinfection), and rRNA degradation was assessed with a Bioanalyzer. The data are from one representative of two experiments. The growth curves were performed in BMM in triplicate and in hepatocytes in duplicate and are shown as the means ± SEM. **, P < 0.01; ***, P < 0.001.
We reasoned that an adequate level of expression of OAS, the producer of 2-5A, would be a requirement for the activation of RNase L and, furthermore, that the level of OAS could vary among cell types. To further investigate the mechanisms underlying the difference in RNase L activation between macrophages and hepatocytes, we compared the basal levels of expression of several genes encoding active OAS proteins and found that OAS mRNA levels were 100- to 1,000-fold higher in macrophages than in hepatocytes (Fig. 4A). Even following MHV infection, at 16 h p.i., OAS1a mRNA levels remained comparatively low in hepatocytes relative to macrophages (Fig. 4B). In addition, while MHV infection of BMM significantly induced IFN-β mRNA expression at 16 h p.i. relative to mock infection, mRNA levels remained low in infected hepatocytes (Fig. 4C). Similar to the pattern of OAS mRNA expression, the basal levels of ISG mRNAs encoding a group of pattern recognition receptors, transcription factors, and antiviral genes involved in the host response to viruses (Fig. 4D) were also higher in BMM than in hepatocytes, which may in part explain the weak induction of IFN-β mRNA in hepatocytes relative to BMM. The levels of RNase L mRNA were not significantly different in BMM and hepatocytes (data not shown).
Fig 4.
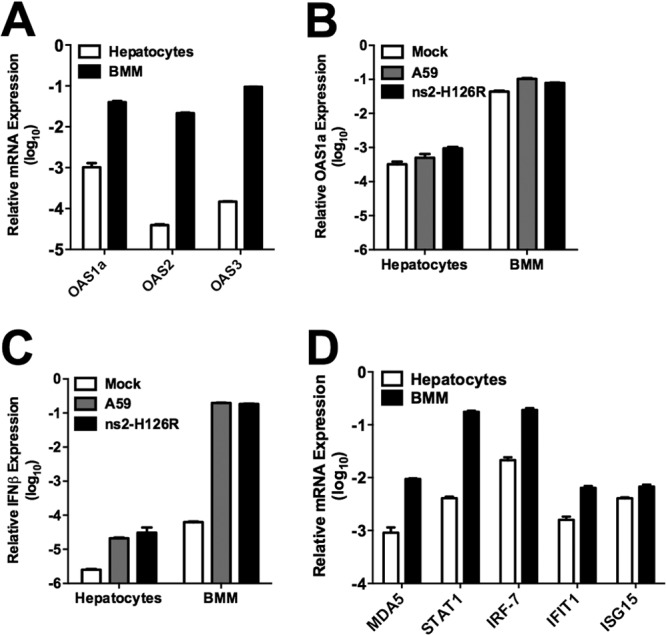
OAS and IFN-β mRNA expression in hepatocytes and BMM. (A) RNA was extracted from BMM and hepatocytes, and basal levels of OAS1a, OAS2, and OAS3 mRNA expression were quantified by qRT-PCR. (B) Hepatocytes and BMM were infected with A59 or ns2-H126R (1 PFU/cell); RNA was extracted at 16 h p.i., and OAS1a mRNA expression was quantified by qRT-PCR. (C) IFN-β mRNA levels were quantified by qRT-PCR using the same RNAs as in panel B. (D) Basal levels of ISG mRNAs, including MDA5, STAT1, IRF-7, IFIT1, and ISG15, were quantified in BMM and hepatocytes by qRT-PCR. mRNA expression levels relative to β-actin mRNA were expressed as 2−ΔCT [ΔCT = CT(gene of interest) − CT(β-actin)]. The data are from one representative of two experiments, performed in triplicate, and are shown as means ± SEM.
MHV replication is restricted by the OAS-RNase L antiviral pathway in microglia but not in neurons, astrocytes, or oligodendrocytes.
As shown above, antagonism of RNase L is not required for efficient MHV replication in the brain. Thus, we investigated the ability of each of the major CNS cell types to restrict A59 and ns2-H126R infection (Fig. 5). Primary cultures of hippocampal neurons, oligodendrocytes, astrocytes, and microglia were each prepared as described in Materials and Methods and previously (3, 19, 20). Cultures were infected at an MOI of 1 PFU/cell, and cells were harvested at optimal times postinfection for assessing the growth kinetics of the virus in each cell type. There were no significant differences in titers between A59 and ns2-H126R in neuron (Fig. 5A), oligodendrocyte (Fig. 5B), or astrocyte (Fig. 5C) cultures at the times examined. However, in microglia (Fig. 5D), the titer of ns2-H126R was 10-fold lower than that of A59 at 24 h postinfection. Thus, like BMM, microglia preferentially restricted ns2-H126R. Furthermore, ns2-H126R replicated to the same extent as A59 in microglia derived from RNase L−/− mice (Fig. 5E), indicating that restriction of ns2 mutant virus in B6 microglia was due to activation of RNase L, as observed previously in BMM (11).
Fig 5.

Infection of primary neural cells with A59 and ns2-H126R. Primary cell cultures of neurons (A), oligodendrocytes (B), astrocytes (C), microglia (D), and RNase L−/− microglia (E) were infected with A59 and ns2-H126R (1 PFU/cell). The virus titer was determined from the supernatant at the times indicated. The data are from one of two replicate experiments (panel E, one experiment) performed in triplicate and are shown as means ± SEM. **, P < 0.01.
Activation of RNase L activity correlates with high basal and induced levels of OAS genes and IFN signaling.
To investigate the possibility that the basal levels of ISGs contribute to and/or predict viral restriction, we quantified the basal mRNA expression levels of OAS genes (Fig. 6A), as well as a selection of other ISGs (Fig. 6B), in the major cell types of the CNS parenchyma. The levels of basal ISG mRNA (Fig. 6A and B) were greater in microglia than in other CNS cell types, as might be expected for cells of macrophage lineage. The basal levels of ISG mRNAs, especially the OAS mRNAs, expressed in neurons and oligodendrocytes (Fig. 6A) were low, near the limit of detection and at least 100- to 1,000-fold lower than in microglia, while astrocytes expressed intermediate mRNA levels. Next, we examined the OAS mRNA levels after viral infection of these CNS cell types (Fig. 6C). At 24 h postinfection with A59 or ns2-H126R, the OAS mRNA levels in neuron and oligodendrocyte cultures were still at or near the limit of detection by qRT-PCR, indicating that MHV does not directly induce the expression of OAS in these two cell types. However, in astrocytes and microglia, infection induced significant upregulation of OAS mRNAs, up to 50-fold for OAS2. However, the levels in astrocytes were still 10- to 100-fold lower than in microglia. A59 and ns2-H126R elicited similar levels of induction.
Fig 6.

OAS expression in primary neural cell cultures. (A) RNA was extracted from neurons, oligodendrocytes, astrocytes, and microglia, and OAS1a, OAS2, and OAS3 mRNA expression levels were quantified by qRT-PCR. (B) MDA5, RIG-I, STAT1, IRF-7, IFIT1, IFIT2, and ISG15 mRNA levels were quantified from the same RNAs as in panel A. (C) Primary cell cultures were infected with A59 or ns2-H126R or mock infected; at 24 h p.i., RNA was extracted and OAS1a mRNA was quantified. mRNA expression levels relative to β-actin mRNA were expressed as 2−ΔCT [ΔCT = CT(gene of interest) − CT(β-actin)]. The data are from one representative of two experiments performed in triplicate and are shown as means and SEM. Dashed lines designate the lower limit of detection.
We reasoned that the lack of preferential restriction of ns2-H126R in CNS cell types other than microglia was due to suboptimal levels of OAS, leading to insufficient production of 2-5A and the consequent lack of activation of RNase L. In order to directly assess the degree of RNase L activity induced during infection of CNS cells, we compared the extents of rRNA cleavage in neurons, oligodendrocytes, astrocytes, and microglia infected at 24 h p.i. with either ns2-H126R or A59 (Fig. 7A). Bioanalyzer analysis of RNA from infected cells showed that neither virus induced detectable rRNA cleavage in neurons, oligodendrocytes, or astrocytes, consistent with the lack of preferential restriction of ns2-H126R in these cell types. In contrast, ns2-H126R, but not A59, induced rRNA cleavage in microglia.
Fig 7.
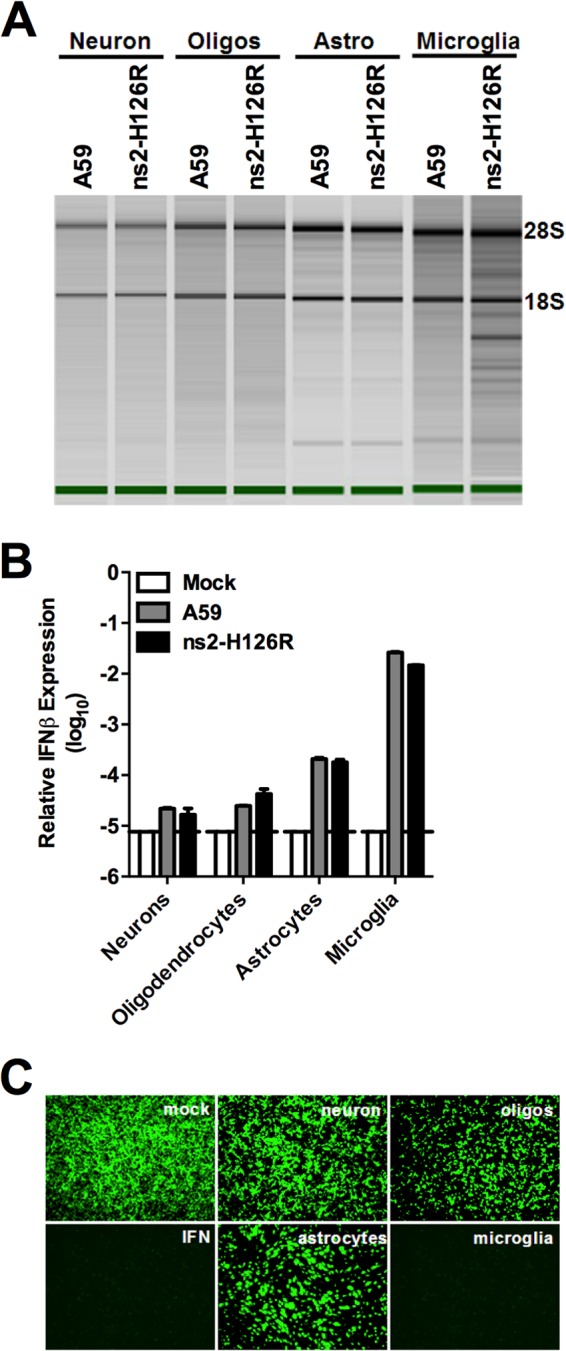
RNase L activity and IFN-β expression following infection of primary neural cells. (A) Primary neurons, oligodendrocytes (Oligos), astrocytes (Astro), and microglia were infected with A59 and ns2-H126R (1 PFU/cell). RNA was harvested 24 h p.i. and analyzed for rRNA degradation with a Bioanalyzer. The positions of 28S and 18S rRNAs are indicated. (B) IFN-β mRNA expression was quantified in RNAs prepared from infected cultures as in panel A. mRNA expression levels relative to β-actin mRNA were expressed as 2−ΔCT [ΔCT = CT(gene of interest) − CT(β-actin)]. Dashed line designates the lower limit of detection. (C) Supernatants from primary CNS cultures, as in panel A, were treated with UV light and incubated with L2 mouse fibroblasts for 24 h, followed by infection with NDV-GFP (1 PFU/cell). As a positive control, L2 cells were treated with IFN-α for 24 h and then infected with NDV-GFP. At 24 h p.i., cells were examined under a Nikon Eclipse 2000E-U-fluorescence microscope.
We had observed that MHV failed to induce expression of type I IFN in many cell types (3, 25) and hypothesized that low OAS mRNA levels in MHV-infected primary cultures (Fig. 6C) could be attributed to insufficient IFN production. Indeed, analysis of IFN-β mRNA levels by qRT-PCR showed that induction by both A59 and ns2-H126R was more than 100-fold greater in microglia than in the other cell types tested (Fig. 7B). A bioassay for antiviral activity (Fig. 7C) confirmed that both A59 and ns2-H126R induced detectable IFN expression in microglia, but not in other cell types. Thus, these data suggest that activation of the OAS-RNase L pathway correlates with the ability of MHV to induce IFN production and underscores the requirement for simultaneous type I IFN signaling and viral infection in cells with low basal OAS mRNA levels.
To assess the effects of IFN pretreatment of cells on ISG mRNA levels and replication of A59 and ns2-H126R, primary CNS cultures were pretreated with IFN-α (100 U/ml). At 24 h posttreatment, OAS mRNA expression, as quantified by qRT-PCR, was significantly induced in all cell types, as expected; the OAS1a and OAS2 mRNA levels in oligodendrocytes were similar to those in microglia (Fig. 8A). We then assessed replication of A59 and ns2-H126R in each cell type following IFN pretreatment. IFN pretreatment severely restricted A59 replication in all neural cell types, up to 1,000-fold in microglia, 100-fold in astrocytes and oligodendrocytes, and 10-fold in neurons (Fig. 8B to E), indicating induction of IFN-dependent antiviral activities. As reported previously (2), ns2-H126R displayed enhanced sensitivity to IFN pretreatment in microglia similar to that in BMM (Fig. 8E), implying that RNase L had been activated. In contrast, ns2-H126R displayed sensitivity to IFN similar to that of A59 in neurons, oligodendrocytes, and astrocytes (Fig. 8B to D), suggesting that RNase L had not been activated despite the increase in OAS levels following IFN treatment.
Fig 8.
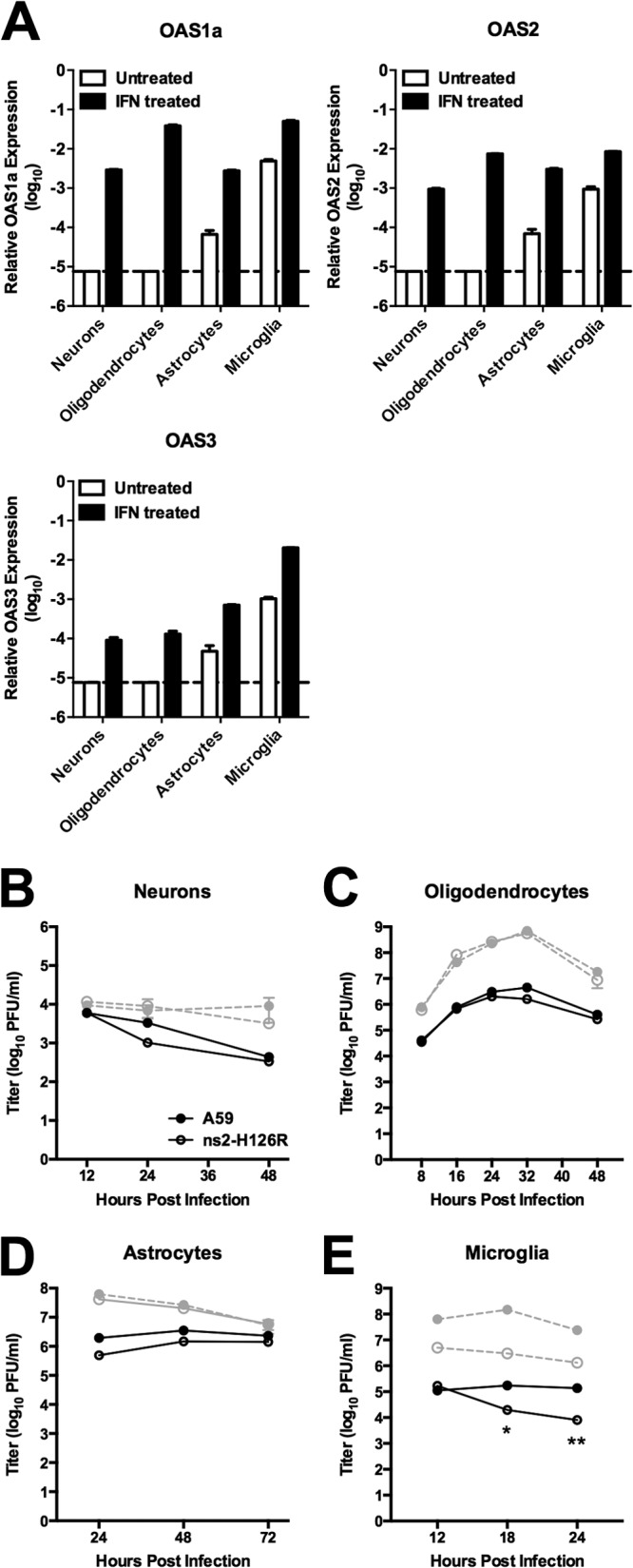
OAS mRNA expression and virus replication in primary neural cell cultures following IFN-α treatment. (A) Primary cell cultures were treated with 100 U/ml of IFN-α (or mock treated) for 24 h. RNA was extracted, and expression levels of OAS1a, OAS2, and OAS3 were assessed by qRT-PCR. mRNA expression levels relative to β-actin mRNA were expressed as 2−ΔCT [ΔCT = CT(gene of interest) − CT(β-actin)]. Dashed lines designate the lower limit of detection. (B to E) Primary cultures were pretreated with IFN and infected with A59 and ns2-H126R. The virus titer was determined from the supernatants at the indicated times (shown in black). Also, shown in gray are the titers from untreated cultures (B to D), with data from Fig. 5. The data are from one representative of two experiments performed in triplicate and are shown as means and SEM. *, P < 0.05; **, P < 0.01.
Several other viruses also fail to activate RNase L in astrocytes, while poly(I·C) activates RNase L in astrocytes and oligodendrocytes.
In order to determine whether the failure to activate RNase L in neurons, astrocytes, and oligodendrocytes was a characteristic of these cell types or specific to MHV, we infected astrocytes with three other viruses, EMCV (16), LaCrosse virus (LACV) (15), and SeV (14) (EMCV and LACV are neurotropic, while SeV is pneumotropic). We had found that RNase L restricted both SeV and EMCV in macrophages (11) and that SeV induced IFN-β in astrocytes (3). However, like A59 and ns2-H126R, EMCV, SeV, and LACV all failed to induce RNase L-mediated degradation of rRNA in astrocytes (Fig. 9A), indicating that the failure to activate RNase L in astrocytes is not specific to MHV.
Fig 9.
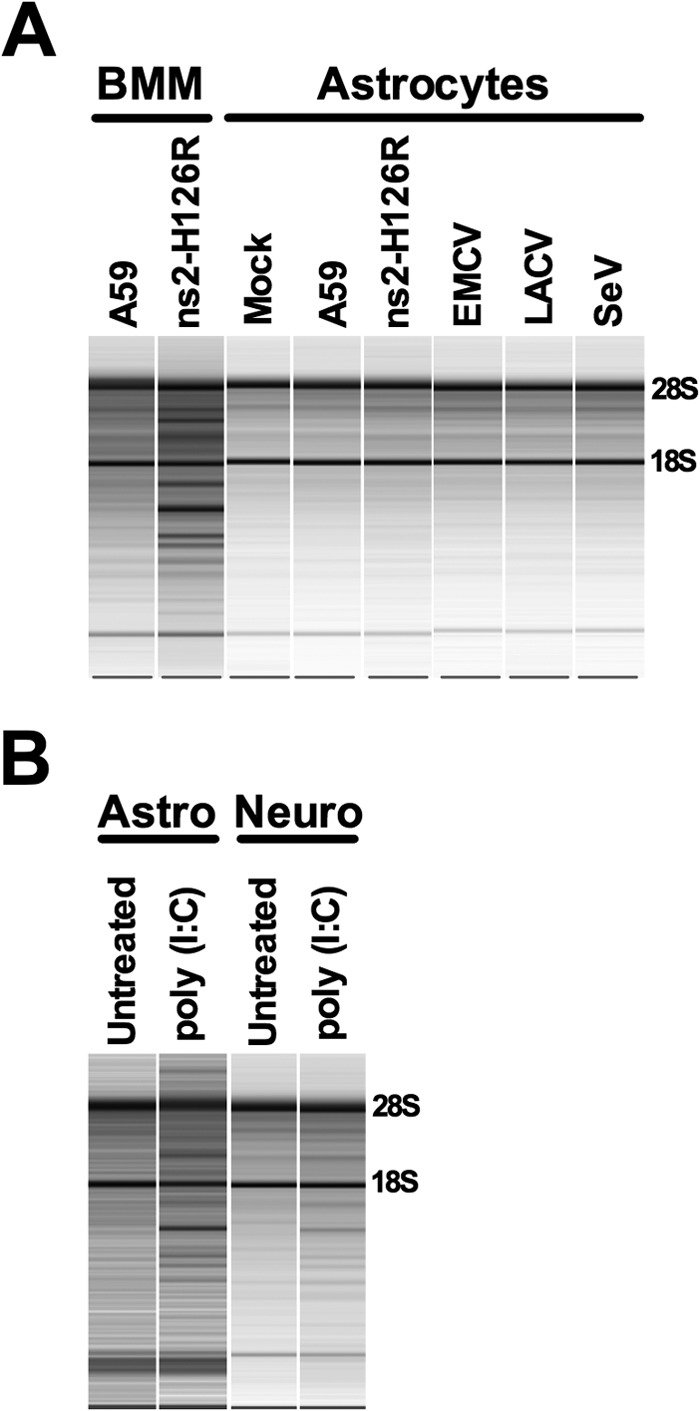
Activation of RNase L by other viruses and poly(I·C). (A) BMM and astrocytes were infected with A59, ns2-H126R, EMCV, LACV, and SeV (1 PFU/cell of SeV and LACV and 0.1 PFU/cell of EMCV). RNA was extracted from infected cells, and rRNA degradation was assessed with a Bioanalyzer. The positions of 28S and 18S rRNAs are indicated. The data shown are taken from three separate chips: (i) BMM RNA; (ii) SeV-infected cell RNA; and (iii) mock-, A59-, ns2-H126R-, EMCV-, and LACV-infected cell RNA. (B) Astrocyte and neuron cultures were treated with 100 U/ml IFN for 24 h and transfected with 10 μg/ml poly(I·C) with Lipofectamine 2000. Six hours later, RNA was extracted, and rRNA degradation was assessed with a Bioanalyzer. Astrocyte and neuron RNAs were analyzed on separate chips. The data are from one representative of two experiments.
We next investigated whether neurons or astrocytes, which did not exhibit RNase L-mediated rRNA degradation in the context of viral infection, can regulate RNase L activity in response to poly(I·C). Poly(I·C) serves as a surrogate for dsRNA for both the induction of type I IFN expression and the activation of OAS. We treated primary neuron and astrocyte cultures with IFN-α and transfected them with poly(I·C). RNA was extracted from treated cells 6 h after poly(I·C) treatment and analyzed for the degradation of rRNA. rRNA degradation was evident in both cultures, indicating that RNase L was activated (Fig. 9B). Thus, astrocytes and neurons have the potential to upregulate RNase L activity, but this pathway is not activated during infection with a variety of viruses, including an ns2-H126R mutant expressing an inactive phosphodiesterase (Fig. 9B).
DISCUSSION
Type I IFN, produced by infected cells, acts in an autocrine and paracrine manner to induce antiviral activities in both virus-infected cells and neighboring uninfected cells. The OAS-RNase L pathway is one such antiviral activity induced by IFN. RNase L mRNA and protein are constitutively expressed in most cell types, and the activity of RNase L is controlled primarily at the posttranscriptional level by the availability of 2-5A (26). Activation of the OAS-RNase L pathway thus depends on whether the level of OAS expression, which is induced at the transcriptional level by type I IFN and activated at the protein level by dsRNA, is sufficient to synthesize adequate amounts of 2-5A (6–9) (Fig. 1). The cellular enzyme 2′-phosphodiesterase (2′-PDE) (also known as PDE12) can cleave 2′,5′-phosphodiesterase linkages. 2′-PDE has been proposed to degrade 2-5A in vivo and thereby downregulate the activity of RNase L, presumably to prevent excessive tissue damage (27). A59 can both activate the OAS-RNase L pathway in certain cell types by recognition of viral RNA by MDA5 and antagonize the pathway by expression of its 2′,5′-phosphodiesterase (ns2) (11), allowing replication in RNase L-activating cells. In contrast, ns2-H126R activates but cannot antagonize the pathway and is restricted in such cells. Thus, the outcome of infection depends on the viral genetics, as well as the balance of activation and antagonism, which in turn is cell type specific.
Previous studies from our laboratory (2, 11) and others (28–30) have shown that basal, as well as virus- and/or IFN-induced, levels of ISG expression are important determinants of permissiveness to viral infection and the resulting cellular and organ tropism. We have reported previously that the basal expression levels of mRNAs encoding ISG proteins, including MDA5, STAT1, and other molecules crucial for recognizing viral invasion and producing an antiviral environment, were greater in the liver than in the CNS and also higher in myeloid cells than in other cell types (2, 11, 31). Similarly, microglia isolated from naive mice or mice infected with an attenuated neurotropic strain of MHV expressed higher basal and induced levels of ISG mRNA than did oligodendrocytes (32). We show here that the expression levels of OAS mRNAs follow similar patterns. The basal mRNA expression levels of the active OAS species OAS1a, -2, and -3 are 100- to 1,000-fold higher in the liver than in the brain, suggesting that the low level of OAS expression in the brain precludes activation of the OAS-RNase L pathway and allows the replication of ns2-H126R. Similarly, among the cell types examined, myeloid cells express the highest basal levels of OAS and are the only cell type thus far tested in which MHV needs an enzymatically active ns2 to ensure efficient replication. Bone marrow dendritic cells, also of myeloid lineage, preferentially restrict ns2-H126R (data not shown).
After infection with A59 or ns2-H126R, neurons and oligodendrocytes showed little increase in OAS mRNA expression, while the OAS expression levels in astrocytes were significantly upregulated, although not to the levels in myeloid cells (Fig. 6C). Virus replication in oligodendrocytes and astrocytes is robust, with titers similar to those produced in microglia or in the L2 cell line. Indeed, A59 causes cytopathic effect (syncytia) in primary microglia and astrocyte cultures. While virus remains more cell associated in neurons, it appears to spread rapidly cell to cell in infected primary neuronal cultures (19). Thus, the lack of activation of RNase L is not due to a low percentage of infected cells. Pretreatment of these cultures with IFN-α to induce the expression of OAS mRNAs, as might occur in vivo (Fig. 7B to E), did not produce activation of RNase L in any nonmyeloid cell type (data not shown), including hepatocytes, and all consequently permitted ns2-H126R to replicate similarly to A59. Interestingly, other interferon-stimulated antiviral activities were clearly effective in all cell types, as the viral titers were lower in pretreated cultures by approximately 100-fold for astrocytes and oligodendrocytes and to a lesser extent for neurons compared to about 1,000-fold in microglia. Similarly, West Nile virus replication was significantly less restricted by IFN pretreatment of primary neuronal cultures than of macrophages, and this correlated with relatively low levels of ISG expression in the neuronal cultures (30, 33).
One possible explanation for the lack of activation of the OAS-RNase L pathway in nonmyeloid cells despite the induction of other antiviral effectors is the requirement for viral dsRNA to be accessible to induce the synthesis of 2-5A by OAS. Indeed, the lack of induction of type I IFN response in nonmyeloid primary cell types (Fig. 7B), as well as in several transformed cell lines, during MHV infection provides further evidence that viral RNA may not be accessible for detection by MDA5 in nonmyeloid cells (25, 34, 35). It has been speculated that MHV (34, 35), as well as other viruses, such as tick-borne encephalitis virus (36), induces a rearrangement of intracellular membranes that could potentially create a compartment that would protect viral dsRNA from detection by cytoplasmic sensors (37–41). However, to our knowledge, no studies of MHV-induced membrane rearrangement or localization of viral transcription complexes and dsRNA have been carried out in myeloid cells. Our future plans include comparing the localization of transcriptional complexes and viral dsRNA between macrophages and other cell types.
The liver is exposed to numerous pathogens and debris flowing though the small blood vessels, or sinusoids. This ongoing antigen exposure may induce type I IFN expression and thus maintain higher basal levels of ISG expression in the liver than in the brain, as described above and previously (2, 42). The liver parenchyma is composed mainly of hepatocytes, while the nonparenchymal Kupffer cells (KC), the resident macrophages of the liver, and liver sinusoidal endothelial cells (LSEC) are located within the sinusoids (43). We have proposed that KC serve as gatekeepers to restrict entrance from the sinusoids into the liver parenchyma to infect hepatocytes (2, 31, 42). We suggest that upon infection of KC, the IFN response, including importantly the OAS-RNase L pathway, creates an antiviral state restricting infection and limiting the spread of virus to the parenchymal hepatocytes, consequently preventing hepatitis. This hypothesis is supported by the following observations. Infection of mice with CD11b+ cell-type-specific ablation of type I interferon receptor expression (IFNAR−/− mice) demonstrated that IFN signaling in macrophages is crucial for control of hepatitis induced by MHV (44), as well as by lymphocytic choriomeningitis virus (45). Expression of an active ns2 phosphodiesterase by A59 facilitates viral replication in macrophages by antagonizing the OAS-RNase L pathway, thus downregulating the IFN response, and this promotes replication in the liver parenchyma, leading to hepatitis (2). Depletion of macrophages promotes replication of ns2-H126R and the consequent development of liver pathology in B6 mice (2), demonstrating that replication in KC is not required for the induction of hepatitis, but rather, that KC protect the host from viral infection. In addition, KC have the highest basal and IFN-induced levels of OAS among liver cell types (46). It is not known whether LSEC also play a role in restricting virus from the liver parenchyma. We are currently investigating the levels of ISG mRNA expression in LSEC, as well as replication of A59 and ns2-H126R in that cell type.
The brain is considered immune privileged and differs from the liver and other peripheral organs in response to infections, in part due to the blood brain barrier and a lack of dendritic cells in the naive brain. Furthermore, as the CNS is composed predominantly of nonrenewable cells, such as neurons, unregulated immune responses could be irreparably destructive to the CNS. Indeed, high levels of IFN-α can be pathogenic in the CNS, as illustrated by individuals with Aicardi-Goutières syndrome, who produce high levels of IFN-α in their cerebrospinal fluid and exhibit symptoms of chronic encephalitis (47, 48). Unregulated RNase L-mediated degradation has been reported to promote apoptosis potentially damaging to nonrenewable cells (49). The failure to respond to ns2-H126R infection by activation of the OAS-RNase L pathway in neurons may reflect the relatively low basal levels of ISG expression, which may limit the damaging effects of excessive IFN signaling (2, 42).
A59 infects all major CNS cell types in vivo (19, 50), as well as in vitro (Fig. 5). Intracranial infection provides immediate access to all cell types. Neurons, the most abundant cell type in the murine brain, generally express low levels of ISG mRNA and minimal basal levels of OAS mRNA, allowing robust virus replication. Astrocytes and oligodendrocytes are similarly susceptible to infection. However, brain-resident macrophages, called microglia, also restrict ns2-H126R replication, albeit less than BMM (Fig. 3B and 5D) (25, 51, 52), and would be expected to limit replication in the brain in vivo. We suggest that microglia are not able to significantly restrict ns2-H126R in the CNS in part because they represent a minority of infected cells (53). In addition, by the time microglia and infiltrating macrophages have secreted IFN, which is late during A59 infection (25, 51, 52), it may be too late to induce OAS-RNase L in other parenchymal cell types already infected by virus. Furthermore, our in vitro data suggest that IFN pretreatment of neurons, astrocytes, and oligodendrocytes also fails to promote an effective RNase L response in MHV-infected cells. Our data, however, do not rule out the possibility that a combination of IFN exposure and infection may trigger RNase L activity in the CNS in vivo. Indeed, expression of OAS was reported for neurons in brains that had been injected with IFN-α (54). Consistent with our results, an attenuated MHV-JHM isolate with tropism restricted to oligodendrocytes replicated to similar titers in the CNSs of RNase L−/− and B6 mice; however, increased foci of infected microglia were observed in the gray matter in RNase L−/− mice, implying OAS-RNase L did provide some protection against viral infection of microglia in that model (55).
In order to investigate whether introduction of MHV through a less artificial route of infection would induce a protective RNase L response, we inoculated virus intranasally. Olfactory bulb neurons may be specialized to control viruses, as they are most exposed to environmental factors. In addition, they continue to replicate in vivo in adult mice (56) and thus may tolerate higher levels of ISG expression. Indeed, ISG induction has been reported in the olfactory bulb, most likely in neurons, in response to vesicular stomatitis virus (VSV) (45, 57). Similarly, VSV infection was restricted by the IFN-stimulated gene product of IFIT2 following intranasal, but not intracranial, inoculation, suggesting that IFN produced during infection of olfactory bulb neurons induced IFIT2 expression throughout the brain (24). However, when inoculated via the intranasal route, ns2-H126R was not preferentially restricted relative to A59 (Fig. 2), implying that the cells of the nasal epithelium and the olfactory neurons were not better able to prevent viral invasion. Consistent with these data, we found no significant differences in the basal levels of OAS mRNA among RNA samples extracted from the olfactory bulbs and from other regions of the brain (data not shown). Interestingly, however, it has recently been reported that cultured granular neurons of the cerebellum express higher basal levels of ISG mRNA than cortical neurons and are consequently more restrictive of infection with several viruses, including MHV-A59 (42).
It is possible that the endothelial cells that line the blood vessels and/or the ependymal cells that line the ventricles of the brain may express relatively high levels of interferon-stimulated genes, including OAS, and thus provide a barrier to viral entry into the brain parenchyma that is bypassed by intracranial and intranasal inoculation. Indeed, while MHV does not spread into the brain after peripheral inoculation in B6 mice, intrahepatic inoculation readily produces encephalitis in mice impaired in type I interferon signaling (either IFNAR−/− or MDA5−/− mice) (unpublished data). Furthermore, ependymal cells have been shown to express higher levels of OAS than other CNS cells in vivo (54). Future studies will be directed at investigating the possibility that one or both of these cell types may play a gatekeeping role similar to the function we propose for KC in the liver and thus prevent the spread of peripheral viral infections into the brain.
ACKNOWLEDGMENTS
This work was supported by NIH grants RO1-NS-081008, R56-AI-095285, and RO1-NS-054695 (S.R.W.) and RO1-CA-044059 (R.H.S.) and the Cellular Neuroscience Core of the Institutional Intellectual and Developmental Disabilities Research Center (HD26979) (J.G.).
Footnotes
Published ahead of print 22 May 2013
REFERENCES
- 1. Navas S, Weiss SR. 2003. Murine coronavirus-induced hepatitis: JHM genetic background eliminates A59 spike-determined hepatotropism. J. Virol. 77:4972–4978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Zhao L, Rose KM, Elliott R, Van Rooijen N, Weiss SR. 2011. Cell-type-specific type I interferon antagonism influences organ tropism of murine coronavirus. J. Virol. 85:10058–10068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Roth-Cross JK, Bender SJ, Weiss SR. 2008. Murine coronavirus mouse hepatitis virus is recognized by MDA5 and induces type I interferon in brain macrophages/microglia. J. Virol. 82:9829–9838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ireland DD, Stohlman SA, Hinton DR, Atkinson R, Bergmann CC. 2008. Type I interferons are essential in controlling neurotropic coronavirus infection irrespective of functional CD8 T cells. J. Virol. 82:300–310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cervantes-Barragan L, Zust R, Weber F, Spiegel M, Lang KS, Akira S, Thiel V, Ludewig B. 2007. Control of coronavirus infection through plasmacytoid dendritic-cell-derived type I interferon. Blood 109:1131–1137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Dong B, Silverman RH. 1995. 2-5A-dependent RNase molecules dimerize during activation by 2-5A. J. Biol. Chem. 270:4133–4137 [DOI] [PubMed] [Google Scholar]
- 7. Malathi K, Dong B, Gale M, Jr, Silverman RH. 2007. Small self-RNA generated by RNase L amplifies antiviral innate immunity. Nature 448:816–819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Silverman RH. 2007. Viral encounters with 2′,5′-oligoadenylate synthetase and RNase L during the interferon antiviral response. J. Virol. 81:12720–12729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhou A, Paranjape J, Brown TL, Nie H, Naik S, Dong B, Chang A, Trapp B, Fairchild R, Colmenares C, Silverman RH. 1997. Interferon action and apoptosis are defective in mice devoid of 2′,5′-oligoadenylate-dependent RNase L. EMBO J. 16:6355–6363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Randall RE, Goodbourn S. 2008. Interferons and viruses: an interplay between induction, signalling, antiviral responses and virus countermeasures. J. Gen. Virol. 89:1–47 [DOI] [PubMed] [Google Scholar]
- 11. Zhao L, Jha BK, Wu A, Elliott R, Ziebuhr J, Gorbalenya AE, Silverman RH, Weiss SR. 2012. Antagonism of the interferon-induced OAS-RNase L pathway by murine coronavirus ns2 protein is required for virus replication and liver pathology. Cell Host Microbe 11:607–616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Roth-Cross JK, Stokes H, Chang G, Chua MM, Thiel V, Weiss SR, Gorbalenya AE, Siddell SG. 2009. Organ-specific attenuation of murine hepatitis virus strain A59 by replacement of catalytic residues in the putative viral cyclic phosphodiesterase ns2. J. Virol. 83:3743–3753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gombold JL, Hingley ST, Weiss SR. 1993. Fusion-defective mutants of mouse hepatitis virus A59 contain a mutation in the spike protein cleavage signal. J. Virol. 67:4504–4512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Basler CF, Mikulasova A, Martinez-Sobrido L, Paragas J, Muhlberger E, Bray M, Klenk HD, Palese P, Garcia-Sastre A. 2003. The Ebola virus VP35 protein inhibits activation of interferon regulatory factor 3. J. Virol. 77:7945–7956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Thompson WH, Kalfayan B, Anslow RO. 1965. Isolation of California Encephalitis Group Virus from a fatal human illness. Am. J. Epidemiol. 81:245–253 [DOI] [PubMed] [Google Scholar]
- 16. Hoskins JM, Sanders FK. 1957. Propagation of mouse encephalomyocarditis virus in ascites tumour cells maintained in vitro. Br. J. Exp. Pathol. 38:268–272 [PMC free article] [PubMed] [Google Scholar]
- 17. Park MS, Shaw ML, Munoz-Jordan J, Cros JF, Nakaya T, Bouvier N, Palese P, Garcia-Sastre A, Basler CF. 2003. Newcastle disease virus (NDV)-based assay demonstrates interferon-antagonist activity for the NDV V protein and the Nipah virus V, W, and C proteins. J. Virol. 77:1501–1511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Caamano J, Alexander J, Craig L, Bravo R, Hunter CA. 1999. The NF-kappa B family member RelB is required for innate and adaptive immunity to Toxoplasma gondii. J. Immunol. 163:4453–4461 [PubMed] [Google Scholar]
- 19. Bender SJ, Phillips JM, Scott EP, Weiss SR. 2010. Murine coronavirus receptors are differentially expressed in the central nervous system and play virus strain-dependent roles in neuronal spread. J. Virol. 84:11030–11044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Feigenson K, Reid M, See J, Crenshaw EB, III, Grinspan JB. 2009. Wnt signaling is sufficient to perturb oligodendrocyte maturation. Mol. Cell Neurosci. 42:255–265 [DOI] [PubMed] [Google Scholar]
- 21. Feigenson K, Reid M, See J, Crenshaw IE, Grinspan JB. 2011. Canonical Wnt signalling requires the BMP pathway to inhibit oligodendrocyte maturation. ASN Neuro. 3:e00061. 10.1042/AN20110004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Xiang Y, Wang Z, Murakami J, Plummer S, Klein EA, Carpten JD, Trent JM, Isaacs WB, Casey G, Silverman RH. 2003. Effects of RNase L mutations associated with prostate cancer on apoptosis induced by 2′,5′-oligoadenylates. Cancer Res. 63:6795–6801 [PubMed] [Google Scholar]
- 23. Weiss SR, Leibowitz JL. 2011. Coronavirus pathogenesis. Adv. Virus Res. 81:85–164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Fensterl V, Wetzel JL, Ramachandran S, Ogino T, Stohlman SA, Bergmann CC, Diamond MS, Virgin HW, Sen GC. 2012. Interferon-induced Ifit2/ISG54 protects mice from lethal VSV neuropathogenesis. PLoS Pathog. 8:e1002712. 10.1371/journal.ppat.1002712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Roth-Cross JK, Martinez-Sobrido L, Scott EP, Garcia-Sastre A, Weiss SR. 2007. Inhibition of the alpha/beta interferon response by mouse hepatitis virus at multiple levels. J. Virol. 81:7189–7199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhou A, Molinaro RJ, Malathi K, Silverman RH. 2005. Mapping of the human RNASEL promoter and expression in cancer and normal cells. J. Interferon Cytokine Res. 25:595–603 [DOI] [PubMed] [Google Scholar]
- 27. Kubota K, Nakahara K, Ohtsuka T, Yoshida S, Kawaguchi J, Fujita Y, Ozeki Y, Hara A, Yoshimura C, Furukawa H, Haruyama H, Ichikawa K, Yamashita M, Matsuoka T, Iijima Y. 2004. Identification of 2′-phosphodiesterase, which plays a role in the 2-5A system regulated by interferon. J. Biol. Chem. 279:37832–37841 [DOI] [PubMed] [Google Scholar]
- 28. Ida-Hosonuma M, Sasaki Y, Toyoda H, Nomoto A, Gotoh O, Yonekawa H, Koike S. 2003. Host range of poliovirus is restricted to simians because of a rapid sequence change of the poliovirus receptor gene during evolution. Arch. Virol. 148:29–44 [DOI] [PubMed] [Google Scholar]
- 29. Ida-Hosonuma M, Iwasaki T, Yoshikawa T, Nagata N, Sato Y, Sata T, Yoneyama M, Fujita T, Taya C, Yonekawa H, Koike S. 2005. The alpha/beta interferon response controls tissue tropism and pathogenicity of poliovirus. J. Virol. 79:4460–4469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Daffis S, Samuel MA, Keller BC, Gale M, Jr, Diamond MS. 2007. Cell-specific IRF-3 responses protect against West Nile virus infection by interferon-dependent and -independent mechanisms. PLoS Pathog. 3:e106. 10.1371/journal.ppat.0030106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhou H, Zhao J, Perlman S. 2010. Autocrine interferon priming in macrophages but not dendritic cells results in enhanced cytokine and chemokine production after coronavirus infection. MBio 1: pii:e00219-10. 10.1128/mBio.00219-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kapil P, Butchi NB, Stohlman SA, Bergmann CC. 2012. Oligodendroglia are limited in type I interferon induction and responsiveness in vivo. Glia 60:1555–1566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Daffis S, Suthar MS, Gale M, Jr, Diamond MS. 2009. Measure and countermeasure: type I IFN (IFN-alpha/beta) antiviral response against West Nile virus. J. Innate Immun. 1:435–445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhou H, Perlman S. 2007. Mouse hepatitis virus does not induce beta interferon synthesis and does not inhibit its induction by double-stranded RNA. J. Virol. 81:568–574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Versteeg GA, Bredenbeek PJ, van den Worm SH, Spaan WJ. 2007. Group 2 coronaviruses prevent immediate early interferon induction by protection of viral RNA from host cell recognition. Virology 361:18–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Overby AK, Popov VL, Niedrig M, Weber F. 2010. Tick-borne encephalitis virus delays interferon induction and hides its double-stranded RNA in intracellular membrane vesicles. J. Virol. 84:8470–8483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hagemeijer MC, Vonk AM, Monastyrska I, Rottier PJ, de Haan CA. 2012. Visualizing coronavirus RNA synthesis in time by using click chemistry. J. Virol. 86:5808–5816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. van den Worm SH, Knoops K, Zevenhoven-Dobbe JC, Beugeling C, van der Meer Y, Mommaas AM, Snijder EJ. 2011. Development and RNA-synthesizing activity of coronavirus replication structures in the absence of protein synthesis. J. Virol. 85:5669–5673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Knoops K, Swett-Tapia C, van den Worm SH, Te Velthuis AJ, Koster AJ, Mommaas AM, Snijder EJ, Kikkert M. 2010. Integrity of the early secretory pathway promotes, but is not required for, severe acute respiratory syndrome coronavirus RNA synthesis and virus-induced remodeling of endoplasmic reticulum membranes. J. Virol. 84:833–846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. van Hemert MJ, van den Worm SH, Knoops K, Mommaas AM, Gorbalenya AE, Snijder EJ. 2008. SARS-coronavirus replication/transcription complexes are membrane-protected and need a host factor for activity in vitro. PLoS Pathog. 4:e1000054. 10.1371/journal.ppat.1000054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Prentice E, McAuliffe J, Lu X, Subbarao K, Denison MR. 2004. Identification and characterization of severe acute respiratory syndrome coronavirus replicase proteins. J. Virol. 78:9977–9986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Cho H, Proll SC, Szretter KJ, Katze MG, Gale M, Jr, Diamond MS. 2013. Differential innate immune response programs in neuronal subtypes determine susceptibility to infection in the brain by positive-stranded RNA viruses. Nat. Med. 19:458–464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Crispe IN. 2009. The liver as a lymphoid organ. Annu. Rev. Immunol. 27:147–163 [DOI] [PubMed] [Google Scholar]
- 44. Cervantes-Barragan L, Kalinke U, Zust R, Konig M, Reizis B, Lopez-Macias C, Thiel V, Ludewig B. 2009. Type I IFN-mediated protection of macrophages and dendritic cells secures control of murine coronavirus infection. J. Immunol. 182:1099–1106 [DOI] [PubMed] [Google Scholar]
- 45. Lang PA, Recher M, Honke N, Scheu S, Borkens S, Gailus N, Krings C, Meryk A, Kulawik A, Cervantes-Barragan L, Van Rooijen N, Kalinke U, Ludewig B, Hengartner H, Harris N, Haussinger D, Ohashi PS, Zinkernagel RM, Lang KS. 2010. Tissue macrophages suppress viral replication and prevent severe immunopathology in an interferon-I-dependent manner in mice. Hepatology 52:25–32 [DOI] [PubMed] [Google Scholar]
- 46. Asada-Kubota M, Tatsumi R, Ueda T, Kobayashi M, Hamada K, Maekawa S, Sokawa Y. 1998. The target cells of injected type I interferons in mouse liver. J. Interferon Cytokine Res. 18:71–74 [DOI] [PubMed] [Google Scholar]
- 47. Stephenson JB. 2008. Aicardi-Goutieres syndrome (AGS). Eur. J. Paediatr. Neurol. 12:355–358 [DOI] [PubMed] [Google Scholar]
- 48. Paul S, Ricour C, Sommereyns C, Sorgeloos F, Michiels T. 2007. Type I interferon response in the central nervous system. Biochimie 89:770–778 [DOI] [PubMed] [Google Scholar]
- 49. Castelli JC, Hassel BA, Maran A, Paranjape J, Hewitt JA, Li XL, Hsu YT, Silverman RH, Youle RJ. 1998. The role of 2′-5′ oligoadenylate-activated ribonuclease L in apoptosis. Cell Death Differ. 5:313–320 [DOI] [PubMed] [Google Scholar]
- 50. Miura TA, Travanty EA, Oko L, Bielefeldt-Ohmann H, Weiss SR, Beauchemin N, Holmes KV. 2008. The spike glycoprotein of murine coronavirus MHV-JHM mediates receptor-independent infection and spread in the central nervous systems of Ceacam1a-/- mice. J. Virol. 82:755–763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Rose KM, Elliott R, Martinez-Sobrido L, Garcia-Sastre A, Weiss SR. 2010. Murine coronavirus delays expression of a subset of interferon-stimulated genes. J. Virol. 84:5656–5669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Rose KM, Weiss SR. 2009. Murine coronavirus cell type dependent interaction with the type I interferon response. Viruses 1:689–712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Phillips JJ, Chua MM, Rall GF, Weiss SR. 2002. Murine coronavirus spike glycoprotein mediates degree of viral spread, inflammation, and virus-induced immunopathology in the central nervous system. Virology 301:109–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Asada-Kubota M, Ueda T, Nakashima T, Kobayashi M, Shimada M, Takeda K, Hamada K, Maekawa S, Sokawa Y. 1997. Localization of 2′,5′-oligoadenylate synthetase and the enhancement of its activity with recombinant interferon-alpha A/D in the mouse brain. Anat. Embryol. 195:251–257 [DOI] [PubMed] [Google Scholar]
- 55. Ireland DD, Stohlman SA, Hinton DR, Kapil P, Silverman RH, Atkinson RA, Bergmann CC. 2009. RNase L mediated protection from virus induced demyelination. PLoS Pathog. 5:e1000602. 10.1371/journal.ppat.1000602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Cowan CM, Roskams AJ. 2002. Apoptosis in the mature and developing olfactory neuroepithelium. Microsc. Res. Tech. 58:204–215 [DOI] [PubMed] [Google Scholar]
- 57. Detje CN, Meyer T, Schmidt H, Kreuz D, Rose JK, Bechmann I, Prinz M, Kalinke U. 2009. Local type I IFN receptor signaling protects against virus spread within the central nervous system. J. Immunol. 182:2297–2304 [DOI] [PubMed] [Google Scholar]