

Abstract
To identify the genetic factors that influence overall survival in never smokers who have non-small cell lung cancer (NSCLC), we performed a consistency meta-analysis study utilizing genome-wide association approaches for overall survival in 327 never smoker NSCLC patients from the MD Anderson Cancer Center and 293 cases from the Mayo Clinic. We then performed a two-pronged validation of the top 25 variants that included additional validation in 1,256 NSCLC patients from Taiwan and assessment of expression quantitative trait loci (eQTL) and differential expression of genes surrounding the top loci in 70 tumors and matched normal tissues. A total of 94 loci were significant for overall survival in both MD Anderson and Mayo studies in the consistency meta-analysis phase, with the top 25 variants reaching a p-value of 10−6. Two variants of these 25 were also significant in the Taiwanese population: rs6901416 (HR:1.44, 95%CI:1.01-2.06) and rs10766739 (HR:1.23, 95%CI:1.00-1.51). These loci resulted in a reduction in median survival time of at least 8 and 5 months in three populations, respectively. An additional six variants (rs4237904, rs7976914, rs4970833, rs954785, rs485411, and rs10906104) were validated through eQTL analysis that identified significant correlations with expression levels of six genes (LEMD3, TMBIM, ATXN7L2, SHE, ITIH2, and NUDT5, respectively) in normal lung tissue. These genes were also significantly differentially expressed between the tumor and normal lung. These findings identify several novel, candidate prognostic markers for NSCLC in never smokers, with eQTL analysis suggesting a potential biological mechanism for a subset of these observed associations.
INTRODUCTION
Lung cancer is the number one cause of cancer-related mortality in the United States with an estimated 157,300 deaths in 2010(1). Non-small cell lung cancer (NSCLC) comprises approximately 80% of all lung cancers with 5 year survival rate for all stages ranging from 11% to 17%(2). It is well-known that the majority of lung cancer deaths are due to tobacco smoking; however 10-20% of lung cancer-related deaths involve people who have never smoked(3). It is increasingly clear that lung cancer in never smokers represents a unique disease entity separate from smoking-related lung cancer, highlighting the need for investigation and discovery of novel genetic factors influencing survival in this population.
Tumor-based studies have shown that lung cancer in never smokers has a distinct profile from that in smokers. Never smokers have very low rates of mutations in K-ras and p53 genes compared with smokers(4-7). Inactivation of p16 by promoter methylation also has been reported to be more frequent in tobacco-associated lung cancer(8-10), whereas EGFR mutations were more common in never smokers(11). Indeed, better efficacy and survival has been reported for EGFR tyrosine kinase inhibitors in the treatment of NSCLC among never smokers(11-13). Furthermore, differences in allelic imbalances have been observed with higher chromosomal aberrations on 3p, 6q, 9p, 17p, and 19p from smokers than never smokers(14). However, these loci were commonly altered in both smoking subgroups in a Chinese population(15). Patients from East Asia, particularly women never smokers, also have a high frequency of EGFR mutations(13). These results indicate that there are also differences in tumor characteristics across ethnic/racial groups that may further influence outcomes.
With these differences between lung cancer in smokers and never smokers in mind, we previously performed a genome-wide association study (GWAS) to identify genetic variants of single nucleotide polymorphisms (SNPs) associated with the risk of developing lung cancer in never smokers(16). In contrast to the candidate genes involving nicotinic receptors and other chromosomal regions (i.e. 5p15, 15q25, and 6p21) previously reported as susceptibility loci for overall lung cancer risk in smoking populations(17-21), we found GPC5 genetic variants were associated with lung cancer in never smokers only and this association was supported by expression QTL analysis (eQTL). Since the GWAS approach itself is only able to map disease risk to specific loci, not necessarily the causal gene, additional clues as to the biological mechanism responsible for the association can be provided by the incorporation of eQTL analysis into GWAS(22-24).
Individual differences in survival among patients treated with identical treatment regimens for same stage tumors strongly implicate a genetic basis also for survival in NSCLC. Identifying the genetic basis that contributes to this variation would be potentially valuable to the clinician for several reasons, including stratification of the population into different prognostic groups to assist in selection of optimal treatment modalities. This may also facilitate risk stratified clinical trials for novel treatments in those patients who are predicted to not respond to standard treatment. In addition, the potential to identify novel targets for therapeutic development to improve treatment response/survival. Few studies have comprehensively examined the influence of germline genetic variants on lung cancer survival. A previous genome-wide study of variation within NSCLC tumors identified polymorphisms within STK39, PCDH7, A2BP1, and EYA2, as well as copy number changes on 3p, 5p, and 8q that were prognostic of overall survival in early stage patients(25). In addition, our group identified a genetic predictor of survival in NSCLC patients treated with platinum-based therapy(26). However, these studies were primarily of smokers and did not explore genetic variation that would predict overall survival in never smoker lung cancer patients. In this study, we conducted the first GWAS among never smoker lung cancer patients to identify germline genetic variants that are associated with overall survival in this distinct population of NSCLC. To increase the sample size and to minimize false discovery, a multi-stage study design using dual genome-wide scan was performed in two independent populations for consistency meta-analysis The top 25 variants were then validated using a two-pronged approach that included genotyping in a large population of never smoker lung cancer patients from Taiwan to better understand the effects of these candidate prognostic loci in a population of non-European descent and eQTL mapping to identify prognostic loci based on potential functional effects within normal lung tissues.
MATERIAL and METHODS
Study Populations
This study involved three independent patient populations from The University of Texas MD Anderson (MDACC) and Mayo Clinic (Mayo) in United States and multiple institutions in Taiwan (Figure 1). All participants included in this study were NSCLC patients that were never smokers. Written informed consent was obtained from all participants and the study was approved by the institutional review boards of each participating institution.
Figure 1.

Schematic of study design involving patient populations from MD Anderson, Mayo Clinic, and Taiwan, and eQTL analysis
MD Anderson study
All cases were newly diagnosed, histologically confirmed NSCLC patients enrolled from 1995 to 2008 in an ongoing epidemiologic lung cancer study at MDACC. Comprehensive epidemiological data were collected, including detailed smoking history. At the end of each interview, blood sample was collected for DNA extraction. Clinical and follow-up data was collected by trained chart reviewers, including date of diagnosis, pathology, grade, clinical stage, surgery, chemotherapy, radiotherapy, chemoradiotherapy, and mortality.
Mayo Clinic study
Never smoker lung cancer patients were identified and enrolled between 1997 and 2007. A detailed subject enrollment process has been reported previously(27, 28). Briefly, new cases diagnosed with lung cancer are identified by a daily electronic pathology reporting system. Once identified, study consents are obtained from the patients for enrollment, their medical records abstracted (from Mayo Clinic and outside records if treated elsewhere), and interviews conducted. Detailed information on demographics and smoking history was collected vital status and cause of death were determined by reviewing the Mayo Clinic registration database and medical records, correspondence from patients’ next of kin, death certificates, obituary documents, the Mayo Clinic Tumor Registry, and the Social Security Death Index.
Taiwan study
All cases were never smokers drawn from the ongoing cooperative study, the Genetic Epidemiological Study of Lung Adenocarcinoma (GELAC) in Taiwan, which includes patients recruited between January 2002 and May 2010 from six hospitals(29). Cases were over the age of 18 with incident primary lung cancer and confirmed by hospital pathologists. Blood sample and epidemiological data on demographic characteristics and smoking history were collected by a trained research nurse at recruitment.
Genotyping
Genotyping and quality control for the MDACC and Mayo datasets have been previously described(16). Briefly, genotyping data for MDACC patients was obtained from Illumina HumanHap 660k BeadChips (San Diego, CA) and genotyping data for Mayo patients was obtained from Illumina HumanHap 370k and 610k BeadChips. All the individuals had call rate ≥95% and SNPs passed the quality control filtering with call rate ≥ 95% and minor allele frequency ≥0.01. Genotyping of the top 25 SNPs was conducted using TaqMan Genotyping assays (Applied Biosystems, Foster City, CA) at the National Health Research Institutes, Taiwan.
Microarray e-QTL Analysis
Total RNA was extracted from a total of 70 tumor tissues and 70 matched normal lung tissues from the Mayo Clinic using miRNeasy Mini Kit (QIAGEN, Valencia, CA) under the manufacturer’s guidelines. All 70 patients were analyzed in the GWAS. Total RNA integrity was assessed using an Agilent 2100 Bioanalyzer. We used the Whole-Genome DASL Assay (Illumina) for gene expression profiling according to the manufacturer’s recommendation. 200ng of RNA from each subject was labeled and hybridized to each array using standard Illumina protocols and scanned on a BeadArray reader. Arrays were normalized using quantile normalization implemented in Bioconductor (www.bioconductor.org) and samples with replicates were averaged. We filtered out probes with median detection p≥0.01 based on reported values from BeadStudio. For the e-QTL analysis, we identified genes within 600 kb surrounding each selected SNP for genotype-phenotype association studies. Gene expression levels in the normal lung tissues were regressed to the number of minor alleles for each SNP representing that region. Genotype-phenotype associations with p<0.05 were considered significant. Differences between tumor and normal tissues were assessed by t-test with false-discovery corrected P-values.
Statistical Analysis
Overall survival time was defined as the time from date of diagnosis to date of death or date of last follow-up, whichever came first. For the US populations, a “never smoker” was defined as an individual who had smoked less than 100 cigarettes during his/her lifetime. “Never smokers” in the Taiwan study were individuals who had never smoked or not smoked 1 cigarette a day or 1 cigarette a week for 6 months at any period during his/her lifetime. MDACC and Mayo independently assessed three genetic models of inheritance (dominant, recessive, and additive) for each SNP using their own GWAS datasets. Hazard ratios (HR) and 95% confidence intervals (95% CI) were estimated using multivariate Cox proportional hazard regression analysis for each SNP. The covariates included were age, gender, stage, treatment information at MDACC and were gender, stage, grade, and treatment information at Mayo. The model with the smallest p-value was used to measure the statistical significance of each SNP. Only dominant model was considered when the rare homozygous genotype was less than five in patients with or without event of interest. Each site identified top 1,000 SNPs with overall survival (excluding SNPs with pairwise R2>0.9) that were overlapping in Illumina HumanHap 660k and HumanHap 370k BeadChips. 991 of the top 1,000 SNPs from the MDACC scan passed quality control measures in the Mayo dataset and 996 of the top 1,000 SNPs from the Mayo scan passed quality control measures in the MDACC dataset. The combined SNP list contained six overlapping SNPs, resulting in a final of 1,981 SNPs in the combined list. Meta-analysis was performed using fixed-effects model and estimated hazards ratio and 95% confidence interval derived from the Cox regression analysis adjusted for the appropriate covariates in the two datasets. Heterogeneity was tested with the Cochran’s Q test. The Conchran’s Q test was calculated by summing the squared deviations of each study’s effect estimate from the overall effect estimate weighted by inverse variance from each study. Kaplan-Meier curves and log-rank tests were used to assess the 3-year survival difference by individual polymorphisms. The quantile–quantile (Q–Q) plot displays the observed test statistic against the expected test statistic. For the MD Anderson population, we also selected the 1st principal component to include in the adjustment based on the scree plot of the top 60 eigenvalues and the Tracy-Widom p-values (data not shown). Only the top eigenvalue was >1.50 and showed clear separation from the other eigenvalues which were all <1.20. The λ(GC) for adjusting the first PC was 1.159 compared to 1.175 without adjusting for PC. The effect of population stratification is predicted to be minimal. Therefore, no principal component was adjusted in the subsequent association analyses. Analyses at MDACC were perform using STATA software (version 10.1, STATA Corporation, College Station, TX) and data manipulations at both MDACC and Mayo were completed using the PLINK and R software packages.
For the top 25 SNPs from the meta-analysis and genotyped in the Taiwan population, one variant (rs4263860) was not polymorphic in the population and was removed from further analysis. Multivariate Cox proportional hazard regression analysis was performed to estimate the HR and 95% CI while adjusting for age, gender, stage, treatment information. For the Taiwan study, analyses were done using R software.
RESULTS
Among all participants, 327 were from MDACC, 293 from Mayo, and 1,256 from Taiwan (Table 1). For the MDACC population, 13% of patients were alive but with follow-up less than three years, 10.4% of alive patients had less than three years of follow-up in Mayo Clinic, and 12.6% of patients are alive but with follow-up less than three years in Taiwan. The median age at diagnosis was ~60 years and a majority of the patients were female, up to 83.7% in the Asian population. The MDACC and Mayo studies were similar in the distributions of stage, while the Taiwan study had an excess of patients with stage IV disease. There were also differences between the US-based studies and the Taiwan study with regard to treatment regimens. These differences were taken into account in the data analysis through multivariable adjustment for sex, stage, and treatment regimens.
Table 1.
Host Characteristics
| MD Anderson N(%) |
Mayo Clinic N(%) |
Taiwan N(%) |
|
|---|---|---|---|
| Total | 327 | 293 | 1,256 |
| Age, mean(sd) | 61.40(13.15) | 62.6(13.2) | 59.6 (11.9) |
| Sex | |||
| Male | 104(31.8) | 98(33.4) | 204 (16.2) |
| Female | 223(68.2) | 195(66.6) | 1,051 (83.7) |
| Clinical stage | |||
| Stage I | 92(28.1) | 105(35.8) | 97 (7.7) |
| Stage II | 15(4.6) | 15(5.1) | 28 (2.2) |
| Stage III | 76(23.2) | 78(26.6) | 271 (21.6) |
| Stage IV | 144(44.0) | 95(32.4) | 753 (60.0) |
| Pathology | |||
| Adenocarcinoma | 244(74.6) | 230(78.5) | 1,011 (80.5) |
| Squamous cell | 26(8.0) | 15(5.1) | 87 (6.9) |
| Non-small cell, not otherwise | |||
| specified | 31(9.5) | 18(11.3) | 99 (7.9) |
| Bronchioloalveolar | 20(6.1) | 15(5.1) | 5 (0.4) |
| Other | 6(1.8) | 15(5.1) | 34 (2.7) |
| Surgery | |||
| Yes | 135(41.3) | 158(53.9) | 291 (23.2) |
| No | 192(58.7) | 135(46.1) | 964 (76.7) |
| Radiation | |||
| Yes | 88(26.9) | 76(25.9) | 519 (41.3) |
| No | 239(73.1) | 217(74.1) | 731 (58.2) |
| Chemotherapy | |||
| Yes | 197(60.2) | 177(60.4) | 1,105 (88.0) |
| No | 130(39.8) | 116(39.6) | 107 (8.5) |
| Chemoradiation | |||
| Yes | 34(10.4) | 69(23.5) | n/a |
| No | 293(89.6) | 224(76.5) | n/a |
| Vital status | |||
| Dead | 217(66.4) | 187(63.8) | 875 (69.7) |
| Alive | 110(33.6) | 106(36.2) | 377 (30.0) |
Percentages my not equal 100% due to missing values
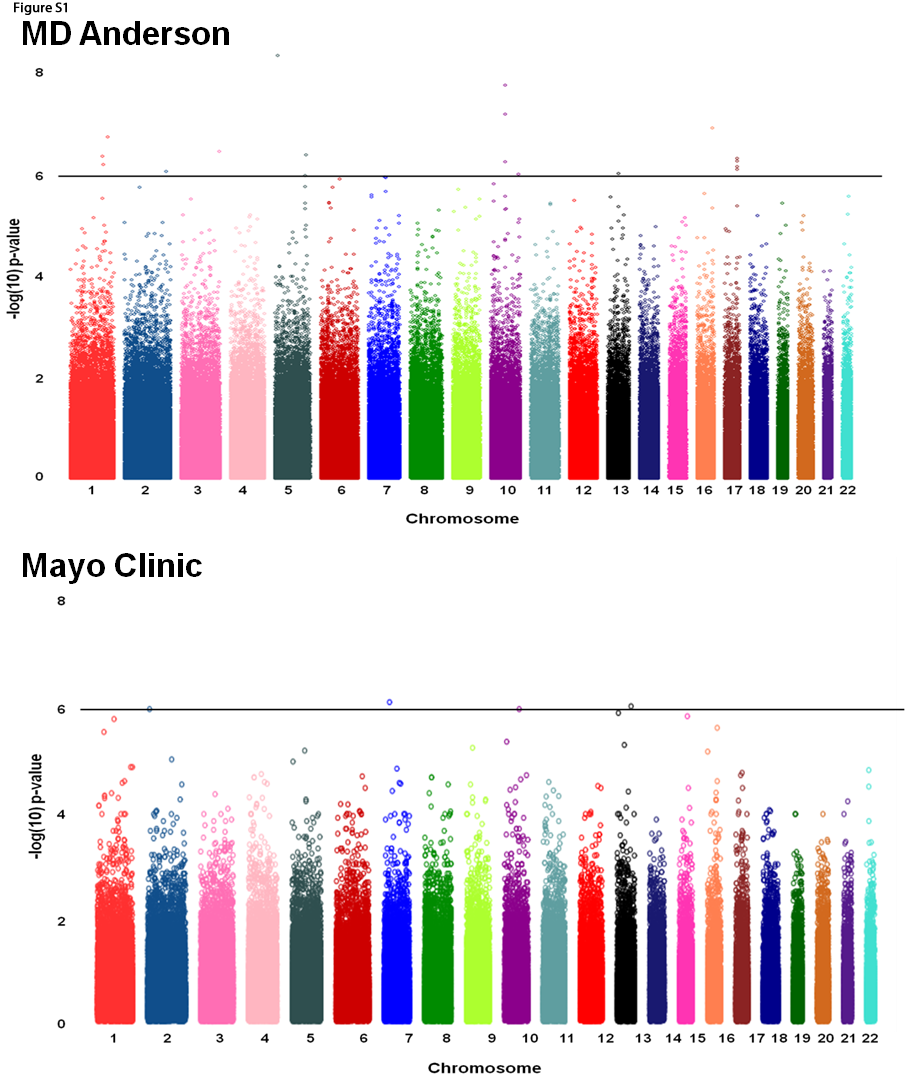
The initial genome-wide scans were done independently by MDACC and Mayo and identified several promising loci (Supplemental Figure 1). From the 1,000 SNPs identified from each site, the number of overlapping SNPs was six, which is more than the 3.17 what would be expected by chance (p=0.10). A meta-analysis was performed for the 1,981 SNPs identified in the combined list (Figure 1). The number of SNPs that showed association with overall survival (p<0.05) in both the MDACC and Mayo studies with a combined p<1.67×10−3 (Supplementary Table 1) was 94, which is more than the 48.4 variants that would be expected by chance alone (p=9.86×10−11). No heterogeneity was detected between MDACC and Mayo study samples (p for heterogeneity>0.05). Of these 94 loci, there were a total of 25 SNPs at a combined p-value of 10−6, including seven reaching 10−7.
These top 25 candidates were genotyped in the Taiwan population for further validation and to explore the effect of these variants in a population with a different genetic background. A SNP on chromosome 6q16 in an intron of NELL1 (Nel-like 1), rs6901416, displayed a similar effect on overall survival in the Taiwanese population as the US populations (HR=2.42, 95% CI: 1.66-3.52, combined p=4.10×10−6). This variant was associated with a 1.44-fold increase risk (95% CI=1.01-2.06, p=0.043) under the recessive model (Table 2) in the Taiwan population and resulted in at least 8 month reduction in median survival time in the three populations (Table 3; Figure 2a). An additional variant, rs10766739 (chromosome 11p15.1, ~58 kb downstream of EPHA7 (EPH receptor A7)), also one of the top locus in the US populations (combined p=3.66×10−7), reached borderline significance with p=0.051 (HR=1.23, 95% CI=1.00-1.51) in the Taiwan population, and had a significant effect on median survival time reducing by at least 6 months in MDACC and Taiwan populations (log-rank p: MDACC=0.029, Taiwan=0.024; Figure 2b). However, it would be expected to have one or two variants reach statistical significance (p<0.05) by chance when testing 25 individual SNPs. Therefore, these candidate variants will need to be further analyzed in other Asian populations to solidify their role in mediating overall survival for never smokers with lung cancer.
Table 2.
Results of two-pronged validation: Taiwan replication and eQTL mapping
| Combined Analysis | Taiwan | eQTL | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SNP | Chr | Location | Host Gene | Model | MAF | HR (95% CI) |
p value |
MAF | HR (95% CI) |
p value |
Top Significant Target Gene |
eQTL p value |
Differential Expression p value (FDR) |
Direction of Differential Expression |
| rs6901416 | 6 | 3′-flanking region |
EPHA7 | rec | 0.254 | 2.42 (1.66-3.52) |
4.10×10−6 | 0.188 | 1.44 (1.01-2.06) |
0.043 | MAP3K7 | 0.28 | 0.74 | no change |
|
| ||||||||||||||
| rs10766739 | 11 | intron | NELL1 | rec | 0.251 | 2.70 (1.84-3.96) |
3.66×10−7 | 0.369 | 1.23 (1.00-1.51) |
0.051 | HTATIP2 | 0.28 | 7.86×10−4 | up in tumor |
|
| ||||||||||||||
| rs4237904* | 12 | intergenic | rec | 0.252 | 2.67 (1.86-3.85) |
1.16×10−7 | 0.527 | 1.00 (0.85-1.17) |
0.96 |
TMBIM4/
LEMD3 |
0.014/ 0.022 |
0.0021/ 0.0031 |
up in tumor | |
|
| ||||||||||||||
| rs7976914* | 12 | intergenic | rec | 0.239 | 2.77 (1.89-4.06) |
1.79×10−7 | 0.288 | 1.10 (0.86-1.42) |
0.44 |
TIMBIM4/
LEMD3 |
0.013/ 0.029 |
0.0021/ 0.0031 |
up in tumor | |
|
| ||||||||||||||
| rs4970833 | 1 | intron | CELSR2 | dom | 0.460 | 1.81 (1.43-2.29) |
9.17×10−7 | 0.475 | 0.99 (0.84-1.15) |
0.85 | ATXN7L2 | 0.037 | 1.62×10−4 | up in tumor |
|
| ||||||||||||||
| rs954785 | 1 | intron | KCNN3 | dom | 0.219 | 1.62 (1.32-1.98) |
3.60×10−6 | 0.006 | 0.46 (0.23-0.92) |
0.029 | SHE | 3.19×10−4 | 1.22×10−6 | down in tumor |
|
| ||||||||||||||
| rs485411 | 10 | nsSNP | FLJ45983 | add | 0.256 | 1.44 (1.23-1.69) |
6.61×10−6 | 0.049 | 1.12 (0.91-1.40) |
0.29 | ITIH2 | 7.10×10−4 | 0.0012 | down in tumor |
|
| ||||||||||||||
| rs10906104 | 10 | intron | CDC123 | rec | 0.246 | 2.37 (1.62-3.47) |
8.84×10−6 | 0.584 | 0.96 (0.83-1.11) |
0.59 | NUDT5 | 0.034 | 7.36×10−7 | up in tumor |
Model: model of inheritance – dom=dominant, rec=recessive, add=additive, MAF: minor allele frequency, Top Significant Target Gene: gene displaying the most significant eQTL within a 600 kb region flanking each SNP.
SNPs in high linkage disequilibrium in US populations (r2=0.88)
Table 3.
Survival durations for significant SNPs by study site
| MDACC | Mayo Clinic | Taiwan | |||||
|---|---|---|---|---|---|---|---|
| SNP | Genotype | MST | p value | MST | p value | MST | p value |
| rs6901416 | TT+TG | 25.3 | >36.0 | 28.1 | |||
| GG | 17.3 | 0.15 | 22.0 | 0.026 | 18.7 | 0.13 | |
|
| |||||||
| rs10766739 | GG+GA | 25.3 | >36.0 | 28.3 | |||
| AA | 9.8 | 0.029 | 31.3 | 0.42 | 22.3 | 0.024 | |
|
| |||||||
| rs4237904 | GG+GT | 26.6 | >36.0 | 27.5 | |||
| TT | 12.4 | 1.52×10−4 | 20.4 | 0.19 | 28.3 | 0.34 | |
|
| |||||||
| rs7976914 | AA+AC | 26 | >36.0 | 27.8 | |||
| CC | 9.9 | 0.0017 | 16.8 | 0.057 | 27.6 | 0.87 | |
|
| |||||||
| rs4970833 | GG | 28.5 | >36.0 | 29.1 | |||
| GC+CC | 22.7 | 0.30 | 34.08 | 0.014 | 27.1 | 0.29 | |
|
| |||||||
| rs954785 | TT | 30.4 | >36.0 | 27.7 | |||
| TG+GG | 18.9 | 0.038 | 32.4 | 0.046 | 33.8 | 0.46 | |
|
| |||||||
| rs485411 | CC | 28.5 | >36.0 | 28.1 | |||
| CT | 22.3 | 35.9 | 25.6 | ||||
| TT | 12.7 | 0.084 | 34.1 | 0.57 | 29.4 | 0.32 | |
|
| |||||||
| rs10906104 | GG+GA | 26.6 | >36.0 | 28.1 | |||
| AA | 16.2 | 0.0033 | 22.0 | 0.088 | 27.7 | 0.65 | |
MST: Median survival time in months
Figure 2.

Kaplan-Meier 3-year survival curves for selected genetic loci in the three study populations. a) rs6901416, b) rs10766739, c) rs4237904, d) rs7976914, and e) rs954785. MST: median survival time in months
To identify potential prognostic loci based on phenotypic validation, we performed cis-eQTL analysis in 600 kb regions flanking each of the 25 candidate SNPs. We observed six SNPs showed a genotype-expression association with nearby genes (Table 2). Three of these variants reached genome-wide significance at 10−7 in the initial consistency meta-analysis (rs7976914, rs4237904, and rs4970833). rs7976914 and rs4237904 are ~48 kb apart and in strong LD (r2=0.88) representing a single unique locus. Subjects with the rare homozygous genotype for either locus had a greater than 2.6-fold significantly increased risk of death in the consistency meta-analysis (HR=2.67, 95% CI=1.86-3.85 and HR:2.77, 95% CI=1.89-4.06, respectively) compared to subjects with the common homozygous/heterozygous genotype. Furthermore, patients with the variant genotype for either SNP had significant reductions in survival times of 14.21 months (p=1.52×10−4) and 16.15 months (p=0.0017), respectively in the MDACC population with similar dramatic reductions in the Mayo population (Figure 2c and 2d; Table 3). Both SNPs are located in a middle of ~300 kb gene desert region on chromosome 12, yet genotypes for these variants were associated with TMBIM4 (p=0.013, p=0.014, respectively) and LEMD3 expression (p=0.027, p=0.022, respectively). The SNPs are over 500 kb from the 3′-UTR of TMBIM4, a gene encoding for transmembrane BAX inhibitor motif containing 4, and over 325 kb to the 3′-UTR of LEMD3 (LEM domain containing 3). The third significant genotype-expression association for a variant that p-value reached 10−7 in the consistency meta-analysis (HR: 1.81, 95% CI: 1.43-2.29) was for rs4970833 located within intron 5 of CELSR2, a gene encoding a receptor with EGF-domains. However, rs4970833 genotypes were associated with ATXN7L2 (ataxin 7-like 2) expression (p=0.037) located 222 kb downstream.
Similarly, an additional three SNPs located within known genes had significant eQTL relationships with distant genes (Table 2). rs954785 was associated with a 1.62-fold increase in risk in the consistency meta-analysis (95% CI: 1.32-1.98) and corresponding significant decreases in median survival time for both the US populations (p=0.038 and p=0.046, respectively; Table 3; Figure 2e). Interestingly, this effect was in the opposite direction for the Taiwan population (HR=0.46, 95% CI:0.23-0.92, p=0.029; Table 3). This variant is located in an intron of KCNN3, a potassium channel, yet a significant genotype-phenotype relationship was identified with SHE (Src homology 2 domain containing E; p=3.19×10−4), located over 195 kb 3′ of KCNN3. FLJ45983 encodes for a gene with unknown function and harbors a non-synonymous variant, rs485411 that results in a His-Arg substitution. This variant resulted in a 1.44-fold increase in risk (95% CI: 1.23-1.69). This SNP was not associated with FLJ45983 expression, but expression of ITIH2 (inter-alpha-trypsin inhibitor heavy chain 2; p=7.10×10−4) nearly 302 kb away on chromosome 10. The final eQTL was identified between rs10906104 (consistency meta-analysis HR: 2.37, 95% CI: 1.62-3.47) and NUDT5 (nudix (nucleoside diphosphate linked moiety X)-type motif 5; p=0.034). rs10906104 is an intronic variant in CDC123, a cell division cycle protein, and located over 42 kb 5′ from NUDT5. Interestingly, all six genes were significantly differentially expressed in 70 tumor tissues compared to adjacent normal tissues (Table 2).
DISCUSSION
Although tobacco use has been established as the main etiologic factor for lung cancer, a substantial fraction of lung cancer cases involves never smokers. A wide range of survival rates seen in patients of this population suggests heterogeneity of prognoses that may be independent of cancer type and disease stage. To identify genetic loci that are associated with overall survival in never smokers with NSCLC, we performed the first dual genome-wide scan, consistency meta-analysis in 327 never smokers recruited from MDACC and 293 never smokers at the Mayo Clinic. Additional validation analysis was performed in a group of 1,256 NSCLC in never smokers from Taiwan, and through exploration of eQTL and differential expression in tumor and adjacent normal lung tissues. This multi-phase approach incorporating a total of 1,876 NSCLC cases and phenotypic analysis identified several novel loci that are associated with overall survival in this distinct patient population.
One of the challenges of GWAS has been the investigation of the genetic effects in non-European populations. It is well established that population substructure can mask true associations that are population specific, necessitating restriction to a single ethnic/racial population to avoid this effect. However, it is also known that NSCLC manifests differently in different populations, particularly in individuals from East Asia(30). Therefore, we assessed the effect of the top 25 candidate variants identified from the European-descent GWAS studies in a large set of never smoker NSCLC cases from Taiwan. Two variants, rs6901416 and rs10766739, were both associated with a 1.2- to 1.5-fold increase in risk and corresponding reduction in survival durations in the Asian population. However, as stated in the results, only these two of the 25 variants genotyped replicated, which is no more than would be expected by chance and these results should be viewed with this in mind. Nevertheless, these two variants are candidates for further consideration. rs6901416 is located at 6q16.1, ~58 kb downstream of EPHA7. Ephrin receptors are receptor tyrosine kinases that are known to play a role in many facets of cancer and many cancer sites(31-34). EPHA7 is highly expressed in lung cancer and a secreted form of EPHA7, generated from a splice variant, was also detected and appeared to be unique to lung cancer(35). rs10766739 was one of the top seven loci in the US populations (combined p=3.66×10−7) and is located in an intron of NELL1 on chromosome 11p15.1. NELL1 encodes a secreted growth factor containing epidermal growth factor (EGF)-like repeats. Significantly, Jin et al. have reported that hypermethylation of NELL1 was a common and early event and was associated with poor prognosis in early-stage esophageal adenocarcinoma(36). Compared to normal tissue, methylation of NELL1 promoter increased with pathologic progression from Barrett’s metaplasia to esophageal adenocarcinoma and correlated with decrease survival in stage I and II patients. A genome-wide scan also identified epigenetic silencing of NELL1 in colon cancer cell lines(37). Taken together, these studies suggest NELL1 as a candidate tumor suppressor gene. No significant eQTL relationships were observed for either of these SNP-gene pairs.
Interestingly, the most significant SNP in the Taiwan replication (rs954785) had an opposite effect as that in the US populations, further emphasizing the differences variants can have in different populations. Together, the findings from the Taiwan population suggest that there may be commonality among risk factors in a European-descent population and Asian population, while also having unique genetic profiles influencing clinical outcomes in never smokers with lung cancer. The results from the Taiwan population will require further validation in an independent Asian population and additional follow-up for confirmation of the findings presented here.
eQTL analysis validated six variants as being associated with overall survival through the identification of genotype-phenotype relationships. rs4237904 and rs7976914 were highly significant in the consistency meta-analysis and associated with 14-20 month reduction in 3-year survival durations. Although they are located in a large gene desert, the expression of two genes in the vicinity of 12q14, LEMD3 and TMBIM4, were found to be significantly correlated with rs4237904 and rs7976914 genotypes (Table 2). Both SNPs are located 3′ distal of these two genes (300-500kb downstream). Since the two SNPs show strong LD and are relatively close to each other, it is possible that they might influence nearby cis-regulatory elements that can affect the transcription of the two surrounding genes from a distance. LEMD3 (also known as MAN1) encodes a LEM domain-containing protein that can antagonize TGF-beta signaling (38). Despite its role in altering TGF-beta signaling, association of this gene with cancer or its alteration in tumorigenesis has not been previously reported. TMBIM4 (or GAAP) is a novel anti-apoptotic regulator that belongs to a family of Bax-inhibitor-1 proteins that can bind to Bcl-2 family members and regulate cell death(39). TMBIM4 is ubiquitously expressed in all tissues, including the lungs. Since the endogenous functions of both aforementioned genes are relatively unstudied, their influence on cancer development and progression remains to be characterized. Moreover, both TMBIM4 and LEMD3 were found to be significantly differentially expressed between lung tumor and adjacent normal tissue, suggesting the potential involvement of this locus in lung tumorigenesis.
eQTL analysis also identified a correlation between genotypes for rs4970833 and ATXN7L2 expression. The variant is an intronic SNP within CELSR2, a receptor with EGF-domains and member of the flamingo subfamily of cadherins, on chromosome 1p21. CELSR2 was previously identified in multiple genome-wide association studies as being a candidate gene for various cardiovascular phenotypes and blood lipid levels(40-42). No studies have investigated CELSR2 in the setting of cancer. However, no eQTL association was evident between CELSR2 and the variant. ATXN7L2 encodes for the ataxin 7-like 2 protein and is a member of the ataxin family. The exact function of ataxin 7 and ataxin 7-like 2 is not clear. The current study provides the first hint of ATXN7L2’s potential role in cancer was provided by the significantly differential expression of the gene in lung cancer tumors and adjacent normal tissue samples (Table 2).
KCNN3 is a small conductance calcium activated potassium channel that has been shown to enhance the migration ability of melanoma cells and thus metastatic ability. An intronic variant in this gene, rs954785, was identified in the consistency meta-analysis as being highly significant for a poor overall survival and significantly reduced median survival time. rs954785 was not associated with KCNN3 expression, but expression of SHE. SHE has not been well characterized and little is known about its function, except for containing a Src homology 2 domain (SH2). This current study is the first to link SHE with cancer.
Another gene with unknown function, FLJ495983, was implicated in mediating overall survival in never smokers due the significant association of a non-synonymous variant, rs485411, with a poor outcome. This gene is close to GATA3, which encodes for a transcription factor that has been implicated in the development of many cancers, particularly luminal breast cancer (reviewed in(43)). eQTL analysis did not identify significant relationships between rs485411 and either of these two genes. The significant association was with ITIH2 located nearly 302 kb telomeric. ITIH2 (inter-alpha-trypsin inhibitor heavy chain 2), also known as SHAP, complexes with hyaluronan (HA) and plays a role in the inflammatory response. Serum levels of SHAP-HA have been associated with a poor outcome in ovarian and endometrial cancers(44, 45). High Glasgow scores, a measure of systemic inflammation, have been shown to be associated with poor outcomes for NSCLC in both smokers and never smokers, although more pronounced in smokers. It is possible that rs485411 is mediating ITIH2 and thus altering the levels of inflammation that would result in a poor outcome.
rs10906104, located in an intron of CDC123, was highly significant in the consistency meta-analysis and was an eQTL for NUDT5 located over 42 kb away and not with CDC123 expression. NUDT5 is a hydrolase that functions to remove toxic nucleotide derivatives from the cell to protect from mutagenesis(46). This includes 8-hydroxy-dGTP, a known mutagen(47), that is produced by reactive oxygen species in the cell. Tumor levels of 8-hydroxy-dGTP incorporated into DNA were found to be predictive of survival in NSCLC with lower 8-hydroxy-dGTP being associated with a better prognosis(48). Studies correlating rs10906104 and 8-hydroxy-dGTP levels would be of interest to determine the link between rs10906104, NUDT5, and 8-hydroxy-dGTP.
The significant eQTL relationships identified in this study were not between the identified SNP and the host gene, instead they were often for genes some distance from the genetic variant. However, the lack of eQTL associations between these host genes and the variants does not preclude them as playing a role in overall survival. eQTL analysis only investigates one mechanism by which a variant can have a phenotypic effect, not taking into consideration potential effects on splicing or allelic imbalance, or more subtle effects through gene regulatory elements that may not reach a significance threshold. Future studies are warranted to explore these relationships in greater detail. Furthermore, although associations were identified between candidate SNPs and both overall survival and cis-gene regulation, we cannot prove causality or functionality of the genotyped SNP in mediating variation in survival. The true causal variant(s) may be in linkage disequilibrium with the observed candidate SNPs. Future experiments are needed to explore this possibility and determine the underling mechanisms responsible for the association. However, the observed associations with cis-gene regulation provides additional support that these genomic regions identified initially through GWAS play a role in variation in overall survival in never smokers with NSCLC.
Previous genome-wide studies of germline genetic variants have generally focused on cancer predisposition rather than the identification of prognostic markers due to the requirement for large sample sizes and the likely presence of subject heterogeneity in clinical characteristics and treatment regimens. In addition, very little overlap between novel loci for susceptibility and clinical outcomes (survival) has been identified through both candidate gene and genome-wide approaches. This suggests that there are distinct biological pathways that mediate the development of disease from response to therapy and clinical outcomes. Recently, three genome-wide studies have been reported with the goal of identifying germline genetic variants associated with survival for NSCLC(25, 26, 49). Candidate loci were found in several genes: STK39, PCDH7, A2BP1, and EYA2 for early-stage lung cancer and CMKLR1, EIF4E2, ETS2, and DSCAM for advanced disease. We did not observe any significant associations with survival for SNPs within the genes implicated in these previous studies, suggesting that the genetic influence on mortality may be different for smokers and never smokers. This would be in line with the hypothesis that lung cancer in never smokers is a distinct disease from that in smokers. Alternatively, variances in population structure and/or clinical characteristics could contribute to differences in risk association.
In this study, we performed a consistency meta-analysis utilizing genome-wide scan data from two independent populations consisting of individuals of European-descent followed by a two prong validation that included replication in a non-European population and analysis of eQTL relationships to minimize potential for false discovery. All populations had detailed clinical information and were similar in clinicopathological characteristics, which minimized study site heterogeneity and allowed us to adjust for potential confounding variables. Moreover, Q-Q plots of the observed and expected associations in the studies indicate little deviation from the identity line except for the tail end, with inflation factors for the studies being 1.175 and 1.094 for MDACC and Mayo, respectively. This provides confidence in the quality and robustness of the consistency meta-analysis. Together, these results identify potential prognostic loci for NSCLC in never smokers and implicate several novel genes as holding a role in influencing lung cancer survival among never smokers.
Supplementary Material
{kind=link}
ACKNOWLEDGEMENTS
This study was supported by US National Institutes of Health grants R01-CA111646 (XW), P50CA070907 (JDM, JAR, XW), R01-CA127615 (XW), R01-CA80127 (PY), R01-CA115857 (PY), and R03-CA77118 (PY). The GELAC study was supported by DOH98-TD-G-111-015 from the National Research Program on Genomic Medicine (NRPGM) in Taiwan and DOH100-TD-PB-111-TM013 from the National Research Program for Biopharmaceuticals (NRPB) in Taiwan. Additional support was provided by the Center for Translational and Public Health Genomics of the Duncan Family Institute for Cancer Prevention and Risk Assessment (XW), MD Anderson Cancer Center Research Trust (XW) and Mayo Foundation Funds (PY). Mayo Genomic Analyses Shared Resources directed by Drs. Julie Cunningham and Jin Jen performed the genotyping and gene expression experiments for the Mayo Clinic study. The authors have declared that no competing interests exist.
Footnotes
Disclosures:
The authors declare that they have no conflicts of interest.
SUPPLEMENTAL DATA DESCRIPTION
Supplemental Data include one figure and one table.
REFERENCES
- 1.Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA Cancer J Clin. 2010;60:277–300. doi: 10.3322/caac.20073. [DOI] [PubMed] [Google Scholar]
- 2.Minna JD. Neoplasms of the Lung. In: S. Fauci Anthony, Braunwald Eugene, L. Kasper Dennis, et al., editors. Harrison’s Principles of Internal Medicine. McGraw-Hill; New York: 2005. pp. 506–15. [Google Scholar]
- 3.Toh CK, Lim WT. Lung cancer in never-smokers. J Clin Pathol. 2007;60:337–40. doi: 10.1136/jcp.2006.040576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Slebos RJ, Hruban RH, Dalesio O, Mooi WJ, Offerhaus GJ, Rodenhuis S. Relationship between K-ras oncogene activation and smoking in adenocarcinoma of the human lung. J Natl Cancer Inst. 1991;83:1024–7. doi: 10.1093/jnci/83.14.1024. [DOI] [PubMed] [Google Scholar]
- 5.Vahakangas KH, Bennett WP, Castren K, Welsh JA, Khan MA, Blomeke B, et al. p53 and K-ras mutations in lung cancers from former and never-smoking women. Cancer Res. 2001;61:4350–6. [PubMed] [Google Scholar]
- 6.Hainaut P, Olivier M, Pfeifer GP. TP53 mutation spectrum in lung cancers and mutagenic signature of components of tobacco smoke: lessons from the IARC TP53 mutation database. Mutagenesis. 2001;16:551–3. doi: 10.1093/mutage/16.6.551. author reply 5-6. [DOI] [PubMed] [Google Scholar]
- 7.Gealy R, Zhang L, Siegfried JM, Luketich JD, Keohavong P. Comparison of mutations in the p53 and K-ras genes in lung carcinomas from smoking and nonsmoking women. Cancer Epidemiol Biomarkers Prev. 1999;8:297–302. [PubMed] [Google Scholar]
- 8.Toyooka S, Suzuki M, Tsuda T, Toyooka KO, Maruyama R, Tsukuda K, et al. Dose effect of smoking on aberrant methylation in non-small cell lung cancers. Int J Cancer. 2004;110:462–4. doi: 10.1002/ijc.20125. [DOI] [PubMed] [Google Scholar]
- 9.Divine KK, Pulling LC, Marron-Terada PG, Liechty KC, Kang T, Schwartz AG, et al. Multiplicity of abnormal promoter methylation in lung adenocarcinomas from smokers and never smokers. Int J Cancer. 2005;114:400–5. doi: 10.1002/ijc.20761. [DOI] [PubMed] [Google Scholar]
- 10.Liu Y, Lan Q, Siegfried JM, Luketich JD, Keohavong P. Aberrant promoter methylation of p16 and MGMT genes in lung tumors from smoking and never-smoking lung cancer patients. Neoplasia. 2006;8:46–51. doi: 10.1593/neo.05586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pao W, Miller V, Zakowski M, Doherty J, Politi K, Sarkaria I, et al. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci U S A. 2004;101:13306–11. doi: 10.1073/pnas.0405220101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350:2129–39. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 13.Paez JG, Janne PA, Lee JC, Tracy S, Greulich H, Gabriel S, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304:1497–500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- 14.Sanchez-Cespedes M, Ahrendt SA, Piantadosi S, Rosell R, Monzo M, Wu L, et al. Chromosomal alterations in lung adenocarcinoma from smokers and nonsmokers. Cancer Res. 2001;61:1309–13. [PubMed] [Google Scholar]
- 15.Wong MP, Lam WK, Wang E, Chiu SW, Lam CL, Chung LP. Primary adenocarcinomas of the lung in nonsmokers show a distinct pattern of allelic imbalance. Cancer Res. 2002;62:4464–8. [PubMed] [Google Scholar]
- 16.Li Y, Sheu CC, Ye Y, de Andrade M, Wang L, Chang SC, et al. Genetic variants and risk of lung cancer in never smokers: a genome-wide association study. Lancet Oncol. 2010;11:321–30. doi: 10.1016/S1470-2045(10)70042-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Amos CI, Wu X, Broderick P, Gorlov IP, Gu J, Eisen T, et al. Genome-wide association scan of tag SNPs identifies a susceptibility locus for lung cancer at 15q25.1. Nat Genet. 2008;40:616–22. doi: 10.1038/ng.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hung RJ, McKay JD, Gaborieau V, Boffetta P, Hashibe M, Zaridze D, et al. A susceptibility locus for lung cancer maps to nicotinic acetylcholine receptor subunit genes on 15q25. Nature. 2008;452:633–7. doi: 10.1038/nature06885. [DOI] [PubMed] [Google Scholar]
- 19.McKay JD, Hung RJ, Gaborieau V, Boffetta P, Chabrier A, Byrnes G, et al. Lung cancer susceptibility locus at 5p15.33. Nat Genet. 2008;40:1404–6. doi: 10.1038/ng.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang Y, Broderick P, Webb E, Wu X, Vijayakrishnan J, Matakidou A, et al. Common 5p15.33 and 6p21.33 variants influence lung cancer risk. Nat Genet. 2008;40:1407–9. doi: 10.1038/ng.273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Landi MT, Chatterjee N, Yu K, Goldin LR, Goldstein AM, Rotunno M, et al. A genome-wide association study of lung cancer identifies a region of chromosome 5p15 associated with risk for adenocarcinoma. Am J Hum Genet. 2009;85:679–91. doi: 10.1016/j.ajhg.2009.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cookson W, Liang L, Abecasis G, Moffatt M, Lathrop M. Mapping complex disease traits with global gene expression. Nat Rev Genet. 2009;10:184–94. doi: 10.1038/nrg2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hsu YH, Zillikens MC, Wilson SG, Farber CR, Demissie S, Soranzo N, et al. An integration of genome-wide association study and gene expression profiling to prioritize the discovery of novel susceptibility Loci for osteoporosis-related traits. PLoS Genet. 2010;6:e1000977. doi: 10.1371/journal.pgen.1000977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chambers JC, Zhang W, Sehmi J, Li X, Wass MN, Van der Harst P, et al. Genome-wide association study identifies loci influencing concentrations of liver enzymes in plasma. Nat Genet. 2011;43:1131–8. doi: 10.1038/ng.970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huang YT, Heist RS, Chirieac LR, Lin X, Skaug V, Zienolddiny S, et al. Genome-wide analysis of survival in early-stage non-small-cell lung cancer. J Clin Oncol. 2009;27:2660–7. doi: 10.1200/JCO.2008.18.7906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu X, Ye Y, Rosell R, Amos CI, Stewart DJ, Hildebrandt MA, et al. Genome-wide association study of survival in non-small cell lung cancer patients receiving platinum-based chemotherapy. J Natl Cancer Inst. 2011;103:817–25. doi: 10.1093/jnci/djr075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang P, Allen MS, Aubry MC, Wampfler JA, Marks RS, Edell ES, et al. Clinical features of 5,628 primary lung cancer patients: experience at Mayo Clinic from 1997 to 2003. Chest. 2005;128:452–62. doi: 10.1378/chest.128.1.452. [DOI] [PubMed] [Google Scholar]
- 28.Yang P, Sun Z, Krowka MJ, Aubry MC, Bamlet WR, Wampfler JA, et al. Alpha1-antitrypsin deficiency carriers, tobacco smoke, chronic obstructive pulmonary disease, and lung cancer risk. Arch Inter Med. 2008;168:1097–103. doi: 10.1001/archinte.168.10.1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hsiung CA, Lan Q, Hong YC, Chen CJ, Hosgood HD, Chang IS, et al. The 5p15.33 locus is associated with risk of lung adenocarcinoma in never-smoking females in Asia. PLoS Genet. 2010:6. doi: 10.1371/journal.pgen.1001051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhou W, Christiani DC. East meets West: ethnic differences in epidemiology and clinical behaviors of lung cancer between East Asians and Caucasians. Chin J Cancer. 2011;30:287–92. doi: 10.5732/cjc.011.10106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang J, Kataoka H, Suzuki M, Sato N, Nakamura R, Tao H, et al. Downregulation of EphA7 by hypermethylation in colorectal cancer. Oncogene. 2005;24:5637–47. doi: 10.1038/sj.onc.1208720. [DOI] [PubMed] [Google Scholar]
- 32.Oricchio E, Nanjangud G, Wolfe AL, Schatz JH, Mavrakis KJ, Jiang M, et al. The Eph-receptor A7 is a soluble tumor suppressor for follicular lymphoma. Cell. 2011;147:554–64. doi: 10.1016/j.cell.2011.09.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Giaginis C, Tsourouflis G, Zizi-Serbetzoglou A, Kouraklis G, Chatzopoulou E, Dimakopoulou K, et al. Clinical significance of ephrin (eph)-A1, -A2, -a4, -a5 and -a7 receptors in pancreatic ductal adenocarcinoma. Pathol Oncol Res. 2010;16:267–76. doi: 10.1007/s12253-009-9221-6. [DOI] [PubMed] [Google Scholar]
- 34.Guan M, Xu C, Zhang F, Ye C. Aberrant methylation of EphA7 in human prostate cancer and its relation to clinicopathologic features. Int J Cancer. 2009;124:88–94. doi: 10.1002/ijc.23890. [DOI] [PubMed] [Google Scholar]
- 35.Tsuboi M, Mori H, Bunai T, Kageyama S, Suzuki M, Okudela K, et al. Secreted form of EphA7 in lung cancer. Int J Oncol. 2010;36:635–40. [PubMed] [Google Scholar]
- 36.Jin Z, Mori Y, Yang J, Sato F, Ito T, Cheng Y, et al. Hypermethylation of the nel-like 1 gene is a common and early event and is associated with poor prognosis in early-stage esophageal adenocarcinoma. Oncogene. 2007;26:6332–40. doi: 10.1038/sj.onc.1210461. [DOI] [PubMed] [Google Scholar]
- 37.Mori Y, Cai K, Cheng Y, Wang S, Paun B, Hamilton JP, et al. A genome-wide search identifies epigenetic silencing of somatostatin, tachykinin-1, and 5 other genes in colon cancer. Gastroenterology. 2006;131:797–808. doi: 10.1053/j.gastro.2006.06.006. [DOI] [PubMed] [Google Scholar]
- 38.Lin F, Morrison JM, Wu W, Worman HJ. MAN1, an integral protein of the inner nuclear membrane, binds Smad2 and Smad3 and antagonizes transforming growth factor-beta signaling. Hum Mol Genet. 2005;14:437–45. doi: 10.1093/hmg/ddi040. [DOI] [PubMed] [Google Scholar]
- 39.Zhou J, Zhu T, Hu C, Li H, Chen G, Xu G, et al. Comparative genomics and function analysis on BI1 family. Comput Biol Chem. 2008;32:159–62. doi: 10.1016/j.compbiolchem.2008.01.002. [DOI] [PubMed] [Google Scholar]
- 40.Willer CJ, Sanna S, Jackson AU, Scuteri A, Bonnycastle LL, Clarke R, et al. Newly identified loci that influence lipid concentrations and risk of coronary artery disease. Nat Genet. 2008;40:161–9. doi: 10.1038/ng.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Teslovich TM, Musunuru K, Smith AV, Edmondson AC, Stylianou IM, Koseki M, et al. Biological, clinical and population relevance of 95 loci for blood lipids. Nature. 2010;466:707–13. doi: 10.1038/nature09270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Aulchenko YS, Ripatti S, Lindqvist I, Boomsma D, Heid IM, Pramstaller PP, et al. Loci influencing lipid levels and coronary heart disease risk in 16 European population cohorts. Nat Genet. 2009;41:47–55. doi: 10.1038/ng.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chou J, Provot S, Werb Z. GATA3 in development and cancer differentiation: cells GATA have it! J Cell Physiol. 2010;222:42–9. doi: 10.1002/jcp.21943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yabushita H, Iwasaki K, Kanyama K, Obayashi Y, Zhuo L, Itano N, et al. Clinicopathological Role of Serum-Derived Hyaluronan-Associated Protein (SHAP)-Hyaluronan Complex in Endometrial Cancer. Obstet Gynecol Int. 2011;2011:739150. doi: 10.1155/2011/739150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Obayashi Y, Yabushita H, Kanyama K, Noguchi M, Zhuo L, Kimata K, et al. Role of serum-derived hyaluronan-associated protein-hyaluronan complex in ovarian cancer. Oncol Rep. 2008;19:1245–51. [PubMed] [Google Scholar]
- 46.Hori M, Satou K, Harashima H, Kamiya H. Suppression of mutagenesis by 8-hydroxy-2′-deoxyguanosine 5′-triphosphate (7,8-dihydro-8-oxo-2′-deoxyguanosine 5′-triphosphate) by human MTH1, MTH2, and NUDT5. Free Radic Biol Med. 2010;48:1197–201. doi: 10.1016/j.freeradbiomed.2010.02.002. [DOI] [PubMed] [Google Scholar]
- 47.Satou K, Kawai K, Kasai H, Harashima H, Kamiya H. Mutagenic effects of 8-hydroxy-dGTP in live mammalian cells. Free Radic Biol Med. 2007;42:1552–60. doi: 10.1016/j.freeradbiomed.2007.02.024. [DOI] [PubMed] [Google Scholar]
- 48.Shen J, Deininger P, Hunt JD, Zhao H. 8-Hydroxy-2′-deoxyguanosine (8-OH-dG) as a potential survival biomarker in patients with nonsmall-cell lung cancer. Cancer. 2007;109:574–80. doi: 10.1002/cncr.22417. [DOI] [PubMed] [Google Scholar]
- 49.Sato Y, Yamamoto N, Kunitoh H, Ohe Y, Minami H, Laird NM, et al. Genome-wide association study on overall survival of advanced non-small cell lung cancer patients treated with carboplatin and paclitaxel. J Thorac Oncol. 2010;6:132–8. doi: 10.1097/JTO.0b013e318200f415. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.