

Abstract
Experimental allergic encephalomyelitis (EAE) can be induced in animal models by injecting the MOG35–55 peptide subcutaneously. Dendritic cells (DCs) that are located at the immunization site phagocytose the MOG35–55 peptide. These DCs mature and migrate into the nearest draining lymph nodes (dLNs), then present antigen, resulting in the activation of naive T cells. T helper type 1 (Th1) and Th17 cells are the primary cells involved in EAE progression. All-trans-retinoic acid (AT-RA) has been shown to have beneficial effects on EAE progression; however, whether AT-RA influences DC maturation or mediates other functions is unclear. In the present study, we showed that AT-RA led to the down-regulation of MHC class II, CD80 (B7-1) and CD86 (B7-2) expressed on the surface of DCs that were isolated from dLNs or spleen 3 days post-immunization in an EAE model. Changes to DC function influenced Th1/Th17 subset polarization. Furthermore, the number of CD44+ monocytes (which might trigger EAE progression) was also significantly decreased in dLNs, spleen, subarachnoid space and the spinal cord parenchyma after AT-RA treatment. These findings are the first to demonstrate that AT-RA impairs the antigen-presenting capacity of DCs, leading to down-regulation of pathogenic Th1 and Th17 inflammatory cell responses and reducing EAE severity.
Keywords: all-trans retinoic acid, dendritic cell, experimental allergic encephalomyelitis, monocyte, T helper type 1/type 17 cells
Introduction
Experimental allergic encephalomyelitis (EAE) is a commonly used animal model for the study of human multiple sclerosis, a disease associated with chronic inflammatory infiltration and demyelination of the central nervous system (CNS). It is generally accepted that both interferon-γ (IFN-γ) -secreting T helper type 1 (Th1) and interleukin-17 (IL-17) -secreting Th17 cells have a pathogenic role during the pathogenesis of EAE.1
Dendritic cells (DCs) are a subset of professional antigen-presenting cells that can drive naive T-cell activation, maturation and polarization into antigen-specific T helper (Th) cell subsets with the potential to cause disease. During the inflammatory phase of EAE, epidermal DCs residing at the immunization site phagocytose the myelin oligodendrocyte glycoprotein peptide located between residues 35 and 55 (MOG35–55), mature (characterized by expression of high levels of MHC class II and co-stimulatory molecules, such as CD80 or CD86), and migrate into the draining lymph nodes (dLNs) where they present antigen to naive T cells.2 For this reason, DCs play a pivotal role in initiating the immune response that can result in the development of EAE. However, most current studies have focused on the DCs that reside within the CNS rather than peripheral DCs located in dLNs and spleen, partly because of the low numbers of peripheral DCs available for study.
In addition to Th1 and Th17 cells, research has recently shown that monocytes have the potential to exacerbate EAE.3,4 Specifically, depletion of monocytes induced a marked suppression in EAE severity.5,6 Typically, blood-derived monocytes are excluded from the CNS; however, once monocytes are recruited into the CNS, they indicate a new phase in the pathology of EAE. It has been reported that the recruitment of monocytes could be triggered by Th1 or Th17 cells and that these events in turn trigger EAE progression.7,8
All-trans retinoic acid (AT-RA), a vitamin A metabolite, plays an essential role in the regulation of immune responses. Several reports have proposed that AT-RA affects immune responses by altering the balance between pro-inflammatory and anti-inflammatory cytokines.9,10 However, the effects of AT-RA on antigen-presenting cell function remain controversial, even though it has been shown that AT-RA enhances murine DC migration into dLNs during tumour immune responses.11 An in vitro study found that AT-RA inhibited the differentiation, maturation and function of human monocyte-derived DCs.12 AT-RA was shown to affect T cells and adaptive immune responses by suppressing lymphocyte proliferation and the production of pro-inflammatory cytokines such as IL-17 and IFN-γ, thereby inhibiting EAE progression.13–17 However, few reports have examined the effects of AT-RA on innate immune cells such as DCs and monocytes in vivo during the pathogenic progression of EAE.
The aim of this study was to investigate in vivo the regulatory effects of AT-RA on immune cells, including peripheral DCs, monocytes and effector Th1 and Th17 cells in the pathogenesis of EAE. We concluded that AT-RA down-regulated DC maturation and decreased the number of pathogenic monocytes and effector T cells, thereby ameliorating EAE disease severity.
Materials and methods
AT-RA preparation
All-trans-retinoic acid (Sigma, St Louis, MO), was stored at −20° and protected from light until used. For in vivo experiments, AT-RA was dissolved in soybean oil (3 mg/ml for intraperitoneal injections). For in vitro experiments, AT-RA was dissolved inDMSO and stored at a concentration of 0·1 m at −20° (protected from light) until used. AT-RA with a concentration of 50 μm was prepared by dilution in RPMI-1640 medium. The AT-RA culture system was protected from light throughout the culture period.
EAE induction and AT-RA treatment
Female C57BL/6 mice that were 6–8 weeks old (Peking Vital River Laboratory Animal Ltd, Beijing, China) were immunized subcutaneously on both sides in the axillary space. The immunization consisted of 200 μg MOG35–55 (MEVGWYRSPFSRVVHLYRNGK) peptide (Bioss, Beijing, China) emulsified in incomplete Freund's adjuvant (Sigma) containing 250 μg Mycobacterium tuberculosis H37Ra (Difco Laboratories, Detroit, MI). Each mouse received 200 ng pertussis toxin in 200 μl PBS (pH 7·4) intravenously on day 0 and day 2 post-immunization.
One day before immunization, mice were divided into two groups and treated with 250 μl soybean oil as a control or with 250 μl AT-RA dissolved in soybean oil intraperiteonally for 4 days (once daily, 300 μg/mice). Mice were monitored daily following immunization. Clinical signs of EAE were assigned using the following scoring system: 0, healthy; 1, limp tail; 2, impaired righting reflex or waddling gait; 3, hind-limb paralysis; 4, hind-limb and forelimb paralysis, and 5, moribund or dead.
Histology
Animals were killed at the peak of EAE, nearly 15 days post-immunization. Spinal cords were removed, immersed in 10% formalin, then embedded in paraffin. Samples were then stained with haemotoxylin & eosin (H&E) to determine degree of inflammatory cell infiltration or luxol fast blue (LFB) to determine the degree of deymelination. For H&E staining, nuclei were stained with alum haematoxylin followed by differentiation with acid alcohol and staining with eosin. For LFB staining, sections were de-paraffinized and hydrated using 95% ethyl alcohol, kept in luxol fast blue solution at 56° overnight, and differentiated sequentially with 95% ethyl alcohol, lithium carbonate solution, and 70% ethyl alcohol, then counterstained with cresyl violet solution. All sections were then dehydrated on a gradient, cleared and mounted.
Isolation, culture and identification of bone mesenchymal DCs
Bone marrow was prepared from the tibia and femur bones of C57BL/6 mice. Erythrocytes were lysed using ammonium chloride erythrocyte-lysing solution (ACK-Lyse). After centrifugation and washing, cells were cultured in 3 ml RPMI-1640 complete medium [10% fetal calf serum (FCS), 100 U/ml penicillin and 100 μg/ml streptomycin; Gibco, Grand Island, NY] at a density of 1 × 106 cells/ml in six-well plates. Murine granulocyte–macrophage colony-stimulating factor (GM-CSF, 4 ng/ml) and murine IL-4 (4 ng/ml) (PeproTech, Rocky Hill, CT) were also added to the culture system. The cells were incubated at 37° in 5% CO2. Two days later, fresh medium with AT-RA at the concentration of 50 μm (dissolved in DMSO) or DMSO alone as a control were added to the culture system for three more days. On day 6, cells in the supernatant were collected, centrifuged and resuspended in fresh complete medium (containing the same concentrations of GM-CSF, IL-4, AT-RA or DMSO), then added back to the culture plates. On day 8, to allow further maturation of DCs, cells were cultured with lipopolysaccharide (LPS; Sigma) at a final concentration of 10 ng/ml for 24 hr before collection. The DCs staining positive for CD11c, CD80, CD86 or MHC-class II were verified using the respective monoclonal antibodies (BD Biosciences, San Jose, CA) and flow cytometric analysis was carried out using a flow cytometer (BD Biosciences).
Isolation of DCs from the lymph nodes and spleen
Three days post-immunization, AT-RA-treated mice and mice from the control group were killed. Spleens and dLNs were harvested, minced and digested in RPMI-1640 supplemented with 10% FCS, 10 U/ml collagenase type D and 5 μg/ml DNAse I (Gibco) for 10 min at 37°. Digested tissues were passed through a 70 μm cell strainer. Cells were collected by centrifugation at 540 g for 5 min and washed twice with Hanks' balanced salts solution (pH 7·2) containing EDTA (5 mm). The cell pellet was then suspended in an iso-osmotic solution consisting of 10·5% (weight/volume) iodixanol (Axis–Shield, Oslo, Norway) at a cell density of 1·5 × 108 cells/ml. Three to four millilitres of the iso-osmotic suspension was transferred into a tube and over-laid with 2 ml FCS. The tube was then centrifuged at 1700 g for 10 min (with a slow acceleration mode and decelerated without brake) and DCs were harvested from the FCS–sample interface. The degree of DC maturation from dLNs and spleen was then assessed by flow cytometry following staining with CD11c-, CD80-, CD86-, and MHC-class II-specific antibodies.
Lymphocyte and monocyte collection and preparation
At different time-points, control and AT-RA-treated mice were killed. Draining LNs and spleens were removed and minced into single cell suspensions. Red blood cells in the splenic cell suspension were lysed following incubation with red blood cell lysing buffer and then cells were suspended in PBS for further analysis. For intracellular cytokine staining, cells were cultured in Dulbecco's modified Eagle's medium (Gibco) supplemented with 10% FCS, 100 U/ml penicillin, 100 μg/ml streptomycin and 292·1 μg/ml l-glutamine (Sigma) in the presence of 25 μg/ml MOG35–55 for 72 hr at 37°. Before staining, cells were incubated overnight with 10 ng/ml IL-2 (PeproTech), ionomycin (50 ng/ml), PMA (1 mg/ml) (Alexis, Farmingdale, NY) and Brefeldin A (1 μl/ml, eBioscience, San Diego, CA) for 4–5 hr.
Generation of mononuclear cells and lymphocytes from spinal cords and the subarachnoid space
Mononuclear cells and lymphocytes were isolated from the CNS by Percoll gradient centrifugation. Briefly, mice were perfused through the left cardiac ventricle with 20 ml cold PBS and 10 mm EDTA, then spinal cords were flushed out from the vertebral canal with PBS. Cells attached to the surface of the spinal cord were obtained by shaking harvested spinal cords in PBS and filtering them through a 70-μm cell strainer. Cells that infiltrated the spinal cord parenchyma were obtained by forcing the spinal cord through a 200-gauge mesh following digestion with collagenase D (2·5 mg/ml) and DNAseI (5 μg/ml) at 37° for 45 min. Cells were harvested from the Percoll interface after gradient centrifugation at 300 g for 30 min.
Co-culture of bone mesenchymal DCs and lymphocytes
Bone mesenchymal DCs (BMDCs) treated with AT-RA or DMSO (as previously stated) were matured by the addition of 10 ng/ml LPS in the presence of 25 μg/ml MOG35–55 peptide for 24 hr. Afterwards, BMDCs were intensively washed and resuspended at a concentration of 1 × 106 cells/ml in complete medium. Lymphocytes were collected from dLNs of EAE mice killed at 5 days post-immunization and 1 × 104 BMDCs were co-cultured with 1 × 105 T cells (ratio 1 : 10) in 200 μl complete medium per well of round-bottomed 96-well plates for 48 hr or 72 hr.
Flow cytometry
All cells were washed and resuspended in wash buffer containing PBS with 0·1% NaN3. Antibodies specific for the respective cell surface markers were diluted in appropriate volumes of FACS buffer containing 1% BSA and incubated with cells for 30 min at 4°. Intracellular staining was performed by fixing cells with 4% paraformaldehyde in PBS for 15 min at 4°, followed by permeabilization with 5% saponin (Sigma) in PBS for 10 min at room temperature. Antibodies specific for intracellular markers were diluted to the appropriate volume in FACS buffer containing 5% saponin and incubated for 20 min at room temperature. All incubations were conducted in the dark and analysis of flow cytometric data was performed using FlowJo software (Treestar, London, MN).
To assess DC maturation, the following panel of monoclonal antibodies was used: FITC-conjugated anti-CD80, phycoerythrin (PE) -conjugated anti-CD11c, peridinin chlorophyll protein (PerCP) -conjugated anti-MHC class II and allophycocyanin (APC) -conjugated anti-CD86 (all from eBioscience). Surface and intracellular staining markers included PE-conjugated anti-IL-17 or anti-IFN-γ, FITC-conjugated anti-CD4 and APC-conjugated anti-CD3 (BD Pharmingen, San Diego, CA). Monocyte surface markers were screened by using PE-conjugated anti-CD44, PerCP-conjugated anti-MHC-class II, PE-conjugated anti-CD36, FITC-conjugated anti-CD40 and FITC-conjugated anti-CD11b (BD Pharmingen).
Apoptosis
The rate of cellular apoptosis was quantified by Annexin-V and propidium iodide (both from eBioscience) double staining. Briefly, cells of dLNs were collected from immunized (EAE) and non-immunized mice with AT-RA or soybean oil treatment for 4 days. Mononuclear cells were seeded onto 96-well plates and incubated overnight in the presence of 25 μg/ml MOG35–55. After intensive washing, cells were resuspended in diluted binding buffer at a density of 5 × 105 cells/ml. About 5 μl Annexin V (1 μg/ml) was added and incubated at room temperature for 10 min, then 10 μl propidium iodide (10 μg/ml) was added and incubated for another 10 min at room temperature in the dark. Finally, cells were analysed by flow cytometry.
Proliferation
The impact of AT-RA-treated or DMSO-treated BMDCs on lymphocytes was determined using a Cell Counting Kit-8 (CCK-8) (Beyotime, Shanghai, China) according to the manufacturer's instructions. Briefly, cells were collected after co-culture with BMDCs for 48 hr by gentle pipetting, then added into another 96-well plate for further incubation for 24 hr. CCK-8 (10 μl) was added into 100 μl medium, and cells were then incubated for 0·5–1 hr at 37°. The absorbance was measured at 450 nm using a microplate spectrophotometer (BIO-RAD, Berkeley, CA).
Statistical analysis
Statistical analysis was performed using GraphPad Prism software (GraphPad Software Inc., San Diego, CA) by one-way analysis of variance and the non-parametric Student's t-test. Results were expressed as the mean ± SE. A difference was considered to be significant when P < 0·05.
Results
AT-RA suppressed EAE severity
EAE was induced by injection of the MOG35–55 peptide. Mice received a total of 300 μg AT-RA (dissolved in soybean oil), administered once daily for 4 days. Control mice were treated with soybean oil only. The clinical scores and disease severity of mice in both the AT-RA-treated group and the control group were monitored daily for 30 days.
The AT-RA-treated mice exhibited mild EAE with reduced severity characterized by later onset and lessened tail paralysis and impaired gait. By contrast, mice in the control group developed significantly more severe disease symptoms measured by obvious tail and limb paralysis, as well as gait impairment. The peak of the disease in the AT-RA treatment group occurred later, around 18 days after immunization, compared with 15 days in the control group. Sick mice rapidly recovered within 4 days of disease presentation (Fig. 1). These data suggested that AT-RA treatment successfully suppressed EAE presentation and progression.
Figure 1.
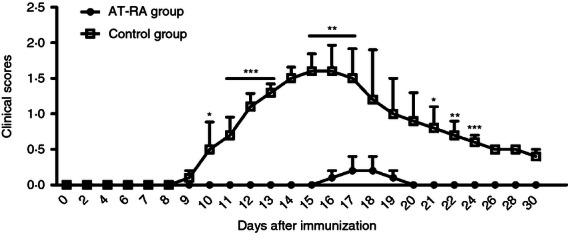
Effects of All-trans retinoic acid (AT-RA) on the severity and clinical scores of experimental autoimmune encephalomyelitis (EAE). EAE was induced in C57BL/6 mice by immunization with the MOG35–55 peptide. Mice were injected intraperitoneally with either AT-RA (300 μg/mice) or the vehicle control. Clinical severity was scored daily. Compared with the control group, EAE presentation in mice treated with AT-RA was delayed and less severe. Data are expressed as the mean ± SE and are representative of a minimum of four experiments. *P < 0·05, **P < 0·01, ***P < 0·001.
To further confirm these results, inflammatory infiltration and the degree of spinal chord demyelination were clarified at the peak phase of the disease (Fig. 2). Microscopic examination revealed that significantly fewer leucocytes had infiltrated into the CNS of AT-RA-treated mice and demyelination was markedly reduced compared with that in control mice. These results demonstrated that AT-RA treatment ameliorated EAE disease severity and development.
Figure 2.
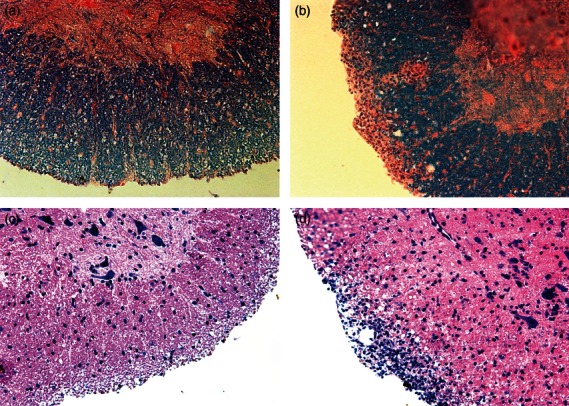
All-trans retinoic acid (AT-RA) treatment developed no obvious spinal cord lymphocyte infiltration and demyelination on 15 days post-immunization (dpi) compared with the experimental autoimmune encephalomyelitis (EAE) control group. Paraffin-embedded spinal cord sections were prepared on 15 dpi and stained with luxol fast blue (LFB) (a, b) or with haemotoxylin & eosin (H&E) (c, d) to determine the degree of demyelination and inflammatory infiltrates. AT-RA-treated mice (a) presented with no obvious deymelination in contrast to control EAE mice (b). AT-RA-treated mice (c) had far less lymphocytic infiltration than control EAE mice (d). Magnification × 200.
AT-RA did not have non-specific toxic effects on mononuclear cells
To study if AT-RA-mediated amelioration of EAE was the result of non-specific toxic effects on mononuclear cells, we isolated mononuclear cells from dLNs from immunized and non-immunized mice with or without AT-RA treatment. Results (Fig. 3) showed that no statistical differences were found in the percentage of cell apoptosis in mice of immunized or non-immunized mice with or without AT-RA treatment.
Figure 3.
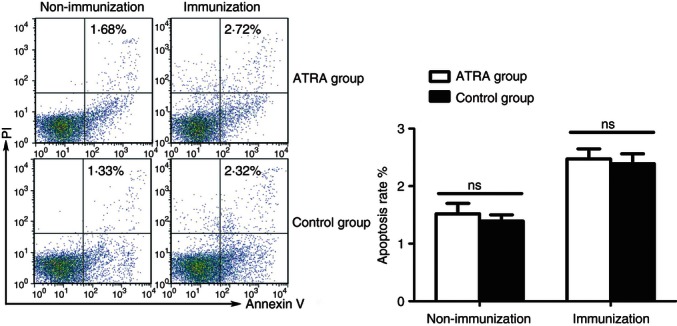
All-trans retinoic acid (AT-RA) administration did not have non-specific toxic effects on mononuclear cells. Immunized and unimmunized mice were given AT-RA or only soybean oil treatment (control group) for 4 days. Mononuclear cells were isolated from draining lymph nodes (dLNs) and the toxic effect of AT-RA was measured by analysing the rate of apoptosis. Apoptotic cells were identified as propidium iodide+ Annexin-V+. Data are representative as mean ± SE of two experiments with five mice in each experiment.
AT-RA inhibited DC maturation in vivo and in vitro
To assess the impact of AT-RA on DC maturation during the priming stage of EAE, we collected DCs from both dLNs and spleens using the density-gradient method as described in the Materials and methods. Conventional DCs (gate M2) had high levels of CD11c expression (Fig. 4a). Hence the percentage of isolated DCs, defined by flow cytometry with CD11chigh expression, was 1·635% ± 0·2497% of dLNs, and 2·629% ± 0·4913% of spleen. The number of purified DCs was 3·82 ± 1·3 × 103 cells in dLNs, and 12·4 ± 2·19 × 103 cells in the spleen of one immunized mouse (Fig. 4b).
Figure 4.

All-trans retinoic acid (AT-RA) treatment suppressed dendritic cell (DC) maturation on 3 days post-immunization (dpi). The DCs were isolated from draining lymph nodes (dLNs) and spleens of experimental autoimmune encephalomyelitis (EAE) mice treated or untreated with AT-RA on 3 dpi. Flow cytometry analysis was used to establish the DC phenotype using the following antibodies: phycoerythrin-conjugated anti-CD11c, FITC-conjugated anti-CD80, peridinin chlorophyll protein-conjugated anti-MHC-class II, and allophycocyanin-conjugated anti-CD86. Cells with DC phenotype in gate M2 were analysed. (a) Left panel, dot plot showed the gating strategy for DC analysis. Right panel, CD11c staining in G1 gated cells was shown. M1: CD11cinterm cells, M2: CD11chigh cells. (b) The percentage and total number of CD11chigh cells in dLNs and spleen of one immunized mice. (c) The effect of AT-RA on the maturation of CD11chigh DCs derived from dLNs. (d) The effect of AT-RA on the maturation of CD11chigh DCs derived from spleen. Data are expressed as mean ± SE and are representative of four independent experiments with eight mice per group. **P < 0·01, ***P < 0·001.
Mature CD11chigh DCs were then identified by flow cytometry based on the expression of co-stimulatory molecules CD80, CD86 and MHC-class II. Results demonstrated that AT-RA efficiently interfered with DC maturation in vivo. AT-RA inhibited DC maturation in dLNs following the same level as in spleens. Three days post-immunization, DCs in the control group expressed significantly higher levels of CD80, CD86 and MHC-class IIhigh in both dLNs and spleens compared with observed expression levels after AT-RA treatment (Fig. 4c,d). These results suggested that AT-RA inhibited DC maturation during the priming stage of EAE.
To investigate whether AT-RA had the same inhibitory effects on DC maturation in vitro, 50 μm AT-RA was added to the culture media of BMDCs. To identify BMDC purity, BMDCs were cultured in the presence of LPS for 24 hr and CD11c staining was performed. Our results showed that the percentage of CD11chigh cells was > 80% (84·762% ± 6·64%) and the mean fluorescence intensity was 296 ± 34·748 (Fig. 5a). CD80, CD86 and MHC-class II surface expression profiles were detected in the CD11chigh BMDCs. Results demonstrated that AT-RA negatively regulated BMDC maturation (shown by statistically lower expression levels of CD80, CD86 and MHC-class II on CD11chigh BMDCs in the AT-RA treatment group compared with the expression profile of cells treated with DMSO only) (Fig. 5b).
Figure 5.
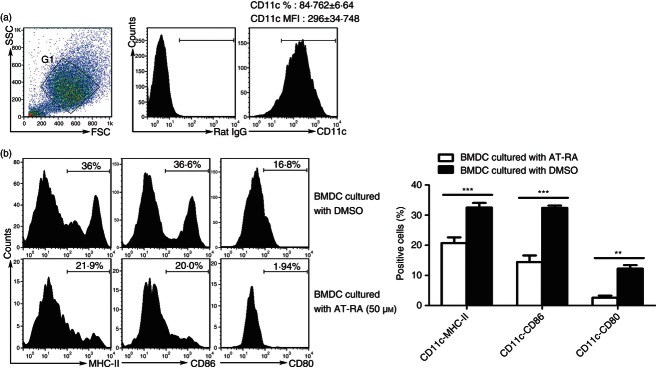
All-trans retinoic acid (AT-RA) treatment decreased the maturation of bone mesenchymal dendritic cells (BMDCs) in vitro. The BMDCs were isolated and cultured in the presence or absence of AT-RA. DMSO used to dissolve AT-RA was used as the diluent control in the untreated group. (a) Phycoerythrin-conjugated anti-CD11c was used to verify the BMDCs subset. Left panel, dot plots show the gating strategy of BMDCs subset. Figure of isotype control antibodies was shown in the middle panel. The percentages of CD11chigh cells and mean fluorescence intensity were shown on the right histogram. Purity of BMDCs was > 80% in our culture system. (b) Analysis of different cell surface markers was performed in the population of CD11chigh cells by flow cytomety using the following antibodies: FITC-conjugated anti-CD80, Peridinin chlorophyll protein-conjugated anti-MHC-class II, and allophycocyanin-conjugated anti-CD86. Data are expressed as the mean ± SE of at least four independent experiments. **P < 0·01, ***P < 0·001.
AT-RA treatment decreased CD44+ mononuclear cells in different tissues at different phases of disease
Using forward scatter channel-side scatter channel (FSC-SSC), we observed that a group of CD44+ monocytes could be identified at different phases of EAE (onset, peak and remission phases) (Fig. 6). Following AT-RA treatment, the numbers of CD44+ monocytes in dLNs, spleen, subarachnoid space and the spinal cord parenchyma were significantly decreased compared with levels observed in the control group. However, the numbers of CD44+ monocytes were identical in both the control and AT-RA treatment group in dLNs and CNS parenchyma 7 days post-immunization (dpi). The decreased CD44+ monocytes, along with reduced EAE clinical scores observed after AT-RA treatment, were consistent with previous reports, suggesting an association between monocyte levels and EAE exacerbation.3–7
Figure 6.

All-trans retinoic acid (AT-RA) treatment decreased the production of CD44+ monocytes. Mononuclear cells were isolated from draining lymph nodes (dLNs), spleen, spinal cord parenchyma (SC), and subarachnoid space (SAS) from mice in the AT-RA and control groups on 7 days post-immunization (dpi) (b), 15 dpi (c), and 21 dpi (d). (a) Dot plots show the gating strategy of phenotype analysis (G1) and further analysis for monocytes (G2). From the left to the right in panel (a), four plots represent data from dLNs, spleen, SC, and SAS, respectively. Cells were stained with allophycocyanin-conjugated anti-CD3 and phycoerythrin-conjugated anti-CD44 antibodies. The majority of CD3− CD44+ cells were mononuclear cells that represented in G2 gate. All the data analysed in (b), (c) and (d) were G1 gated cells. Data are expressed as the mean ± SE of three experiments with five or six mice per group at each time-point. *P < 0·05, **P < 0·01.
It is well accepted that inflammatory cells first accumulate within the subarachnoid space and then infiltrate the spinal cord parenchyma at the onset of EAE. Unexpectedly, during the T-cell priming stage of EAE (7 dpi), elevated numbers of CD44+ monocytes were already present in the subarachnoid space in the control group with few detectable CD3+ T cells present. The CD44+ monocyte population remained at a stable level during the peak and recovery phases of EAE. Although only a few CD44+ monocytes continued to infiltrate the spinal cord parenchyma, there was a statistical difference between the levels of monocyte infiltration in the AT-RA treatment group and the control group.
To further characterize this population of monocytes, multi-colour staining was performed on monocytes isolated from dLNs, spleen, subarachnoid space and spinal cord parenchyma using anti-MHC-class II, anti-CD40, anti-CD11b and anti-CD36 antibodies (see Supplementary material, Fig. S1). Results showed that this population of monocytes was CD11b+ CD36+ in all tissues, partially expressed CD40 and MHC-class IIhigh in dLNs, but were CD40– and MHC-class IIlow in spleen, subarachnoid space and the spinal cord parenchyma. Based on this staining profile, these monocytes were considered to be members of the mononuclear phagocytic system, indicating that this population might participate in EAE pathogenesis. The reason why this population of monocytes decreased dramatically in all related tissues and at all phases of the disease in the AT-RA treatment group might be that AT-RA directly suppressed the development of these cells through mechanisms that were still undefined.
AT-RA treatment decreased the numbers of pathogenic Th1 and Th17 cells
Both Th1 and Th17 cells are major players in the mediation of pathogenic processes associated with EAE. We therefore examined the numbers of peripheral Th1 and Th17 cells during EAE progression. Lymphocytes collected from dLNs and spleens at different disease phases (0, 7, 15 and 21 dpi) were cultured for 72 hr, stimulated with 10 ng/ml IL-2 overnight, ionomycin (50 ng/ml), PMA (1 mg/ml) and Brefeldin A (1 μl/ml) for 4–5 hr and then stained with anti-CD3, anti-CD4, anti-IFN-γ and anti-IL-17 antibodies. The percentages of Th1/Th17 cells in the dLNs were significantly greater in the control group than in the AT-RA treatment group at the peak and remission phases of EAE (Fig. 7). During the onset phase, the numbers of Th1 and Th17 cells in the control group were also greater (although not significantly) than in the AT-RA treatment group. In the spleen, the Th1 and Th17 profiles were similar (Fig. 8). In general, the numbers of Th1 and Th17 cells were reduced in the AT-RA treatment group compared with the control group, even though there were no significant differences in Th17 numbers between groups at 8 and 14 dpi. These data demonstrated that AT-RA treatment reduced the numbers of Th1 and Th17 cells at all time-points compared with the control group.
Figure 7.
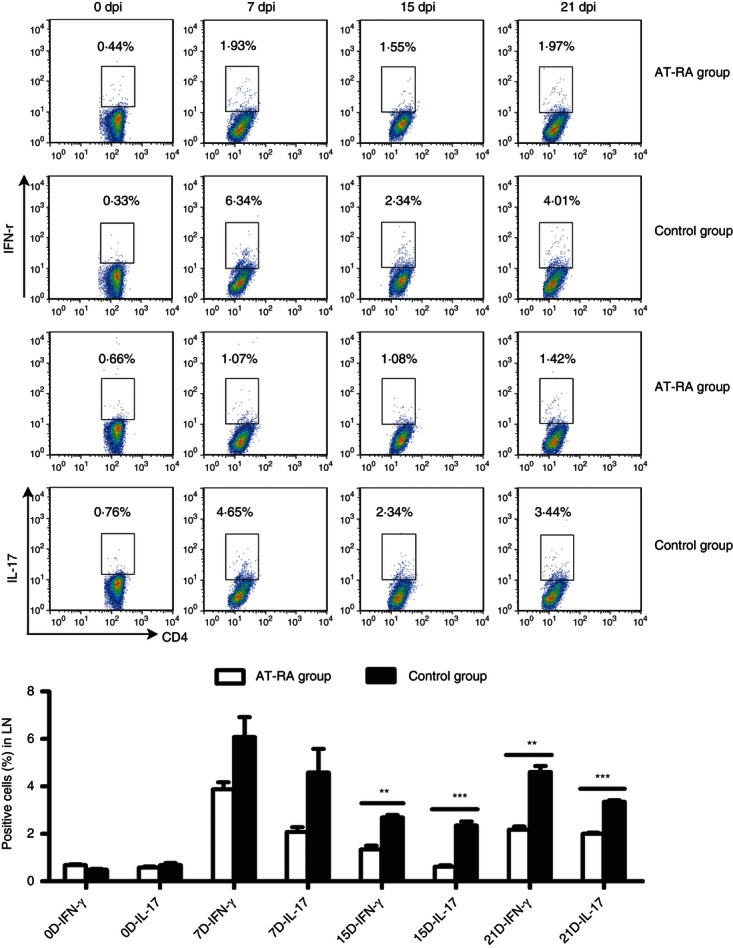
All-trans retinoic acid (AT-RA) abrogated the induction of T helper type 1 (Th1) and Th17 cells in lymph nodes in vivo. Lymphocytes from draining lymph nodes (dLNs) of animals in both the AT-RA and control group were harvested on 0, 7, 15 and 21 dpi. Cells were stained with FITC-conjugated anti-CD4 and allophycocyanin-conjugated anti-CD3, then fixed, permeabilized, and stained intracellularly with phycoerythrin-conjugated anti-interleukin-17 (IL-17) or anti-interferon-γ (IFN-γ). Data shown are expressed as the mean ± SE of three experiments with five or six mice per group at each time-point. **P < 0·01, ***P < 0·001.
Figure 8.
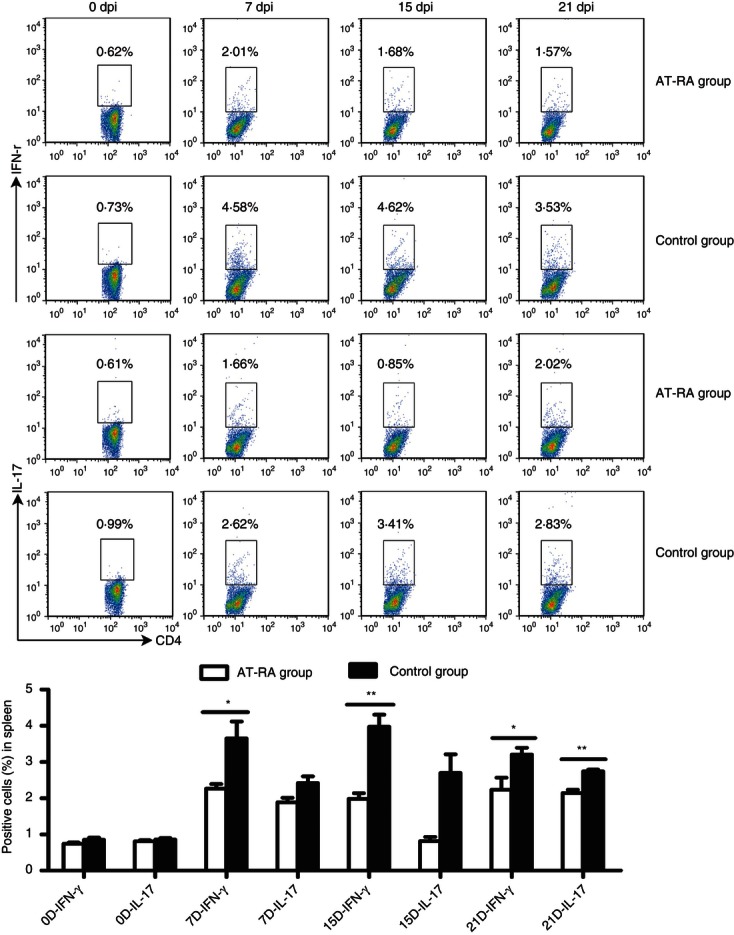
All-trans retinoic acid (AT-RA) abrogated the induction of T helper type 1 (Th1) and Th17 cells in the spleen in vivo. Lymphocytes collected from the spleen were cultured and analysed as described in Fig. 7. Data are expressed as the mean ± SE. Results are representative of three experiments with five or six mice per group at each time point. *P < 0·05, **P < 0·01.
To assess whether the diminished maturation of AT-RA pre-treated BMDCs was less efficient to drive the differentiation of Th cells in vitro, we co-incubated lymphocytes with AT-RA or DMSO pre-treated BMDCs loaded with MOG35–55 peptide and detected the levels of T-cell proliferation and production of cytokines in vitro (Fig. 9). In contrast to the control group (lymphocytes only), T-cell proliferation was greatly increased when co-cultured with BMDCs. Moreover, AT-RA pre-treated BMDCs suppressed T-cell proliferation to a significantly lower level compared with DMSO pre-treated BMDCs (Fig. 9a). In addition, cytokine production seemed to present as the same tendency, that the percentages of IL-17-secreting CD4+ T cells and IFN-γ-secreting CD4+ T cells were higher in co-cultured groups than in the AT-RA pre-treated BMDCs co-cultured group (Fig. 9b,c). Collectively, these findings suggested that AT-RA-treated BMDCs were inefficient at fully activating T cells in vitro.
Figure 9.
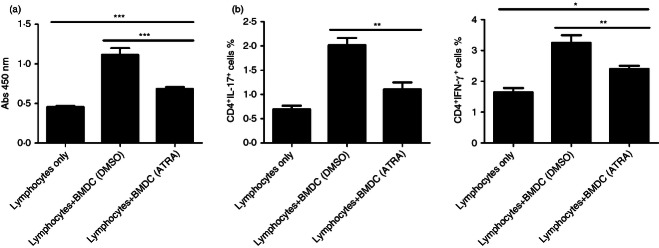
All-trans retinoic acid (AT-RA) pre-treated bone mesenchymal dendritic cells (BMDCs) abrogated the proliferation of lymphocytes and the differentiation of T helper type 1 (Th1)/Th17 in vitro. AT-RA/DMSO pre-treated BMDCs were incubated with lymphocytes isolated form draining lymph nodes (dLNs) of experimental autoimmune encephalomyelitis (EAE) mice 5 days post-immunization (dpi). Proliferation of lymphocytes (a) was measured by CCK-8 and cytokine production in the population of CD4-positive cells (b) was analysed with flow cytometry. Data, shown as mean ± SE, are representative of two experiments with five mice per experiment. *P < 0·05, **P < 0·01, ***P < 0·001.
The observations of decreased levels of pathogenic cells, as well as attenuated clinical symptoms and rapid recovery in the AT-RA treatment group, were consistent with the fact that peripheral polarization of lymphocyte responses contributed to CNS inflammation. Our study suggested that AT-RA treatment ameliorated EAE disease progression and severity by down-regulating Th1 and Th17 responses. Based on results demonstrating that AT-RA could suppress DC maturation at the T-cell priming stage, we proposed that AT-RA could also down-regulate Th1- and Th17-cell-mediated responses. However, additional studies will be needed to further define the mechanisms by which AT-RA influences T-cell responses in EAE.
Discussion
In general, naive T cells recirculate between the circulation system, the lymphatic system, and secondary lymphoid tissues as a means of surveying the antigenic milieu. Naive T cells are activated only when stimulated by mature DCs which have migrated into the dLNs or spleen and expressed the appropriate antigen/MHC complex and co-stimulatory molecules. The amount and the quality of peptide-MHC ligands and co-stimulatory molecules are likely to determine the nature of the T-cell–DC interaction.18,19 Our results demonstrated that AT-RA suppressed the number of mature DCs (defined as MHC-class IIhigh CD80+ CD86+on CD11chigh cells) compared with levels observed in the control group (Fig. 4). In addition, it was shown in vitro that AT-RA negatively modulated DC maturation (Fig. 5). Because the high MHC-class II levels and co-stimulatory signal expression on DCs favour the occurrence of stable interactions between antigen-specific T cells and DCs, AT-RA might inhibit T-cell activation indirectly. Given the association of EAE amelioration with T-cell inefficiency, one could hypothesize that AT-RA may extenuate disease through a non-specific toxic effect on lymphocytes. However, the apoptosis rate of mononuclear cells was found to have no obvious difference between the AT-RA-treated group and the control group both in immunized and non-immunized mice after 4 days of administration (Fig. 3). Hence, other mechanisms by which AT-RA induced disease amelioration might exist. The most likely explanation is immature DCs leading to T-cell inefficiency. These results support previously published results demonstrating that AT-RA reduced the capacity of umbilical cord blood monocyte-derived DCs to activate alloreactive T cells.20
Unexpectedly, we found that a population of monocytes with a CD44high CD11b+ CD36+ CD40+/− MHC-class II+/− surface phenotype was abrogated after AT-RA treatment. Generally, monocytes possess the potential to exert both positive and negative effects, depending on their cytokine secretion profile.21 It has been shown that monocytes are involved in the exacerbation of EAE,3,4 and increased numbers of circulating pro-inflammatory monocytes correlate with relapses in EAE.22 In our study, there was a significant decrease in monocytes, in both the dLNs and spleen after AT-RA treatment (Fig. 6). Fewer monocytes indicated fewer drivers, pro-inflammatory cytokines and chemokines, to recruit additional immune cells to enhance the host autoimmune response. Moreover, a previous study has shown that AT-RA down-regulates interferon-inducible protein 10,23 tumour necrosis factor-α and other central pro-inflammatory cytokines and chemokines in monocytes at the level of transcription.24,25 These findings may explain our observation that AT-RA treatment decreased the level of monocytes associated with an attenuation of clinical symptoms and an increased recovery rate.
The CNS is considered to be an immune-privileged tissue defined by a minimal number of lymphocytes infiltrating under physiological conditions. However, inflammatory cells are capable of infiltrating the CNS parenchyma during EAE progression. It was generally thought that inflammatory responses associated with EAE initially occurred under the subarachnoid space. Our results showed that a significant number of pro-inflammatory monocytes (CD44high CD11b+ CD36+ CD40− MHC-class II+/−) accumulated in the subarachnoid space during both the early and late stages of EAE in the control group (Fig. S1). In contrast, the number of monocytes in the AT-RA treatment group was significantly lower in these tissues at similar time-points. Because CD11b is an integrin that plays an important role in mediating leucocyte adhesion, enhanced numbers of CD11b+ cells might facilitate the invasion of pro-inflammatory monocytes into the CNS.26 CD44 is also involved in maintaining monocytes in circulation during inflammatory responses and mediating their homing to inflammatory sites.27 EAE progression can be exacerbated by large numbers of CD44high CD11b+ cells that infiltrate into the subarachnoid space. Although our study did not find many CD44high CD11b+ cells that infiltrated into the spinal cord parenchyma, statistical differences were still observed between the AT-RA treatment and control groups. Further investigation is needed to find the reason why this population of monocytes do not infiltrate the parenchyma of CNS. T cells infiltrating the spinal cord at the onset of EAE produced cytokines and chemokines that up-regulated trafficking across the blood–brain barrier and blood–spinal cord barrier. This traffic was associated with the initial recruitment of additional inflammatory cells including Th1 and Th17 cells, neutrophils and pro-inflammatory monocytes.28 These data further suggested that recruitment of inflammatory monocytes represented a kind of inflammatory trigger, regardless of their minority, probably acting as a modulator by secreting pro-inflammatory cytokines. Collectively, our results suggested that the AT-RA-induced decrease of monocytes was associated with the exacerbation of EAE. However, the exact mechanism by which AT-RA affects the function of circulating monocytes remains to be determined.
The chronic demyelination observed during EAE progression is associated with pro-inflammatory responses mediated by infiltrating T cells. Both IFN-γ-secreting Th1 cells and IL-17-secreting Th17 cells have been shown to have a pathogenic role in EAE progression and presentation. In this study, we observed significant decreases in Th1/Th17 cells following AT-RA treatment, particularly during the peak phase of disease (Figs 7 and 8). In addition, we have shown in vitro that the diminished percentages of pathogenic Th1/Th17 cells and decreased proliferating ability of lymphocytes were closely associated with the AT-RA-treated BMDCs (Fig. 9).
Although lineage-specific transcription factors or effector cytokines may contribute to the suppression of RA on Th-cell subsets,29–32 accumulating studies indicate that RA can influence T-cell development based on the underlying innate immune responses. In the intestinal immune system, RA was found to facilitate the production of CD103+ DC cells, which disrupted the balance between regulatory T cells and Th17 cells, resulting in extenuated ileitis.33 RA deficiency preferred Th1 responses to Th2 responses by altering the lymphoid/myeloid DC population in spleen.34 In this study, we could conservatively conclude that AT-RA decreased the number of mature DCs and so down-regulated the potential of activating cognate T cells during the priming stage of EAE, resulting in decreased Th1/Th17 responses during the later phases of disease.
However, T-cell polarization is a complex process initiated in secondary lymphoid organs. Pro-inflammatory and anti-inflammatory signals derived from other effector cells, such as natural killer cells or macrophages, are also involved in determining the degree of T-cell polarization. Whether AT-RA treatment could also affect these cells (or act directly on T cells) remains to be determined. Additionally, AT-RA could bind to its nuclear receptor, retinoic acid receptors, and activate the transduction of target genes via complex genetic and epigenetic mechanisms.35 Additional research will be necessary to further define the anti-inflammatory effects of AT-RA on inflammatory cells.
This study demonstrates that AT-RA treatment successfully inhibits the development of EAE. The immune regulatory effects of AT-RA on the immune response include suppression of mature DCs, the decrease of inflammatory monocytes, and the diminished polarization of Th1/Th17 cells. Further research is needed to understand the exact mechanisms by which AT-RA treatment prevents EAE progression. More importantly, the inhibitory effect of AT-RA on the professional antigen-presenting capacity of DCs may be the most specific and effective approach for modulating immune responses. In this regard, our study describes a strategy for effectively targeting DC maturation for the prevention or amelioration of autoimmune inflammation.
Acknowledgments
Thanks are due to Prof. Barbara Gastel and Michelle Yeoman in Texas A&M University for their revision of the English. This research was supported by Harbin Science & Technology Bureau Fund (2009RFXXS009;2008RFQXS085), Natural Science Foundation for Youth of China (30901330; 81000512; 81000511), Natural Science Foundation of China (81171121), Heilongjiang Provincial Natural Science Foundation (LC2011C14), Harbin Medical University Cell Biological Engineering Centre (1151gzx05), Open Fund of Key Laboratory of Myocardial Ischemia Mechanism and Treatment (KF201013), New Century Pioneer Foundation of Ministry of Education of Hei Longjiang Province (1251-NCET-010), and Master Innovation Research Foundation of Hei Longjiang Province (YJSCX2011-347HLJ).
Disclosures
The authors declare no conflict of interest.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Figure S1. Identification of monocytes.
References
- 1.Stromnes IM, Cerretti LM, Liggitt D, Harris RA, Goverman JM. Differential regulation of central nervous system autoimmunity by Th1 and Th17 cells. Nat Med. 2008;14:337–42. doi: 10.1038/nm1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zozulya AL, Clarkson BD, Ortler S, Fabry Z, Wiendl H. The role of dendritic cells in CNS autoimmunity. J Mol Med (Berl) 2010;88:535–44. doi: 10.1007/s00109-010-0607-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ponomarev ED, Shriver LP, Maresz K, Dittel BN. Microglial cell activation and proliferation precedes the onset of CNS autoimmunity. J Neurosci Res. 2005;81:374–89. doi: 10.1002/jnr.20488. [DOI] [PubMed] [Google Scholar]
- 4.Heppner FL, Greter M, Marino D, et al. Experimental autoimmune encephalomyelitis repressed by microglial paralysis. Nat Med. 2005;11:146–52. doi: 10.1038/nm1177. [DOI] [PubMed] [Google Scholar]
- 5.Huitinga I, van Rooijen N, de Groot CJ, Uitdehaag BM, Dijkstra CD. Suppression of experimental allergic encephalomyelitis in Lewis rats after elimination of macrophages. J Exp Med. 1990;172:1025–33. doi: 10.1084/jem.172.4.1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brosnan CF, Bornstein MB, Bloom BR. The effects of macrophage depletion on the clinical and pathologic expression of experimental allergic encephalomyelitis. J Immunol. 1981;126:614–20. [PubMed] [Google Scholar]
- 7.Swanborg RH. Experimental autoimmune encephalomyelitis in rodents as a model for human demyelinating disease. Clin Immunol Immunopathol. 1995;77:4–13. doi: 10.1016/0090-1229(95)90130-2. [DOI] [PubMed] [Google Scholar]
- 8.Ajami B, Bennett JL, Krieger C, McNagny KM, Rossi FM. Infiltrating monocytes trigger EAE progression, but do not contribute to the resident microglia pool. Nat Neurosci. 2011;14:1142–9. doi: 10.1038/nn.2887. [DOI] [PubMed] [Google Scholar]
- 9.Iwata M, Hirakiyama A, Eshima Y, Kagechika H, Kato C, Song SY. Retinoic acid imprints gut-homing specificity on T cells. Immunity. 2004;21:527–38. doi: 10.1016/j.immuni.2004.08.011. [DOI] [PubMed] [Google Scholar]
- 10.Mora JR, von Andrian UH. Role of retinoic acid in the imprinting of gut-homing IgA-secreting cells. Semin Immunol. 2009;21:28–35. doi: 10.1016/j.smim.2008.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Darmanin S, Chen J, Zhao S, et al. All-trans retinoic acid enhances murine dendritic cell migration to draining lymph nodes via the balance of matrix metalloproteinases and their inhibitors. J Immunol. 2007;179:4616–25. doi: 10.4049/jimmunol.179.7.4616. [DOI] [PubMed] [Google Scholar]
- 12.Jin CJ, Hong CY, Takei M, et al. All-trans retinoic acid inhibits the differentiation, maturation, and function of human monocyte-derived dendritic cells. Leuk Res. 2011;34:513–20. doi: 10.1016/j.leukres.2009.10.006. [DOI] [PubMed] [Google Scholar]
- 13.Racke MK, Burnett D, Pak SH, Albert PS, Cannella B, Raine CS, McFarlin DE, Scott DE. Retinoid treatment of experimental allergic encephalomyelitis. IL-4 production correlates with improved disease course. J Immunol. 1995;154:450–8. [PubMed] [Google Scholar]
- 14.Massacesi L, Castigli E, Vergelli M, Olivotto J, Abbamondi AL, Sarlo F, Amaducci L. Immunosuppressive activity of 13-cis-retinoic acid and prevention of experimental autoimmune encephalomyelitis in rats. J Clin Invest. 1991;88:1331–7. doi: 10.1172/JCI115438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xu J, Drew PD. 9-Cis-retinoic acid suppresses inflammatory responses of microglia. J Neuroimmunol. 2006;171:135–44. doi: 10.1016/j.jneuroim.2005.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang T, Niwa S, Bouda K, Matsuura S, Homma T, Shudo K, Nagai H. The effect of Am-80, one of retinoids derivatives on experimental allergic encephalomyelitis in rats. Life Sci. 2000;67:1869–79. doi: 10.1016/s0024-3205(00)00776-1. [DOI] [PubMed] [Google Scholar]
- 17.Abtahi Froushani SM, Delirezh N, Hobbenaghi R, Mosayebi Gh. Therapeutic effects of all-trans retinoic acid on experimental autoimmune encephalomyelitis and its role in T-helper lymphocyte responses. Tehran Univ Med J. 2012;69:710–7. [Google Scholar]
- 18.Bousso P. T-cell activation by dendritic cells in the lymph nodes: lessons from the movies. Immunology. 2008;8:675–84. doi: 10.1038/nri2379. [DOI] [PubMed] [Google Scholar]
- 19.Mempel TR, Henrickson SE, Von Andrian UH. T-cell priming by dendritic cells in lymph nodes occurs in three distinct phases. Nature. 2004;427:154–9. doi: 10.1038/nature02238. [DOI] [PubMed] [Google Scholar]
- 20.Tao Y, Yang Y, Wang W. Effect of all-trans-retinoic acid on the differentiation, maturation and functions of dendritic cells derived from cord blood monocytes. FEMS Immunol Med Microbiol. 2006;47:444–50. doi: 10.1111/j.1574-695X.2006.00108.x. [DOI] [PubMed] [Google Scholar]
- 21.Yona S, Jung S. Monocytes: subsets, origins, fates and functions. Curr Opin Hematol. 2010;17:53–9. doi: 10.1097/MOH.0b013e3283324f80. [DOI] [PubMed] [Google Scholar]
- 22.King IL, Dickendesher TL, Segal BM. Circulating Ly-6C+ myeloid precursors migrate to the CNS and play a pathogenic role during autoimmune demyelinating disease. Blood. 2009;113:3190–7. doi: 10.1182/blood-2008-07-168575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tsai YC, Chang HW, Chang TT, Lee MS, Chu YT, Hung CH. Effects of all-trans retinoic acid on Th1- and Th2-related chemokines production in monocytes. Inflammation. 2008;31:428–33. doi: 10.1007/s10753-008-9095-x. [DOI] [PubMed] [Google Scholar]
- 24.Kolseth IB, Agren J, Sundvold-Gjerstad V, Lyngstadaas SP, Wang JE, Dahle MK. 9-cis retinoic acid inhibits inflammatory responses of adherent monocytes increases their ability to induce classical monocyte migration. J Innate Immun. 2012;4:176–86. doi: 10.1159/000332375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mathew JS, Sharma RP. Effect of all-trans-retinoic acid on cytokine production in a murine macrophage cell line. Int J Immunopharmacol. 2000;2:693–706. doi: 10.1016/s0192-0561(00)00032-1. [DOI] [PubMed] [Google Scholar]
- 26.Abbas AK, Lichtman AH. Cellular and Molecular Immunology. 5th edn. Philadelphia, PA: Saunders; 2003. [Google Scholar]
- 27.Xu H, Manivannan A, Crane I, Dawson R, Liversidge J. Critical but divergent roles for CD62L and CD44 in directing blood monocyte trafficking in vivo during inflammation. Blood. 2008;112:1166–74. doi: 10.1182/blood-2007-06-098327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Piccio L, Rossi B, Scarpini E, et al. Molecular mechanisms involved in lymphocyte recruitment in inflamed brain microvessels: critical roles for P-selectin glycoprotein ligand-1 and heterotrimeric G(i)-linked receptors. J Immunol. 2002;168:1940–9. doi: 10.4049/jimmunol.168.4.1940. [DOI] [PubMed] [Google Scholar]
- 29.Jeon EJ, Yoon BY, Lim JY, et al. Adoptive transfer of all-trans-retinal-induced regulatory T cells ameliorates experimental autoimmune arthritis in an interferon-γ knockout model. Autoimmunity. 2012;45:460–9. doi: 10.3109/08916934.2012.682666. [DOI] [PubMed] [Google Scholar]
- 30.Elas KM, Laurence A, Davidson TS, Stephens G, Kanno Y, Shevach EM, O'Shea JJ. Retinoic acid inhibits Th17 polarization and enhances FoxP3 expression through a Stat-3/Stat-5 independent signaling pathway. Blood. 2008;111:1013–20. doi: 10.1182/blood-2007-06-096438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xiao S, Jin H, Korn T, Liu SM, Oukka M, Lim B, Kuchroo VK. Retinoic acid increases Foxp regulatory T cells and inhibits development of Th17 cells by enhancing TGF-β-driven Smad3 signaling and inhibiting IL-6 and IL-23 receptor expression. J Immunol. 2008;181:2277–84. doi: 10.4049/jimmunol.181.4.2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhou L, Ivanov, Spolski R, et al. IL-6 programs Th17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat Immunol. 2007;8:967–74. doi: 10.1038/ni1488. [DOI] [PubMed] [Google Scholar]
- 33.Collins CB, Aherne CM, Kominsky D, McNamee EN, Lebsack MD, Eltzschig H, Jedlicka P, Rivera-Nieves J. Retinoic acid attenuates ileitis by restoring the balance between T-helper 17 and T regulatory cells. Gastroenterology. 2011;141:1821–31. doi: 10.1053/j.gastro.2011.05.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Duriancik DM, Hoag KA. Vitamin A deficiency alters splenic dendritic cell subsets and increases CD8+Gr-1+ memory T lymphocytes in C57BL/6J mice. Cell Immunol. 2010;265:156–63. doi: 10.1016/j.cellimm.2010.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hall JA, Grainger JR, Spencer SP, Belkaid Y. The role of retinoic acid in tolerance and immunity. Immunity. 2011;35:13–22. doi: 10.1016/j.immuni.2011.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.