

Abstract
High mobility group box 1 (HMGB1) is an established inflammatory mediator when released from cells. Recent studies have implicated extracellular HMGB1 in the pathogenesis of various autoimmune diseases. The objective of this study was to determine whether HMGB1 could be a therapeutic target for experimental autoimmune encephalomyelitis (EAE). In this study, an anti-HMGB1 monoclonal antibody was injected intraperitoneally into a mouse model of EAE. We also measured serum cytokines levels in EAE and anti-HMGB1 monoclonal antibody-treated EAE. As a result, intraperitoneal injection of an anti-HMGB1 monoclonal antibody ameliorated the clinical and pathological severity of EAE and attenuated interleukin-17 up-regulation in serum. In conclusion, HMGB1 is involved in EAE pathogenesis and could trigger inflammation in the central nervous system. The novel aspect of this study is the demonstration that anti-HMGB1 ameliorates EAE. HMGB1 may be a novel therapeutic strategy for multiple sclerosis.
Keywords: cytokine, experimental autoimmune encephalomyelitis, high mobility group box 1, monoclonal antibody, multiple sclerosis
Introduction
Multiple sclerosis (MS) is an autoimmune-mediated inflammatory disease of the central nervous system (CNS) [1,2]. Although treatment with fingolimod [3], rituximab [4], alemtuzumab [5] and natalizumab [6] has been newly introduced for preventing MS relapses, therapies for acute MS exacerbations are limited.
High mobility group box 1 (HMGB1) was identified recently as a damage- or pathogen-associated molecular pattern [7]. Within a cell, HMGB1 binds to DNA and participates in many nuclear functions [8], and when released from a cell it acts as an alarmin and is involved in inflammatory processes [9]. HMGB1 is released from activated immune cells or damaged, dying cells during necrosis and during the late phase of cellular apoptosis [10,11]. Extracellular HMGB1 exerts its biological actions by binding to cell-surface receptors, such as the receptor of advanced glycation end-products (RAGE), Toll-like receptor (TLR)-2, TLR-4 and intracellular receptor TLR-9 [12–16]. It has also been reported that HMGB1 can activate autoreactive B cells [17] and that the interaction of HMGB1 with RAGE on endothelial cells up-regulates vascular cell adhesion molecules and intracellular adhesion molecules, leading to the recruitment of macrophages and monocytes for promotion of cell migration [9,18]. Purified recombinant HMGB1 added to cultures of human monocytes stimulated the release of inflammatory cytokines, such as tumour necrosis factor (TNF)-α, interleukin (IL)-1a, IL-1b, IL-1 receptor antagonist, IL-6, IL-8, macrophage inflammatory protein (MIP)-1a and MIP-1b, which amplify inflammation [19].
Recent studies have shown associations between HMGB1 and autoimmune diseases. High HMGB1 levels have been found in rheumatoid arthritis (RA), Sjögren's syndrome (SS), Churg–Strauss syndrome and systemic lupus erythematosus (SLE) [20–23]. Although important roles of HMGB1 in some autoimmune diseases have been examined, and one study reported that HMGB1 and its receptors RAGE, TLR-2 and TLR-4 were up-regulated in active lesions of patients with MS and experimental autoimmune encephalomyelitis (EAE) [24], the treatment potential of anti-HMGB1 monoclonal antibody for EAE has not been studied extensively.
The objective of this study was to determine whether HMGB1 can be a therapeutic target for EAE. For this objective, we administered an anti-HMGB1 monoclonal antibody to a mouse model of EAE which is a widely used animal model for MS.
Materials and methods
EAE induction in mice
Wild-type C57BL/6 mice were purchased from Japan SLC, Inc. (Shizuoka, Japan). The mice were housed in specific pathogen-free facilities at Chiba University with a maximum of four animals per cage, with free access to water and standard rodent chow. EAE was induced using immunization with myelin oligodendrocyte glycoprotein (MOG). C57BL/6 mice were injected subcutaneously at two sites, with a total of 200 μg MOG peptide 35–55 in complete Freund's adjuvant containing 400 μg of killed Mycobacterium tuberculosis H37Ra. They were also injected intraperitoneally with 500 ng pertussis toxin on the day of immunization (days 1 and 2) using Hooke kits (EK-0115; Hooke Laboratories, Lawrence, MA, USA). EAE was scored on the following scale: 0 = no clinical signs; 1 = partial paralysis of tail; 2 = flaccid tail; 3 = limp tail and partial weakness of hind legs; 4 = limp tail and complete weakness of hind legs; 5 = limp tail, complete weakness of hind legs and partial of front legs; and 6 = complete hind and front legs paralysis. All experimental animal procedures were approved by the Institutional Animal Care and Use Committee of Chiba University.
Treatment with anti-HMGB1 monoclonal antibody
We evaluated the effects of an anti-HMGB1 mouse monoclonal antibody (Abnova Corporation, Taipei, Taiwan) on EAE development. For comparison, we used mouse immunoglobulin (Ig)G (Abcam, Cambridge, UK). Both anti-HMBG1 antibody and IgG were prepared in sterile phosphate-buffered saline (PBS) and 200 μl was injected intraperitoneally at each administration. Mice immunized with MOG were administered either 20 μg anti-HMGB1 monoclonal antibody [EAE + anti-HMGB1(20) group; n = 8] or 5 μg anti-HMGB1 monoclonal antibody [EAE + anti-HMGB1(5) group; n = 6] on days 11–15 after immunization with MOG. For comparison, mice received 20 μg mouse IgG [EAE + IgG(20) group; n = 6] on days 11–15 after immunization with MOG. Control EAE mice were administered 200 μl PBS alone (EAE + PBS group; n = 8). Two of eight mice in each EAE + anti-HMGB1(20) group and each EAE + PBS group were for autopsy, and they were excluded for the analysis of cytokines or clinical score.
Serum cytokines in mice
To examine possible mechanisms by which the anti-HMGB1 monoclonal antibody could attenuate EAE, we determined serum IL-4, IL-6, IL-10, IL-17, interferon (IFN)-γ and TNF-α levels in mice serum. Determinations were performed for EAE-induced mice on day 1 (before immunization with MOG) and day 18 after EAE induction using a multiplexed fluorescent magnetic bead-based immunoassay (Bio-Rad Laboratories, Hercules, CA, USA), according to the manufacturer's instructions. In brief, serum samples were centrifuged and supernatants were collected and analysed simultaneously for the above-mentioned cytokines. All serum samples were diluted fourfold with specific Bio-Plex sample diluents. Anti-cytokine-conjugated beads (50 μl) were added to wells of a 96-well filter plate and washed twice. Next, 50 μl of either sample or cytokine standard was added to wells and incubated for 30 min. After three washes, detection antibody (25 μl) was added to each well and incubated for 30 min. The plate was washed three times and 50 μl of streptavidin–phycoerythrin was added to each well, followed by another 10 min of incubation. Finally, 125 μl of assay buffer was added and analysed used a Bio-Plex array reader (Bio-Rad). Cytokine levels were calculated with reference to a standard curve for each cytokine.
Pathological analysis
Mice spinal cords were removed on day 18 after EAE induction. Mice that had median severity scores in the EAE + anti-HMGB1(20) and EAE + PBS groups as well as normal (untreated) mice were killed. Pathological examinations were performed using formalin-fixed sections of spinal cords. Spinal cord tissue was processed as follows: after initial fixation in formalin, the spinal cord tissue was cut at 10 μm in the axial plane from the cervical to lumbar spinal cord and stained with haematoxylin and eosin (H&E) and Luxol Fast Blue (LFB). Immunohistochemical staining of spinal cord sections was performed by the avidin–biotin complex method, using a rabbit monoclonal antibody against HMGB1 (Abnova Corporation; species reactivity: human, mouse, rat). After section deparaffinization with xylene and gradual dehydration, endogenous peroxidase activity was blocked with 0·5% H2O2 for 15 min. Tissue sections were incubated with 10% normal goat serum (G9023; Sigma-Aldrich, Tokyo, Japan) in PBS and diluted primary antibody (rabbit monoclonal antibody against mouse HMGB1, 1:1000) at 4°C overnight. The sections were washed in PBS containing 0·05% Tween-20 (PBST), followed by incubation with the secondary antibody biotinylated goat anti-rabbit IgG (BA-1000, diluted 1:1000; Vector Laboratories, Burlingame, CA, USA) at 4°C overnight. The sections were then washed in PBST and incubated with Vectastain ABC reagent (PK-6100, diluted 1:1000; Vector Laboratories) for 2 h, and then washed in PBST. Finally, staining was visualized using 3,3-diaminobenzidine tetrahydrochloride (Sigma-Aldrich) and 0·03% hydrogen peroxide in Tris-buffered saline for 10 min. They were then examined under a light microscope.
For the histological evaluation, we examined 10 transverse sections from the cervical to lumbar spinal cord per mouse and scored inflammation (inflammatory index) as follows: 0, no inflammation; 1, cellular infiltration only in the perivascular areas and meninges; 2, mild cellular infiltration in the parenchyma; 3, moderate cellular infiltration in the parenchyma; and 4, severe cellular infiltration in parenchyma.
Statistical analyses
All data were analysed according to the intention-to-treat principle. For baseline variables, groups were compared using Wilcoxon's signed-rank test for paired continuous measures and the Mann–Whitney U-test for unpaired continuous measures, as appropriate. All comparisons were planned, statistical tests were two-sided and P < 0·05 was considered statistically significant.
Results
Anti-HMGB1 monoclonal antibody ameliorates the severity of EAE
In EAE-induced mice, the EAE + anti-HMGB1(20) group reduced the severity of EAE significantly at days 16–21, 26 and 27 (P < 0·05) compared with the EAE + PBS group (Fig. 1a). The clinical score of the EAE + anti-HMGB1(20) group [mean ± standard error of the mean (s.e.m.); 2·67 ± 0·49] was still lower than the EAE + PBS group (3·17 ± 0·40) at day 45. There were no significant differences between the EAE + PBS and EAE + anti-HMGB1(5) groups or between the EAE + PBS and EAE + IgG(20) groups. The mean ± s.e.m. maximum EAE scores and cumulative EAE scores of each group (from days 1 to 30) are shown in Fig. 1b. The cumulative EAE scores for the EAE + anti-HMGB1(20) group were significantly lower than those for the EAE + PBS group (P = 0·010) and the same tendency (P = 0·096) was seen in the maximum EAE scores, but not significant.
Figure 1.
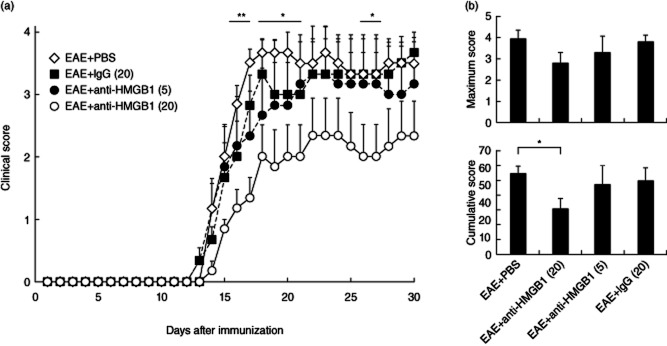
Effects of administering an anti-high mobility group box 1 (HMGB1) monoclonal antibody on experimental autoimmune encephalomyelitis (EAE) clinical scores of mice. (a) Clinical EAE disease scores in EAE-induced mice. On days 11–15 after EAE induction in mice, mice were injected intraperitoneally with either 20 μg/day of anti-HMGB1 monoclonal antibody [EAE + anti-HMGB1(20); n = 6], 5 μg/day anti-HMGB1 monoclonal antibody [EAE + anti-HMGB1(5); n = 6] or 20 μg/day of control mouse immunoglobulin (Ig)G [EAE + IgG(20); n = 6]. Untreated control EAE mice were administered phosphate-buffered saline (PBS) [EAE + PBS; n = 6]. Clinical scores for the EAE + anti-HMGB1(20) group were reduced compared with those for the EAE + PBS group (days 16–21, 26 and 27). Results are mean ± standard error of the mean (s.e.m.). **P < 0·01 and *P < 0·05 by Mann–Whitney U-test between EAE + anti-HMGB1(20) and EAE + PBS groups. (b) The mean ± s.e.m. of the maximum EAE scores and cumulative EAE scores of each group. The cumulative EAE scores for the EAE + anti-HMGB1(20) group were significantly lower than those for the EAE + PBS group (P = 0·010).
Anti-HMGB1 monoclonal antibody attenuates serum IL-17 up-regulation in EAE
Serum IL-6 (P = 0·046) and IL-17 (P = 0·023) levels in the EAE + PBS group (n = 6) were increased significantly on day 18 compared with those on day 1. In contrast, serum IL-17 (P = 0·463) levels in the EAE + anti-HMGB1(20) group (n = 6) at day 18 did not show any increase compared with those on day 1, although there were increased IL-6 (P = 0·046) levels in this group (Fig. 2) (Wilcoxon's signed-rank test). Other cytokines, including IL-4, IL-10, IFN-γ and TNF-α, were not significantly different between the EAE + PBS group and the EAE + anti-HMGB1(20) group. There is no significant difference in cytokine levels between EAE + anti-HMGB1(20) group and EAE + PBS (P = 0·046) group on days 1 and 18 (Mann–Whitney U-test).
Figure 2.
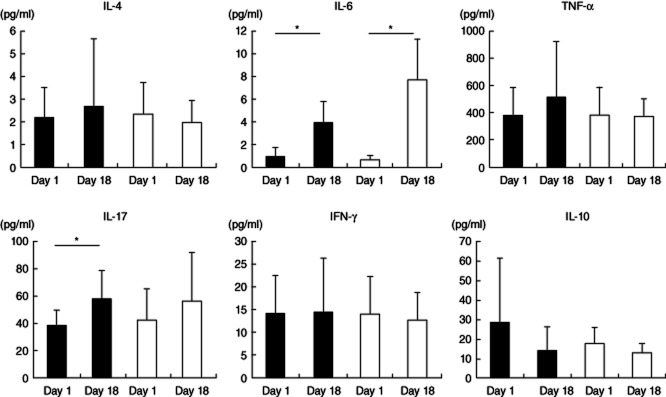
Serum cytokine changes in anti-high mobility group box 1 (HMGB1) monoclonal antibody-treated experimental autoimmune encephalomyelitis (EAE) mice and untreated EAE mice. Serum cytokine levels were determined in EAE mice that received 20 μg/day anti-HMGB1 monoclonal antibody [EAE + anti-HMGB1(20), n = 6] (white bars) or phosphate-buffered saline (PBS) [EAE + PBS, n = 6] (black bars). Serum interleukin (IL)-6 levels in the EAE + anti-HMGB1(20) (P = 0·046) and EAE + PBS (P = 0·046) groups were increased on day 18 compared with those on day 1, while serum IL-17 levels were increased only in the EAE + PBS group (P = 0·023) on day 18 (Wilcoxon's signed-rank test). There are no significant differences in cytokine levels between the EAE + anti-HMGB1(20) group and EAE + PBS (P = 0·046) group on days 1 and 18 (Mann–Whitney U-test).
Anti-HMGB1 antibody attenuates CNS inflammation and demyelination in EAE
Median EAE scores for the EAE + anti-HMGB1(20) and EAE + PBS groups were 3 and 4, respectively. Mice were killed on day 18 after EAE induction. Anti-HMGB1 monoclonal antibody treatment (20 μg/day × 5 days/mouse) reduced infiltration of cells and demyelination in the spinal cord of EAE mice, as well as the inflammatory index [EAE + PBS group = 2·80 ± 0·13 (mean ± s.e.); EAE + anti-HMGB1(20) group = 1·50 ± 0·17; P < 0·001] (Fig. 3). Immunohistochemical study demonstrated the reduced nuclear HMGB1 staining in the EAE + PBS and EAE + anti-HMGB1(20) groups; some nuclear HMGB1 staining was left in the EAE + anti-HMGB1(20) group and was lost completely in the EAE + PBS group (Fig. 4).
Figure 3.
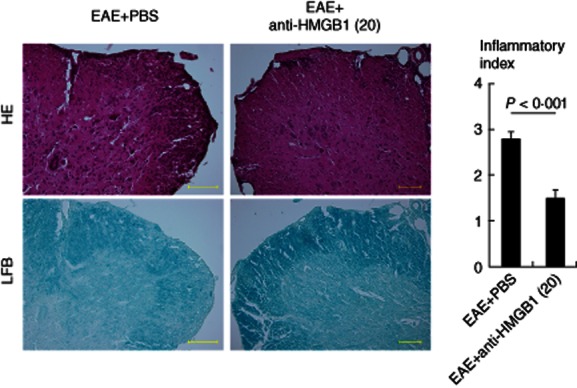
Pathological findings of anti-high mobility group box 1 (HMGB1) monoclonal antibody-treated experimental autoimmune encephalomyelitis (EAE) and phosphate-buffered saline (PBS)-treated EAE. Haematoxylin and eosin (H&E) and Luxol Fast Blue (LFB) staining of the spinal cord of anti-HMGB1 monoclonal antibody (20 μg × 5 days)-treated EAE mouse [EAE + anti-HMGB1(20)] and PBS-treated EAE mouse (EAE + PBS). The EAE + anti-HMGB1(20) mouse showed diminished infiltration of cells and demyelination compared with the EAE + PBS group. Inflammatory index of EAE + anti-high mobility group box 1 (HMGB1)(20) was significantly lower than that of EAE + PBS. Yellow bars = 200 μm.
Figure 4.
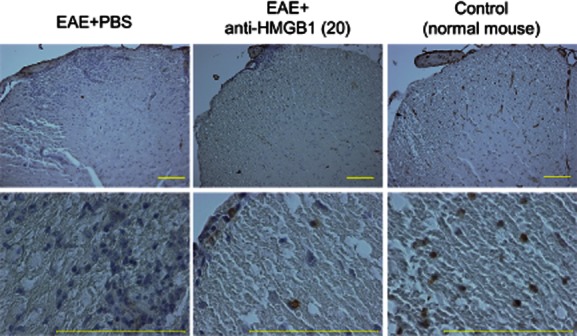
Immunohistochemical staining for HMGB1. HMGB1 staining of the spinal cord of anti-HMGB1 monoclonal antibody (20 μg × 5 days)-treated experimental autoimmune encephalomyelitis (EAE) mouse [EAE + anti-HMGB1(20)], phosphate-buffered saline (PBS)-treated EAE mouse (EAE + PBS) and normal control mouse. Nuclear HMGB1 staining was confirmed in the normal mouse, but not in the EAE + PBS mouse. Some nuclear HMGB1 staining was observed in the EAE + anti-HMGB1(20) mouse. Yellow bars = 200 μm.
Discussion
It has been reported previously that HMGB1 played important roles in some autoimmune-mediated diseases such as SLE, RA and SS [20,22–25]. However, there has been only one report to date regarding a possible correlation between HMGB1 and EAE or MS. HMGB1 was secreted by macrophages and microglia in CNS, and its receptors RAGE, TLR-2 and TLR-4 were expressed highly in the active lesions of EAE and patients with MS [24]. Recently, we also identified protein CSF HMGB1 levels in MS patients were significantly higher than those in non-inflammatory neurological diseases patients, and these levels in MS patients correlated with CSF cell counts [26]. Thus, there is a potential interaction among these molecules in the inflammatory processes involved in EAE and MS pathogenesis, and HMGB1 in the CNS may be a useful biomarker for CNS inflammation.
To the best of our knowledge, this is the first study to demonstrate that HMGB1 inhibition by a specific monoclonal antibody could be used for EAE treatment. Intraperitoneal anti-HMGB1 monoclonal antibody administration (20 μg × 5 days) reduced EAE clinical score and ameliorated EAE pathology, such as infiltration of inflammatory cells and demyelination. Interestingly, our immunohistochemical studies revealed the presence of HMGB1 immunoreactivity in the nuclei of spinal cord cells in normal mice, whereas these immunoreactive nuclei were lost completely in EAE and were decreased (but remained) in anti-HMGB1 monoclonal antibody-treated EAE mice. These results are essentially consistent with a previous study: extranuclear (cytoplasmic) HMGB1 immunoreactivity was exhibited in active MS lesions and, conversely, nuclear HMGB1 immunoreactivity was exhibited in inactive lesions [24]. Distribution of HMGB1 immunoreactivity suggests that HMGB1 is released from the cell nucleus when CNS inflammation occurs. Complete loss of nuclear HMGB1 immunoreactivity in the EAE + PBS group would indicate that all nuclear HMGB1 induced inflammation then it was drained. Neutralizing extracellular HMGB1 in CNS with an anti-HMGB1 monoclonal antibody alleviated inflammation, which may have resulted in residual nuclear HMGB1 staining in anti-HMGB1 monoclonal antibody-treated EAE mice spinal cord sections.
It has been reported that prophylactic treatment with intravenous immunoglobulin at the time of EAE induction reduced disease symptoms and the underlying CNS pathology in EAE [27]. However, in this study, control IgG administration in the same amount as the anti-HMGB1 monoclonal antibody (20 μg × 5 days) did not reduce EAE scores, which suggests that specific attenuation of HMGB1 and not IgG administration can ameliorate EAE pathology.
Some studies have described that IL-17 had a predominant role in MS pathogenesis [28,29]. However, most evidence was derived from EAE [30]. In addition to IL-17, IL-6 plays important roles in EAE [31,32]. Our serum cytokine analyses for EAE mice also revealed significant up-regulation of IL-17 and IL-6. We have demonstrated that treatment with an anti-HMGB1 monoclonal antibody (20 μg/day for 5 days) reduced IL-17 production in the peripheral circulation, which may contribute to amelioration of the clinical and pathological severity of EAE. However, these changes in cytokine levels were in the peripheral circulation; thus, CNS cytokine expression analysis in EAE and anti-HMGB1 monoclonal antibody-treated EAE will be needed.
Recently, a few reports have described therapeutic anti-HMGB1 monoclonal antibody intervention. Specific inhibition of endogenous HMGB1 reversed the lethality of established sepsis therapeutically, presumably by abrogating HMGB1-induced IL-6 and TNF-α release from macrophage-like cells [19]. Anti-HMGB1 monoclonal antibody therapy could be effective in the treatment of brain ischaemia as it inhibited the permeability of the blood–brain barrier, the activation of microglia, the expression of TNF-α and inducible nitric oxide synthase and suppressed the activity of matrix metalloproteinase-9 by efficient clearance of circulating HMGB1 [33,34]. Anti-HMGB1 monoclonal antibody therapy partially prevented joint destruction and exhibited beneficial anti-arthritic effects in models of arthritis [35], and attenuated cardiac pathological changes and reduced the number of infiltrating inflammatory cells in the heart in experimental autoimmune myocarditis by suppressing T helper type 17 (Th17) cells [36]. Prevention of CNS inflammation observed in the anti-HMGB1 monoclonal antibody-treated EAE mice in this study may also have derived from the above-mentioned mechanisms, such as attenuation of permeability of the blood–brain barrier, activation of microglia, suppression of the activity of matrix metalloproteinase-9 and an immune response and inflammatory cytokine release by immune cells such as IL-17, through neutralization of CNS HMGB1 with a specific antibody. Anti-HMGB1 monoclonal antibody therapy may be potentially useful during acute disease exacerbations to ameliorate pathology when the blood–brain barrier at a lesion is disrupted, because IgG antibodies are large molecules. This monoclonal antibody therapy would be novel from the perspective that it could be applied during acute disease exacerbations. Thus, analysis of anti-HMGB1 monoclonal antibody effects in human MS is required.
In conclusion, our results show that anti-HMGB1 monoclonal antibody administration ameliorated the clinical severity of EAE significantly by ameliorating EAE pathology in the CNS, and presumably attenuating the immune response via cytokine release and permeability of the blood–brain barrier. HMGB1 may play a key role in controlling autoimmune responses by stimulating the release of inflammatory cytokines. The control of HMGB1 is considered to be a critical factor in the pathogenesis of autoimmune diseases, including MS. Anti-HMGB1 monoclonal antibody therapy could have therapeutic potential not only for MS, but also for other autoimmune diseases.
Acknowledgments
This study was funded partly by research grants from the Ministry of Education, Science and Technology (grant number 24790873) and the Japan Multiple Sclerosis Society (A.U.).
Disclosures
The authors declare that they have no conflicts of interest.
References
- 1.Compston A, Coles A. Multiple sclerosis. Lancet. 2008;372:1502–1517. doi: 10.1016/S0140-6736(08)61620-7. [DOI] [PubMed] [Google Scholar]
- 2.Polman CH, Reingold SC, Edan G, et al. Diagnostic criteria for multiple sclerosis: 2005 revisions to the ‘McDonald Criteria’. Ann Neurol. 2005;58:840–846. doi: 10.1002/ana.20703. [DOI] [PubMed] [Google Scholar]
- 3.Kappos L, Antel J, Comi G, et al. Oral fingolimod (FTY720) for relapsing multiple sclerosis. N Engl J Med. 2006;355:1124–1140. doi: 10.1056/NEJMoa052643. [DOI] [PubMed] [Google Scholar]
- 4.Hawker K, O'Connor P, Freedman MS, et al. Rituximab in patients with primary progressive multiple sclerosis: results of a randomized double-blind placebo-controlled multicenter trial. Ann Neurol. 2009;66:460–471. doi: 10.1002/ana.21867. [DOI] [PubMed] [Google Scholar]
- 5.Coles AJ, Fox E, Vladic A, et al. Alemtuzumab more effective than interferon β-1a at 5-year follow-up of CAMMS223 clinical trial. Neurology. 2012;78:1069–1078. doi: 10.1212/WNL.0b013e31824e8ee7. [DOI] [PubMed] [Google Scholar]
- 6.Polman CH, O'Connor PW, Havrdova E, et al. A randomized, placebo-controlled trial of natalizumab for relapsing multiple sclerosis. N Engl J Med. 2006;354:899–910. doi: 10.1056/NEJMoa044397. [DOI] [PubMed] [Google Scholar]
- 7.Harris HE, Raucci A. Alarmin(g) news about danger: workshop on innate danger signals and HMGB1. EMBO Rep. 2006;7:774–778. doi: 10.1038/sj.embor.7400759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bustin M, Reeves R. High-mobility-group chromosomal proteins: architectural components that facilitate chromatin function. Prog Nucleic Acid Res Mol Biol. 1996;54:35–100. doi: 10.1016/s0079-6603(08)60360-8. [DOI] [PubMed] [Google Scholar]
- 9.Fiuza C, Bustin M, Talwar S, et al. Inflammation-promoting activity of HMGB1 on human microvascular endothelial cells. Blood. 2003;101:2652–2660. doi: 10.1182/blood-2002-05-1300. [DOI] [PubMed] [Google Scholar]
- 10.Bell CW, Jiang W, Reich CF, III, et al. The extracellular release of HMGB1 during apoptotic cell death. Am J Physiol Cell Physiol. 2006;291:C1318–1325. doi: 10.1152/ajpcell.00616.2005. [DOI] [PubMed] [Google Scholar]
- 11.Scaffidi P, Misteli T, Bianchi ME. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature. 2002;418:191–195. doi: 10.1038/nature00858. [DOI] [PubMed] [Google Scholar]
- 12.Kokkola R, Andersson A, Mullins G, et al. RAGE is the major receptor for the proinflammatory activity of HMGB1 in rodent macrophages. Scand J Immunol. 2005;61:1–9. doi: 10.1111/j.0300-9475.2005.01534.x. [DOI] [PubMed] [Google Scholar]
- 13.Yang H, Hreggvidsdottir HS, Palmblad K, et al. A critical cysteine is required for HMGB1 binding to Toll-like receptor 4 and activation of macrophage cytokine release. Proc Natl Acad Sci USA. 2010;107:11942–11947. doi: 10.1073/pnas.1003893107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Park JS, Svetkauskatie D, He Q, et al. Involvement of Toll-like receptors 2 and 4 in cellular activation by high mobility group box 1 protein. J Biol Chem. 2004;279:7370–7377. doi: 10.1074/jbc.M306793200. [DOI] [PubMed] [Google Scholar]
- 15.Tian J, Avalos AM, Mao SY, et al. Toll-like receptor 9-dependent activation by DNA-containing immune complexes is mediated by HMGB1 and RAGE. Nat Immunol. 2007;8:487–496. doi: 10.1038/ni1457. [DOI] [PubMed] [Google Scholar]
- 16.Park JS, Gamboni-Robertson F, He Q, et al. High mobility group box 1 protein interacts with multiple Toll-like receptors. Am J Physiol Cell Physiol. 2006;290:C917–924. doi: 10.1152/ajpcell.00401.2005. [DOI] [PubMed] [Google Scholar]
- 17.Avalos AM, Kiefer K, Tian J, et al. RAGE-independent autoreactive B cell activation in response to chromatin and HMGB1/DNA immune complexes. Autoimmunity. 2010;43:103–110. doi: 10.3109/08916930903384591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rouhiainen A, Kuja-Panula J, Wilkman E, et al. Regulation of monocyte migration by amphoterin (HMGB1) Blood. 2004;104:1174–1182. doi: 10.1182/blood-2003-10-3536. [DOI] [PubMed] [Google Scholar]
- 19.Andersson U, Wang H, Palmblad K, et al. High mobility group 1 protein (HMG-1) stimulates proinflammatory cytokine synthesis in human monocytes. J Exp Med. 2000;192:565–570. doi: 10.1084/jem.192.4.565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ek M, Popovic K, Harris HE, et al. Increased extracellular levels of the novel proinflammatory cytokine high mobility group box chromosomal protein 1 in minor salivary glands of patients with Sjogren's syndrome. Arthritis Rheum. 2006;54:2289–2294. doi: 10.1002/art.21969. [DOI] [PubMed] [Google Scholar]
- 21.Taira T, Matsuyama W, Mitsuyama H, et al. Increased serum high mobility group box-1 level in Churg–Strauss syndrome. Clin Exp Immunol. 2007;148:241–247. doi: 10.1111/j.1365-2249.2007.03347.x. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 22.Taniguchi N, Kawahara K, Yone K, et al. High mobility group box chromosomal protein 1 plays a role in the pathogenesis of rheumatoid arthritis as a novel cytokine. Arthritis Rheum. 2003;48:971–981. doi: 10.1002/art.10859. [DOI] [PubMed] [Google Scholar]
- 23.Urbonaviciute V, Furnrohr BG, Meister S, et al. Induction of inflammatory and immune responses by HMGB1-nucleosome complexes: implications for the pathogenesis of SLE. J Exp Med. 2008;205:3007–3018. doi: 10.1084/jem.20081165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Andersson A, Covacu R, Sunnemark D, et al. Pivotal advance: HMGB1 expression in active lesions of human and experimental multiple sclerosis. J Leukoc Biol. 2008;84:1248–1255. doi: 10.1189/jlb.1207844. [DOI] [PubMed] [Google Scholar]
- 25.Abdulahad DA, Westra J, Bijzet J, et al. High mobility group box 1 (HMGB1) and anti-HMGB1 antibodies and their relation to disease characteristics in systemic lupus erythematosus. Arthritis Res Ther. 2011;13:R71. doi: 10.1186/ar3332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Uzawa A, Mori M, Masuda S, et al. CSF high-mobility group box 1 is associated with intrathecal inflammation and astrocytic damage in neuromyelitis optica. J Neurol Neurosurg Psychiatry. 2012 doi: 10.1136/jnnp-2012-304039. doi: 10.1136/jnnp-2012-304039. [DOI] [PubMed] [Google Scholar]
- 27.Humle Jorgensen S, Sorensen PS. Intravenous immunoglobulin treatment of multiple sclerosis and its animal model, experimental autoimmune encephalomyelitis. J Neurol Sci. 2005;233:61–65. doi: 10.1016/j.jns.2005.03.005. [DOI] [PubMed] [Google Scholar]
- 28.Ishizu T, Osoegawa M, Mei FJ, et al. Intrathecal activation of the IL-17/IL-8 axis in opticospinal multiple sclerosis. Brain. 2005;128:988–1002. doi: 10.1093/brain/awh453. [DOI] [PubMed] [Google Scholar]
- 29.Lock C, Hermans G, Pedotti R, et al. Gene-microarray analysis of multiple sclerosis lesions yields new targets validated in autoimmune encephalomyelitis. Nat Med. 2002;8:500–508. doi: 10.1038/nm0502-500. [DOI] [PubMed] [Google Scholar]
- 30.Komiyama Y, Nakae S, Matsuki T, et al. IL-17 plays an important role in the development of experimental autoimmune encephalomyelitis. J Immunol. 2006;177:566–573. doi: 10.4049/jimmunol.177.1.566. [DOI] [PubMed] [Google Scholar]
- 31.Quintana A, Müller M, Frausto RF, et al. Site-specific production of IL-6 in the central nervous system retargets and enhances the inflammatory response in experimental autoimmune encephalomyelitis. J Immunol. 2009;183:2079–2088. doi: 10.4049/jimmunol.0900242. [DOI] [PubMed] [Google Scholar]
- 32.Eugster HP, Frei K, Kopf M, et al. IL-6-deficient mice resist myelin oligodendrocyte glycoprotein-induced autoimmune encephalomyelitis. Eur J Immunol. 1998;28:2178–2187. doi: 10.1002/(SICI)1521-4141(199807)28:07<2178::AID-IMMU2178>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 33.Liu K, Mori S, Takahashi HK, et al. Anti-high mobility group box 1 monoclonal antibody ameliorates brain infarction induced by transient ischemia in rats. FASEB J. 2007;21:3904–3916. doi: 10.1096/fj.07-8770com. [DOI] [PubMed] [Google Scholar]
- 34.Zhang J, Takahashi HK, Liu K, et al. Anti-high mobility group box-1 monoclonal antibody protects the blood–brain barrier from ischemia-induced disruption in rats. Stroke. 2011;42:1420–1428. doi: 10.1161/STROKEAHA.110.598334. [DOI] [PubMed] [Google Scholar]
- 35.Schierbeck H, Lundbäck P, Palmblad K, et al. Monoclonal anti-HMGB1 (high mobility group box chromosomal protein 1) antibody protection in two experimental arthritis models. Mol Med. 2011;17:1039–1044. doi: 10.2119/molmed.2010.00264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Su Z, Sun C, Zhou C, et al. HMGB1 blockade attenuates experimental autoimmune myocarditis and suppresses Th17-cell expansion. Eur J Immunol. 2011;41:3586–3595. doi: 10.1002/eji.201141879. [DOI] [PubMed] [Google Scholar]