

Abstract
Bats are increasingly recognized as reservoir species for a variety of zoonotic viruses that pose severe threats to human health. While many RNA viruses have been identified in bats, little is known about bat retroviruses. Endogenous retroviruses (ERVs) represent genomic fossils of past retroviral infections and, thus, can inform us on the diversity and history of retroviruses that have infected a species lineage. Here, we took advantage of the availability of a high-quality genome assembly for the little brown bat, Myotis lucifugus, to systematically identify and analyze ERVs in this species. We mined an initial set of 362 potentially complete proviruses from the three main classes of ERVs, which were further resolved into 13 major families and 86 subfamilies by phylogenetic analysis. Consensus or representative sequences for each of the 86 subfamilies were then merged to the Repbase collection of known ERV/long terminal repeat (LTR) elements to annotate the retroviral complement of the bat genome. The results show that nearly 5% of the genome assembly is occupied by ERV-derived sequences, a quantity comparable to findings for other eutherian mammals. About one-fourth of these sequences belong to subfamilies newly identified in this study. Using two independent methods, intraelement LTR divergence and analysis of orthologous loci in two other bat species, we found that the vast majority of the potentially complete proviruses identified in M. lucifugus were integrated in the last ∼25 million years. All three major ERV classes include recently integrated proviruses, suggesting that a wide diversity of retroviruses is still circulating in Myotis bats.
INTRODUCTION
With 1,116 known extant species in 202 genera, bats (order Chiroptera) constitute more than 20% of living mammal species (1). The family Vespertilionidae, which contains about one-third of all bat species and more than 100 species in the genus Myotis, ranks among the most species rich of all mammal families. Bats display many exceptional developmental and physiological characteristics, including the extreme elongation of digits to form webbed wings enabling powered flight, the capacity of several species to undergo extended hibernation, and extraordinary life spans for their size and metabolic rate (up to 34 years in the wild for Myotis), making them emerging models for research in limb development (2, 3) and aging (4). Bats have also gained attention in biomedical research because a number of bat species have been identified as zoonotic reservoirs for some of the most sinister viruses infecting humans, such as rabies, Ebola, Marburg, Hendra, Nipah, and SARS-like viruses (5–10). A recent study suggests that bats host almost twice as many zoonotic viruses per species as rodents, another important reservoir of zoonotic viruses (11).
The growing notoriety of bats as reservoirs for zoonotic viruses has generated considerable interest in the scientific community and prompted a broad effort to characterize the viruses naturally infecting bats, including recent metagenomic surveys of the “virome” of several bat species (12–15). Together, these studies have led to the detection of a large number of viruses affiliated with diverse mammalian families of (mostly) RNA viruses, as well as insect and plant viruses (12–15).
Retroviruses are unique among vertebrate viruses in that they possess an obligatory chromosomal integration stage in their replication cycle. Integration may occasionally occur in the germ line, which can result in vertical inheritance and fixation in the host population (16–18). Such endogenous retroviruses (ERVs) have been identified in nearly all vertebrate genomes examined (16–18), and they often occupy a substantial fraction of mammalian genomes, accounting for about 8% of human (19) and 10% of mouse nuclear genome sequences (20). The infiltration and amplification of ERVs in vertebrate genomes are pervasive and represent a source of genetic variation thought to have had a strong impact on the biology and evolution of host species (18, 21, 22). Furthermore, because ERV integration events can often be dated, they provide a precious fossil record of past retroviral infections that have afflicted the host species or its ancestors (22–25).
Despite the prevalence of ERVs in mammalian genomes and their biological relevance, relatively few bat retroviral sequences have been reported in the literature (26–29). Initially, these were only short ERV fragments isolated by PCR with degenerate primers designed to amplify conserved pol domains of retroviruses (26, 27). More recently, traces of foamy viruses (spumaviruses) were identified in bat viromes (15), and Cui et al. reported an apparently complete sequence for an exogenous gammaretrovirus (Rhinolophus ferrumequinum retrovirus [RfRV]) in the greater horseshoe bat, as well as defective gammaretroviral sequences in other bat species (30). Lastly, the same group identified ∼50 copies of endogenous gammaretroviruses in the draft genome sequences of M. lucifugus and of the megabat Pteropus vampyrus and were able to recover a total of 16 proviruses with both of the long terminal repeats (LTRs) but apparently defective coding capacity (28). These results suggested that bats are host to a large diversity of gammaretroviruses, including endogenous elements (28, 30). Early this year, endogenous betaretroviruses were also identified and reported in megabats and microbats (29). However, the overall diversity and evolution of ERVs in bat genomes remain largely unexplored.
In this study, we take advantage of the recent public release of a high-quality, 7× genome assembly (http://www.genome.gov/25521745) of the little brown bat Myotis lucifugus, one of the most common species in North America, to perform a comprehensive mining and analysis of ERVs in a bat species. We found that the amount and diversity of ERVs in M. lucifugus rival those observed in other mammalian genomes and include both ancient and recent integration events. Our study suggests that the vespertilionid bats have been subject to considerable levels of retroviral infections over the last ∼25 million years (My) and that diverse retroviruses are likely still circulating among natural populations of M. lucifugus.
MATERIALS AND METHODS
ERV mining.
The M. lucifugus 7× genome assembly Myoluc 2.0 was downloaded from NCBI (NCBI accession number AAPE02000000) and used as the input for two ERV identification pipelines (Fig. 1). First, we identified pairs of putative LTRs separated by 1 to 15 kb and flanked by target site duplications (TSD) by using LTRharvest (31). The LTR nucleotide similarity threshold used in LTRharvest was >80%, with other parameters set to their defaults. Internal retroviral sequence features of ERV candidates, including protein domains, primer-binding sites (PBS), and polypurine tracts (PPT), were predicted using LTRdigest (32). The M. lucifugus tRNA library used for PBS annotation was generated for the Myoluc 2.0 genome assembly using tRNAscan-SE (33), and 32 retroviral-related protein domain profiles (see File S1 in the supplemental material) used for putative domain annotation were downloaded from the Pfam database (34). To remove false positives and arrive at a list of high-confidence full-length ERVs, we applied two additional filters. First, we performed a tblastn search against all repeat libraries in Repbase (version 17.11) (45) to remove candidates whose reverse transcriptase (RT) domains were most closely related to those of non-LTR retrotransposons. Second, we required each candidate to contain at least 3 of the 5 canonical retroviral protein domains (Gag, PR, RT, IN, and RH) identified by LTRharvest. We also observed that some of the predicted LTR boundaries were truncated, so we manually refined the LTR termini for each of the filtered full-length ERVs using genomic alignments with blastn. The second ERV identification pipeline employed MGEScan-LTR (36) with the default parameters. The outputs from LTRdigest and MGEScan-LTR were also submitted to CENSOR (37) to systematically identify any other known repetitive elements inserted within the candidate ERVs. This approach was also used to eliminate several false positives where a pair of short interspersed elements (SINEs) flanking putative retroviral domains was misidentified as LTRs.
Fig 1.
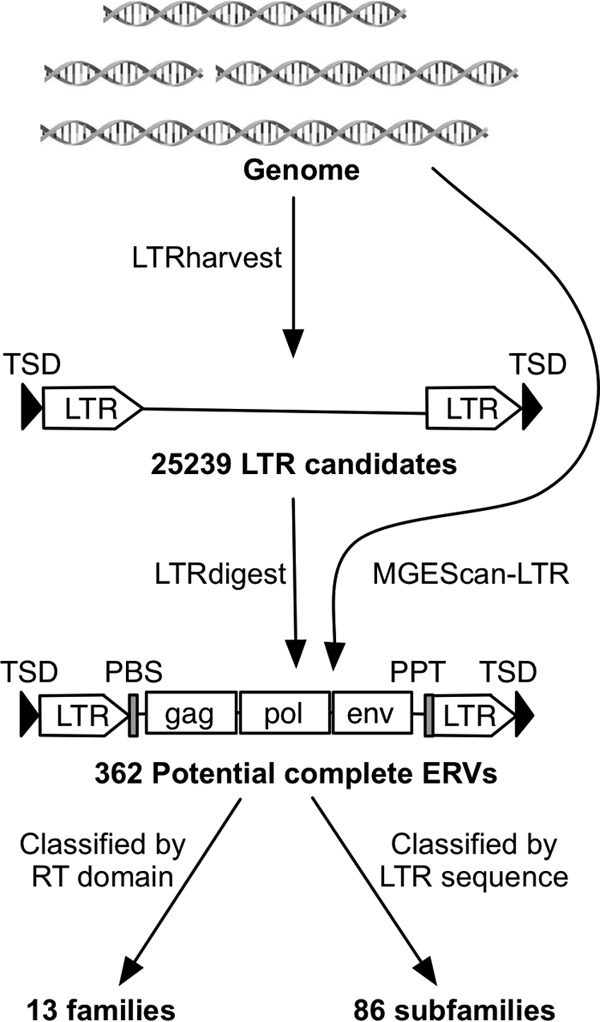
Potential complete ERV identification pipeline. We identified 25,239 LTR candidates with pairs of putative LTRs and TSD using LTRharvest, and all of them were annotated using LTRdigest. LTR candidates with canonical retroviral features were extracted as potential complete ERVs. We also used the independent pipeline MGEScan-LTR to identify potential complete ERVs. By combining the two independent pipelines, we identified 362 potential complete ERVs. They were further classified into 13 families based on RT domain phylogeny and into 86 subfamilies based on LTR sequence similarity.
ERV classification and phylogenetic analysis.
We used MUSCLE (38), complemented by manual refinements, to build an amino acid multiple alignment of the RT domain from 177 full-length bat ERVs and 20 known exogenous and endogenous retroviruses (see File S2 in the supplemental material). A neighbor-joining phylogeny was built from the RT domain alignment using MEGA5 (39) with 1,000 bootstrap replicates, applying the pairwise deletion option and using JTT as the amino acid substitution model (40). A Bayesian phylogenetic reconstruction was built using MrBayes 3.1.2 (41) with two runs of 5 million generations, employing a mixed-rate model. The tree was sampled every 100 generations. Posterior probabilities supporting family clustering are summarized in Table 1. For subfamily clustering of LTR sequences, we used Vmatch with parameters set according to the LTRdigest protocol (32).
Table 1.
Summary of 13 MLERV families
| Family | Class | Neighbor-joining bootstrap | Bayesian posterior probability | Age (Mya) | LTR length (bp) | Internal length (bp) | Copy no. |
|---|---|---|---|---|---|---|---|
| MLERV1 | I | 99.5 | 1 | 4.2 | 442 | 6,554 | 33 |
| MLERV2 | I | 100 | 1 | 6.9 | 418 | 7,531 | 5 |
| MLERV3 | I | 86.8 | 0.99 | 6.8 | 433 | 5,961 | 71 |
| MLERV4 | I | 100 | 0.99 | 15.8 | 339 | 7,098 | 48 |
| MLERV5 | I | 0.75 | 1 | 15.0 | 358 | 7,380 | 16 |
| MLERV6 | I | 100 | 1 | 13.0 | 425 | 9,007 | 3 |
| MLERV7 | III | 100 | 1 | 3.0 | 868 | 10,596 | 2 |
| MLERV8 | II | 100 | 0.99 | 4.8 | 334 | 5,042 | 23 |
| MLERV9 | II | 98.3 | 0.99 | 13.1 | 393 | 5,216 | 19 |
| MLERV10 | II | 76 | 0.99 | 7.6 | 444 | 4,344 | 25 |
| MLERV11 | II | 100 | 0.99 | 10.5 | 546 | 5,515 | 48 |
| MLERV12 | II | 0.54 | 0.93 | 8.7 | 432 | 6,529 | 20 |
| MLERV13 | II | 100 | 0.87 | 9.6 | 421 | 7,170 | 41 |
My, million years.
Dating ERV insertions by using LTR divergence.
LTR pairs from 362 full-length ERVs were aligned using the Smith-Waterman algorithm (42). CpG sites in all LTR sequences were removed, and the pairwise evolutionary distance K of LTR pairs was corrected using the Jukes-Cantor model (43). A previously estimated substitution rate (r) of 2.692 × 109 for the M. lucifugus lineage (44) was used for dating each insertion. The date of ERV integration was calculated as K/2r.
RepeatMasker analysis.
To generate a systematic annotation of ERVs in the M. lucifugus genome assembly, we first collected the LTR sequences and internal regions of potentially complete proviruses identified ab initio. We first separate the LTR sequences and internal regions from complete elements and extracted representative or consensus sequences from each of the 86 subfamilies. These 172 sequences formed our M. lucifugus ERV (MLERV) library. To remove any repetitive elements nested within the MLERV library, we screened this library using RepeatMasker (version 3.3.0) (3.0.1996-2010 [http://www.repeatmasker.org]) with a library of non-ERV repetitive elements from Repbase (version 17.11) (45). We then combined our 172 MLERV entries with a Repbase ERV library (version 17.11) (45) to build a custom ERV library. This library was used to subsequently run RepeatMasker on the M. lucifugus genome assembly, using the sensitive Crossmatch alignment program with the default parameters.
Estimation of full-length ERVs and solitary LTRs.
A Perl script was used to parse the RepeatMasker output to systematically identify LTR pairs flanking internal ERV regions masked on the same DNA strand. A potential full-length ERV was considered when a pair of similar LTR fragments were separated by less than 20 kb and the alignment of the pair of LTR fragments spanned at least 100 bp. We also required that at least 500 bp of internal region were masked as internal ERV sequences.
To estimate solitary LTR numbers, we parsed the RepeatMasker output to map solo LTRs. In many cases, we found LTRs to be fragmented. To better estimate solitary LTR copy numbers from fragmented pieces in the genome, we aligned fragmented LTR sequence to their consensus sequence and calculated the occurrence of each base in the alignment. Theoretically, the abundance of each base should be the same. In reality, it fluctuates because of genome rearrangements. Therefore, we used the median occurrence of an LTR as a proxy for its genomic copy number. Paired LTRs from full-length elements are included in the genomic copy number as well, so we subtracted twice the full-length ERV copy number from the genomic copy number and used it as the solitary LTR number.
Identification of orthologous MLERV insertion sites in other bat genomes.
A Perl script was designed to find orthologous loci of MLERVs in other bat genome assemblies. The first and last 100 bp of each MLERV plus 300 bp of their flanking sequences were extracted from the M. lucifugus genome assembly and used as queries to search other genome assemblies using blastn. Genome sequences matching only the flanking region in the queries were labeled “empty sites,” while sequences matching both the flanking and repeat regions were labeled “occupied.” A given MLERV was considered present or absent at an orthologous locus when at least one end could be unambiguously labeled an occupied or empty site. All of the orthologous loci were validated by manual inspection.
RESULTS
De novo detection of ERVs in the M. lucifugus genome.
We used two different ERV mining pipelines (Fig. 1). The first strategy relies on the combination of LTRharvest (31) and LTRdigest (32). We used LTRharvest to define ERV candidates by scanning the genome sequence for putative LTR pairs (100 to 1,000 bp) separated by 1,000 to 15,000 bp and flanked by target site duplications (TSD). LTRdigest then screens and annotates each internal sequence of the ERV candidates for putative protein-coding domains (e.g., reverse transcriptase, integrase, etc.), primer-binding sites (PBS), and polypurine tracts (PPT), characteristic of complete proviruses. A filter is then applied to retain complete or nearly complete ERVs based on the presence of a subset of these features (see Materials and Methods). To complement this approach, we applied a second computational tool, MGEScan-LTR, designed to identify full-length LTR retrotransposons, including ERVs (36). LTRdigest and MGEScan-LTR both use HMMER (46) to identify protein domains; however, LTRdigest outputs all retroviral protein domains with an E value of <1−6 for further identification, while MGEScan-LTR retains candidates with a set of protein domains with a combined E value of <1−10 or a longest open reading frame (ORF) length of >700 bp.
With LTRharvest (31), we identified 25,239 ERV candidates with a pair of predicted LTRs in the M. lucifugus genome. This large output was filtered down to 217 ERV candidates by LTRdigest (32). Applying MGEScan-LTR (36) revealed 245 putative full-length ERVs (Fig. 1). While the total numbers of ERVs identified by the two pipelines were similar, only a small subset of elements were identified by both programs, as determined based on their location in the genome assembly. After removing redundant elements and false positives (see Materials and Methods), we arrived at a total of 362 distinct and potentially complete proviruses identified in the M. lucifugus genome assembly (hereinafter referred to as potentially complete ERVs) (Fig. 1). The LTR lengths of these 362 ERVs vary from 154 to 840 bp, and their total internal lengths range from 2,291 to 12,503 bp after removing secondary transposon insertions (see Table S1 in the supplemental material). Of the 362 ERVs, 252 (∼70%) have perfect TSD, and 27 have identifiable TSD with 1 or 2 mutations.
Phylogenetic analysis and classification of ERVs from M. lucifugus.
Of the 362 ERVs identified as described above, 177 had a reverse transcriptase (RT) domain conserved enough to be aligned confidently for phylogenetic analysis. We used this conserved RT domain (47) to build a multiple alignment and compute phylogenetic trees using the neighbor-joining method implemented in MEGA (39) and the Bayesian method implemented in MrBayes (48). Both methods produced trees with nearly identical topologies, allowing us to classify bat ERVs into 13 major families, denoted MLERV1 to -13 (Fig. 2 and Table 1). We defined all families as monophyletic groups of closely related branches with bootstrap support of at least 75% in neighbor joining and posterior probability of at least 0.75 in Bayesian trees (except for the MLERV12 family, which was supported by 54% bootstrap but a posterior probability of 0.93). Representatives of the known retroviral classes were included in our phylogenetic analysis in order to assign the MLERV families to one of the three major ERV classes. We were able to identify 6 MLERV families (MLERV1 to -6) comprised of 145 elements as class I ERVs (gammaretroviruses), 6 MLERV families (MLERV7 to -12) accounting for 157 elements as class II ERVs (betaretroviruses), and one family (MLERV13) represented by two elements as class III ERVs (spumaretroviruses).
Fig 2.

Phylogeny of 13 MLERV families and reference retroviral sequences. Class I, class II, and class III ERVs are illustrated with blue, yellow, and green, respectively, and reference retroviral sequences are shown in red. MLERV families with neighbor joining bootstrap values higher than 95 are labeled with an asterisk at the root, and those with a bootstrap value of between 75 and 95 are labeled with a dot at the root. KoRV, koala retrovirus; GaLV, gibbon ape leukemia virus; MDEV, Mus dunni endogenous virus; PERV, porcine endogenous retrovirus; MuLV, murine leukemia virus; FELV, feline leukemia virus; BaEV, baboon endogenous virus; HERV, human endogenous retrovirus; ZFERV, zebrafish endogenous retrovirus; WDSV, walleye dermal sarcoma virus; SnRV, snakehead fish retrovirus; FeFV, feline foamy virus; HFV, human foamy virus; BLV, bovine leukemia virus; RSV, Rous sarcoma virus; GH-G18, Golden hamster intracisternal A-particle H18; RERV, rabbit endogenous retrovirus; HML, human MMTV-like; SRV, simian type D retrovirus; MMTV, mouse mammary tumor virus.
To further classify MLERVs into subfamilies, we compared their LTR sequences, which are among the most rapidly evolving sequences in retroviruses (49, 50). Based on a 75% interelement LTR nucleotide similarity cutoff, the program Vmatch (www.vmatch.de) clustered the 362 potential complete ERVs into 86 subfamilies (including 40 singletons) (see Table S2 in the supplemental material). Although families and subfamilies were defined independently, we found that the two classification levels were congruent in that ERVs falling within a given subfamily also belonged to the same family. One advantage of the classification based on LTR sequences is that we could generally assign elements with highly diverged, partial or missing RT domains to one of the families defined upon RT phylogeny. By combining these different classification methods, we were able to assign 354 ERVs to one of the 13 families defined in Table 1, leaving only 8 ERVs presently unclassified.
Census of the ERV population in the M. lucifugus genome assembly.
To comprehensively assess the abundance of ERV-derived sequences in M. lucifugus, we ran RepeatMasker to annotate the 7× genome assembly using a custom library combining consensus or representative sequences for each of the 86 MLERV subfamilies defined above and all nonredundant ERV sequences deposited in Repbase (45) (see Materials and Methods). The total length of ERV-related sequences annotated by RepeatMasker amounted to 89 Mb, which represents 4.9% of the 1.8-Gb genome assembly after removing gaps.
To further delineate the ERV composition of the bat genome, we implemented custom scripts (available upon request) to parse the RepeatMasker output and estimate the numbers of full-length ERVs (as defined by the presence of a pair of LTRs flanking a sequence masked as an internal ERV region) and solitary LTRs for each major class of ERVs (see Materials and Methods). Solitary LTRs typically arise as a result of intraelement recombination between the 5′ and 3′ LTRs of a full-length provirus.
For class I ERVs, the approach identified 464 full-length proviruses and 35,404 solitary LTRs in the genome assembly. The sizes of full-length class I ERVs in M. lucifugus typically range from 6 to 9 kb (Table 1). MLERV3 is the most diverse family in this class, including 15 distinct subfamilies (Fig. 2 and Table 1). Together, the total genomic length occupied by class I elements is estimated at 31.5 Mb (1.66% of the genome assembly).
Class II ERVs were represented by 638 full-length proviruses and 10,858 solitary LTRs. The lengths of full-length class II ERVs range from 4.5 to 9.5 kb. The most abundant class II family is MLERV11 (Fig. 2 and Table 1). Notably, subfamily MLERV11_2 includes 123 potentially full-length copies, more than any other MLERV subfamily. In total, class II ERVs occupy 9.1 Mb (0.48%) of the genome assembly.
Covering 49.2 Mb (or 2.6%) of DNA, class III ERVs account for the largest amount of ERV-derived sequences in the genome assembly. This result was somewhat surprising in light of our initial ab initio mining of ERVs, which had retrieved a single class III family (MLERV7) represented by only 2 complete canonical copies (Fig. 2 and Table 1). Nonetheless, our parsing of the RepeatMasker output identified 571 full-length and 81,967 solitary LTRs affiliated with class III ERVs. Manual inspection of a subset of these sequences revealed that they represent relatively ancient and often nonautonomous class III elements previously identified in other mammalian genomes, such as mammalian apparent LTR retrotransposons (MaLRs) (51). Thus, the discrepancy between the results of the ab initio search and the RepeatMasker annotation can be explained by the fact that most class III ERVs are represented by highly decayed copies and nonautonomous elements, as well as abundant solitary LTRs derived from ancient families (see below). By design, such incomplete or highly diverged copies cannot be identified by the two pipelines used for our ab initio mining (31, 32, 36). The difficulty in identifying class III ERVs using ab initio approaches has been reported for other mammals (52–58).
Overall, the ERV coverage of the bat genome (89 Mb, 4.9%) is less than that in the human (261 Mb, 9.0%) and mouse (285 Mb, 10.9%) genomes but similar to the ERV coverage of the dog genome (115 Mb, 4.8%) (RepeatMasker) (Fig. 3a). However, the bat genome assembly is less complete and of poorer quality than the mouse and human genome assemblies. Because ERVs and other repeats tend to be overrepresented in nonassembled regions of sequenced genomes (gaps), our estimate of ERV abundance in M. lucifugus should be viewed as a conservative estimate.
Fig 3.

Comparison of ERV abundance and dynamics in different genomes. Different ERV classes and DNA transposons are labeled with different colors. (a) Comparison of percentages of genomes derived from different classes of ERVs in little brown bat and other mammals. (b) ERV and DNA transposon dynamics in little brown bat, human, and mouse genomes. Distance to consensus was corrected using the Jukes-Cantor model. Older elements are more distant from the consensus. The abundance is illustrated also, using the percentage of the genome.
Comparative demography of ERVs in bat, human, and mouse.
The RepeatMasker output provides a measure of sequence divergence for each DNA segment annotated to its closest consensus sequence in our ERV library, enabling us to examine the tempo and evolutionary dynamics of ERV invasions in M. lucifugus in comparison to those in human and mouse (Fig. 3b). Overall, the demographic profile of M. lucifugus ERVs is more similar to that of human ERVs: class III ERVs are the most abundant and the most diverged (ancient), class II ERVs are the least abundant but the most recent, while class I ERVs occupy an intermediate position both in abundance and divergence. The similar histories of ERV accumulation in the bat and human (and to some extent mouse) lineages are to be contrasted with the dramatic differences in DNA transposon activity, which is strikingly elevated in the M. lucifugus lineage (Fig. 3b), consistent with previous reports (59, 60).
Recent ERV infiltrations in the M. lucifugus lineage.
The 5′ and 3′ LTR sequences from a given provirus are typically identical upon chromosomal integration and are expected to diverge subsequently by accumulating substitutions at the neutral rate of the host species. Thus, if the host neutral substitution rate is known, the age of an individual ERV integration event can be estimated by measuring the pairwise distance between LTR sequences (61). We applied this method and a rate of neutral substitution previously estimated for the M. lucifugus lineage (44) to date the integration of the 362 potential complete ERVs predicted ab initio, as we expected these to represent some of the youngest elements in the genome (Fig. 4a).
Fig 4.
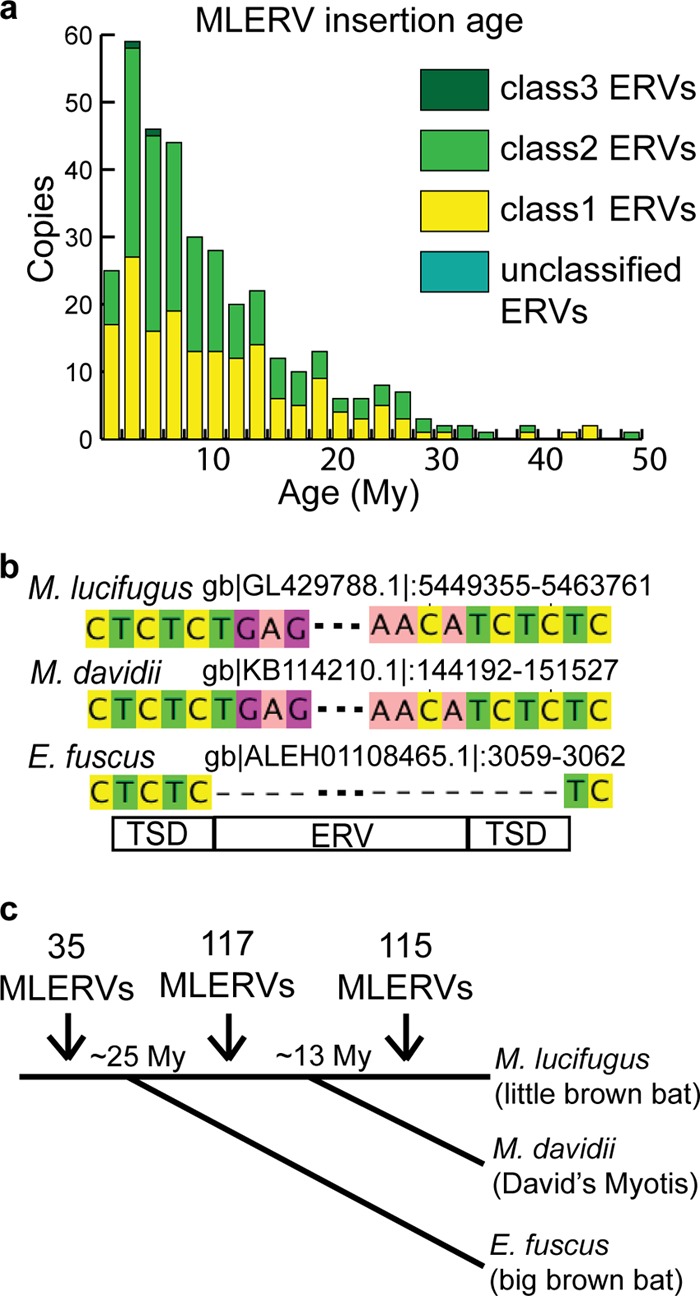
Recent ERV invasion in M. lucifugus genome. (a) Most of the 362 complete ERVs invaded the M. lucifugus genome recently. Copy numbers of different ERV classes are shown in different colors; age was estimated by LTR pair divergence. (b) An example of an MLERV integration event (TG…CA) specific to the Myotis lineage. The MLERV is present in the M. davidii genome with target site duplication (TCTC), but a precise empty site is found in E. fuscus. (c) Orthologous loci of MLERVs in E. fuscus and M. davidii indicate that most of the ERVs invaded after divergence from E. fuscus, and ERVs were active before and after speciation of M. lucifugus from M. davidii. The speciation times between M. lucifugus and M. davidii and between M. lucifugus and E. fuscus were around 13 My and around 25 My, respectively. Numbers of ERV insertions are labeled between time points.
The results show that, indeed, the vast majority of the ERVs surveyed integrated relatively recently, with 232 of 362 (64%) proviral integrations estimated to be less than 10 My old according to this analysis. Twenty-three of these elements have strictly identical LTR pairs, and another 58 elements have LTRs that are >99% identical, indicating that all these ERVs have inserted very recently, probably within the past 2 My (Fig. 4a). The most recently active subfamily according to this analysis is MLERV3_15 of the ERV I class. We estimated that each of the 12 copies of MLERV3_15 have inserted within the last 2.5 My, including 4 copies with identical LTRs (see Table S2 in the supplemental material). These data suggest that the M. lucifugus lineage has been subject to many recent ERV infiltrations.
We measured the age of MLERV integration events alternatively by assessing their presence or absence at orthologous genomic positions in closely related bat species. Recently, draft genome assemblies of two additional vespertilionid bats, Eptesicus fuscus and Myotis davidii, were released (NCBI accession number ALEH01000000 and ALWT01000000, respectively) (62). E. fuscus has been estimated to share an ancestor with Myotis bats at ∼25 My ago (63, 64), and the time of divergence of M. lucifugus and M. davidii is predicted to be around 10 to 15 My (64–66). We used BLAST with queries representing the termini of each of the individual full-length MLERV copies plus 300 bp of flanking genomic sequences to identify orthologous regions in the E. fuscus and M. davidii genomes (Fig. 4b shows an example). After combining information from these two other bat genomes and manually inspecting each locus, about 35% of orthologous MLERV loci could be unambiguously identified in E. fuscus and about 70% in M. davidii (see Tables S1 and S3 in the supplemental material). Among these, we found evidence for 137 MLERVs present at orthologous positions in M. davidii, while 115 MLERVs were missing at the orthologous site in this species (Fig. 4c) (52 of these loci are precisely missing the MLERV and have only one copy of the TSD). In the E. fuscus draft genome assembly, we identified 35 MLERVs present at orthologous loci, while 94 MLERVs were missing at orthologous positions (see Tables S1 and S3 in the supplemental material). Together, these data indicate that the vast majority of potential complete ERVs detected in M. lucifugus integrated after speciation of E. fuscus and Myotis, and many ERVs continued to accumulate during Myotis evolution and integrated after the divergence of M. lucifugus and M. davidii (Fig. 4c and Table 2).
Table 2.
Ortholog status of identifiable complete ERVs in M. davidii and E. fuscus
| Ortholog status in other genomes | Copy no. | Avg age (My)a | Oldest ERV infiltration (My) | Latest ERV infiltration (My) |
|---|---|---|---|---|
| M. davidii | ||||
| Empty site | 115 | 4.2 | 24.3 | 0.0 |
| Occupied site | 137 | 15.9 | 54.8 | 1.4 |
| E. fuscus | ||||
| Empty site | 94 | 7.1 | 27.5 | 0.0 |
| Occupied site | 35 | 23.1 | 52.3 | 5.4 |
Age is estimated using LTR pair comparison.
Our age estimates based on these cross-species genomic comparisons were largely concordant with the age of ERV integrations calculated by LTR divergence. Indeed, MLERVs with orthologous empty sites in E. fuscus were on average much younger (7.2 My) than those with occupied sites (23.1 My). The oldest MLERV insertion with an empty site in E. fuscus was predicted to be 27 My old according to LTR divergence, which is roughly consistent with the divergence time of ∼25 My estimated between these two bat species (63). However, we note that the age of the youngest MLERV insertions with occupied orthologous sites in E. fuscus was significantly underestimated by LTR divergence (5.4 My). Similar trends were found in M. davidii (summarized in Table 2). This discrepancy between the results of the two dating methods could be caused by gene conversion homogenizing LTR sequences, leading to underestimation of the timing of integration, as previously reported in other genomes (67). These data emphasize the need to apply multiple methods to confidently date ERV integration events.
DISCUSSION
Census of ERVs in the M. lucifugus genome.
By combining two different ab initio mining strategies, we identified 362 potentially complete proviruses in the M. lucifugus genome. Nearly all of these elements fall within 86 subfamilies that enabled us to identify a multitude of related sequence fragments using RepeatMasker, including nearly 1,700 full-length ERVs and 130,000 solitary LTRs in the M. lucifugus genome assembly. When used in conjunction with mammalian ERV sequences catalogued in Repbase, our collection allowed us to estimate that ERVs occupy 4.9% of the bat genome, a substantial fraction comparable to that observed in other eutherian genomes (Fig. 3a) (19, 20, 69).
Our data complement previous findings by Cui et al. (28), who identified 3 major groups (A, B, and C) of gammaretroviruses in the M. lucifugus genome by BLAST searches. Our approach identified these three groups as the MLERV2, MLERV1, and MLERV3 families, respectively. We discovered three additional gammaretrovirus families (MLERV4 to -6) (Fig. 2 and Table 1). The total length of sequences derived from the MLERV4 family alone is 9.2 Mb, or ∼0.5% of the genome assembly. At the time of this study, there were 5 entries of internal (coding) ERV regions and 132 entries of LTR sequences for M. lucifugus in Repbase, a comprehensive database for transposable element sequences, including ERVs (45). We identified both LTR and internal sequences for 13 families and 86 subfamilies of ERVs, most of which were not reported in Repbase (see Table S2 in the supplemental material). Furthermore, through manual examination, we found that several of the M. lucifugus LTR sequences deposited in Repbase were actually truncated at their 5′ end (data not shown). Thus, our manually curated collection of 86 reference ERV sequences will be useful to replace or complement existing Repbase entries. Overall, the coverage of MLERV families newly identified in this study amounts to 23 Mb of the genome assembly, thereby substantially improving the census of ERVs in this bat species.
Comparison of ERV diversity in M. lucifugus with that of other mammals.
With regard to ERV diversity within M. lucifugus, we found that class I (gammaretroviruses) and class II (betaretroviruses) ERVs are similarly diverse (each composed of 6 major families), but the total amount of genomic DNA derived from class II ERVs (9.1 Mb) is considerably smaller than that derived from class I ERVs (31.5 Mb). Class III (spumaviruses) ERVs are the most abundant (49.2 Mb) in the genome, but they are generally older and more degraded than class I and II elements, which hampered the identification of full-length class III ERVs using ab initio methods, as reported for other mammalian genomes (52–58, 70). Using RepeatMasker, we identified 571 apparently full-length class III ERVs, but we observed that a large fraction of these elements are nonautonomous MaLR-like elements that are comparable to those abundantly populating the human and mouse genomes (19, 20). Nonetheless, we note that the only class III family we detected ab initio in M. lucifugus (MLERV7) is a relatively young family, with an age estimated at ∼4 My (Table 1). Thus, all three major ERV classes are represented by relatively recent insertions in the M. lucifugus genome.
Overall, the demographic profile of the three ERV classes in M. lucifugus was more similar to that seen in the human genome (Fig. 3b). While the bulk of class III ERVs likely predate the radiation of eutherian mammals and, thus, have essentially been inherited through vertical decent, the amplification of class I and II ERVs is much more recent and largely lineage specific (Fig. 3). We conclude that there was a parallel invasion and expansion of these two classes of ERV in the human and bat lineages.
An important motivation for our analysis of ERVs in M. lucifugus relates to recent findings of massive lineage-specific DNA transposon activity in M. lucifugus (Fig. 3b) (59, 60). There is strong evidence that several of these DNA transposons have been acquired horizontally (71, 72), possibly reflecting a peculiar sensitivity of the germ line of this group of bats to lateral infiltration of mobile elements. Because retroviral endogenization also represents a form of horizontal transfer to the germ line, it was of interest to see whether these bats also display a greater vulnerability to ERV invasions. While we found clear evidence of recent ERV colonization in the genome of M. lucifugus, neither the diversity nor the sheer amount of ERV sequences depart dramatically from the diversity or amount observed in other mammalian genomes (Fig. 3). Thus, while the mobile element landscape of M. lucifugus is exceptional in terms of recent DNA transposon invasions, M. lucifugus does not appear to be an outlier among eutherian mammals in terms of its ERV population. We conclude that the apparent vulnerability of vespertilionid bats to horizontal transfer of DNA transposons is not generalizable to all types of mobile elements.
Superspreader hypothesis.
Recently, Magiorkinis et al. (73) proposed the “superspreader” hypothesis, which postulates that ERVs lacking coding capacity for an envelope (env-less ERVs) amplify more efficiently within the genome than those encoding an intact envelope. The hypothesis was supported by a detailed phylogenetic analysis of intracisternal A-type particles (IAPs) from several mammalian genomes (73) and for several primate ERV families (74). In M. lucifugus, we classified MLERVs to 13 families and 86 subfamilies. At the family level, we found no clear relationship between the presence of an envelope domain and family copy number; however, at the subfamily level, we observed that the most successful subfamilies are predominantly composed of env-less elements (see Table S2 in the supplemental material). For example, the two largest subfamilies in our data set (MLERV4_6 and MLERV11_2) are entirely composed of copies lacking an identifiable envelope domain. Thus, the pattern of MLERV subfamily expansion brings further support to the superspreader hypothesis.
Bats as possible zoonotic reservoirs of retroviruses.
We found several clear examples of very recent ERV families in M. lucifugus. A good illustration is MLERV3_15, a subfamily of class I elements. Four of the 12 copies identified in the genome have identical LTR pairs, while the other eight have LTR pairs that are >99% identical, indicative of nearly contemporary integration events (see Table S2 in the supplemental material). All 12 copies are also absent at orthologous positions in M. davidii (see Table S3). Nonetheless, none of the MLERV3_15 copies identified appear to retain intact coding capacity, suggesting that they are currently incapable of replicating autonomously.
However, in a recently active class I ERV subfamily, MLERV2_2 (0 to 3 My old), we identified one copy (entry 74) with apparently intact gag, pro, pol, and env coding regions, suggesting that this copy might be replication competent. In addition, another apparently intact and functional class II ERV was recently identified in M. lucifugus (29). Together, these results suggest that both class I and II ERVs in M. lucifugus are potentially capable of autonomous replication and of producing infectious viral particles.
Among the most recently integrated (<10 My ago) potentially complete proviruses supported by both LTR-LTR divergence and cross-species analysis, we were able to detect members of all three main retroviral classes (see Table S2 in the supplemental material). Our finding of recently integrated spumaretroviruses and gammaretroviruses is consistent with the identification of exogenous members of these retroviral taxa in several bat species, including microbats (15, 30). We also identified proviral copies of betaretroviruses (e.g., MLERV12_4) that have retained identical LTRs flanked by perfect TSD and are absent in M. davidii (see Tables S2 and S3), which suggests that M. lucifugus was also infected by exogenous betaretroviruses in the recent past. Together, these data indicate that a wide diversity of retroviruses have recently infected these bats and are likely still circulating in natural populations of M. lucifugus. Given the apparent propensity of bats to act as reservoir species for zoonotic viruses that are highly pathogenic to humans, these observations raise concerns that these animals may also be capable of transmitting zoonotic retroviruses to humans.
Supplementary Material
ACKNOWLEDGMENTS
We thank Aurelie Kapusta, Ellen Pritham, and Claudia Marquez for helpful discussions and Ray Malfavon-Borja for critical reading of the manuscript.
X.Z. and C.F. were supported by grant R01-GM077582 from the National Institutes of Health.
Footnotes
Published ahead of print 29 May 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.00892-13.
REFERENCES
- 1. Simmons NB. 2005. Order Chiroptera, p 312–529 Wilson DE, Reeder DM. (ed), Mammal species of the world: a taxonomic and geographic reference, 3rd ed, vol 1 Johns Hopkins University Press, Baltimore, MD [Google Scholar]
- 2. Hockman D, Mason MK, Jacobs DS, Illing N. 2009. The role of early development in mammalian limb diversification: a descriptive comparison of early limb development between the Natal long-fingered bat (Miniopterus natalensis) and the mouse (Mus musculus). Dev. Dyn. 238:965–979 [DOI] [PubMed] [Google Scholar]
- 3. Behringer RR, Rasweiler JJ, Chen CH, Cretekos CJ. 2009. Genetic regulation of mammalian diversity. Cold Spring Harbor Symp. Quant. Biol. 74:297–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Munshi-South J, Wilkinson GS. 2010. Bats and birds: exceptional longevity despite high metabolic rates. Ageing Res. Rev. 9:12–19 [DOI] [PubMed] [Google Scholar]
- 5. Calisher CH, Childs JE, Field HE, Holmes KV, Schountz T. 2006. Bats: important reservoir hosts of emerging viruses. Clin. Microbiol. Rev. 19:531–545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Dobson AP. 2005. What links bats to emerging infectious diseases? Science 310:628–629 [DOI] [PubMed] [Google Scholar]
- 7. Lau SKP, Woo PCY, Li KSM, Huang Y, Tsoi H-W, Wong BHL, Wong SSY, Leung S-Y, Chan K-H, Yuen K-Y. 2005. Severe acute respiratory syndrome coronavirus-like virus in Chinese horseshoe bats. Proc. Natl. Acad. Sci. U. S. A. 102:14040–14045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Li W, Shi Z, Yu M, Ren W, Smith C, Epstein JH, Wang H, Crameri G, Hu Z, Zhang H, Zhang J, McEachern J, Field H, Daszak P, Eaton BT, Zhang S, Wang L-F. 2005. Bats are natural reservoirs of SARS-like coronaviruses. Science 310:676–679 [DOI] [PubMed] [Google Scholar]
- 9. Wong S, Lau S, Woo P, Yuen K-Y. 2007. Bats as a continuing source of emerging infections in humans. Rev. Med. Virol. 17:67–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Drexler JF, Corman VM, Wegner T, Tateno AF, Zerbinati RM, Gloza-Rausch F, Seebens A, Müller MA, Drosten C. 2011. Amplification of emerging viruses in a bat colony. Emerg. Infect. Dis. 17:449–456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Luis AD, Hayman DTS, O'Shea TJ, Cryan PM, Gilbert AT, Pulliam JRC, Mills JN, Timonin ME, Willis CKR, Cunningham AA, Fooks AR, Rupprecht CE, Wood JLN, Webb CT. 2013. A comparison of bats and rodents as reservoirs of zoonotic viruses: are bats special? Proc. Biol. Sci. 280:20122753. 10.1098/rspb.2012.2753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Li L, Victoria JG, Wang C, Jones M, Fellers GM, Kunz TH, Delwart E. 2010. Bat guano virome: predominance of dietary viruses from insects and plants plus novel mammalian viruses. J. Virol. 84:6955–6965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Donaldson EF, Haskew AN, Gates JE, Huynh J, Moore CJ, Frieman MB. 2010. Metagenomic analysis of the viromes of three North American bat species: viral diversity among different bat species that share a common habitat. J. Virol. 84:13004–13018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ge X, Li Y, Yang X, Zhang H, Zhou P, Zhang Y, Shi Z. 2012. Metagenomic analysis of viruses from bat fecal samples reveals many novel viruses in insectivorous bats in China. J. Virol. 86:4620–4630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wu Z, Ren X, Yang L, Hu Y, Yang J, He G, Zhang J, Dong J, Sun L, Du J, Liu L, Xue Y, Wang J, Yang F, Zhang S, Jin Q. 2012. Virome analysis for identification of novel mammalian viruses in bat species from Chinese provinces. J. Virol. 86:10999–11012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gifford R, Tristem M. 2003. The evolution, distribution and diversity of endogenous retroviruses. Virus Genes 26:291–315 [DOI] [PubMed] [Google Scholar]
- 17. Blikstad V, Benachenhou F, Sperber GO, Blomberg J. 2008. Evolution of human endogenous retroviral sequences: a conceptual account. Cell. Mol. Life Sci. 65:3348–3365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jern P, Coffin JM. 2008. Effects of retroviruses on host genome function. Annu. Rev. Genet. 42:709–732 [DOI] [PubMed] [Google Scholar]
- 19. Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, Funke R, Gage D, Harris K, Heaford A, Howland J, Kann L, Lehoczky J, LeVine R, McEwan P, McKernan K, Meldrim J, Mesirov JP, Miranda C, Morris W, Naylor J, Raymond C, Rosetti M, Santos R, Sheridan A, Sougnez C, Stange-Thomann N, Stojanovic N, Subramanian A, Wyman D, Rogers J, Sulston J, Ainscough R, Beck S, Bentley D, Burton J, Clee C, Carter N, Coulson A, Deadman R, Deloukas P, Dunham A, Dunham I, Durbin R, French L, Grafham D, et al. 2001. Initial sequencing and analysis of the human genome. International Human Genome Sequencing Consortium. Nature 409:860–921 [DOI] [PubMed] [Google Scholar]
- 20. Waterston RH, Lindblad-Toh K, Birney E, Rogers J, Abril JF, Agarwal P, Agarwala R, Ainscough R, Alexandersson M, An P, Antonarakis SE, Attwood J, Baertsch R, Bailey J, Barlow K, Beck S, Berry E, Birren B, Bloom T, Bork P, Botcherby M, Bray N, Brent MR, Brown DG, Brown SD, Bult C, Burton J, Butler J, Campbell RD, Carninci P, Cawley S, Chiaromonte F, Chinwalla AT, Church DM, Clamp M, Clee C, Collins FS, Cook LL, Copley RR, Coulson A, Couronne O, Cuff J, Curwen V, Cutts T, Daly M, David R, Davies J, Delehaunty KD, Deri J, Dermitzakis ET, et al. 2002. Initial sequencing and comparative analysis of the mouse genome. Mouse Genome Sequencing Consortium. Nature 420:520–562 [DOI] [PubMed] [Google Scholar]
- 21. Kurth R, Bannert N. 2010. Beneficial and detrimental effects of human endogenous retroviruses. Int. J. Cancer 126:306–314 [DOI] [PubMed] [Google Scholar]
- 22. Feschotte C, Gilbert C. 2012. Endogenous viruses: insights into viral evolution and impact on host biology. Nat. Rev. Genet. 13:283–296 [DOI] [PubMed] [Google Scholar]
- 23. Katzourakis A, Gifford RJ, Tristem M, Gilbert MTP, Pybus OG. 2009. Macroevolution of complex retroviruses. Science 325:1512. [DOI] [PubMed] [Google Scholar]
- 24. Emerman M, Malik HS. 2010. Paleovirology—modern consequences of ancient viruses. Plos Biol. 8:e1000301. 10.1371/journal.pbio.1000301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Holmes EC. 2011. The evolution of endogenous viral elements. Cell Host Microbe 10:368–377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tristem M, Kabat P, Lieberman L, Linde S, Karpas A, Hill F. 1996. Characterization of a novel murine leukemia virus-related subgroup within mammals. J. Virol. 70:8241–8246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Baillie GJ, van de Lagemaat LN, Baust C, Mager DL. 2004. Multiple groups of endogenous betaretroviruses in mice, rats, and other mammals. J. Virol. 78:5784–5798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cui J, Tachedjian G, Tachedjian M, Holmes EC, Zhang S, Wang L-F. 2012. Identification of diverse groups of endogenous gammaretroviruses in mega- and microbats. J. Gen. Virol. 93:2037–2045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hayward JA, Tachedjian M, Cui J, Field H, Holmes EC, Wang L-F, Tachedjian G. 2013. Identification of diverse full-length endogenous betaretroviruses in megabats and microbats. Retrovirology 10:35. 10.1186/1742-4690-10-35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cui J, Tachedjian M, Wang L, Tachedjian G, Wang L-F, Zhang S. 2012. Discovery of retroviral homologs in bats: implications for the origin of mammalian gammaretroviruses. J. Virol. 86:4288–4293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ellinghaus D, Kurtz S, Willhoeft U. 2008. LTRharvest, an efficient and flexible software for de novo detection of LTR retrotransposons. BMC Bioinform. 9:18. 10.1186/1471-2105-9-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Steinbiss S, Willhoeft U, Gremme G, Kurtz S. 2009. Fine-grained annotation and classification of de novo predicted LTR retrotransposons. Nucleic Acids Res. 37:7002–7013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lowe TM, Eddy SR. 1997. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25:955–964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Punta M, Coggill PC, Eberhardt RY, Mistry J, Tate J, Boursnell C, Pang N, Forslund K, Ceric G, Clements J, Heger A, Holm L, Sonnhammer ELL, Eddy SR, Bateman A, Finn RD. 2012. The Pfam protein families database. Nucleic Acids Res. 40:D290–D301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Reference deleted.
- 36. Rho M, Choi J-H, Kim S, Lynch M, Tang H. 2007. De novo identification of LTR retrotransposons in eukaryotic genomes. BMC Genomics 8:90. 10.1186/1471-2164-8-90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kohany O, Gentles AJ, Hankus L, Jurka J. 2006. Annotation, submission and screening of repetitive elements in Repbase: RepbaseSubmitter and Censor. BMC Bioinform. 7:474. 10.1186/1471-2105-7-474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32:1792–1797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. 2011. MEGA5: Molecular Evolutionary Genetics Analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28:2731–2739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Jones DT, Taylor WR, Thornton JM. 1992. The rapid generation of mutation data matrices from protein sequences. Comput. Appl. Biosci. 8:275–282 [DOI] [PubMed] [Google Scholar]
- 41. Ronquist F, Huelsenbeck JP. 2003. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19:1572–1574 [DOI] [PubMed] [Google Scholar]
- 42. Smith TF, Waterman MS. 1981. Identification of common molecular subsequences. J. Mol. Biol. 147:195–197 [DOI] [PubMed] [Google Scholar]
- 43. Jukes TH, Cantor CR. 1969. Evolution of protein molecules, p 21–132 Munro HN, Mammalian protein metabolism, vol III Academic Press, San Diego, CA [Google Scholar]
- 44. Pace JK, Gilbert C, Clark MS, Feschotte C. 2008. Repeated horizontal transfer of a DNA transposon in mammals and other tetrapods. Proc. Natl. Acad. Sci. U. S. A. 105:17023–17028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Jurka J, Kapitonov VV, Pavlicek A, Klonowski P, Kohany O, Walichiewicz J. 2005. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet. Genome Res. 110:462–467 [DOI] [PubMed] [Google Scholar]
- 46. Eddy SR. 2011. Accelerated profile HMM searches. PLoS Comput. Biol. 7:e1002195. 10.1371/journal.pcbi.1002195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Xiong Y, Eickbush TH. 1990. Origin and evolution of retroelements based upon their reverse transcriptase sequences. EMBO J. 9:3353–3362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Höhna S, Larget B, Liu L, Suchard MA, Huelsenbeck JP. 2012. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61:539–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Slattery JP, Franchini G, Gessain A. 1999. Genomic evolution, patterns of global dissemination, and interspecies transmission of human and simian T-cell leukemia/lymphotropic viruses. Genome Res. 9:525–540 [PubMed] [Google Scholar]
- 50. Fernández-Medina RD, Ribeiro JMC, Carareto CMA, Velasque L, Struchiner CJ. 2012. Losing identity: structural diversity of transposable elements belonging to different classes in the genome of Anopheles gambiae. BMC Genomics 13:272. 10.1186/1471-2164-13-272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Smit AF. 1993. Identification of a new, abundant superfamily of mammalian LTR-transposons. Nucleic Acids Res. 21:1863–1872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. McCarthy EM, McDonald JF. 2004. Long terminal repeat retrotransposons of Mus musculus. Genome Biol. 5:R14. 10.1186/gb-2004-5-3-r14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Polavarapu N, Bowen NJ, McDonald JF. 2006. Identification, characterization and comparative genomics of chimpanzee endogenous retroviruses. Genome Biol. 7:R51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Polavarapu N, Bowen NJ, McDonald JF. 2006. Newly identified families of human endogenous retroviruses. J. Virol. 80:4640–4642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Garcia-Etxebarria K, Jugo BM. 2010. Genome-wide detection and characterization of endogenous retroviruses in Bos taurus. J. Virol. 84:10852–10862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Martínez Barrio Á, Ekerljung M, Jern P, Benachenhou F, Sperber GO, Bongcam-Rudloff E, Blomberg J, Andersson G. 2011. The first sequenced carnivore genome shows complex host-endogenous retrovirus relationships. PLoS One 6:e19832. 10.1371/journal.pone.0019832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Garcia-Etxebarria K, Jugo BM. 2012. Detection and characterization of endogenous retroviruses in the horse genome by in silico analysis. Virology 434:59–67 [DOI] [PubMed] [Google Scholar]
- 58. Brown K, Moreton J, Malla S, Aboobaker AA, Emes RD, Tarlinton RE. 2012. Characterisation of retroviruses in the horse genome and their transcriptional activity via transcriptome sequencing. Virology 433:55–63 [DOI] [PubMed] [Google Scholar]
- 59. Ray DA, Feschotte C, Pagan HJT, Smith JD, Pritham EJ, Arensburger P, Atkinson PW, Craig NL. 2008. Multiple waves of recent DNA transposon activity in the bat, Myotis lucifugus. Genome Res. 18:717–728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Pritham EJ, Feschotte C. 2007. Massive amplification of rolling-circle transposons in the lineage of the bat Myotis lucifugus. Proc. Natl. Acad. Sci. U. S. A. 104:1895–1900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Dangel AW, Baker BJ, Mendoza AR, Yu CY. 1995. Complement component C4 gene intron 9 as a phylogenetic marker for primates: long terminal repeats of the endogenous retrovirus ERV-K(C4) are a molecular clock of evolution. Immunogenetics 42:41–52 [DOI] [PubMed] [Google Scholar]
- 62. Zhang G, Cowled C, Shi Z, Huang Z, Bishop-Lilly KA, Fang X, Wynne JW, Xiong Z, Baker ML, Zhao W, Tachedjian M, Zhu Y, Zhou P, Jiang X, Ng J, Yang L, Wu L, Xiao J, Feng Y, Chen Y, Sun X, Zhang Y, Marsh GA, Crameri G, Broder CC, Frey KG, Wang L-F, Wang J. 2013. Comparative analysis of bat genomes provides insight into the evolution of flight and immunity. Science 339:456–460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Miller-Butterworth CM, Murphy WJ, O'Brien SJ, Jacobs DS, Springer MS, Teeling EC. 2007. A family matter: conclusive resolution of the taxonomic position of the long-fingered bats, miniopterus. Mol. Biol. Evol. 24:1553–1561 [DOI] [PubMed] [Google Scholar]
- 64. Lack JB, Roehrs ZP, Stanley CE, Jr, Ruedi M, Van Den Bussche RA. 2010. Molecular phylogenetics of Myotis indicate familial-level divergence for the genus Cistugo (Chiroptera). J. Mammal. 91:976–992 [Google Scholar]
- 65. Agnarsson I, Zambrana-Torrelio CM, Flores-Saldana NP, May-Collado LJ. 2011. A time-calibrated species-level phylogeny of bats (Chiroptera, Mammalia). PLoS Curr. 3:RRN1212. 10.1371/currents.RRN1212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Stadelmann B, Lin LK, Kunz TH, Ruedi M. 2007. Molecular phylogeny of New World Myotis (Chiroptera, Vespertilionidae) inferred from mitochondrial and nuclear DNA genes. Mol. Phylogenet. Evol. 43:32–48 [DOI] [PubMed] [Google Scholar]
- 67. Kijima TE, Innan H. 2010. On the estimation of the insertion time of LTR retrotransposable elements. Mol. Biol. Evol. 27:896–904 [DOI] [PubMed] [Google Scholar]
- 68. Reference deleted.
- 69. Lindblad-Toh K, Wade CM, Mikkelsen TS, Karlsson EK, Jaffe DB, Kamal M, Clamp M, Chang JL, Kulbokas EJ, Zody MC, Mauceli E, Xie X, Breen M, Wayne RK, Ostrander EA, Ponting CP, Galibert F, Smith DR, deJong PJ, Kirkness E, Alvarez P, Biagi T, Brockman W, Butler J, Chin C-W, Cook A, Cuff J, Daly MJ, DeCaprio D, Gnerre S, Grabherr M, Kellis M, Kleber M, Bardeleben C, Goodstadt L, Heger A, Hitte C, Kim L, Koepfli K-P, Parker HG, Pollinger JP, Searle SMJ, Sutter NB, Thomas R, Webber C, Baldwin J, Abebe A, Abouelleil A, Aftuck L, Ait-zahra M, et al. 2005. Genome sequence, comparative analysis and haplotype structure of the domestic dog. Nature 438:803–819 [DOI] [PubMed] [Google Scholar]
- 70. Han K, Konkel MK, Xing J, Wang H, Lee J, Meyer TJ, Huang CT, Sandifer E, Hebert K, Barnes EW, Hubley R, Miller W, Smit AFA, Ullmer B, Batzer MA. 2007. Mobile DNA in Old World monkeys: a glimpse through the rhesus macaque genome. Science 316:238–240 [DOI] [PubMed] [Google Scholar]
- 71. Gilbert C, Schaack S, Pace JK, Brindley PJ, Feschotte C. 2010. A role for host-parasite interactions in the horizontal transfer of transposons across phyla. Nature 464:1347–1350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Thomas J, Schaack S, Pritham EJ. 2010. Pervasive horizontal transfer of rolling-circle transposons among animals. Genome Biol. Evol. 2:656–664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Magiorkinis G, Gifford RJ, Katzourakis A, De Ranter J, Belshaw R. 2012. Env-less endogenous retroviruses are genomic superspreaders. Proc. Natl. Acad. Sci. U. S. A. 109:7385–7390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Belshaw R, Katzourakis A, Paces J, Burt A, Tristem M. 2005. High copy number in human endogenous retrovirus families is associated with copying mechanisms in addition to reinfection. Mol. Biol. Evol. 22:814–817 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.