

Abstract
Herpes simplex virus 1 (HSV-1) replication initiates inflammation and angiogenesis responses in the cornea to result in herpetic stromal keratitis (HSK), which is a leading cause of infection-induced vision impairment. Chemokines are secreted to modulate HSK by recruiting leukocytes, which affect virus growth, and by influencing angiogenesis. The present study used a murine infection model to investigate the significance of the chemokine CXC chemokine ligand 10 (CXCL10; gamma interferon-inducible protein 10 [IP-10]) in HSK. Here, we show that HSV-1 infection of the cornea induced CXCL10 protein expression in epithelial cells. The corneas of mice with a targeted disruption of the gene encoding CXCL10 displayed decreases in levels of neutrophil-attracting cytokine (interleukin-6), primary neutrophil influx, and viral clearance 2 or 3 days postinfection. Subsequently, absence of CXCL10 aggravated HSK with elevated levels of interleukin-6, chemokines for CD4+ T cells and/or neutrophils (macrophage inflammatory protein-1α and macrophage inflammatory protein-2), angiogenic factor (vascular endothelial growth factor A), and secondary neutrophil influx, as well as infiltration of CD4+ T cells to exacerbate opacity and angiogenesis in the cornea at 14 and up to 28 days postinfection. Our results collectively show that endogenous CXCL10 contributes to recruit the primary neutrophil influx and to affect the expression of cytokines, chemokines, and angiogenic factors as well as to reduce the viral titer and HSK severity.
INTRODUCTION
Herpes simplex virus 1 (HSV-1) infects more than 80% of adults worldwide (1). HSV-1 infection of the cornea can result in herpetic stromal keratitis (HSK), which is the most common cause of infection-induced corneal blindness, especially in the elderly population of the Western world. In the United States, it is estimated that 400,000 persons are affected, with 20,000 new cases per year (1).
Studies using the murine model have revealed multiple events, including viral replication, chemokine and cytokine production, leukocyte influx, and neovascularization, during HSK progression (1, 2). Viral replication initiates all complex events to positively affect the severity of HSK (3). Viral replication is eventually terminated by the host immune response about 1 week postinfection (p.i.). However, neovascularization and inflammation may intensify, in part because neovessels bring in more inflammatory infiltrates.
Abundant neutrophils are detected in the stroma of infected cornea and display a biphasic influx (1, 4, 5). The primary neutrophil influx reaches a peak at 2 to 3 days p.i., declines to a basal level at 5 days p.i., and participates in virus clearance directly or indirectly by activating other inflammatory cells, such as monocytes (1, 4–8). The secondary neutrophil influx is more intense, begins at 8 to 9 days p.i., and reaches a peak at about 2 to 3 weeks p.i. when HSK is evident (1, 5, 9). It is generally believed that the secondary neutrophil influx contributes to lesions by serving as an activator for T cell-mediated inflammatory responses (1, 5). Among T cells present in the infected cornea, CD4+ T cells outnumber CD8+ T cells and have been shown to orchestrate chronic inflammatory lesions (1, 2).
Chemokines recruiting CD4+ T cells and/or neutrophils, such as macrophage inflammatory protein-1α (MIP-1α) and macrophage inflammatory protein-2 (MIP-2/CXCL2), are induced in the HSV-1-infected cornea and have been shown to affect HSK development (6, 9). The recruited leukocytes can further enhance inflammation by increasing cytokine expression (1). In addition to inflammation, neovascularization also contributes to lesions. HSV-1 infection has been shown to increase the expression of potent angiogenic factors, including fibroblast growth factor 2 (FGF-2; also known as basic fibroblast growth factor) and vascular endothelial growth factor A (VEGF-A), to cause neovascularization in the cornea (1, 10). Neovascularization and inflammation, two hallmarks of HSK, start to develop about 1 week p.i. and result in evident lesions with a substantial leukocyte influx brought in via neovessels at 2 to 3 weeks p.i (1).
CXC chemokine ligand 10 (CXCL10; gamma interferon [IFN-γ]-inducible protein 10 [IP-10]) is a chemokine which attracts mainly T cells and NK cells through binding to the receptor CXCR3 (11, 12). However, CXCL10 has been reported to recruit neutrophils or to bind to other cells by an unknown mechanism independent of CXCR3 (11, 13, 14). CXCL10 expression can be upregulated in a variety of cells, including epithelial cells and neutrophils, by stimulation with IFN-α, IFN-β, IFN-γ, or viruses (12, 15–21). CXCL10 is a fascinating chemokine because of its controversial roles in viral infections. Endogenous CXCL10 is known to protect mice infected with coxsackievirus B3, mouse hepatitis virus, or dengue virus, mostly by decreasing tissue viral loads (16, 19, 22). However, CXCL10 has been implicated in aggravating virus-induced diseases, such as demyelination in the mouse brain induced by mouse hepatitis virus, meningoencephalitis in mice induced by lymphocytic choriomeningitis virus, liver damage in humans induced by chronic hepatitis C virus infection, and neurological disorders induced by human immunodeficiency virus (19, 23–25). Additionally, CXCL10 serves as an angiostatic factor to inhibit the expression of VEGF-A or the angiogenic response induced by FGF-2 or VEGF-A (26–29).
HSV-1 infection of the cornea has been shown to induce CXCL10 (18, 30, 31). Previous reports investigating the role of endogenous CXCL10 in HSK were not conclusive because mice died about 1 week after infection with a virulent HSV-1 strain before the corneas could develop evident lesions (18, 31). The present study used an HSV-1 strain which is proficient in inducing HSK but not encephalitis to infect wild-type mice and mice with a targeted disruption of the gene encoding CXCL10. Our results provide the first evidence that endogenous CXCL10 reduces the severity of HSK.
MATERIALS AND METHODS
The cell and virus.
African green monkey kidney (Vero) cells were maintained and propagated according to the instructions of the American Type Culture Collection. Wild-type HSV-1 strains RE and McKrae were propagated and titrated via plaque assay on Vero cell monolayers. RE, which has been used extensively for studying HSK since 1976 (32–34), was kindly provided by Robert Lausch.
Infection of mice.
All mouse experiment protocols were approved by the Laboratory Animal Committee of National Cheng Kung University. Five- to six-week-old male and female C57BL/6J mice and C57BL/6J-derived mice deficient in CXCL10 (B6.129S4-Cxcl10tm1Adl/J) due to a targeted disruption of the gene were used for study. Mice were purchased from The Jackson Laboratory, bred, and maintained under specific-pathogen-free conditions in the Laboratory Animal Center of our university. Mice were anesthetized and infected with 5 × 104 PFU of strain RE or mock infected with lysates of uninfected Vero cells topically on the right eye following scarification (20 times) of the cornea with a needle. In addition, C57BL/6J mice were infected with 1 × 105 PFU of strain McKrae or mock infected on the right eye without scarifying the cornea. RE-infected mouse eyes were examined weekly after infection to monitor corneal opacity and angiogenesis as described in our previous report (35). The corneal opacity was graded on a scale of 0 to 5 as follows: 0, normal cornea; 1, mild corneal haze; 2, moderate corneal opacity or scarring; 3, severe corneal opacity, iris visible; 4, opaque cornea, iris invisible; and 5, necrotizing cornea with vascularization. Corneal angiogenesis was scored by measuring the length of neovessels. Briefly, the cornea was divided into four quadrants. The length of the longest neovessel in each quadrant was graded between 0 (no neovessel) and 4 (neovessel in the corneal center) in increments of about 0.4 mm (the radius of the cornea is about 1.5 mm). The angiogenesis score for each cornea (ranging from 0 to 16) was the sum of the four quadrants. In separate experiments, mice were sacrificed, and the eyes and trigeminal ganglia were harvested to determine viral titers via plaque assay.
Quantification of cytokines, chemokines, and angiogenic factors.
Three mouse eyeballs were pooled into one sample in a volume of 1 ml of phosphate-buffered saline to measure CXCL10. In addition, four to six corneas were harvested and pooled into one sample in a volume of 0.5 ml of RPMI 1640 medium for measurement of interleukin-6 (IL-6) (R&D Systems), IFN-γ (R&D Systems), MIP-2 (R&D Systems), MIP-1α (R&D Systems), VEGF-A (R&D Systems), and FGF-2 (Signosis). Samples were frozen at −80°C and homogenized. The homogenates were centrifuged to obtain supernatants for enzyme-linked immunosorbent assays (ELISAs) using commercially available kits according to the manufacturer's instructions.
Histological, immunofluorescence, and immunohistochemical staining.
Briefly, mouse eyes were fixed in 10% neutral buffered formalin, embedded in paraffin, and sectioned. Sections (6 μm) were deparaffinized and stained with hematoxylin and eosin. In addition, deparaffinized sections were treated with 1% fetal bovine serum to block nonspecific binding before incubation with antibodies against HSV-1 (Dako), mouse CXCL10 (R&D Systems), keratin K3 (clone AE5; Millipore), or Ly6G (clone 1A8; BD Pharmingen) or with isotype-matched control antibodies overnight at 4°C. Subsequently, bound anti-HSV-1 antibody was detected by donkey anti-rabbit immunoglobulin G Alexa Fluor 488 (Invitrogen), bound anti-CXCL10 antibody was detected by donkey anti-goat Alexa Fluor 594, and bound anti-keratin K3 antibody was detected by donkey anti-mouse immunoglobulin G Alexa Fluor 488. For detection of Ly6G signals, sections were treated with horseradish peroxidase-labeled donkey anti-rat immunoglobulin G (Jackson ImmunoResearch Laboratories, Inc.) followed by 3-amino-9-ethylcarbazole (AEC kit; Zymed Laboratories, Inc.) before they were counterstained with hematoxylin. Mouse eyes were also embedded in optimal cutting temperature (OCT) medium (Thermo Fisher Scientific, Inc.), snap-frozen in liquid nitrogen, and sectioned. Sections (6 μm) were air dried, fixed in cold acetone, and incubated with antibodies against mouse CD4 (clone RM4-5; Santa Cruz Biotechnology, Inc.), CD8 (clone JXYT8; Santa Cruz Biotechnology, Inc.), or CD31 (BD Pharmingen) or with isotype-matched control antibodies overnight at 4°C. The resulting sections were treated with horseradish peroxidase-labeled donkey anti-rat immunoglobulin G (Jackson ImmunoResearch Laboratories, Inc.) and 3-amino-9-ethylcarbazole (AEC kit) before counterstaining with hematoxylin. Antibodies against HSV-1 antigens, mouse CXCL10, keratin K3, CD31, Ly6G, CD4, or CD8 detected signals, whereas isotype-matched control antibodies failed to detect specific signals. Images were photographed using an Olympus DP12 digital microscope camera (Olympus).
Flow cytometry.
Three to five corneas were pooled and incubated in phosphate-buffered saline containing 20 mM EDTA at 37°C for 15 min to separate the epithelial sheet from the stroma. The stroma was treated with 82 units/stroma of type I collagenase at 37°C for 1 h to release cells. Subsequently, cells were stained with phycoerythrin-conjugated antibody against mouse CD4 (clone GK1.5; eBioscience) or fluorescein isothiocyanate-conjugated antibody against mouse CD8a (clone 53-6.7; eBioscience) or the isotype-matched control antibody. Additionally, cells were stained with the antibody against mouse Ly6G (clone 1A8) or the isotype-matched control antibody before incubation with the fluorescein isothiocyanate-conjugated secondary antibody. In separate experiments, two to four corneas were pooled and incubated with 82 units/cornea of type I collagenase at 37°C for 80 min to release cells from corneas. Then cells were stained with the antibody against mouse CD31 (BD Pharmingen) or the isotype-matched control antibody before incubation with the fluorescein isothiocyanate-conjugated secondary antibody. The stained cells were analyzed by a FACSCalibur instrument (BD Biosciences) using WinMDI software.
Assaying the anti-HSV-1 activity of CXCL10.
Corneas removed from wild-type mice and mice deficient in CXCL10 were infected with strain RE (2 × 105 PFU/cornea). In addition, the corneas removed from wild-type mice were treated with recombinant murine CXCL10 (10 μg/ml) 30 min before and also during infection.
Chemotaxis assay.
Neutrophils were isolated from the peritoneal fluid of >8-week-old ICR mice as described previously (36). For chemotaxis assays, 5 × 105 neutrophils were resuspended in RPMI 1640 medium containing 1% bovine serum albumin and placed in the upper chamber of a 24-transwell plate with a 5-μm-pore-size filter (Corning Life Sciences). The lower chamber contained medium with or without 100 ng/ml of recombinant murine CXCL10 protein (PeproTech, Inc.). The cultures were incubated at 37°C for 4 h before the cells in the upper chamber were fixed in methanol, stained with Giemsa (Merck), and washed with water. The numbers of cells migrating through filters were examined and counted under a microscope.
Statistical analyses.
Data are expressed as means ± standard errors (SE) unless noted otherwise. For statistical comparison, chemokine and cytokine levels as well as numbers of CD31+ cells, neutrophils, and CD4+ T cells determined by flow cytometric or chemotaxis assay were analyzed by a Student t test. Corneal opacity scores and angiogenesis scores were analyzed by a Wilcoxon signed-rank test. HSK incidences were analyzed by Fisher's exact test. Tissue viral loads were analyzed by a Mann-Whitney U test. All P values are for two-tailed significance tests. A P value of <0.05 is considered statistically significant.
RESULTS
HSV-1 infection of the cornea induces CXCL10 expression in epithelial cells.
We first investigated the kinetics of viral growth and CXCL10 induction in the HSV-1-infected mouse eye. Mice were infected in the right eye with HSV-1 strain RE (5 × 104 PFU/eye) or mock infected with lysates of uninfected Vero cells topically on the cornea following scarification as inoculation of HSV-1 via the abraded eye mimics human infection in some individuals. Mouse eyes were harvested after infection and processed to assay for viral titers and CXCL10 protein. Eye viral titers were high at day 1 p.i. and declined to a very low level by day 7 p.i. (Fig. 1A). ELISAs failed to detect CXCL10 protein in the mock-infected eye (Fig. 1B). In the infected eye, CXCL10 displayed biphasic induction, with high quantities at days 2 and 6 p.i. and undetectable levels by days 14 to 28 p.i. Notably, CXCL10 levels in the infected eye were significantly higher than those in the mock-infected eye (P < 0.05) from days 1 to 8 p.i.
Fig 1.

HSV-1 infection of the cornea induces CXCL10 expression in epithelial cells. Viral titers (A) and CXCL10 levels (B) in the eyes of mice mock infected (Mock) or infected with strain RE (HSV-1) on scarified corneas at the indicated times postinfection are shown. Viral titers (C) and CXCL10 levels (D) in the eyes of mice mock infected (Mock) or infected with strain McKrae (HSV-1) on intact corneas without scarification at the indicated times postinfection are shown. Data represent means plus SE (error bars) of more than three samples per group. *, P < 0.05; **, P < 0.01; ***, P < 0.001, via a Student t test, compared with the mock-infected group. BD, below detection. The mock-infected and RE-infected eyes harvested 2 days postinfection were processed and stained with Hoechst and antibodies against HSV-1 or CXCL10 (E) or antibodies against keratin K3 or CXCL10 (F). The corneal portion is shown. Data are representative of at least three samples from two independent experiments. E, epithelium. S, stroma.
We also investigated the kinetics of viral growth and CXCL10 induction in the mouse eye infected with another HSV-1 strain (McKrae) without scarifying the cornea. Eye viral titers increased from days 2 to 7 p.i. (Fig. 1C). CXCL10 was detected in the infected eye at days 5 and 7 p.i. but not in the mock-infected eye (Fig. 1D). The CXCL10 level in the infected eye at day 7 p.i. was significantly higher than that in the mock-infected eye (P < 0.05).
We performed immunofluorescence staining to detect the cells in the infected cornea expressing CXCL10. In the mock-infected cornea, neither viral antigen nor CXCL10 was detected (Fig. 1E). In the RE-infected cornea harvested at day 2 p.i., abundant viral antigen was detected in the anterior epithelium. Copious CXCL10 was detected in the region of epithelium immediately adjacent to the region with viral antigen. We also costained corneal sections for CXCL10 and keratin K3, which is the intermediate filament cytoskeleton specifically expressed in corneal epithelial cells (37), and found that cells in the infected cornea were dually positive (Fig. 1F). Theses results show that epithelial cells (keratinocytes) express CXCL10 in the infected cornea.
Absence of CXCL10 aggravates HSK.
We studied the influence of CXCL10 on HSK development using C57BL/6J mice and C57BL/6J-derived mice with a targeted disruption of the gene encoding CXCL10. Mice were infected with strain RE and monitored for corneal opacity and angiogenesis, two important features of HSK. All infected wild-type mice and CXCL10 gene knockout (Cxcl10−/−) mice survived. The corneal opacity scores of both wild-type and Cxcl10−/− mice reached peaks at day 14 p.i. and persisted thereafter (Fig. 2A). Notably, the mean corneal opacity scores of Cxcl10−/− mice were significantly higher than those of wild-type mice from days 14 to 28 p.i. (P < 0.05). About 82% of Cxcl10−/− mice displayed severe HSK with evident inflammation, neovascularization, opaque corneas, and invisible irises (an opacity score of ≥4) by day 28 p.i. (Fig. 2B). In contrast, only 38% of wild-type mice displayed severe HSK, which was significantly lower than the proportion of Cxcl10−/− mice (P < 0.05) at day 28 p.i.
Fig 2.

Absence of CXCL10 aggravates HSK. (A) The corneal opacity scores of wild-type (WT) mice (n = 13) and Cxcl10−/− mice (n = 11) at the indicated times after infection with strain RE are shown. Data represent means ± SE (error bars). *, P < 0.05, via a Wilcoxon signed-rank test, compared with wild-type mice. (B) The incidences of HSK lesions scored ≥4 in wild-type and Cxcl10−/− mice at 28 days postinfection are shown. Data represent means plus SE (error bars). *, P < 0.05, via Fisher's exact test. The eyes of wild-type and Cxcl10−/− mice harvested at 28 days postinfection were processed and stained with hematoxylin-eosin (C) or the antibody against CD31 (E). The corneal portion is shown. The reddish-brown in panel E denotes a positive reaction. Data are representative of at least three samples from three independent experiments. (D) The angiogenesis scores of wild-type mice (n = 25) and Cxcl10−/− mice (n = 19) at 22 days postinfection are shown. Each point on the scattergram represents an individual sample, and horizontal lines represent the mean values of each group. **, P < 0.01, via a Wilcoxon signed-rank test. (F) The numbers of CD31+ cells in the corneas of infected wild-type and Cxcl10−/− mice harvested at 23 days postinfection are shown. Data represent means plus SE (error bars) of more than four samples per group. *, P < 0.05, via a Student t test.
We performed hematoxylin-eosin staining to examine the pathological change in the cornea harvested at day 28 p.i. Histologically, the mock-infected corneas from wild-type and Cxcl10−/− mice were similar in morphology (Fig. 2C). However, the infected corneas from Cxcl10−/− mice were much thicker, with profound edema and inflammatory infiltrate, especially in the stroma, than the infected corneas from wild-type mice. The results of opacity scores and hematoxylin-eosin staining analysis are consistent and collectively show that absence of CXCL10 aggravates HSK.
Abundant and extended neovessels were found in the infected corneas of Cxcl10−/− mice, with a mean angiogenesis score significantly higher than that in the infected corneas of wild-type mice (P < 0.01) by day 22 p.i. (Fig. 2D). Since endothelial cells constituting the newly formed blood vessels express CD31 (38), we performed CD31 immunohistochemical staining on the corneas harvested at day 28 p.i. CD31 was not detected in the mock-infected corneas of wild-type and Cxcl10−/− mice (Fig. 2E). Abundant CD31 was detected in the corneal stroma of infected Cxcl10−/− mice, but minimal CD31 was detected in the corneal stroma of infected wild-type mice. To further assess neovascularization, we quantified the number of cells expressing CD31 in the infected corneas harvested at day 23 p.i. using flow cytometry as previously described (38). The mean number of cells expressing CD31 in the infected corneas of Cxcl10−/− mice was significantly higher than that in the infected corneas of wild-type mice (P < 0.05) by 3.3-fold (Fig. 2F). The results of immunohistochemical staining and flow cytometric analyses of CD31 as well as angiogenesis scores are consistent and collectively show that absence of CXCL10 exacerbates the angiogenesis of HSK.
Absence of CXCL10 increases viral loads in the eye and trigeminal ganglion.
We investigated the effect of CXCL10 deficiency on tissue viral loads. The mean viral titers in eyes of Cxcl10−/− mice were higher than those of wild-type mice at days 1, 3, 5, and 7 p.i. with a significant difference at day 3 p.i. (P < 0.05) of 1.3 log (Fig. 3A). The mean viral titers in trigeminal ganglia of Cxcl10−/− mice were also higher than those of wild-type mice from days 3 to 7 p.i. (Fig. 3B) with significant differences found at both days 3 and 5 p.i. (P < 0.01).
Fig 3.

Absence of CXCL10 increases tissue viral loads. The viral loads in the right eyes (A) and trigeminal ganglia (B) of wild-type (WT) mice and Cxcl10−/− mice at the indicated times after infection with strain RE are shown. Data represent means ± SE (error bars) of three to nine samples per data point. *, P < 0.05; **, P < 0.01, via a Mann-Whitney U test.
Exogenous CXCL10 treatment or endogenous CXCL10 fails to reduce HSV-1 growth in excised mouse corneas.
We next investigated how CXCL10 could contribute to viral clearance. Previous in vitro studies showed that treatment with CXCL10 (0.1 to 10 μg/ml) reduced the yields of HSV-1 in neurons cultured from the human fetal brain and of dengue virus (17, 39). Therefore, we studied the anti-HSV-1 activity of exogenous CXCL10 treatment in the excised mouse cornea. Treatment with recombinant murine CXCL10 (10 μg/ml) before and also during infection with strain RE failed to affect viral titers in mouse corneas removed from wild-type mice at 24 and 48 h p.i. This result is consistent with previous findings that CXCL10 pretreatment failed to affect HSV-1 replication in mouse splenocytes, the mouse fibroblast cell line (L-929), and microglial cells cultured from the human fetal brain (18, 39). Additionally, comparable viral titers were detected in corneas removed from wild-type or Cxcl10−/− mice 24 and 48 h after infection with strain RE, suggesting that endogenous CXCL10 fails to affect HSV-1 yields. In the mouse cornea, as ex vivo results showed that both exogenous CXCL10 treatment and endogenous CXCL10 failed to directly reduce HSV-1 titers, our further in vivo investigation focused on the chemokine function of CXCL10.
Absence of CXCL10 decreases the primary neutrophil influx during acute infection but subsequently increases the secondary neutrophil influx and infiltration of CD4+ T cells in the infected cornea at the stage with evident HSK.
We analyzed the effect of CXCL10 on leukocyte influx in the cornea at day 3 p.i. when there was a significant difference in eye viral titers between wild-type and Cxcl10−/− mouse groups. Hematoxylin-eosin staining analysis showed fewer polymorphonuclear cells in the infected corneas of Cxcl10−/− mice than in the infected corneas of wild-type mice (data not shown). Thus, we performed immunohistochemical staining and detected fewer Ly6G+ neutrophils in the infected corneas of Cxcl10−/− mice than in the infected corneas of wild-type mice (Fig. 4A, top panel). Additionally, flow cytometric analysis showed that the mean number of Ly6G+ neutrophils in infected corneas of Cxcl10−/− mice was significantly lower than that in infected corneas of wild-type mice (P < 0.05) by 2.3-fold (Fig. 4B). Taken together, these results indicate that the absence of CXCL10 decreases the primary influx of neutrophils, which have been shown to participate in HSV-1 clearance directly or indirectly by activating other inflammatory cells, such as monocytes (1, 4–8).
Fig 4.

The effect of CXCL10 deficiency on leukocyte influx in the infected mouse cornea. The eyes of infected wild-type (WT) mice and Cxcl10−/− mice harvested at the indicated days postinfection (dpi) with strain RE were processed and stained with antibodies against Ly6G (A), CD4 (C), and CD8 (E). The corneal portion is shown. Reddish-brown denotes a positive reaction. In separate experiments, the numbers of Ly6G+ cells at 3 and 28 days p.i. (B), CD4+ T cells at 28 days p.i. (D), and CD8+ T cells at 28 days p.i. (F) in the corneas of infected wild-type and Cxcl10−/− mice were determined by flow cytometry. Data represent means plus SE or means ± SE (error bars) from three independent experiments. *, P < 0.05; **, P < 0.01, by a Student t test between wild-type and Cxcl10−/− mouse groups at the same time point.
As hematoxylin-eosin staining analysis detected more inflammatory infiltrate in the infected corneas of Cxcl10−/− mice with severe HSK at day 28 p.i. than in the infected corneas of wild-type mice (Fig. 2C), we monitored the leukocyte influx at this time point. Immunohistochemical staining analysis detected more Ly6G+ neutrophils and CD4+ T cells, mostly in the corneal stroma, in infected Cxcl10−/− mice than in infected wild-type mice (Fig. 4A, bottom panels, and C). Flow cytometric analysis also showed that the mean numbers of Ly6G+ neutrophils and CD4+ T cells in infected corneas of Cxcl10−/− mice were significantly higher than those in infected corneas of wild-type mice (P < 0.05) by 2.8- and 2.9-fold, respectively (Fig. 4B and D). Immunohistochemical staining and flow cytometric analyses detected low levels of CD8+ T cells in the infected corneas of wild-type and Cxcl10−/− mice (Fig. 4E and F). The difference in levels of CD8+ T cells in the infected corneas of wild-type and Cxcl10−/− mice was not statistically significant (P > 0.05) (Fig. 4F). These results reveal that the absence of CXCL10 increases the secondary neutrophil influx and infiltration of CD4+ T cells in the cornea at the stage when HSK is evident.
In vitro chemotaxis assay shows that CXCL10 recruits mouse neutrophils.
One in vitro study reported that treatment with CXCL10 (100 ng/ml) significantly increased the migration of human neutrophils (40). We performed chemotaxis assays on mouse neutrophils isolated from the peritoneal fluid as few reports have investigated the recruitment of mouse neutrophils by CXCL10. The purity of neutrophils used for the assay was >99% based on polymorphonuclear morphology after Giemsa staining (Fig. 5A). The chemotaxis assay showed that recombinant murine CXCL10 at a concentration of 100 ng/ml significantly increased the migration of mouse neutrophils (Fig. 5B) (P < 0.001).
Fig 5.
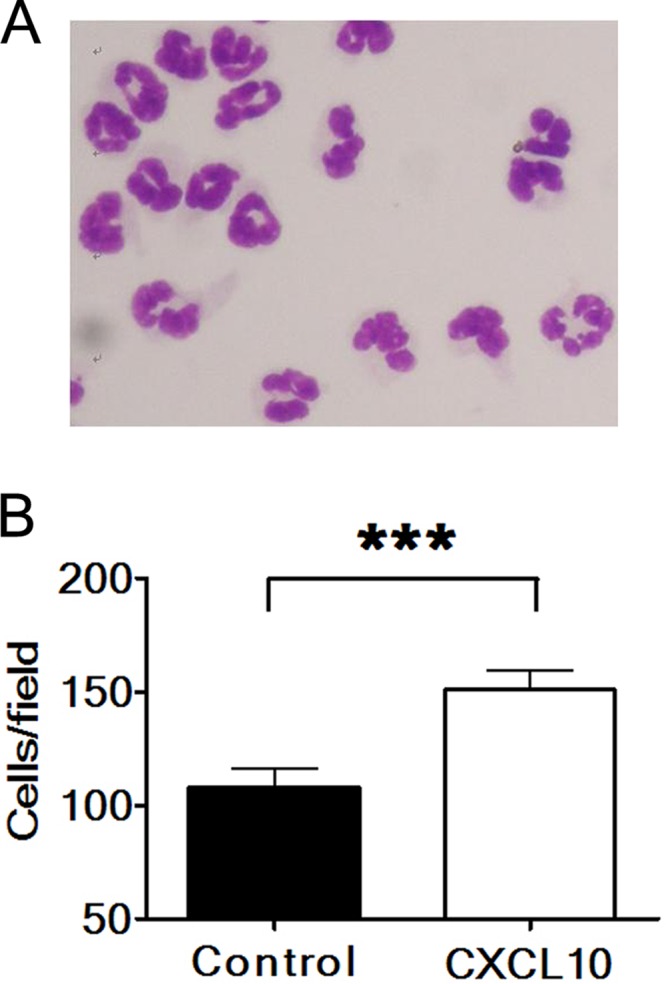
CXCL10 recruits neutrophils. (A) More than 99% of cells used for chemotaxis assays exhibited the morphology of polymorphonuclear neutrophils after Giemsa staining. (B) Neutrophils migrating through transwells in response to medium without (Control) or with CXCL10 protein were counted under a microscope. Five fields were counted for each well. Data represent means plus SE (error bars) of five wells from three independent experiments. ***, P < 0.001, via a Student t test.
Absence of CXCL10 reduces IL-6 expression during acute infection but increases IL-6, MIP-2, and MIP-1α levels in the infected cornea at the stage with evident HSK.
As absence of CXCL10 affects the influx of CD4+ T cells and, especially, neutrophils, we further investigated the influence of CXCL10 deficiency on the expression of other chemoattractants attracting T cells and particularly neutrophils. HSV-1 infection of the mouse cornea has been shown to induce three factors to recruit neutrophils. MIP-2 promotes the primary neutrophil influx, MIP-1α directs the secondary neutrophil influx as well as infiltration of CD4+ T cells, and IL-6 increases the primary neutrophil influx by enhancing the expression of both MIP-2 and MIP-1α (6, 9, 41). Additionally, endogenous IL-6 can regulate neutrophil trafficking in the murine model of peritoneal inflammation (42). Few studies have linked IFN-γ with neutrophil influx, so we used IFN-γ levels as a control. Therefore, we measured IL-6, IFN-γ, MIP-2, and MIP-1α in the cornea from days 2 to 28 p.i. using ELISAs.
The corneal IL-6 levels of infected Cxcl10−/− mice were low at day 2 p.i., similar at day 6 p.i., high at day 14 p.i., and similar at day 28 p.i. compared with those of infected wild-type mice (Fig. 6A), with significant differences found at days 2 and 14 p.i. (P < 0.05) of about 5- and 6-fold, respectively. The corneal IFN-γ levels of infected Cxcl10−/− and wild-type mice were not significantly different from days 2 to 28 p.i. (Fig. 6B). The profile of corneal MIP-2 levels was similar to that of corneal IL-6 levels in infected Cxcl10−/− and wild-type mice (Fig. 6C). The corneal MIP-2 level of infected Cxcl10−/− mice was significantly higher than that of infected wild-type mice at day 14 p.i. (P < 0.01) by 27-fold. The corneal MIP-1α levels of infected Cxcl10−/− mice were high at days 6 and 14 p.i. and similar at days 2 and 28 p.i. compared with those of infected wild-type mice, with a significant difference of 30-fold found at day 14 p.i. (P < 0.05) (Fig. 6D). The results of leukocyte infiltration, cytokines, and chemokines collectively show that absence of CXCL10 reduces IL-6 expression during the primary neutrophil influx but subsequently increases the expression in the infected cornea of IL-6, MIP-2, and MIP-1α at the stage with severe HSK and massive influx of neutrophils and T cells.
Fig 6.

The effect of CXCL10 deficiency on the expression of cytokine, chemokines, and angiogenic factors in the infected mouse cornea. The levels of IL-6, IFN-γ, MIP-2, MIP-1α, VEGF-A, and FGF-2 in the corneas of wild-type (WT) mice and Cxcl10−/− mice at the indicated times after infection with strain RE are shown. Data represent means ± SE (error bars) of three to five samples per data point. *, P < 0.05; **, P < 0.01, by a Student t test between wild-type and Cxcl10−/− mouse groups at the same time point.
Absence of CXCL10 increases the expression of angiogenic factor VEGF-A in the infected cornea.
HSV-1 infection of the mouse cornea is shown to increase the expression of potent angiogenic factors, VEGF-A and FGF-2, to enhance HSK severity (1, 10). CXCL10 is a known angiostatic factor capable of suppressing the expression of VEGF-A and the angiogenic responses induced by both VEGF-A and FGF-2 (26–29). As absence of CXCL10 enhances the angiogenesis of HSK, we examined the influence of CXCL10 deficiency on the expression of the angiogenic factors VEGF-A and FGF-2. ELISA results showed that the corneal VEGF-A levels of infected Cxcl10−/− mice at day 14 p.i. were significantly higher than those of infected wild-type mice (P < 0.05) by 5-fold (Fig. 6E). The corneal FGF-2 levels of infected Cxcl10−/− mice were slightly lower from days 2 to 6 p.i. but were slightly higher from days 14 to 40 p.i. than those of infected wild-type mice (Fig. 6F) (P > 0.05).
DISCUSSION
The present study is the first to show that HSV-1-infection of the cornea induces epithelial cells to express CXCL10, which reduces HSK, with increases in the primary neutrophil influx and viral clearance. These findings provide a better understanding of the induction and function of CXCL10 in HSK.
We detected two peaks of CXCL10 expression in the RE-infected eye at days 2 and 6 p.i. Few reports have used immunofluorescence staining to investigate the cells expressing CXCL10 in the HSV-1-infected cornea in vivo (18, 31, 43). In this study, we detected abundant CXCL10 in epithelial cells. Type I IFN (IFN-α/β) and type II IFN (IFN-γ) have been shown to be major CXCL10 inducers in primary epithelial cells cultured from humans and in leukocytes (20, 21, 44, 45). In the HSV-1-infected cornea, IFN-α is detected in epithelial cells 2 to 3 days p.i. (46), and one investigation has reported that the type I IFN signaling pathway is required for CXCL10 expression (47). IFN-γ is detected in the RE-infected cornea at day 6 p.i. (Fig. 6B). Accordingly, type I and II IFNs are likely to stimulate CXCL10 production at days 2 and 6 p.i., respectively.
In mice, endogenous CXCL10 recruits both CD4+ and CD8+ T cells to decrease the titer of mouse hepatitis virus in the brain, directs CD8+ T cells to reduce the titer of West Nile virus in the brain, and attracts NK cells to diminish the titer of coxsackievirus B3 in the heart (16, 19, 48). In HSV-infected mice, endogenous CXCL10 promotes both CD8+ T cells and NK cells to decrease the titer of a virulent HSV-1 strain in the brain (49). Endogenous CXCL10 protects mice from HSV-2 genital infection by decreasing neuropathogenesis induced by tumor necrosis factor alpha (50). One previous report comparing Cxcl10−/− and wild-type mice showed that CXCL10 expression initially orchestrated the inflammatory response to reduce acute HSV-1 titers in the cornea (31). However, this report failed to identify the effector as comparable numbers of NK cells were detected in the corneas of Cxcl10−/− and wild-type mice during acute HSV-1 infection (31). Here, we reveal that endogenous CXCL10 significantly reduces CD4+ cells and slightly decreases CD8+ T cells after acute infection. Surprisingly, CXCL10 enhances HSV-1 clearance with an increase of the primary neutrophil influx during acute infection. We are not aware of any report showing that CXCL10 affects neutrophil influx to influence the progression of virus-induced disease. It thus appears that the effectors recruited by CXCL10 to affect viral disease progression are diverse and may depend on the particular tissue that is infected and/or on the virus.
Our in vitro and in vivo results show that CXCL10 increases the migration of mouse neutrophils. These results conform with previous in vivo reports showing that the Cxcl10 transgene in astrocytes induced neutrophil accumulation in the mouse brain (13) and that endogenous CXCL10 recruited neutrophils into the mouse lung upon oxidative stress (51). While CXCL10 can act on T cells via the receptor CXCR3 (12), it also can interact with neutrophils and other cells through glycosaminoglycan, an unidentified receptor, or other unknown mechanisms (11, 13, 14). Similar to the report of the Cxcl10 transgene in astrocytes (13), our unpublished results showed minimal CXCR3 expression on mouse neutrophils, suggesting that the recruitment of neutrophils by CXCL10 might be independent of CXCR3. Exactly how CXCL10 recruits neutrophils requires further elucidation.
In the mouse cornea, HSV-1 infection can induce two neutrophil influxes. Three endogenous factors have been shown to enhance neutrophil influxes, with IL-6 and MIP-2 recruiting the primary one and MIP-1α directing the secondary one (6, 9, 41). Here, we add CXCL10 to the list. CXCL10 is unique because it contributes to reduce the eye viral titer and HSK severity, whereas IL-6, MIP-2, and MIP-1α aggravate corneal opacity and fail to affect the eye viral titer (6, 9, 41). In addition, our results of high IL-6 and MIP-2 levels detected in the infected cornea of Cxcl10−/− mice with massive neutrophils 2 weeks p.i. suggest that these two factors may associate with the secondary neutrophil influx, in addition to the primary neutrophil influx shown previously (6, 41). Neutrophils can secrete IL-6, MIP-2, and MIP-1α (41, 52). In the HSV-1-infected cornea, CXCL10 deficiency may decrease IL-6 expression by reducing the primary neutrophil influx during acute infection and may increase IL-6, MIP-2, and MIP-1α levels by enhancing the secondary neutrophil influx at the stage with evident HSK.
Previous reports comparing Cxcl10−/− and wild-type mice or using antibody to neutralize CXCL10 in mice failed to address the significance of endogenous CXCL10 in HSK because mice were infected with a neurovirulent HSV-1 strain (McKrae) and succumbed to death at 7 to 9 days p.i. before developing evident lesions (18, 31). Although these reports observe a transient increase of viral titers in the eyes of CXCL10-deficient mice, they did not identify how CXCL10 contributes to decrease virus. Our study provides the first evidence of how endogenous CXCL10 decreases HSK severity and eye viral titers in mice.
ACKNOWLEDGMENTS
We thank Robert Anderson for critical reading of the manuscript and helpful suggestions.
This work was supported by a grant from the National Science Council in Taiwan (NSC-100-2320-B-006-014).
Footnotes
Published ahead of print 29 May 2013
REFERENCES
- 1. Biswas PS, Rouse BT. 2005. Early events in HSV keratitis—setting the stage for a blinding disease. Microbes Infect. 7:799–810 [DOI] [PubMed] [Google Scholar]
- 2. Hendricks RL. 1997. An immunologist's view of herpes simplex keratitis: Thygeson lecture 1996, presented at the Ocular Microbiology and Immunology Group meeting, October 26, 1996. Cornea 16:503–506 [PubMed] [Google Scholar]
- 3. Babu JS, Thomas J, Kanangat S, Morrison LA, Knipe DM, Rouse BT. 1996. Viral replication is required for induction of ocular immunopathology by herpes simplex virus. J. Virol. 70:101–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tumpey TM, Chen SH, Oakes JE, Lausch RN. 1996. Neutrophil-mediated suppression of virus replication after herpes simplex virus type 1 infection of the murine cornea. J. Virol. 70:898–904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Thomas J, Gangappa S, Kanangat S, Rouse BT. 1997. On the essential involvement of neutrophils in the immunopathologic disease: herpetic stromal keratitis. J. Immunol. 158:1383–1391 [PubMed] [Google Scholar]
- 6. Yan XT, Tumpey TM, Kunkel SL, Oakes JE, Lausch RN. 1998. Role of MIP-2 in neutrophil migration and tissue injury in the herpes simplex virus-1-infected cornea. Invest. Ophthalmol. Vis. Sci. 39:1854–1862 [PubMed] [Google Scholar]
- 7. Frank GM, Buela KA, Maker DM, Harvey SA, Hendricks RL. 2012. Early responding dendritic cells direct the local NK response to control herpes simplex virus 1 infection within the cornea. J. Immunol. 188:1350–1359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Conrady CD, Zheng M, Mandal NA, van Rooijen N, Carr DJ. 2013. IFN-alpha-driven CCL2 production recruits inflammatory monocytes to infection site in mice. Mucosal Immunol. 6:45–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tumpey TM, Cheng H, Cook DN, Smithies O, Oakes JE, Lausch RN. 1998. Absence of macrophage inflammatory protein-1α prevents the development of blinding herpes stromal keratitis. J. Virol. 72:3705–3710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zheng M, Deshpande S, Lee S, Ferrara N, Rouse BT. 2001. Contribution of vascular endothelial growth factor in the neovascularization process during the pathogenesis of herpetic stromal keratitis. J. Virol. 75:9828–9835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Campanella GS, Lee EM, Sun J, Luster AD. 2003. CXCR3 and heparin binding sites of the chemokine IP-10 (CXCL10). J. Biol. Chem. 278:17066–17074 [DOI] [PubMed] [Google Scholar]
- 12. Liu MT, Chen BP, Oertel P, Buchmeier MJ, Armstrong D, Hamilton TA, Lane TE. 2000. The T cell chemoattractant IFN-inducible protein 10 is essential in host defense against viral-induced neurologic disease. J. Immunol. 165:2327–2330 [DOI] [PubMed] [Google Scholar]
- 13. Boztug K, Carson MJ, Pham-Mitchell N, Asensio VC, DeMartino J, Campbell IL. 2002. Leukocyte infiltration, but not neurodegeneration, in the CNS of transgenic mice with astrocyte production of the CXC chemokine ligand 10. J. Immunol. 169:1505–1515 [DOI] [PubMed] [Google Scholar]
- 14. Soejima K, Rollins BJ. 2001. A functional IFN-gamma-inducible protein-10/CXCL10-specific receptor expressed by epithelial and endothelial cells that is neither CXCR3 nor glycosaminoglycan. J. Immunol. 167:6576–6582 [DOI] [PubMed] [Google Scholar]
- 15. Luster AD, Ravetch JV. 1987. Biochemical characterization of a gamma interferon-inducible cytokine (IP-10). J. Exp. Med. 166:1084–1097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yuan J, Liu Z, Lim T, Zhang H, He J, Walker E, Shier C, Wang Y, Su Y, Sall A, McManus B, Yang D. 2009. CXCL10 inhibits viral replication through recruitment of natural killer cells in coxsackievirus B3-induced myocarditis. Circ. Res. 104:628–638 [DOI] [PubMed] [Google Scholar]
- 17. Chen JP, Lu HL, Lai SL, Campanella GS, Sung JM, Lu MY, Wu-Hsieh BA, Lin YL, Lane TE, Luster AD, Liao F. 2006. Dengue virus induces expression of CXC chemokine ligand 10/IFN-gamma-inducible protein 10, which competitively inhibits viral binding to cell surface heparan sulfate. J. Immunol. 177:3185–3192 [DOI] [PubMed] [Google Scholar]
- 18. Carr DJ, Chodosh J, Ash J, Lane TE. 2003. Effect of anti-CXCL10 monoclonal antibody on herpes simplex virus type 1 keratitis and retinal infection. J. Virol. 77:10037–10046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dufour JH, Dziejman M, Liu MT, Leung JH, Lane TE, Luster AD. 2002. IFN-gamma-inducible protein 10 (IP-10; CXCL10)-deficient mice reveal a role for IP-10 in effector T cell generation and trafficking. J. Immunol. 168:3195–3204 [DOI] [PubMed] [Google Scholar]
- 20. Gottlieb AB, Luster AD, Posnett DN, Carter DM. 1988. Detection of a gamma interferon-induced protein IP-10 in psoriatic plaques. J. Exp. Med. 168:941–948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Molesworth-Kenyon SJ, Oakes JE, Lausch RN. 2005. A novel role for neutrophils as a source of T cell-recruiting chemokines IP-10 and Mig during the DTH response to HSV-1 antigen. J. Leukoc. Biol. 77:552–559 [DOI] [PubMed] [Google Scholar]
- 22. Hsieh MF, Lai SL, Chen JP, Sung JM, Lin YL, Wu-Hsieh BA, Gerard C, Luster A, Liao F. 2006. Both CXCR3 and CXCL10/IFN-inducible protein 10 are required for resistance to primary infection by dengue virus. J. Immunol. 177:1855–1863 [DOI] [PubMed] [Google Scholar]
- 23. Christensen JE, Simonsen S, Fenger C, Sorensen MR, Moos T, Christensen JP, Finsen B, Thomsen AR. 2009. Fulminant lymphocytic choriomeningitis virus-induced inflammation of the CNS involves a cytokine-chemokine-cytokine-chemokine cascade. J. Immunol. 182:1079–1087 [DOI] [PubMed] [Google Scholar]
- 24. Harvey CE, Post JJ, Palladinetti P, Freeman AJ, French RA, Kumar RK, Marinos G, Lloyd AR. 2003. Expression of the chemokine IP-10 (CXCL10) by hepatocytes in chronic hepatitis C virus infection correlates with histological severity and lobular inflammation. J. Leukoc. Biol. 74:360–369 [DOI] [PubMed] [Google Scholar]
- 25. Kolb SA, Sporer B, Lahrtz F, Koedel U, Pfister HW, Fontana A. 1999. Identification of a T cell chemotactic factor in the cerebrospinal fluid of HIV-1-infected individuals as interferon-gamma inducible protein 10. J. Neuroimmunol. 93:172–181 [DOI] [PubMed] [Google Scholar]
- 26. Angiolillo AL, Sgadari C, Taub DD, Liao F, Farber JM, Maheshwari S, Kleinman HK, Reaman GH, Tosato G. 1995. Human interferon-inducible protein 10 is a potent inhibitor of angiogenesis in vivo. J. Exp. Med. 182:155–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bodnar RJ, Yates CC, Wells A. 2006. IP-10 blocks vascular endothelial growth factor-induced endothelial cell motility and tube formation via inhibition of calpain. Circ. Res. 98:617–625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Strieter RM, Kunkel SL, Arenberg DA, Burdick MD, Polverini PJ. 1995. Interferon gamma-inducible protein 10 (IP-10), a member of the C-X-C chemokine family, is an inhibitor of angiogenesis. Biochem. Biophys. Res. Commun. 210:51–57 [DOI] [PubMed] [Google Scholar]
- 29. Aronica SM, Raiber L, Hanzly M, Kisela C. 2009. Antitumor/antiestrogenic effect of the chemokine interferon inducible protein 10 (IP-10) involves suppression of VEGF expression in mammary tissue. J. Interferon Cytokine Res. 29:83–92 [DOI] [PubMed] [Google Scholar]
- 30. Araki-Sasaki K, Tanaka T, Ebisuno Y, Kanda H, Umemoto E, Hayashi K, Miyasaka M. 2006. Dynamic expression of chemokines and the infiltration of inflammatory cells in the HSV-infected cornea and its associated tissues. Ocul. Immunol. Inflamm. 14:257–266 [DOI] [PubMed] [Google Scholar]
- 31. Wuest T, Farber J, Luster A, Carr DJ. 2006. CD4+ T cell migration into the cornea is reduced in CXCL9 deficient but not CXCL10 deficient mice following herpes simplex virus type 1 infection. Cell. Immunol. 243:83–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Metcalf MF, McNeill JI, Kaufman HE. 1976. Experimental disciform edema and necrotizing keratitis in the rabbit. Invest. Ophthalmol. 15:979–985 [PubMed] [Google Scholar]
- 33. Hendricks RL, Tumpey TM. 1990. Contribution of virus and immune factors to herpes simplex virus type I-induced corneal pathology. Invest. Ophthalmol. Vis. Sci. 31:1929–1939 [PubMed] [Google Scholar]
- 34. Hendricks RL, Janowicz M, Tumpey TM. 1992. Critical role of corneal Langerhans cells in the CD4- but not CD8-mediated immunopathology in herpes simplex virus-1-infected mouse corneas. J. Immunol. 148:2522–2529 [PubMed] [Google Scholar]
- 35. Yao HW, Chen SH, Li C, Tung YY, Chen SH. 2012. Suppression of transcription factor early growth response 1 reduces herpes simplex virus 1-induced corneal disease in mice. J. Virol. 86:8559–8567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Luo Y, Dorf ME. 2001. Isolation of mouse neutrophils. Curr. Protoc. Immunol. Chapter 3:Unit 3.20. 10.1002/0471142735.im0320s22 [DOI] [PubMed] [Google Scholar]
- 37. Irvine AD, Corden LD, Swensson O, Swensson B, Moore JE, Frazer DG, Smith FJ, Knowlton RG, Christophers E, Rochels R, Uitto J, McLean WH. 1997. Mutations in cornea-specific keratin K3 or K12 genes cause Meesmann's corneal dystrophy. Nat. Genet. 16:184–187 [DOI] [PubMed] [Google Scholar]
- 38. Biswas PS, Banerjee K, Kim B, Smith J, Rouse BT. 2005. A novel flow cytometry based assay for quantification of corneal angiogenesis in the mouse model of herpetic stromal keratitis. Exp. Eye Res. 80:73–81 [DOI] [PubMed] [Google Scholar]
- 39. Lokensgard JR, Hu S, Sheng W, vanOijen M, Cox D, Cheeran MC, Peterson PK. 2001. Robust expression of TNF-α, IL-1β, RANTES, and IP-10 by human microglial cells during nonproductive infection with herpes simplex virus. J. Neurovirol. 7:208–219 [DOI] [PubMed] [Google Scholar]
- 40. Taub DD, Lloyd AR, Conlon K, Wang JM, Ortaldo JR, Harada A, Matsushima K, Kelvin DJ, Oppenheim JJ. 1993. Recombinant human interferon-inducible protein 10 is a chemoattractant for human monocytes and T lymphocytes and promotes T cell adhesion to endothelial cells. J. Exp. Med. 177:1809–1814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Fenton RR, Molesworth-Kenyon S, Oakes JE, Lausch RN. 2002. Linkage of IL-6 with neutrophil chemoattractant expression in virus-induced ocular inflammation. Invest. Ophthalmol. Vis. Sci. 43:737–743 [PubMed] [Google Scholar]
- 42. Fielding CA, McLoughlin RM, McLeod L, Colmont CS, Najdovska M, Grail D, Ernst M, Jones SA, Topley N, Jenkins BJ. 2008. IL-6 regulates neutrophil trafficking during acute inflammation via STAT3. J. Immunol. 181:2189–2195 [DOI] [PubMed] [Google Scholar]
- 43. Molesworth-Kenyon SJ, Popham N, Milam A, Oakes JE, Lausch RN. 2012. Resident corneal cells communicate with neutrophils leading to the production of IP-10 during the primary inflammatory response to HSV-1 infection. Int. J. Inflam. 2012:810359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gasperini S, Marchi M, Calzetti F, Laudanna C, Vicentini L, Olsen H, Murphy M, Liao F, Farber J, Cassatella MA. 1999. Gene expression and production of the monokine induced by IFN-γ (MIG), IFN-inducible T cell α chemoattractant (I-TAC), and IFN-γ-inducible protein-10 (IP-10) chemokines by human neutrophils. J. Immunol. 162:4928–4937 [PubMed] [Google Scholar]
- 45. Spurrell JC, Wiehler S, Zaheer RS, Sanders SP, Proud D. 2005. Human airway epithelial cells produce IP-10 (CXCL10) in vitro and in vivo upon rhinovirus infection. Am. J. Physiol. Lung Cell. Mol. Physiol. 289:L85–95 [DOI] [PubMed] [Google Scholar]
- 46. Conrady CD, Jones H, Zheng M, Carr DJ. 2011. A functional type I interferon pathway drives resistance to cornea herpes simplex virus type 1 infection by recruitment of leukocytes. J. Biomed. Res. 25:111–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wuest T, Austin BA, Uematsu S, Thapa M, Akira S, Carr DJ. 2006. Intact TRL 9 and type I interferon signaling pathways are required to augment HSV-1 induced corneal CXCL9 and CXCL10. J. Neuroimmunol. 179:46–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Klein RS, Lin E, Zhang B, Luster AD, Tollett J, Samuel MA, Engle M, Diamond MS. 2005. Neuronal CXCL10 directs CD8+ T-cell recruitment and control of West Nile virus encephalitis. J. Virol. 79:11457–11466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wuest TR, Carr DJ. 2008. Dysregulation of CXCR3 signaling due to CXCL10 deficiency impairs the antiviral response to herpes simplex virus 1 infection. J. Immunol. 181:7985–7993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Thapa M, Carr DJ. 2008. Herpes simplex virus type 2-induced mortality following genital infection is blocked by anti-tumor necrosis factor alpha antibody in CXCL10-deficient mice. J. Virol. 82:10295–10301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Michalec L, Choudhury BK, Postlethwait E, Wild JS, Alam R, Lett-Brown M, Sur S. 2002. CCL7 and CXCL10 orchestrate oxidative stress-induced neutrophilic lung inflammation. J. Immunol. 168:846–852 [DOI] [PubMed] [Google Scholar]
- 52. Lin YJ, Lai MD, Lei HY, Wing LY. 2006. Neutrophils and macrophages promote angiogenesis in the early stage of endometriosis in a mouse model. Endocrinology 147:1278–1286 [DOI] [PubMed] [Google Scholar]