

Abstract
In this study, we investigated the efficacy of new bifunctional peptide inhibitors (BPIs) in suppressing experimental autoimmune encephalomyelitis (EAE) in an animal model. BPI [e.g. proteolipid protein–cyclo(1,8)-CPRGGSVC-NH2 (PLP-cIBR)] is a conjugate between the PLP139–151 peptide derived from proteolipid protein (PLP) and the cIBR7 peptide derived from domain-1 (D1) of intercellular adhesion molecule-1 (ICAM-1). PLP–cIBR is designed to bind to major histocompatibility complex (MHC)-II and leucocyte function-associated antigen-1 (LFA-1) simultaneously to inhibit the formation of the immunological synapse and alter the differentiation and activation of a subpopulation of T cells, thus inducing immunotolerance. The results show that PLP–cIBR is highly potent in ameliorating EAE, even at low concentrations and less frequent injections. Mice treated with PLP–cIBR had a higher secretion of cytokines related to regulatory and/or suppressor cells compared to phosphate-buffered saline (PBS)-treated mice. In contrast, T helper type 1 (Th1) cytokines were higher in mice treated with PBS compared to PLP–cIBR, suggesting that it suppressed Th1 proliferation. Also, we observed significantly less demyelination in PLP-cIBR-treated mice compared to the control, further indicating that PLP–cIBR promoted protection against demyelination.
Keywords: antigen-presenting cell, bifunctional peptide inhibitor, EAE, leucocyte infiltration, lovastatin
Introduction
Multiple sclerosis (MS) is a progressive inflammatory disease of the central nervous system (CNS) characterized by perivascular inflammation, demyelination and axonal damage [1]. The pathophysiology of MS appears to be heterogeneous, and it has been suggested that molecular mimicry, bystander activation and epitope spreading could be possible mechanisms that initiate and perpetuate the disease. Progression of this disease includes breaching of the blood–brain barrier (BBB) and infiltration of inflammatory leucocytes (e.g. autoreactive CD4+ and CD8+ T cells and monocytes) into the CNS [2,3]. The BBB leakiness and leucocyte infiltration further sustain the inflammatory response through activation of resident glial cells and secretion of inflammatory mediators [4]. Thus, the leucocytes can cause demyelination and axonal loss, which lead to neurological deficits in MS patients.
Most currently approved drugs for the treatment of MS include anti-inflammatory agents [e.g. interferon (IFN)-β and mitoxanthrone], natalizumab and glatiramer acetate. Glatiramer acetate was designed as a decoy for the myelin basic protein (MBP) to alter the immune response from T helper type 1 (Th1) to Th2 phenotype [5]. Natalizumab, a humanized monoclonal antibody (mAb), is an antagonist of the α4-subunit of α4β1 or very late antigen 4 (VLA-4), which blocks leucocyte trafficking to the CNS and inhibits T cell activation. Although some of these drugs were effective in some patients, many individuals experienced poor response or adverse side effects [6,7]. Some of the current drugs suppress the general immune response without selectively suppressing a subpopulation(s) of autoreactive T cells, and lower the defence mechanism against pathogenic infections. Hence, there is a need to develop a new therapy that selectively suppresses a subpopulation of autoreactive T cells that causes the autoimmune disease without attenuating the ability to fight infections. Many attempts have been made to induce antigen-specific tolerance by interfering with antigen presentation by antigen-presenting cells (APC) to suppress T cell activation [2,8]. One of the strategies is by delivering altered-peptide ligands (APL) to generate immune tolerance (i.e. mucosal or nasal-induced tolerance and coupled-cell-induced tolerance). The proposed mechanism of action of APL molecules is that they compete with the native peptide ligands for binding to major histocompatibility complex II (MHC-II) on APC for T cell receptor (TCR) recognition on T cells and, as a result, alter the signalling cascade necessary for full T cell activation. APL molecules contain one or more amino acid substitutions and may function either as antagonists or partial agonists in the TCR/MHC-II-antigen-binding process. It was proposed that the antagonistic action of APL induces T cell anergy, while a partial-agonist action induces incomplete activation of T cells. Partial activation can generate tolerance via Th2-cell- and Th3-cell-dependent responses or bystander suppression through regulatory T cells (Treg) cells [9,10]. Although APL molecules have suppressed experimental autoimmune encephalomyelitis (EAE) successfully in animals, these strategies have encountered several challenges in human clinical trials of MS; these challenges include the lack of efficacy, exacerbation of the disease and generation of hypersensitivity reactions. The failed clinical trials of APL molecules have stimulated the evaluation of native antigen peptides that target subpopulations of T cells that recognize myelin epitopes as well as the evaluation of new treatment regimens (i.e. less stringent dosage and frequency of administration) to reduce side effects.
Our approach to suppress or prevent the development of autoimmune diseases is by controlling the activation of immune cells in an antigen-specific manner using bifunctional peptide inhibitor (BPI) molecules [2,8]. BPI molecules are constructed by conjugating a disease-specific antigenic peptide and a cell adhesion peptide via a linker. Our previous studies have shown that proteolipid protein (PLP)–BPI derivatives constructed from PLP139–151 antigenic peptide [11] and LFA-alpha blocker left (LABL) peptide from the I-domain of αL integrin (CD11a237–246) can suppress effectively the onset and progression of experimental EAE in mice [12–14]. In addition, the glutamic acid decarboxylase (GAD)–BPI molecule, a conjugate between GAD208–217 antigenic peptide and LABL peptide, can inhibit type-1 diabetes in non-obese diabetic (NOD) mice [15]. The hypothesis is that the BPI molecule inhibits the formation of the immunological synapse by binding to both intercellular adhesion molecule-1 (ICAM-1) and MHC-II on the surface of APC [8]. Co-capping experiments showed that I-Ag7 and ICAM-1 receptors were co-localized on the surface of GAD–BPI-treated APC, and binding of GAD–BPI on the APC was blocked by either anti-I-Ag7 or anti-ICAM-1, suggesting that the BPI molecule can bridge MHC-II and ICAM-1 on the surface of APC [15]. The inhibition of immunological synapse formation alters the differentiation of T cells from inflammatory to regulatory cells [2,16].
In this work, we investigated new PLP–cIBR derivatives that were conjugates between PLP139–151 peptide and cyclic peptides (derivatives of cIBR7 = cyclo(1,8)-CPRGGSVC-NH2 [17]) linked via a spacer. In contrast to PLP–BPI, the cell adhesion peptide (i.e. cIBR peptide derivatives) in PLP–cIBR was derived from the sequence of domain-1 (D1) of ICAM-1, which binds to the I-domain of leucocyte function-associated antigen-1 (LFA-1) [18–20]. The proposed mechanism of activity of PLP–cIBR is that both PLP and cIBR peptides bind to MCH-II and LFA-1, respectively, to prevent the formation of immunological synapse and to alter T cell differentiation and activation. The effect of different residues in cIBR peptide on the activity of PLP–cIBR was evaluated to determine whether adding Leu residue to the N-terminal of cIBR7 or adding Leu residue to the N-terminal with elimination of the Val residue from the C-terminal of cIBR7 (Table 1) has effects on biological activity [17]. Because lovastatin and simvastatin have been evaluated in clinical trials for the treatment of multiple sclerosis [21–24], and have been studied widely in mouse models, lovastatin was used as a standard to evaluate the efficacy of PLP–cIBR derivatives in suppressing EAE and to provide a perspective on the dose–efficacy relationship of these two potential drugs for MS. Lovastatin is an ideal choice for comparison studies for two reasons. First, lovastatin has been shown to inhibit cellular infiltration into the CNS [25]. Secondly, lovastatin can inhibit ICAM-1/LFA-1-mediated immune cell adhesion via binding to the I-domain of CD11a of LFA-1 [26]. As shown previously with BPI molecules, the PLP–cIBR molecule could prevent the infiltration of leucocytes by blocking ICAM-1/LFA-1-mediated immune cell adhesion to the vascular endothelial cells of the BBB as well as preventing BBB leakiness [16]. Finally, the underlying mechanisms of in-vivo activity of Ac-PLP-cIBR-NH2-1 and lovastatin were investigated by determining cytokine secretion phenotypes and their effect on pathologies of the brain.
Table 1.
List of peptides used in the present study
| Peptide | Sequence |
|---|---|
| PLP139–151 | HSLGKWLGHPDKF |
| Ac-PLP-cIBR-NH2-1 | Ac-HSLGKWLGHPDKF-(AcpGAcpGAcp)2-CPRGGSVC-NH2 |
| Ac-PLP-cIBR-NH2-2 | Ac-HSLGKWLGHPDKF-(AcpGAcpGAcp)2-CLPRGGSVC-NH2 |
| Ac-PLP-BPI-NH2-3 | Ac-HSLGKWLGHPDKF-(AcpGAcpGAcp)2-CLPRGGSC-NH2 |
| Ac-PLP-BPI-PEG6 | Ac-HSLGKWLGHPDKF-(C2H5O)3-G-(C2H5O)3-ITDGEATDSG-NH2 |
| Ac-PLPsc-cIBR-NH2-1 | Ac-SLKHGGLWPHKDF-(AcpGAcpGAcp)2-CPRGGSVC-NH2 |
BPI is composed of antigen epitope peptide [proteolipid protein (PLP)139–151], varying spacers and cyclo(1,8)-CPRGGSVC-NH2 (cIBR) peptide, where Acp in the spacer represents ε-aminocaproic acid. The peptides are capped at both ends, i.e. N-terminal acetylated (Ac-) and C-terminal amidated (-NH2).
Materials and methods
Peptides
Peptides (Table 1) were synthesized in an automated peptide synthesis system (Pioneer; Perceptive Biosystems, Framingham, MA, USA) using 9-fluorenyl-methoxy-carbonyl-protected amino acid chemistry on appropriate polyethylene glycol–polystyrene (PEG-PS™) resin (GenScript Corporation, Piscataway, NJ, USA). Each peptide was removed from the resin and deprotected with trifluoroacetic acid in the presence of scavengers. The crude peptides were purified by high-performance liquid chromatography (HPLC) using a C18 semi-preparative reversed-phase column (Varian, Palo Alto, CA, USA). Cyclization of the cIBR peptide fragment from the pure linear peptide was accomplished by air oxidation in ammonium bicarbonate buffer (0·05 M, pH 8·5) overnight in high dilution (0·06 mM). After cyclization, the peptide solution was concentrated using a rotary evaporator followed by lyophilization. The lyophilized powder containing peptide and ammonium bicarbonate was subjected to purification by semi-preparative HPLC using a C18 column. The pure fractions of the peptide were pooled and lyophilized. The identity of the pure final product was confirmed by electrospray ionization mass spectrometry.
Animals
SJL/J (H-2S) female mice were purchased from Charles River (Wilmington, MA, USA) and housed under specific pathogen-free conditions at the Association for Assessment and Accreditation of Laboratory Animal Care-approved facility at the University of Kansas. The Institutional Animal Care and Use Committee at The University of Kansas approved all experimental procedures for using live animals.
Induction of EAE and clinical evaluation
SJL/J female mice (5–7 weeks old) were immunized subcutaneously (s.c.) with 200 μg of PLP139–151 in a 0·2-ml emulsion composed of equal volumes of phosphate-buffered saline (PBS) and complete Freund's adjuvant (CFA) containing killed Mycobacterium tuberculosis strain H37RA (at a final concentration of 4 mg/ml; Difco, Detroit, MI, USA). The PLP139–151 in CFA was administered to regions above the shoulder and the flanks (total of four sites; 50 μl at each injection site). Then, 200 ng of pertussis toxin (List Biological Laboratories Inc., Campbell, CA, USA) was injected intraperitoneally (i.p.) on the day of immunization (day 0) and 2 days post-immunization. The disease progress was evaluated in a blinded fashion using clinical scoring as follows: 0, no clinical signs of the disease; 1, tail weakness or limp tail; 2, paraparesis (weakness or partial paralysis of one or two hind limbs); 3, paraplegia (complete paralysis of two hind limbs); 4, paraplegia with forelimb weakness or paralysis; and 5, moribund (mice were euthanized once they were found to be moribund). Body weights were also measured daily.
BPI and lovastatin treatments
Study I
This study was performed to evaluate the efficacy of different PLP–cIBR derivatives in suppressing EAE. After immunization with PLP139–151/CFA, the immunized mice received intravenous injections of Ac-PLP-cIBR-NH2-1, -2 or -3 at a concentration of 100 nmol/injection/day on days 4, 7 and 10. The control mice received three injections of PBS. The efficacy of each peptide was evaluated by monitoring the clinical score and the change in body weight.
The potency of Ac-PLP-cIBR-NH2-1 was compared to that of positive controls (Ac-PLP-BPI-PEG6 and PLP) and a negative control (PBS). In this case, the immunized animals received intravenous (i.v.) injections of 50 nmol/injection/day of Ac-PLP-cIBR-NH2-1, Ac-PLP-BPI-PEG6 and PLP on days 4 and 7. Finally, the efficacy of Ac-PLP-cIBR-NH2-1 was also compared to that of a scrambled peptide Ac-PLPsc-cIBR and PBS by injecting the peptides at on days 4, 7 and 10 (50 nmol/injection/day). Then, the clinical scores and changes in body weight were monitored and compared.
Study II
The efficacy of three injections on days 4, 7 and 10 of Ac-PLP-cIBR-NH2-1 at a low dose (50 nmol/injection/day) was compared to 20 daily injections of lovastatin from day 0 at a higher dose (495 nmol/injection/day equivalent to 10 mg/kg body weight). Because pure lovastatin (Sigma Aldrich, St Louis, MO, USA) is not soluble in PBS, stock solution was dissolved initially in 100 μl of dimethylsulphoxide (DMSO) and finally suspended in PBS (pH 7·4). The clinical scores and body weight of the mice were monitored daily.
Study III
For the therapeutic study, the efficacy of Ac-PLP-cIBR-NH2-1 was compared to that of PBS. In the peptide-treated group, the mice were left untreated until they had an EAE clinical score of 1 or more for the first time. When a mouse had a clinical score of 1, it received i.v. injections of peptides (50 nmol/injection) daily for a maximum of three injections; however, peptide injection was discontinued once the disease score returned to below 1. The clinical scores and body weights were monitored.
Cytokine production
SJL/J mice were immunized with PLP139–151/CFA and pertussis toxin as described above. The mice were then treated with Ac-PLP-cIBR-NH2-1 (50 nmol/mouse on days 4, 7 and 10), lovastatin (495 nmol/mouse daily from days 0 to 20) or PBS (control). Representative spleens from each group were harvested on days 13 (peak of disease severity) and 35 (period of disease remission). Splenocytes were isolated by gently dispersing the spleen using the coarse portion of a 1·0-ml syringe in a Petri dish containing RPMI-1640 medium [10% fetal bovine serum (FBS), 0·05 M Eagle's basal medium (BME)]. The cells were then filtered through a 40-μm strainer. After centrifugation, the red blood cells were lysed using ammonium chloride (ACK) lysis buffer. The remaining white blood cells were washed three times with medium. The splenocytes (5 × 106 cells) were cultured in parallel in the absence or presence of 20 μM PLP in RPMI-1640 medium (10% FBS, 0·05 M BME). Supernatants were collected at the 72-h time-point and analysed for cytokine levels using a fully quantitative enzyme-linked immunosorbent assay (ELISA)-based Q-Plex™ Mouse Cytokine-Screen (Quansys Biosciences, Logan, UT, USA).
Histological analysis
SJL/J mice were immunized with PLP139–151/CFA and pertussis toxin as described above. The mice were then treated with Ac-PLP-cIBR-NH2-1 (50 nmol/mouse on days 4, 7 and 10), lovastatin (495 nmol/mouse daily from days 0 to 20) or PBS (control). On day 35, three mice from each treatment group were anaesthetized and perfused transcardially with saline followed by ice-cold 4% paraformaldehyde in 0·1 M PBS (pH = 7·4). Whole brains were sectioned in a sagittal manner, fixed in 4% paraformaldehyde for 48 h and paraffin-embedded. Sagittal sections (5 μm thick) were stained with haematoxylin and eosin (H&E) or Luxol Fast Blue (LFB) for evidence of infiltrating inflammatory cells and demyelination. All H&E histological scores were evaluated blindly by a board-certified pathologist (IHC World LLC, Woodstock, MD, USA). All LFB slides were analysed using the area fraction method (IHC World LLC). Stained tissue sections were examined under a Nikon Eclipse 90i light microscope, and images were captured with a Nikon Digital Camera DXM 1200c and NIS Elements software (Nikon, Tokyo, Japan) for light microscopy. Representative digital micrographs of the sections were obtained for each treatment group.
Statistical analysis
Statistical comparisons among the groups in EAE clinical scores were performed by calculating the average score for each mouse during the peak of the disease (i.e. days 10–20) and performing a Mann–Whitney U-test. Statistical differences in percentage loss in body weight were determined by calculating the average for each mouse during the peak of the disease (i.e. days 10–20) and performing a Mann–Whitney U-test. Comparison of cytokine production in the spleen and differences in the myelin-covered area fraction in LFB-stained brain sections were performed by one-way analysis of variance. All analyses were carried out with StatView (SAS Institute, Cary, NC, USA).
Results
Suppression of EAE by PLP–cIBR derivatives
In study I, three injections of PLP–cIBR derivatives (Ac-PLP-cIBR-NH2-1, Ac-PLP-cIBR-NH2-2 and Ac-PLP-cIBR-NH2-3) at 100 nmol/injection/day on days 4, 7 and 10 were extremely efficacious in suppressing EAE compared with PBS treatment (Fig. 1a). At this dose and dosing regimen, all three PLP–cIBR derivatives suppressed completely the progression of EAE, and it was difficult to differentiate the efficacies among these PLP–cIBR derivatives. However, PBS-treated animals exhibited severe signs of EAE, particularly at the peak of the disease on days 11–17, with clinical scores as high as 4 (Fig. 1a). Mice treated with all three PLP–cIBR derivatives did not show significant (P < 0·001) loss in body weight compared to PBS-treated mice (Fig. 1b). At the peak of disease (days 11–17), PBS-treated mice had as much as a 15% loss in body weight compared to less than 1% loss in body weight among the PLP–cIBR-treated mice. Additionally, more than 95% of the mice receiving any of the PLP–cIBR derivatives did not develop clinical signs of the disease during the course of the study; even the EAE-positive mice in this group showed delayed onset with attenuated severity of the disease. In contrast, only 8% of the mice receiving PBS did not develop severe disease.
Figure 1.
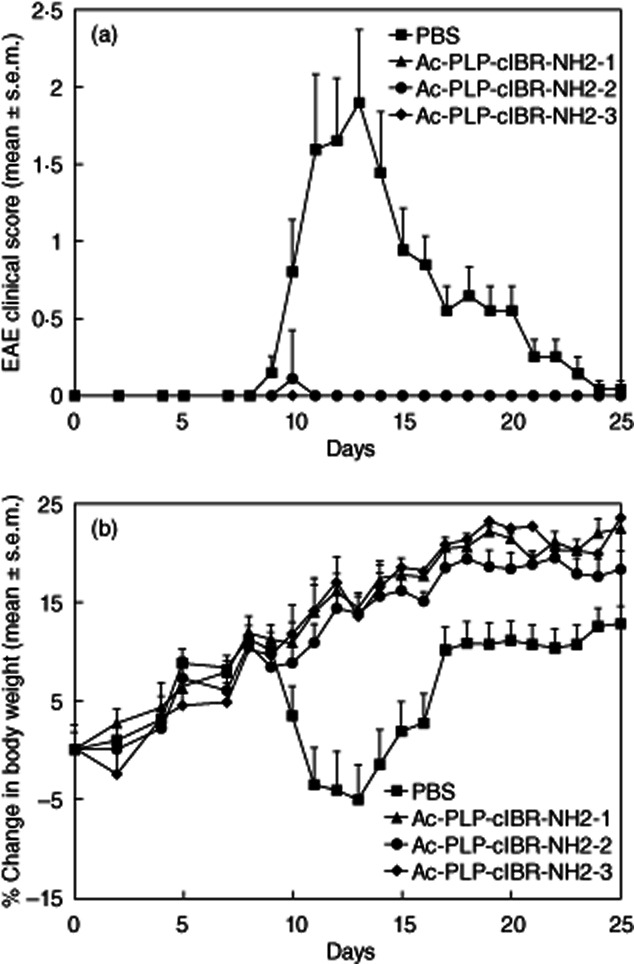
In-vivo efficacies of proteolipid protein–cyclo(1,8)-CPRGGSVC-NH2 (PLP–cIBR) derivatives (Ac-PLP-cIBR-NH2-1, -2 and -3) in suppressing experimental autoimmune encephalomyelitis (EAE) in the mouse model upon treatment with the peptide after immunization with PLP139–151/complete Freund's adjuvant (CFA) on day 0. The immunized mice received intravenous injections of the indicated PLP–cIBR peptide (100 nmol/injection/day) on days 4, 7 and 10. The efficacy of the peptide was determined by (a) clinical disease score of EAE and (b) percentage change in body weight. Results are expressed as the mean ± standard error of the mean (n = 10). EAE scores from all PLP–bifunctional peptide inhibitors (BPI)-treated mice were significantly lower (P < 0·001) than those of phosphate-buffered saline (PBS)-treated mice. Loss of body weight was significantly lower (P < 0·001) in PLP–cIBR-treated mice than in those treated with PBS.
Next, the efficacy of Ac-PLP-cIBR-NH2-1 was compared to that of positive control peptides, including Ac-PLP-BPI-PEG6, PLP and PBS. Previously, Ac-PLP-BPI-PEG6 had shown excellent ability to suppress EAE and was more efficient than PLP peptide alone [14]. To be able to differentiate the activities of Ac-PLP-cIBR-NH2-1 and positive controls, the frequency of injections and dose of the peptides were reduced to two injections on days 4 and 7 at 50 nmol/injection/day. It is clear that Ac-PLP-cIBR-NH2-1 has significantly better efficacy in suppressing the progress of EAE, having lower EAE clinical scores (P < 0·01) than those of Ac-PLP-BPI-PEG6, PLP and PBS (Fig. 2). Ac-PLP-cIBR-NH2-1-treated mice had a significantly lower loss of body weight than those treated with Ac-PLP-BPI-PEG6, PLP or PBS (P < 0·01, Fig. 2). At this dose, although Ac-PLP-BPI-PEG6 was not as potent as Ac-PLP-cIBR-NH2-1, it could delay onset of the disease more effectively than PBS and PLP alone. These results indicate that Ac-PLP-cIBR-NH2-1 is the most potent BPI molecule found to date for suppressing EAE.
Figure 2.
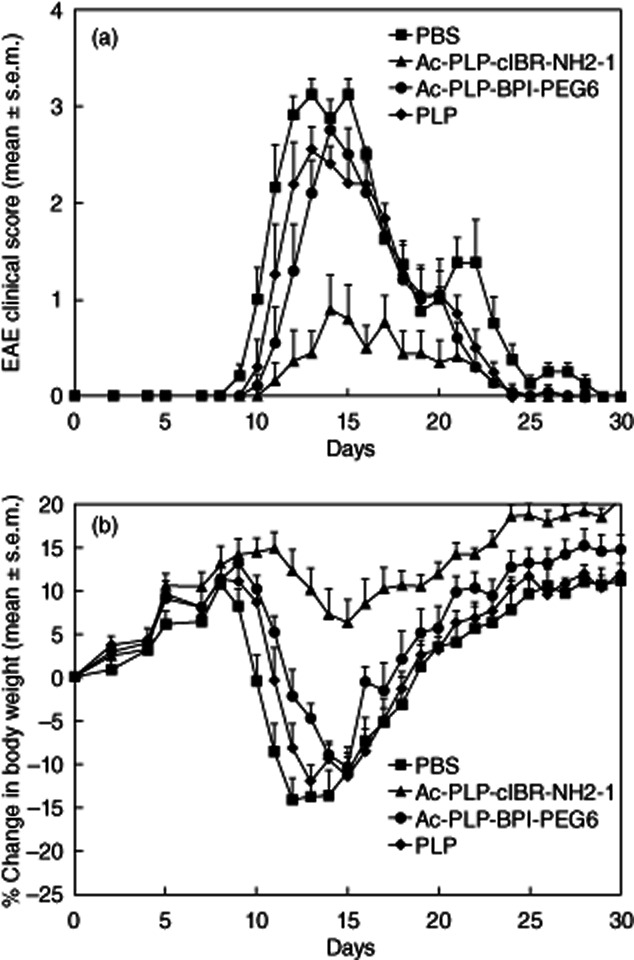
In-vivo experimental autoimmune encephalomyelitis (EAE)-suppressive activity of Ac-PLP-cIBR-NH2-1 and control peptides in the EAE model to study the effects of a reduced dose and the importance of bifunctional peptide inhibitors (BPI) structure. proteolipid protein (PLP)139–151/complete Freund's adjuvant (CFA)-immunized mice received intravenous injections of vehicle or indicated peptides (50 nmol/injection/day on days 4 and 7). (a) Clinical EAE disease score. (b) Change in body weight. Results are expressed as the mean ± standard error of the mean (n = 10). Statistical values for EAE clinical scores and loss in body weight compared with phosphate-buffered saline (PBS)-treated mice were as follows: Ac-PLP-cIBR-NH2-1, P < 0·01; Ac-PLP-BPI-PEG6, P > 0·05; and PLP, P > 0·05.
To evaluate further the specificity of Ac-PLP-cIBR-NH2-1, its EAE-suppressive activity was compared with that of Ac-PLPSC-cIBR-NH2-1 (negative control), which contains a scrambled PLP sequence. Ac-PLP-cIBR-NH2-1-treated animals had significantly lower EAE scores (P < 0·0001) with minimal loss of body weight (P < 0·0001) compared to the Ac-PLPSC-cIBR-NH2-1-treated groups (Fig. 3). Mice receiving Ac-PLPSC-cIBR-NH2-1 injections exhibited severe EAE symptoms similar to those of PBS-treated mice (P > 0·05 for clinical scores). There was also no significant difference observed in loss of body weight between mice treated with Ac-PLPSC-cIBR-NH2-1 and PBS. However, the disease scores show some of the residual EAE-suppressive activity of Ac-PLPSC-cIBR-NH2-1 (Fig. 3a); this activity is due to the cIBR fragment in the BPI molecule, because the cIBR portion can bind to the I-domain of LFA-1 and block ICAM-1/LFA-1 interactions as Signal-2. As a result of blocking Signal-2, T cell activation was suppressed. Although it is less likely, it is possible that scrambling the PLP sequence (PLPSC) did not abolish completely its binding property to MHC-II; thus, the residual peptide binding exhibits EAE-suppressive activity. However, it is important to note that scrambling of the PLP sequence diminishes the activity of BPI significantly; thus, the correct sequence of PLP is necessary for Ac-PLP-cIBR-NH2-1 activity to suppress EAE.
Figure 3.
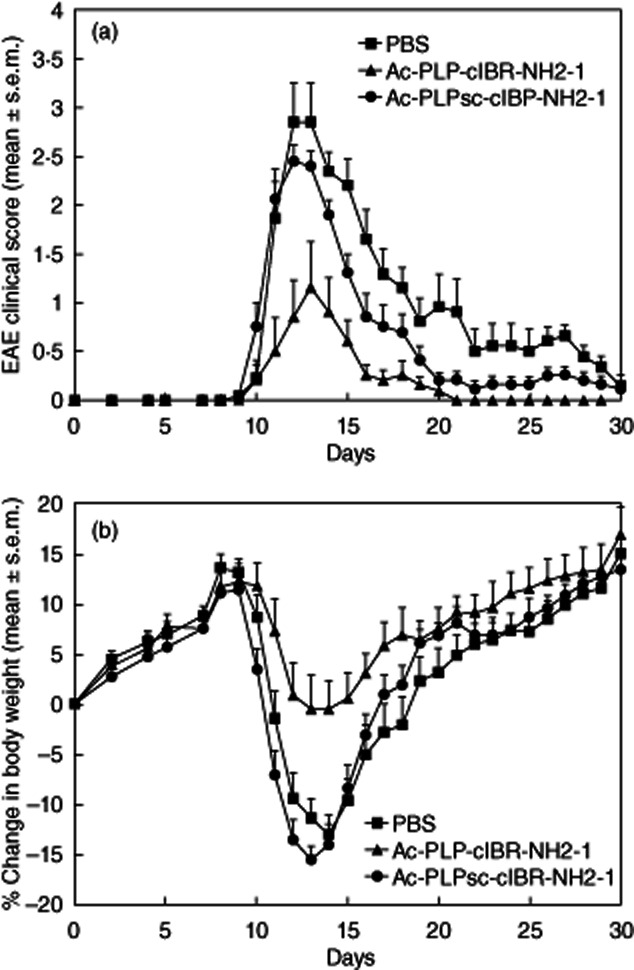
In-vivo experimental autoimmune encephalomyelitis (EAE)-suppressive activity of Ac-PLP-cIBR-NH2-1 and Ac-PLPsc-cIBR-NH2-1 (scrambled sequence) in the EAE model to evaluate the importance of bifunctional peptide inhibitors (BPI) structure. Proteolipid protein (PLP)139–151/complete Freund's adjuvant (CFA)-immunized mice received intravenous injections of vehicle or indicated peptides (50 nmol/injection/day on days 4, 7 and 10). (a) Clinical EAE disease score. (b) Change in body weight. Results are expressed as the mean ± standard error of the mean (n = 10). Statistical values for EAE clinical scores and loss in body weight compared with phosphate-buffered saline (PBS)-treated mice were as follows: Ac-PLP-cIBR-NH2-1, P < 0·01; and Ac-PLPsc-cIBR-NH2-1, P > 0·05.
Evaluating the therapeutic efficacy of Ac-PLP-cIBR-NH2-1 with lovastatin as a standard
The in-vivo study II was designed to evaluate the potency of Ac-PLP-cIBR-NH2-1. Lovastatin was used as a standard. In this study, the dosage and dosing regimen were chosen based on our preliminary work and the literature. Our initial studies showed that lovastatin must be injected at higher concentrations than BPI molecules to realize any appreciable suppression of EAE. Similarly, Greenwood et al. reported that mice treated with lovastatin prior to the onset of the disease had less severe clinical scores compared to those treated with vehicle only, and the benefits were sustained as long as daily injections were maintained. However, when lovastatin treatments were discontinued, EAE developed rapidly [27]. As shown in Fig. 4, daily injections (495 nmol/injection) of lovastatin for 20 days significantly (P < 0·001) decreased disease severity compared to PBS-treated controls. However, it is remarkable that fewer injections (three injections) and a lower dose (50 nmol/injection) of Ac-PLP-cIBR-NH2-1 were far more potent than daily injections of lovastatin. All aspects of EAE, including severity, onset and duration of the disease, were significantly (P < 0·001) lower in Ac-PLP-cIBR-NH2-1-treated mice than in lovastatin-treated mice, suggesting that Ac-PLP-cIBR-NH2-1 was more potent than lovastatin in suppressing EAE.
Figure 4.
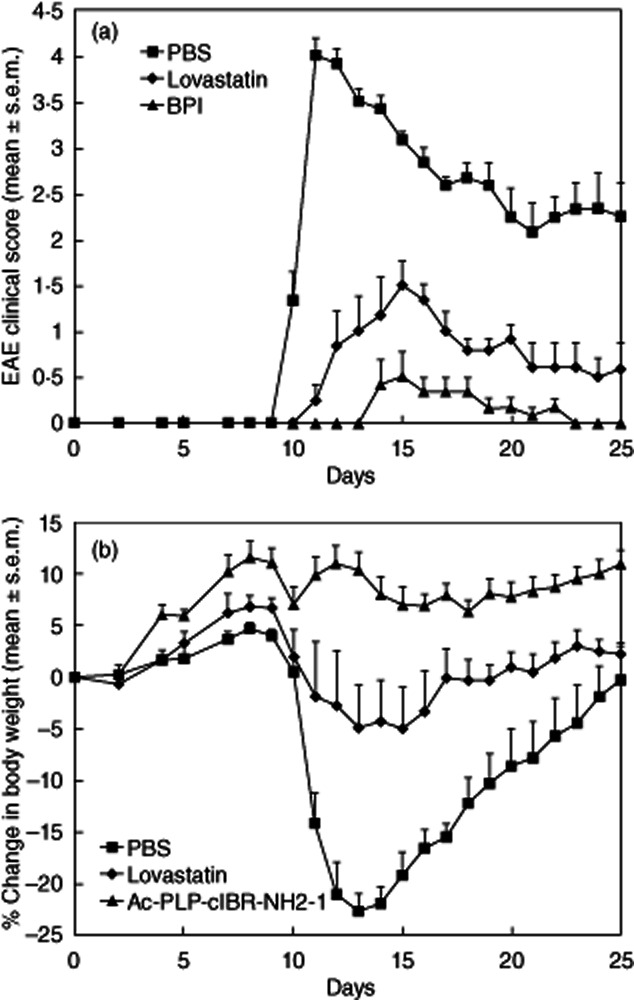
In-vivo comparison of Ac-PLP-cIBR-NH2-1 and lovastatin in the treatment of experimental autoimmune encephalomyelitis (EAE) in the mouse model. The mice were immunized with proteolipid protein (PLP)139–151/complete Freund's adjuvant (CFA) and were treated with Ac-PLP-cIBR-NH2-1 (50 nmol/injection/day) on days 4, 7 and 10 or received daily injections of lovastatin for 20 days from day 0 (495 nmol/injection/day). (a) Clinical EAE disease score. (b) Change in body weight. Results are expressed as the mean ± standard error of the mean (n = 10). There are significant differences (P < 0·001) in EAE clinical scores and loss of body weight between Ac-PLP-BPI-NH2-2- and lovastatin-treated mice.
Clinical applications of BPI
To mimic the clinical situation in which MS patients would have shown apparent signs of the disease prior to treatment, the mice were treated with a maximum of three injections of Ac-PLP-cIBR-NH2-1 to reverse the disease progression after they showed clinical scores of 1 or higher. However, almost all the PLP139–151/CFA-immunized mice showed an appreciable loss of body weight with corresponding worsening of neurological symptoms, as indicated by increasing clinical scores during days 10–12. After injection of Ac-PLP-cIBR-NH2-1, there was a rapid decline in the clinical scores, particularly on days 14–20 (P < 0·001; Fig. 5a). PBS-treated animals had a strong relapse from day 20 (scores around 0·5) to day 45 (scores around 2). In contrast, Ac-PLP-cIBR-NH2-1-treated mice showed very low relapse scores from day 20 (scores around 0) to day 45 (scores around 0·3). These mice regained their body weight faster than the control mice on days 14–20 (P < 0·001, Fig. 5b). During the relapse, Ac-PLP-cIBR-NH2-1-treated mice gained weight from days 20 to 45 while the body weights of PBS-treated mice on day 45 were similar to those on day 20 (Fig. 5b). For all except a few treated mice, two injections of Ac-PLP-cIBR-NH2-1 were sufficient to reverse the disease, and their clinical disease scores returned to a normal level (0·5 or less) within 2–3 days. In contrast, clinical scores of the control group (PBS-treated) remained fairly high for a longer period of time.
Figure 5.
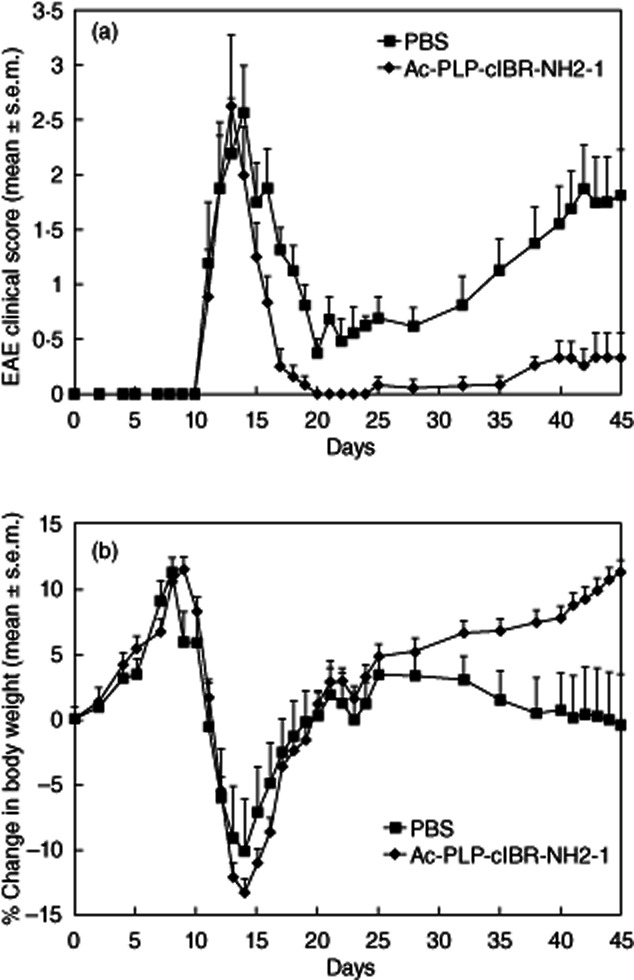
In-vivo therapeutic activity of Ac-PLP-cIBR-NH2-1 in reversing experimental autoimmune encephalomyelitis (EAE) in the mouse model. The mice were immunized with proteolipid protein (PLP)139–151/complete Freund's adjuvant (CFA) followed by intravenous treatments of Ac-PLP-cIBR-NH2-1 (50 nmol/injection/day) up to a maximum of three injections starting on the day of disease onset. (a) Clinical EAE disease score. (b) Change in body weight. Results are expressed as the mean ± standard error of the mean (n = 10). There are significant differences (P < 0·001) in EAE clinical scores and loss of body weight between Ac-PLP-cIBR-NH2-1- and phosphate-buffered saline (PBS)-treated mice.
Cytokine production by splenocytes
To elucidate the mechanisms underlying EAE suppression by Ac-PLP-cIBR-NH2-1 and lovastatin treatments, the phenotype of cytokine secretion was characterized on day 13 at the peak of disease and on day 35 during disease remission. In this study, the production of inflammatory cytokines such as interleukin (IL)-6, IL-12, IL-17, tumour necrosis factor (TNF)-α and IFN-γ was evaluated. It is clear that both Ac-PLP-cIBR-NH2-1 and lovastatin suppress IL-6 production at day 13 and that their effects were less pronounced on day 35 (Fig. 6a). It is remarkable that Ac-PLP-cIBR-NH2-1 suppresses IL-12 production almost completely at both days 13 and 35, while lovastatin treatment does not produce any suppression compared to PBS treatment (Fig. 6b). During the adverse disease state on day 13, mice treated with Ac-PLP-cIBR-NH2-1 produced lower IL-17 than lovastatin- and PBS-treated mice (Fig. 6c); on day 35, however, the IL-17 production was higher in PLP–cIBR- and lovastatin-treated mice than in those treated with PBS. Mice treated with either Ac-PLP-cIBR-NH2-1 or lovastatin had lower levels of IFN-γ on day 13 (Fig. 6d).
Figure 6.

Characterization of T helper type 1 (Th1)-like cytokine secretion phenotypes. Lymphocytes were isolated from the spleens of immunized mice that were treated with either Ac-PLP-cIBR-NH2-1 or phosphate-buffered saline (PBS) on days 4 and 7. The pooled lymphocytes in triplicate were stimulated with mitomycin-treated syngeneic splenocytes (1:10) and proteolipid protein (PLP)139–151 for 48 h. Following activation and cytokine production, the supernatants were analysed for various cytokines. Statistical significance values compared to those for PBS are as follows: (a) Ac-PLP-cIBR-NH2-1 (P < 0·001, day 13; P > 0·05, day 35), lovastatin (P < 0·01, day 13; P < 0·05, day 35), (b) Ac-PLP-cIBR-NH2-1 (P < 0·05, day 13; P < 0·05, day 35), lovastatin (P > 0·05, day 13; P > 0·05, day 35), (c) Ac-PLP-cIBR-NH2-1 (P < 0·0001, day 13; P < 0·05, day 35), lovastatin (P < 0·0001, day 13; P < 0·001, day 35) and (d) Ac-PLP-cIBR-NH2-1 (P < 0·0001, day 13; P < 0·05, day 35) and lovastatin (P < 0·0001, day 13; P > 0·05, day 35).
Next, the effect of these molecules in shifting the balance of lymphocyte subpopulations from Th17 and Th1 to Treg and/or Th2 T cells was evaluated by determining IL-2, IL-4, IL-5 and IL-10 production upon treatments. On day 13, lovastatin-treated mice produced significantly higher IL-2 than those treated with PBS; however, Ac-PLP-cIBR-NH2-1-treated mice had IL-2 similar to PBS-treated mice (Fig. 7a). In contrast, the IL-2 secretions from lymphocytes isolated from Ac-PLP-cIBR-NH2-1- and lovastatin-treated mice were lower than those from PBS-treated mice on day 35 (Fig. 7a). It is interesting to find that Ac-PLP-cIBR-NH2-1 increased production of IL-4 on day 13 compared to PBS (Fig. 7b); in contrast, lovastatin suppressed production of IL-4 compared to PBS. No detectable level of IL-4 at day 35 was seen in any group. Mice treated with either Ac-PLP-cIBR-NH2-1 or lovastatin had significantly lower levels of IL-5 on day 13. On day 35, Ac-PLP-cIBR-NH2-1-treated mice had higher IL-5 than PBS- and lovastatin-treated animals. In Ac-PLP-cIBR-NH2-1-treated mice, the IL-5 production was higher in day 35 than on day 13. On days 13 and 35, Ac-PLP-cIBR-NH2-1-treated mice had higher IL-10 than those treated with PBS, and the IL-10 production was higher at day 35 than at day 13 in peptide-treated mice (Fig. 7d). Although there was no significant difference in the secretion of IL-10 in lovastatin-treated mice on day 13, it is interesting to note that IL-10 production on day 35 was significantly higher than in PBS-treated mice (Fig. 7d).
Figure 7.

Characterization of regulatory T cells (Treg) and T helper type 2 (Th2)-like cytokine secretion phenotypes. Lymphocytes were isolated from the spleens of immunized mice that were treated with either Ac-PLP-cIBR-NH2-1 or phosphate-buffered saline (PBS) on days 4 and 7. The pooled lymphocytes in triplicate were stimulated with mitomycin-treated syngeneic splenocytes (1:10) and proteolipid protein (PLP)139–151 for 48 h. Following activation and cytokine production, the supernatants were analysed for various cytokines. Statistical significance values compared to those for PBS are as follows: (a) Ac-PLP-cIBR-NH2-1 (P > 0·05, day 13; P < 0·0001, day 35), lovastatin (P < 0·0001, day 13; P < 0·0001, day 35), (b) Ac-PLP-cIBR-NH2-1 (P < 0·01, day 13; P < 0·0001, day 35), lovastatin (P < 0·0001, day 13; P < 0·001, day 35), (c) Ac-PLP-cIBR-NH2-1 (P < 0·0001, day 13; P < 0·0001, day 35), lovastatin (P < 0·0001, day 13; P < 0·001, day 35), and (d) Ac-PLP-cIBR-NH2-1 (P < 0·01, day 13; P < 0·05, day 35), and lovastatin (P > 0·05, day 13; P < 0·01, day 35).
Histological analysis of brain sections
To investigate whether clinical improvement after treatment with Ac-PLP-cIBR-NH2-1 and lovastatin was accompanied by decreased neuropathology, the infiltration of inflammatory mononuclear cells into the CNS and demyelination at the end of the study (day 35) were evaluated. Histological analysis was conducted on day 35 because, at this time-point, the disease should have progressed into the different parts of the brain. It has been shown that the disease initiates upon inflammation and damage of the spinal cord and the disease progresses from the spinal cord to the brain. Studies investigating the time–course of BBB breakdown in EAE mice support the common notion that EAE progresses from the spinal cord to more, and deeper, regions of the brain [28]. However, others have also shown that leucocyte infiltration is more dependent upon inherent local susceptibility than on the presence of the disease in adjacent caudal regions [3]. In brain sections stained with H&E (Fig. 8a–c), no significant difference in leucocyte infiltration was detected in the three different groups. Further evaluation revealed that the cortex and dentate nucleus appeared intact in all the three groups, including the control. Vasculature appeared unremarkable, with mild gliosis throughout. Overall, purkinje cells were normal in number and no neoplasm was observed. However, area fraction analysis in brain sections stained with LFB (Fig. 8d–f) revealed significant (P < 0·001) demyelination in the control group compared to mice treated with either Ac-PLP-cIBR-NH2-1 or lovastatin (Table 2). There was no difference in the area fraction of myelin between Ac-PLP-cIBR-NH2-1- and lovastatin-treated mice (Table 2), indicating that both Ac-PLP-cIBR-NH2-1 and lovastatin suppressed the demyelination process.
Figure 8.

Histochemical study of brain sections isolated from mice on day 35. Brain sections were stained with haematoxylin and eosin in order to evaluate general cell infiltration [(a) phosphate-buffered saline (PBS)-, (b) lovastatin- and (c) Ac-PLP-cIBR-NH2-1-treated mice]. There was no significant difference in the level of leucocyte infiltration into the central nervous system (CNS) in all the groups. Brain sections were stained with Luxol Fast Blue (LFB) in order to evaluate the extent of demyelination in the CNS [(d) PBS-, (e) lovastatin- and (f) Ac-PLP-cIBR-NH2-1-treated mice]. Lovastatin- and Ac-PLP-cIBR-NH2-1-treated mice had significantly (P < 0·0001) less demyelination than PBS-treated mice.
Table 2.
Summary of area fraction results on LFB stained brain sections isolated from experimental autoimmune encephalomyelitis (EAE)-induced and treated mice
| Group | Dose | Average area* (%) |
|---|---|---|
| PBS | 100 μl/mouse on days 4, 7, and 10 | 3·32 ± 0·6 |
| Ac-PLP-cIBR-NH2-1 | 100 nmol/mouse on days 4, 7, and 10 | 18·04 ± 1·25 |
| Lovastatin | 495 nmol/mouse on days 4, 7, and 10 | 20·34 ± 1·29 |
Average area of Luxol Fast Blue (LFB)-stained brain sections expressed as the mean ± s.d. PBS: phosphate-buffered saline.
Discussion
This study showed that all three PLP–cIBR derivatives were excellent in suppressing EAE upon three injections at 100 nmol/injection, but it was difficult to differentiate the efficacies of these peptides. Minor changes in the sequence of the cIBR7 peptide may not change the binding properties of the cIBR7 fragment to the I-domain of LFA-1 receptor [17]. The major finding is that substituting the cell adhesion peptide, called LABL, that binds to ICAM-1 on PLP–BPI for cIBR-7 peptide that binds I-domain of LFA-1 in PLP–cIBR enhances the activity of BPI molecules. In this case, Ac-PLP-cIBR-NH2-1 was more potent than Ac-PLP-BPI-PEG6 and PLP peptides in suppressing EAE, suggesting that Ac-PLP-cIBR-NH2-1 is an efficient modulator of immunological synapse formation. Because the PLP–cIBR molecule has significantly higher efficacy than the parent PLP peptide, it is proposed that PLP–cIBR has different mechanisms of action compared to the mechanism of action of the parent PLP peptide.
There are several proposed mechanisms of action of PLP–cIBR in suppressing the EAE in animal models. First, the activity of PLP–cIBR molecules could be due to a mechanism similar to that of soluble antigen-specific immunotherapy [29–31]. As in the soluble antigenic peptide, the PLP portion of PLP–cIBR binds to empty MHC-II on the surface of immature dendritic cells [32], and the presence of the cIBR peptide serves merely to increase the probability of PLP–cIBR-binding to dendritic cells via LFA-1 receptors. Recognition of the PLP–MHC-II complex by TCR on naive T cells in the absence of co-stimulatory signal (Signal-2 from CD80 and CD86) on immature dendritic cells causes the naive T cells to differentiate to Treg cells [31–35]. The splenocytes from PLP–cIBR-treated mice produced higher IL-10 than that from PBS-treated mice; the IL-10 could indicate the possibility of the presence of Treg cells (Fig. 7d, P < 0·001) [36,37]. The PLP–cIBR-treated mice also produced higher transforming growth factor (TGF-β) at the peak of disease compared to PBS-treated animals (data not shown). High secretion of IL-10 and TGF-β indicates the differentiation and proliferation of Treg cells that suppress the proliferation of inflammatory Th17 and Th1 cells in PLP–cIBR-treated mice [38,39]. In future, we will evaluate the production of forkhead box protein 3 (FoxP3)-positive CD4+ T cells to determine whether BPI molecules generate Treg cell expansion. Ac-PLP-cIBR-NH2-1 could also work by inducing anergy. It is known that injection of a high dose of soluble peptide or DNA vaccination to produce antigenic peptides blocks T cell proliferation and/or IL-2 production, leading to a state of T cell unresponsiveness also referred to as anergy. Studies of patients with relapsing–remitting multiple sclerosis (RR–MS) prove that long-term treatment with glatiramer acetate resulted in T cell tolerance to peripheral antigens. The tolerance arises from a significant loss of the proliferative response in peripheral blood mononuclear cells (PBMCs) [40]. In our case, we observed significantly lower IL-2 production at the end of the study (day 35) compared to day 13, suggesting that anergy could contribute to the observed tolerance. More studies are needed to ascertain this phenomenon.
The second and third proposed mechanisms involved simultaneous binding of PLP–cIBR to MHC-II and LFA-1 to inhibit immunological synapse formation at the interface between APC and T cells. The second mechanism is due possibly to simultaneous binding of PLP–cIBR to MHC-II and LFA-1 on the surface of APC, especially mature dendritic cells. Although the majority of LFA-1 receptors are found on T cells, LFA-1 receptors are also found on APC. This binding process prevents the translocation of Signal-1 and Signal-2 during interaction between T cells and APC [8]. As a result, it prevents the differentiation or proliferation of inflammatory Th17 and Th1 cells. The third possible mechanism is that PLP–cIBR first binds to empty MHC-II on mature dendritic cells and then, upon interaction between T cells to mature dendritic cells, the cIBR portion of PLP–cIBR binds to LFA-1 on the surface of T cells. This cross-cellular receptor binding also prevents the translocation of Signal-1 and Signal-2 in forming the immunological synapse. As a result, both mechanisms suppress differentiation and proliferation of inflammatory T cells such as Th17 and Th1 and promote the differentiation of Th2 cells [41]. IL-17 production was suppressed in PLP–cIBR-treated mice compared to those treated with PBS, indicating suppression of production of Th17 cells [42]. Suppression of the Th1 cell population is reflected by lower production of IFN-γ, IL-12 and IL-6. IL-12 has a role in stimulating IFN-γ production, suggesting that there is a relationship between lowering of IL-12 and IFN-γ. Finally, treatment with PLP–cIBR also shifts the balance to Th2 cell proliferation, which is supported by the increase in IL-4-producing cells.
Our finding demonstrates that three low doses (50 nmol/injection) of Ac-PLP-cIBR-NH2-1 are more potent than 20 high doses (495 nmol/injection) of lovastatin in suppressing EAE (Fig. 4). Lovastatin has been evaluated in clinical trials for the treatment of MS as well as in suppressing EAE in the animal model [22,23,27]. Greenwood et al. reported that treatment of EAE animals with lovastatin prior to onset of disease inhibited the disease effectively compared to the control treatment [27]. The disease was well controlled as long as the daily injections were administered; however, most of the animals experienced EAE relapse rapidly upon cessation of treatment [27]. Additionally, lovastatin administration after disease onset had little therapeutic effect [27]. In contrast, treatment with BPI molecules after disease manifestation resulted in rapid recovery as well as inhibition of any relapse of EAE, suggesting that BPI molecules have different mechanisms of action than does lovastatin in suppressing EAE [12].
Evaluation of cytokine production at the peak of the disease on day 13 and at the remission of the disease on day 35 was carried out to differentiate between the mechanisms of action of PLP–cIBR and lovastatin. Unlike PLP–cIBR, lovastatin does not enhance IL-10 production on day 13. However, during remission on day 35, the lovastatin-treated mice produced more IL-10 than did mice treated with PBS.
Both PLP–cIBR- and lovastatin-treated mice had lower IFN-γ and IL-6 production on days 13 and 35, suggesting that both molecules suppress Th1 proliferation. IL-6 promotes inflammatory response and modulates body temperature control in the hypothalamus to generate fever. IL-6 is produced by macrophages, monocytes and Th2 cells, which can stimulate antibody production by B cells. On day 13, a higher secretion of IL-2 was found in lovastatin-treated mice than in those treated with PBS and PLP–cIBR (Fig. 7a), indicating a major difference in mechanism of action between lovastatin and PLP–cIBR. Activated Th1 cells produce IL-2 upon engagement of the TCR and the CD28 co-stimulatory molecule to differentiate a cytotoxic T cell (CTL) precursor to CTL [43]. Although IL-2 plays a crucial part in the development of FoxP3+ Treg [43], the inability of Treg cells to produce IL-2 is related directly to the suppressive activity of FoxP3+. In the absence of a time-dependent study on cytokine recall responses, it is hard to know the appropriate time at which to conduct cytokine recall responses in the periphery and correlate them with the disease progression, as cytokines in the periphery do not reflect that in the brain. Thus, days 13 and 35 were chosen based on the severity of the disease and the potential relapse time, respectively. Floris et al. showed that a few or no cellular infiltrates migrate to the spinal cord or cerebellum in the early stage of the disease (days 0–13); however, they observed a more than 10-fold increase of cell infiltrates at the peak of the disease (days 14–17) [25]. This result suggests that prior to day 14 these autoreactive cells were still in the periphery. McGeachy et al. showed that the proportion of CD4+CD25+ cells in the spleen remained fairly constant from days 13 to 20, but they found a marked increase in the percentage of these CD4+CD25+ cells in the CNS as EAE progressed from days 13 to 20 [44]. In addition, Fissolo et al. conducted a cytokine recall study on day 14 and showed that myelin oligodendrocyte glycoprotein (MOG)–DNA vaccination inhibited the progression of EAE through expansion of Treg cells in the periphery [45]. These periphery Treg cells could then migrate and accumulate in the CNS [45]. Thus, evaluating cytokine recall responses in splenic cell cultures on day 13 should provide an insight into cytokine production just prior to disease flares in treated and untreated mice. Conversely, day 35 should provide information into what occurs before relapse or the long-term effect of BPI treatment.
Another dramatic difference between lovastatin and PLP–cIBR is in the production of IL-4; lovastatin lowered IL-4 production compared to PBS, while PLP–cIBR increased the production of IL-4. This suggests that lovastatin decreases the population of Th2 cells, while PLP–cIBR proliferates these cells. PLP–cIBR-treated mice had higher levels of IL-5 compared to PBS- and lovastatin-treated mice at the peak and remission of the disease. This supports the idea that PLP–cIBR increases the population of Th2 cells. Lovastatin had the same levels of IL-5 at the peak and remission of the disease. Lovastatin suppressed the Th17 differentiation and proliferation to a lesser extent than did PLP–cIBR on the peak of the disease on day 13; however, IL-17 production in lovastatin- and PLP–cIBR-treated mice was higher than in PBS-treated mice at the disease remission (day 35). While lovastatin did not suppress the production of IL-12, PLP–cIBR caused significant suppression of IL-12 produced normally by macrophages and dendritic cells; there is a relationship between IL-12 and IFN-γ production in which increased production of IFN-γ by Th1 cells stimulates macrophages to produce IL-12. Hence, the result suggests that PLP–cIBR is a more dramatic suppressor of Th1 differentiation than lovastatin.
In contrast to the differences in various cytokine profiles, there was no significant infiltration of leucocytes found in the brain in Ac-PLP-cIBR-NH2-1- and lovastatin-treated mice on day 35 when compared to PBS-treated mice, as demonstrated by H&E staining (Fig. 8a–c). Pifarre et al. observed that EAE mice treated with sildefanil had dramatically decreased T cell infiltration and smaller infiltrates compared to vehicle-treated mice on day 21 post-immunization; however, there was no significant level of T cell infiltration after 26 days post-immunization [46]. This is one of the possible reasons why there was no observable difference in brain infiltration of leucocytes in PLP–cIBR-, lovastatin- and PBS-treated animals after the remission of the disease on day 35 (i.e. during remission). This is consistent with the thought that T cells infiltrating the CNS are important only as the initiator and early effector cells in the development of EAE, while infiltrated macrophages, dendritic cells and resident microglia constitute the ultimate effector cells that amplify late-stage neuroinflammation and tissue damage. Also, the fact that we saw extensive demyelination in untreated mice compared to treated mice indicates that inflammatory infiltrates were present prior to day 35, but had been cleared away. We observed less demyelination in Ac-PLP-cIBR-NH2-1- and lovastatin-treated mice because these molecules inhibited cellular infiltration into the CNS. In the future, we would like to investigate time-dependent infiltration of leucocytes as well as immunostaining to determine periods of severe infiltration and what types of cells infiltrate the CNS at a particular stage of the disease. Finally, less demyelination of CNS in Ac-PLP-cIBR-NH2-1- and lovastatin-treated mice compared to PBS-treated mice suggests that both Ac-PLP-cIBR-NH2-1- and lovastatin-treated mice received protection from demyelination. It is also observed that fewer and lower doses of Ac-PLP-cIBR-NH2-1 afforded the same protection as frequent and high doses of lovastatin.
One of the major challenges in developing newer therapies is safety. For example, lovastatin is thought to be a membrane disruptor, and long-term treatment with lovastatin may affect several aspects of remyelination. Maier et al. reported that, although lovastatin promoted the formation of membrane sheets by primary oligodendrocytes (OLGs), the membranes lacked major myelin proteins (e.g. MBP and PLP) and showed an abnormal microtubule cytoskeleton [47]. Although lovastatin could be a potential agent for the treatment of MS, it might be a good idea to conduct extended studies on its effects in the CNS, particularly in the targeted patient population. This is because cholesterol is a major component of myelin [48], and inhibition of cholesterol synthesis as well as inhibition of protein isoprenylation by statins may interfere with the desired remyelination of axons in MS patients. Taking this into consideration, the hope is that designing molecules that are more specific and potent at low doses will help to minimize some of the adverse side effects. Although the in-vivo efficacy of PLP–cIBR is excellent at low doses, more studies are needed to ascertain its safety.
In conclusion, PLP–cIBR has excellent efficacy in inhibiting EAE and is a potential therapeutic agent for the treatment of MS. The EAE-suppressive activity of PLP–cIBR molecules could be attributed to inhibition of the immunological synapse, leading possibly to suppression of Th1 and Th17 differentiation and proliferation. In the future, the effect of PLP–cIBR on Treg differentiation and proliferation will be evaluated. Also, data obtained from LFB-stained brain sections in Ac-PLP-cIBR-NH2-1-treated mice suggest that PLP–cIBR molecules could promote protection against demyelination. The effect of PLP–cIBR in preventing spinal cord inflammation and damage will be studied in future. Further studies in structural modification and proper dosing regimens (e.g. dose and dosing schedule) are needed to improve the efficacy and safety of PLP–cIBR molecules.
Acknowledgments
We acknowledge the National Institutes of Health (R01-AI-063002 and R56-AI-063002) and National Multiple Sclerosis Society for supporting this work. We also thank Nancy Harmony for proofreading the manuscript.
Disclosure
The authors do not have any conflict of interests related to this work.
References
- 1.Lassmann H. Classification of demyelinating diseases at the interface between etiology and pathogenesis. Curr Opin Neurol. 2001;14:253–258. doi: 10.1097/00019052-200106000-00001. [DOI] [PubMed] [Google Scholar]
- 2.Badawi AH, Siahaan TJ. Immune modulating peptides for the treatment and suppression of multiple sclerosis. Clin Immunol. 2012;144:127–138. doi: 10.1016/j.clim.2012.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tonra JR, Reiseter BS, Kolbeck R, et al. Comparison of the timing of acute blood–brain barrier breakdown to rabbit immunoglobulin G in the cerebellum and spinal cord of mice with experimental autoimmune encephalomyelitis. J Comp Neurol. 2001;430:131–144. [PubMed] [Google Scholar]
- 4.Hafler DA. Multiple sclerosis. J Clin Invest. 2004;113:788–794. doi: 10.1172/JCI21357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Johnson KP. Management of relapsing/remitting multiple sclerosis with copolymer 1 (Copaxone) Mult Scler. 1996;1:325–326. doi: 10.1177/135245859600100606. [DOI] [PubMed] [Google Scholar]
- 6.Castelli-Haley J, Oleen-Burkey MA, Lage MJ, Johnson KP. Glatiramer acetate and interferon beta-1b: a study of outcomes among patients with multiple sclerosis. Adv Ther. 2009;26:552–562. doi: 10.1007/s12325-009-0028-3. [DOI] [PubMed] [Google Scholar]
- 7.Dhib-Jalbut S, Chen M, Henschel K, Ford D, Costello K, Panitch H. Effect of combined IFNbeta-1a and glatiramer acetate therapy on GA-specific T-cell responses in multiple sclerosis. Mult Scler. 2002;8:485–491. doi: 10.1191/1352458502ms862oa. [DOI] [PubMed] [Google Scholar]
- 8.Manikwar P, Kiptoo P, Badawi AH, Buyuktimkin B, Siahaan TJ. Antigen-specific blocking of CD4-specific immunological synapse formation using BPI and current therapies for autoimmune diseases. Med Res Rev. 2012;32:727–764. doi: 10.1002/med.20243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Young DA, Lowe LD, Booth SS, et al. IL-4, IL-10, IL-13, and TGF-beta from an altered peptide ligand-specific Th2 cell clone down-regulate adoptive transfer of experimental autoimmune encephalomyelitis. J Immunol. 2000;164:3563–3572. doi: 10.4049/jimmunol.164.7.3563. [DOI] [PubMed] [Google Scholar]
- 10.Nicholson LB, Murtaza A, Hafler BP, Sette A, Kuchroo VK. A T cell receptor antagonist peptide induces T cells that mediate bystander suppression and prevent autoimmune encephalomyelitis induced with multiple myelin antigens. Proc Natl Acad Sci USA. 1997;94:9279–9284. doi: 10.1073/pnas.94.17.9279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tuohy VK, Lu Z, Sobel RA, Laursen RA, Lees MB. Identification of an encephalitogenic determinant of myelin proteolipid protein for SJL mice. J Immunol. 1989;142:1523–1527. [PubMed] [Google Scholar]
- 12.Kobayashi N, Kiptoo P, Kobayashi H, Ridwan R, Brocke S, Siahaan TJ. Prophylactic and therapeutic suppression of experimental autoimmune encephalomyelitis by a novel bifunctional peptide inhibitor. Clin Immunol. 2008;129:69–79. doi: 10.1016/j.clim.2008.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kobayashi N, Kobayashi H, Gu L, Malefyt T, Siahaan TJ. Antigen-specific suppression of experimental autoimmune encephalomyelitis by a novel bifunctional peptide inhibitor. J Pharmacol Exp Ther. 2007;322:879–886. doi: 10.1124/jpet.107.123257. [DOI] [PubMed] [Google Scholar]
- 14.Ridwan R, Kiptoo P, Kobayashi N, et al. Antigen-specific suppression of experimental autoimmune encephalomyelitis by a novel bifunctional peptide inhibitor: structure optimization and pharmacokinetics. J Pharmacol Exp Ther. 2010;332:1136–1145. doi: 10.1124/jpet.109.161109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Murray JS, Oney S, Page JE, et al. Suppression of type 1 diabetes in NOD mice by bifunctional peptide inhibitor: modulation of the immunological synapse formation. Chem Biol Drug Des. 2007;70:227–236. doi: 10.1111/j.1747-0285.2007.00552.x. [DOI] [PubMed] [Google Scholar]
- 16.Badawi AH, Kiptoo P, Wang WT, et al. Suppression of EAE and prevention of blood–brain barrier breakdown after vaccination with novel bifunctional peptide inhibitor. Neuropharmacology. 2012;62:1874–1881. doi: 10.1016/j.neuropharm.2011.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Iskandarsyah, Tejo BA, Tambunan US, Verkhivker G, Siahaan TJ. Structural modifications of ICAM-1 cyclic peptides to improve the activity to inhibit heterotypic adhesion of T cells. Chem Biol Drug Des. 2008;72:27–33. doi: 10.1111/j.1747-0285.2008.00676.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Anderson ME, Siahaan TJ. Mechanism of binding and internalization of ICAM-1-derived cyclic peptides by LFA-1 on the surface of T cells: a potential method for targeted drug delivery. Pharm Res. 2003;20:1523–1532. doi: 10.1023/a:1026188212126. [DOI] [PubMed] [Google Scholar]
- 19.Anderson ME, Tejo BA, Yakovleva T, Siahaan TJ. Characterization of binding properties of ICAM-1 peptides to LFA-1: inhibitors of T-cell adhesion. Chem Biol Drug Des. 2006;68:20–28. doi: 10.1111/j.1747-0285.2006.00407.x. [DOI] [PubMed] [Google Scholar]
- 20.Zimmerman T, Oyarzabal J, Sebastian ES, et al. ICAM-1 peptide inhibitors of T-cell adhesion bind to the allosteric site of LFA-1. An NMR characterization. Chem Biol Drug Des. 2007;70:347–353. doi: 10.1111/j.1747-0285.2007.00566.x. [DOI] [PubMed] [Google Scholar]
- 21.Youssef S, Stuve O, Patarroyo JC, et al. The HMG-CoA reductase inhibitor, atorvastatin, promotes a Th2 bias and reverses paralysis in central nervous system autoimmune disease. Nature. 2002;420:78–84. doi: 10.1038/nature01158. [DOI] [PubMed] [Google Scholar]
- 22.Paintlia AS, Paintlia MK, Singh AK, et al. Regulation of gene expression associated with acute experimental autoimmune encephalomyelitis by lovastatin. J Neurosci Res. 2004;77:63–81. doi: 10.1002/jnr.20130. [DOI] [PubMed] [Google Scholar]
- 23.Paintlia AS, Paintlia MK, Singh I, Singh AK. Combined medication of lovastatin with rolipram suppresses severity of experimental autoimmune encephalomyelitis. Exp Neurol. 2008;214:168–180. doi: 10.1016/j.expneurol.2008.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vollmer T, Key L, Durkalski V, et al. Oral simvastatin treatment in relapsing-remitting multiple sclerosis. Lancet. 2004;363:1607–1608. doi: 10.1016/S0140-6736(04)16205-3. [DOI] [PubMed] [Google Scholar]
- 25.Floris S, Blezer EL, Schreibelt G, et al. Blood–brain barrier permeability and monocyte infiltration in experimental allergic encephalomyelitis: a quantitative MRI study. Brain. 2004;127:616–627. doi: 10.1093/brain/awh068. [DOI] [PubMed] [Google Scholar]
- 26.Kallen J, Welzenbach K, Ramage P, et al. Structural basis for LFA-1 inhibition upon lovastatin binding to the CD11a I-domain. J Mol Biol. 1999;292:1–9. doi: 10.1006/jmbi.1999.3047. [DOI] [PubMed] [Google Scholar]
- 27.Greenwood J, Walters CE, Pryce G, et al. Lovastatin inhibits brain endothelial cell Rho-mediated lymphocyte migration and attenuates experimental autoimmune encephalomyelitis. FASEB J. 2003;17:905–907. doi: 10.1096/fj.02-1014fje. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cross AH, O'Mara T, Raine CS. Chronologic localization of myelin-reactive cells in the lesions of relapsing EAE: implications for the study of multiple sclerosis. Neurology. 1993;43:1028–1033. doi: 10.1212/wnl.43.5.1028. [DOI] [PubMed] [Google Scholar]
- 29.Yamazaki S, Inaba K, Tarbell KV, Steinman RM. Dendritic cells expand antigen-specific Foxp3+ CD25+ CD4+ regulatory T cells including suppressors of alloreactivity. Immunol Rev. 2006;212:314–329. doi: 10.1111/j.0105-2896.2006.00422.x. [DOI] [PubMed] [Google Scholar]
- 30.Sabatos-Peyton CA, Verhagen J, Wraith DC. Antigen-specific immunotherapy of autoimmune and allergic diseases. Curr Opin Immunol. 2010;22:609–615. doi: 10.1016/j.coi.2010.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Larche M, Wraith DC. Peptide-based therapeutic vaccines for allergic and autoimmune diseases. Nat Med. 2005;11:S69–76. doi: 10.1038/nm1226. [DOI] [PubMed] [Google Scholar]
- 32.Santambrogio L, Sato AK, Fischer FR, Dorf ME, Stern LJ. Abundant empty class II MHC molecules on the surface of immature dendritic cells. Proc Natl Acad Sci USA. 1999;96:15050–15055. doi: 10.1073/pnas.96.26.15050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Metzler B, Anderton SM, Manickasingham SP, Wraith DC. Kinetics of peptide uptake and tissue distribution following a single intranasal dose of peptide. Immunol Invest. 2000;29:61–70. doi: 10.3109/08820130009105145. [DOI] [PubMed] [Google Scholar]
- 34.Menges M, Rossner S, Voigtlander C, et al. Repetitive injections of dendritic cells matured with tumor necrosis factor alpha induce antigen-specific protection of mice from autoimmunity. J Exp Med. 2002;195:15–21. doi: 10.1084/jem.20011341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dhodapkar MV, Steinman RM, Krasovsky J, Munz C, Bhardwaj N. Antigen-specific inhibition of effector T cell function in humans after injection of immature dendritic cells. J Exp Med. 2001;193:233–238. doi: 10.1084/jem.193.2.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gabrysova L, Wraith DC. Antigenic strength controls the generation of antigen-specific IL-10-secreting T regulatory cells. Eur J Immunol. 2010;40:1386–1395. doi: 10.1002/eji.200940151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mazza G, Sabatos-Peyton CA, Protheroe RE, Herman A, Campbell JD, Wraith DC. Isolation and characterization of human interleukin-10-secreting T cells from peripheral blood. Hum Immunol. 2010;71:225–234. doi: 10.1016/j.humimm.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yu P, Gregg RK, Bell JJ, et al. Specific T regulatory cells display broad suppressive functions against experimental allergic encephalomyelitis upon activation with cognate antigen. J Immunol. 2005;174:6772–6780. doi: 10.4049/jimmunol.174.11.6772. [DOI] [PubMed] [Google Scholar]
- 39.Fu S, Zhang N, Yopp AC, et al. TGF-beta induces Foxp3+ T-regulatory cells from CD4+ CD25− precursors. Am J Transplant. 2004;4:1614–1627. doi: 10.1111/j.1600-6143.2004.00566.x. [DOI] [PubMed] [Google Scholar]
- 40.Fransson ME, Liljenfeldt LS, Fagius J, Totterman TH, Loskog AS. The T-cell pool is anergized in patients with multiple sclerosis in remission. Immunology. 2009;126:92–101. doi: 10.1111/j.1365-2567.2008.02881.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hedegaard CJ, Krakauer M, Bendtzen K, Lund H, Sellebjerg F, Nielsen CH. T helper cell type 1 (Th1), Th2 and Th17 responses to myelin basic protein and disease activity in multiple sclerosis. Immunology. 2008;125:161–169. doi: 10.1111/j.1365-2567.2008.02837.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 cells. Annu Rev Immunol. 2009;27:485–517. doi: 10.1146/annurev.immunol.021908.132710. [DOI] [PubMed] [Google Scholar]
- 43.Cheng G, Yu A, Malek TR. T-cell tolerance and the multi-functional role of IL-2R signaling in T-regulatory cells. Immunol Rev. 2011;241:63–76. doi: 10.1111/j.1600-065X.2011.01004.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McGeachy MJ, Stephens LA, Anderton SM. Natural recovery and protection from autoimmune encephalomyelitis: contribution of CD4+CD25+ regulatory cells within the central nervous system. J Immunol. 2005;175:3025–3032. doi: 10.4049/jimmunol.175.5.3025. [DOI] [PubMed] [Google Scholar]
- 45.Fissolo N, Costa C, Nurtdinov RN, et al. Treatment with MOG–DNA vaccines induces CD4(+)CD25(+)FoxP3(+) regulatory T cells and up-regulates genes with neuroprotective functions in experimental autoimmune encephalomyelitis. J Neuroinflammation. 2012;9:1–13. doi: 10.1186/1742-2094-9-139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pifarre P, Prado J, Baltrons MA, et al. Sildenafil (Viagra) ameliorates clinical symptoms and neuropathology in a mouse model of multiple sclerosis. Acta Neuropathol. 2011;121:499–508. doi: 10.1007/s00401-010-0795-6. [DOI] [PubMed] [Google Scholar]
- 47.Maier O, De Jonge J, Nomden A, Hoekstra D, Baron W. Lovastatin induces the formation of abnormal myelin-like membrane sheets in primary oligodendrocytes. Glia. 2009;57:402–413. doi: 10.1002/glia.20769. [DOI] [PubMed] [Google Scholar]
- 48.Morell P, Jurevics H. Origin of cholesterol in myelin. Neurochem Res. 1996;21:463–470. doi: 10.1007/BF02527711. [DOI] [PubMed] [Google Scholar]