

Abstract
Although adjuvants are critical vaccine components, their modes of action are poorly understood. Here, we investigated the mechanisms by which the heat-killed mycobacteria in complete Freund’s adjuvant (CFA) promote T helper 17 (Th17) CD4+ T cell responses. We found that IL-17 secretion by CD4+ T cells following CFA immunization requires MyD88 and IL-1β/IL-1 receptor (IL-1R) signaling. Through measurement of antigen-specific responses after adoptive transfer of OTII cells, we confirmed that MyD88-dependent signaling controls Th17 differentiation rather than simply production of IL-17. Additional experiments showed that CFA-induced Th17 differentiation involves IL-1β processing by the inflammasome, as mice lacking caspase 1, ASC, or NLRP3 exhibit partially defective responses after immunization. Biochemical fractionation studies further revealed that peptidoglycan is the major component of heat-killed mycobacteria responsible for inflammasome activation. By assaying Il1b transcripts in the injection site skin of CFA-immunized mice, we found that signaling through the adaptor molecule CARD9 plays a major role in triggering pro-IL-1β expression. Moreover, we demonstrated that recognition of the mycobacterial glycolipid trehalose dimycolate (cord factor) by the C type lectin receptor mincle partially explains this CARD9 requirement. Importantly, purified peptidoglycan and cord factor administered in mineral oil synergized to recapitulate the Th17-promoting activity of CFA, and, as expected, this response was diminished in caspase 1-and CARD9-deficient mice. Taken together, these findings suggest a general strategy for the rational design of Th17-skewing adjuvants by combining agonists of the CARD9 pathway with inflammasome activators.
INTRODUCTION
The choice of adjuvants is often a critical factor in the success of vaccines, but the number of adjuvants available for clinical use is very limited. Effective adjuvants are known to act on the innate immune system to not only increase the magnitude of vaccine-induced immune responses, but also to direct the appropriate class of effector response (1). However, the innate immune pathways that must be targeted in order to elicit particular types of adaptive immunity are poorly understood, thus hampering the development of rationally-designed adjuvants.
Historically, mycobacteria and their components have been the basis of numerous adjuvants and immunotherapies. For instance, BCG instillation is widely used to treat superficial bladder cancer and BCG has been employed as an adjuvant in experimental vaccines against Leishmania spp. and other pathogens (2). However, the best-known use of mycobacteria for stimulating the immune system is in complete Freund’s adjuvant (CFA), a water-in-oil emulsion containing heat-killed M. tuberculosis strain H37Ra or M. butyricum. Although not appropriate for human use because of its adverse inflammatory side-effects, CFA has for decades been the adjuvant of choice for inducing strong humoral and cellular immune responses in experimental animals(3). Moreover, administering self antigens (e.g. myelin basic protein) in CFA breaks self tolerance and results in autoimmunity (1, 3). Previous studies have shown that CFA promotes strong T helper 1 (Th1) and T helper 17 (Th17) CD4+ T cell responses, and this is a likely explanation for the potency of the adjuvant in inducing experimental autoimmune disease (3, 4).
Although Th1 and Th17 responses cause autoimmunity under certain circumstances, they are also protective against many pathogens, and adjuvants triggering their differentiation could be useful tools for vaccination. While Th1 responses have long been appreciated for their role in cell-mediated immunity against intracellular pathogens, Th17 responses are now known to be protective against fungi and some extracellular bacteria (5). This is clearly evident in Job’s Syndrome patients, who mount defective Th17 responses as a result of mutations in the STAT3 gene, and are highly susceptible to Candida albicans and other fungal infections (6). Moreover, it has recently been shown that Th17 and Th1 cells can cooperate in host defense against two major intracellular pathogens—M. tuberculosis and Francisella tularensis (7, 8).
T cell subset differentiation is largely directed by the innate immune system. Recognition of pathogen-associated molecular patterns and danger signals by germline-encoded innate immune receptors leads to cellular activation and production of T cell-polarizing cytokines (3). However, in the case of complex microbial stimuli, it is not clear how activation of particular combinations of pattern recognition receptors causes innate immune cells to promote CD4+ T cell differentiation into specific subsets. For instance, the mycobacteria in CFA are known to activate several Toll-like receptors, the intracellular NOD1 and NOD2 receptors, and multiple C type lectin receptors (9–16), but the respective contributions of these innate immune pathways in triggering Th17 differentiation in response to CFA immunization are poorly understood.
In the present study, we have undertaken a systematic investigation of innate immune receptors activated by CFA to understand their respective roles in promoting Th17 polarization. We demonstrate a major role for IL-1β/IL-1R signaling on both T cells and the non-T cell compartment in driving CFA-induced Th17 responses. Moreover, in investigating the mechanisms involved in IL-1β production in response to CFA, we have elucidated important roles for mincle/CARD9-dependent signaling and the inflammasome, a molecular complex that proteolytically activates pro-IL-1β and pro-IL-18 (17). Finally, we have assigned functions for the mycobacterial cord factor and peptidoglycan components of CFA in triggering IL-1β induction and processing respectively. Together, these findings elucidate a major pathway for the generation of IL-17-producing CD4+ T cells in response to mycobacterial products, which could be utilized in the design of novel Th17-promoting adjuvants.
MATERIALS AND METHODS
Mice
C57BL/6 mice were purchased from Taconic Farms. Il18r1 −/− mice were purchased from The Jackson Laboratory. CD45.1 congenic Rag1 −/− OTII mice and Il1r1 −/− mice backcrossed to B6 for 10 generations were supplied by Taconic Farms via a contract with NIAID. Myd88 −/− mice, backcrossed to B6 for 10 generations, were obtained from S. Akira (Osaka University, Osaka, Japan). Tlr2 −/− Tlr9 −/−, Tlr4 −/−, and Myd88 −/− Trif −/− mice were generously provided by D. Golenbock and E. Lien (University of Massachusetts, Worcester, MA). Nod1 −/− Nod2 −/− mice (18), backcrossed to B6 for 10 generations, were originally obtained from G. Nunez (University of Michigan, Ann Arbor, MI). Il1a −/− and Il1b −/− mice were generated by Y. Iwakura (University of Tokyo, Tokyo, Japan) and generously provided by T. Merkel (Food and Drug Administration, Bethesda, MD). Card9 −/− mice (19) were generated by X. Lin (MD Anderson, Houston, TX). Clec4e −/− (mincle −/−) mice (20), backcrossed at least 7 generations to B6, were kindly provided by S. Yamasaki (Kyushu University, Kyushu, Japan). Pycard −/− (Asc −/−) mice were generated by Millenium Pharmaceuticals (Cambridge, MA). Casp1 −/− mice were generously supplied by R. Flavell (Yale University, New Haven, CT) and subsequently backcrossed to B6 until 10 generations. Nlrp3 −/− and Nlrc4 −/− mice were kindly provided by V. Dixit (Genentech, San Francisco, CA). All animals were maintained in an AALAC-accredited animal facility at the National Institute of Allergy and Infectious Diseases (NIAID), National Institutes of Health (Bethesda, MD). Mice were used according to animal study proposals approved by the NIAID Animal Care and Use Committee.
Bone marrow from P2xr7 −/− mice was kindly provided by F. Tam (Imperial College, London, UK).
Reagents
CFA, IFA, ATP, Brefeldin A, cytochalasin D, propyl gallate, butylated hydroxyanisole, NaCl, and KCl were purchased from Sigma-Aldrich. Trehalose dimycolate (TDM) purified from M. tuberculosis was from Sigma-Aldrich or Enzo Life Sciences, Imject OVA and alum were from Pierce/Thermo Fisher Scientific. OVA323-339 was from the NIAID Research Technologies Branch. Ultrapure LPS, N-glycolyl muramyl dipeptide (MDP), TriDAP, and flagellin were from Invivogen. Heat-killed M. tuberculosis H37Ra was from Difco/BD Biosciences.
Immunizations
For IFA and CFA immunizations, mice were injected subcutaneously at four sites along the back (100 μl per site) with a total of 100 μg OVA protein or OVA323-339 peptide. CFA emulsions contained 0.5 mg/ml heat-killed M. tuberculosis H37Ra. For IFA supplementation experiments, TDM was dissolved in IFA and heated to 55°C and sonicated prior to preparing emulsions with OVA323-339 peptide. TDM was used at a concentration of 0.125 mg/ml in these emulsions. H37Ra peptidoglycan was suspended in IFA and used in emulsions at a concentration of 0.5 mg/ml.
Adoptive transfer and ex vivo T cell restimulation
To measure endogenous polyclonal CD4+ T cell responses, draining (inguinal, axillary, and brachial) LNs were harvested 14 days after CFA/OVA immunization. LNs were dissociated through 100 μm cell strainers and LN cells were plated at 1x10^6 cells per well in a flat-bottom 96 well plate. The cells were incubated with 100 μg/ml OVA protein for 1.5 h at 37°C. Brefeldin A (5 μg/ml) was then added and the cells were incubated for an additional 5–6 hours at 37°C. For adoptive transfer experiments, mice received 3×10^5 CD45.1 congenic Rag1 −/− OTII cells intravenously and were then immunized with CFA/OVA323-339 peptide the next day. Draining LNs were harvested 10 days later and cells were restimulated with 25 μg/ml OVA323-339 as above.
Antibodies and flow cytometry
Antibodies against the following molecules were purchased from eBioscience, Biolegend, or BD Biosciences: CD4 (RM4-4), CD45.1 (A20), CD45.2 (104), CD44 (IM7), IFN-γ (XMG1.2), IL-17 (eBio17B7), Foxp3 (FJK-16s), and RORγt (B2D). Blue fixable live/dead cell stain and streptavidin-Qdot605 were from Molecular Probes/Invitrogen. All samples were acquired on an LSRII flow cytometer (Becton Dickinson) and analyzed using Flowjo software (Tree Star).
Tissue harvest and quantitative PCR
Skin was harvested from the injection site of CFA/OVA immunized mice at various times after immunization. Subcutaneous fat was removed and the skin was homogenized in Trizol (Invitrogen). Total RNA was extracted according to the manufacturer’s protocol and cDNA was reverse transcribed with Superscript III reverse transcriptase and random primers (Invitrogen). Quantitative PCR was performed on an ABI Prism 7900 HT Sequence Detection System using Power SYBR Green Master Mix (Applied Biosystems/Life Technologies) for detection. Fold induction of gene expression was calculated using the ΔΔCT method, normalizing mRNA levels for each sample to levels of hypoxanthine guanine phosphoribosyl transferase (HPRT) and comparing to mRNA levels in unimmunized controls. The following primer pairs, derived from the literature or designed with ProbeFinder software (Roche), were used:
Hprt F: GCCCTTGACTATAATGAGTACTTCAGG, Hprt R: TTCAACTTGCGCTCATCTTAGG
Il1b F: TGTAATGAAAGACGGCACACC, Il1b R: TCTTCTTTGGGTATTGCTTGG
Il6 F: ACAACCACGGCCTTCCCTACTT, Il6 R: CACGATTTCCCAGAGAACATGTG
Il23a F: CACCAGCGGGACATATGAA, Il23a R: CCTTGTGGGTCACAACCAT
Macrophage and dendritic cell stimulation and immunoblotting
Bone marrow-derived macrophages (BMDM) were prepared by growing bone marrow cells in 30% L929 cell-conditioned medium for 6–8 days. Bone marrow-derived dendritic cells (BMDC) were prepared as previously described (21). BMDM and BMDC were then harvested, primed where indicated for 3–4 hours with 20 ng/ml LPS, and then stimulated for 1 hr with 5 mM ATP or overnight with heat-killed H37Ra (500 μg/ml), alum (400 μg/ml), or flagellin (1 μg/ml) in OptiMEM serum-free media. DOTAP liposomal transfection reagent (Roche) was used according to the manufacturer’s instructions to deliver flagellin intracellularly. For inhibition of potassium efflux, KCl or NaCl were added to the OptiMEM medium. For experiments with cytochalasin D or reactive oxygen species (ROS) scavengers, the inhibitors were present both during LPS priming and during the subsequent inflammasome stimulation step. For stimulation with heat-killed H37Ra fractions, all fractions except for polar and nonpolar lipids were used at a concentration of 250 μg/ml. Lipids were used at 50 μg/ml because of toxicity at higher concentrations.
After stimulation, supernatants were harvested and cells were lysed with cell lysis buffer (Cell Signaling Technology) supplemented with 2 mM phenylmethanesulfonyl fluoride (Sigma-Aldrich) and a protease inhibitor cocktail (EMD Chemicals). Supernatants were precipitated with 1 volume methanol and 0.25 volumes chloroform and the protein pellets were resuspended in reducing sample buffer (Pierce/Thermo Fisher Scientific). The samples were then separated on 15% polyacrylamide gels and transferred to Hybond-P PVDF membranes (GE Healthcare). IL-1β was detected with a goat anti-IL-1β antibody (AF-401; R&D systems) and caspase 1 was detected with rabbit anti-mouse caspase 1 (sc-514; Santa Cruz Biotechnology). For loading controls, equal amounts of cell lysates were separated on 12% polyacrylamide gels and blotted as above or with mouse anti-GAPDH antibody (sc-32233; Santa Cruz Biotechnology).
Polar and nonpolar lipid extraction from heat-killed H37Ra
Polar and nonpolar lipids were extracted as described (22). Briefly, freeze-dried M. tuberculosis H37Ra cells were treated in 22 ml of methanolic saline (20 ml 0.3% NaCl and 20 ml CH3OH) and 22 ml of petroleum ether for 2 h. The suspension was centrifuged and the upper layer containing nonpolar lipids was separated. An additional 22 ml of petroleum ether was added, mixed and centrifuged as described above. The two upper petroleum ether fractions were combined and dried. For polar lipids, 26 ml CHCl3/CH3OH/0.3% NaCl (9:10:3, v/v/v) was added to the lower aqueous phase and stirred for 4 h. The mixture was filtered and the filter cake re-extracted twice with 8.5 ml of CHCl3/CH3OH/0.3% NaCl (5:10:4, v/v/v). The delipidated cells were retained for further extraction and purification of lipoglycans as described below. Equal amounts of CHCl3 and 0.3% NaCl (14.5 ml each) were added to the combined filtrates and stirred for 1 h. The mixture was allowed to settle, and the lower layer containing the polar lipids recovered and dried. The polar and nonpolar lipid extracts were examined by two dimensional thin-layer chromatography (2D-TLC) on aluminum backed plates of silica gel 60 F254 (Merck 5554) using solvent systems A–E with the appropriate staining solution to detect the presence of lipids, glycolipids or phospholipids as described (22).
Isolation and extraction of lipoglycans
Lipoglycans were extracted from the above delipidated cells as previously described (23). Briefly, cells were broken by sonication (MSE Soniprep 150, 12 micron amplitude, 60s ON, 90s OFF for 10 cycles, on ice) and the cell debris refluxed 5 times with 50% C2H5OH at 68°C, for 12 h intervals. The cell debris (mAGP) was removed by centrifugation and the supernatant containing lipoglycans, neutral glycans, and proteins was dried. This dried extract was then treated with hot phenol–H2O. The aqueous phase was dialyzed and dried, followed by extensive treatments with α-amylase, DNase, RNase chymotrypsin and trypsin. This fraction was dialyzed to remove the low MW break-down products formed after the enzyme treatment, thus yielding the crude lipoglycan fraction, containing lipoarabinomannan, lipomannan and glucans.
Preparation of the mycolyl-arabinogalactan-peptidoglycan (mAGP) complex and release of mycolic acid methyl esters (MAMES), soluble arabinogalactan (AG), and peptidoglycan (PGN) from the mAGP complex
The crude insoluble material from the above lipoglycan extraction was extracted three times with 2% SDS in phosphate buffered saline at 95°C for 1 h, washed with water, 80% (v/v) acetone in water, and acetone, and finally lyophilized to yield a highly purified cell wall mAGP preparation (24). Mycolic acid methyl esters were prepared initially from the mAGP by treatment with 0.5 % (w/v) KOH in methanol at 37°C for 4 days (25). The treated mAGP was collected by centrifugation at 27,000 g for 20 min, and then washed repeatedly with methanol, and re-centrifuged at 27,000 g and the pellet recovered. The mycolic acid methyl esters were then extracted from the treated mAGP using diethyl ether, re-centrifuged at 27, 000 g and the pellet (AGP) and soluble mycolic acid methyl esters recovered in the diethyl ether supernatant. The extraction process using diethyl ether was repeated thrice. The combined ether fractions were evaporated to dryness to provide the mycolic acid methyl esters. The AGP complex was hydrolyzed with 0.2 M H2SO4 at 85°C for 30 min and neutralized with BaCO3. The supernatant, which contained the solubilized AG, was recovered after centrifugation at 27,000 g for 30 min and was dialyzed (MWCO 3,500). The residual pellet was the PGN fraction. The supernatant was then made up to 80 % cold ethanol and left at −20°C overnight to precipitate polysaccharides. The pellet was then recovered by centrifugation, at 27,000 g for 30 min, and freeze-dried to afford AG, which was stored at −20°C. The recovered PGN fraction was finally extensively washed with water, freeze dried, and stored at −20°C.
Statistical analysis
The statistical significance of differences between groups was analyzed via unpaired Student’s t test using Graphpad Prism software version 5.0c. The p values are shown as follows: * p < 0.05, ** p < 0.01, *** p < 0.001
RESULTS
MyD88 is required for CFA-induced Th17 polarization
MyD88 signaling has previously been shown to play an important role in the adjuvant activity of CFA (26). To specifically investigate the requirement for this adaptor molecule in CFA-induced Th17 responses, we immunized WT and Myd88 −/− mice subcutaneously with ovalbumin (OVA) in CFA and measured OVA-specific interleukin-17 (IL-17) and interferon-γ (IFN-γ) production by CD4+ T cells in the draining lymph nodes (LNs) two weeks later. As expected, we found that CFA immunization induced robust differentiation of IL-17- and IFN-γ-single producing (SP) CD4+ T cells, as well as a population of IL-17/IFN-γ double-producing (DP) cells (Fig. 1a). However, in Myd88 −/− mice, the Th17 and IL-17/IFN-γ DP responses were almost completely abrogated, although there was still a residual IFN-γ response.
Figure 1.

MyD88-dependent IL-1β/IL-1R signaling is critical for CFA-induced Th17 polarization. (a) Draining LN cells were harvested from WT or Myd88 −/− mice 14 days after CFA/OVA immunization and intracellular cytokine staining (ICS) for IL-17 and IFN-γ was performed after ex vivo OVA restimulation. Plots are gated on CD4+ CD44hi Foxp3− cells. (b–d) The indicated mice were immunized and draining LN responses were measured as in a. Frequencies of cytokine-producing cells within the CD4+ CD44hi Foxp3− population are shown. Data are representative of at least three independent experiments (a, b) or are pooled from three to four experiments (c, d). Each point represents data from an individual mouse.
To test whether there was a specific impairment in IL-17 production or a more general defect in Th17 differentiation in Myd88 −/− mice, we adoptively transferred CD45.1 congenic Rag1 −/− OTII cells into CD45.2 congenic WT or Myd88 −/− mice and then immunized the mice with OVA323-339 peptide in CFA. We found that Myd88 −/− recipients contained markedly reduced frequencies of IL-17-producing and IL-17/IFN-γ DP OTII T cells compared to WT recipients (Supplementary Fig. 1a, b). Importantly, this correlated with a major loss in expression of the Th17 master regulator RORγt in the OTII cells isolated from Myd88 −/− recipients, suggesting that MyD88 is required for Th17 differentiation, rather than simply secretion of IL-17 (Supplementary Fig. 1c, d).
The requirement for MyD88 in Th17 responses reflects a role for IL-1β/IL-1R signaling rather than TLRs or IL-18R
MyD88 is required for signaling via most TLRs as well as IL-1R family members. To understand the requirement for MyD88 in CFA-induced CD4+ T cell polarization, we thus investigated the role of TLRs, IL-1R, and IL-18R in the response to CFA. The mycobacteria in CFA contain the TLR2 agonists 19 kDa lipoprotein and arabinose-capped lipoarabinomannan, the TLR2/TLR4-activating phosphatidyl-myo-inositols (PIMs), and unmethylated CpG DNA (recognized by TLR9) (13). Therefore, we examined the response of Tlr2 −/− Tlr9 −/− and Tlr4 −/− mice to immunization. OVA-specific IL-17 production by LN CD4+ T cells was not significantly reduced in these mice (Fig. 1b), suggesting that recognition of CFA by these TLRs is dispensable for adjuvant-induced T cell polarization. In contrast, using Il1r1 −/− and Il18r1 −/− mice, we found that IL-1R is required for CFA-driven Th17 and IL-17/IFN-γ DP responses, whereas IL-18R is dispensable (Fig. 1c).
Two IL-1 species—IL-1α and IL-1β—signal through IL-1R. We found only a partial defect in IL-17 production in immunized Il1a −/− mice, compared to a marked reduction in Il1b −/− mice (Fig. 1d), suggesting that IL-1β is the primary IL-1 cytokine required for CFA-induced Th17 responses. Taken together, these findings argue that the role of MyD88 in CFA-triggered Th17 and IL-17/IFN-γ DP cell differentiation is primarily downstream of IL-1β/IL-1R signaling.
Activation of the inflammasome by M. tuberculosis peptidoglycan contributes to CFA-induced Th17 differentiation
IL-1β is initially produced as an immature cytokine that must be proteolytically processed to its bioactive form. The cleavage of pro-IL-1β to its mature form is usually thought to be accomplished by the caspase 1 inflammasome (17), so we asked whether activation of this complex contributed to CFA-induced Th17 responses. To address this question, we immunized Casp1 −/−, Pycard (Asc) −/−, Nlrp3 −/−, and Nlrc4 −/− mice with CFA/OVA. We observed a substantial defect in Th17 responses in Casp1 −/− and Asc −/− mice, but only a slight reduction in Nlrp3 −/− mice and unimpaired responses in Nlrc4 −/− animals (Fig. 2a, b).
Figure 2.

Activation of the inflammasome by mycobacterial peptidoglycan contributes to CFA-induced Th17 polarization. (a, b) The indicated mice were immunized with CFA/OVA and draining LN cells were harvested 14 days after CFA/OVA immunization. IL-17 and IFN-γ production were analyzed by ICS after OVA restimulation and frequencies of cytokine-producing cells within the CD4+ CD44hi Foxp3− population are shown. (c) LPS-primed bone marrow-derived macrophages (BMDM) were stimulated with heat-killed M. tuberculosis H37Ra (20 μg/ml, 100 μg/ml, or 500 μg/ml), alum, or ATP. Supernatants and cell lysates were harvested and immunoblotted for IL-1β, caspase 1, and GAPDH. (d) LPS-primed BMDM generated from WT, Nlrp3 −/−, or Nlrc4 −/− mice were stimulated with heat-killed H37Ra or flagellin and supernatants and cell lysates were immunoblotted for IL-1β and caspase 1. (e) Polar lipids, nonpolar lipids, lipoglycans, mycolic acid methyl esters (MAMES), and arabinogalactan (AG) were sequentially extracted from the cell wall of heat-killed H37Ra, as described in the Materials and Methods, and used to stimulate LPS-primed BMDM. Supernatants and cell lysates were then immunoblotted as above. (f) The insoluble H37Ra fractions remaining after each extraction described in e (mAGP = mycolyl-arabinogalactan-peptidoglycan, AGP = arabinogalactan-peptidoglycan, PGN = peptidoglycan) were used to stimulate LPS-primed BMDM and supernatants and cell lysates were immunoblotted as above. Data are pooled from three experiments (a) or representative of at least three independent experiments (b–f). SN = supernatant, CL = cell lysate.
To further analyze the inflammasome’s role in the response to CFA, we stimulated bone marrow-derived macrophages (BMDM) in vitro and assayed their activation of caspase 1 and ability to secrete mature IL-1β. We found that stimulation of LPS-primed BMDM with heat-killed M.tb. H37Ra was sufficient to induce caspase 1 cleavage and IL-1β secretion, and that this required NLRP3 as well as phagocytosis of the M.tb., potassium efflux, and reactive oxygen species (Fig. 2c, d and Supplementary Fig. 2). NLRP3-dependent IL-1β production and caspase 1 cleavage in response to heat-killed H37Ra were also observed in the absence of LPS priming, indicating that H37Ra itself is sufficient for inducing pro-IL-1β and components of the inflammasome (data not shown). Similar results were obtained with bone marrow-derived dendritic cells (data now shown).
Infection of macrophages with live M.tb. is known to activate the NLRP3 inflammasome, and this is thought to occur in response to the mycobacterial ESX-1 secretion system (27). Since heat-killed mycobacteria do not secrete proteins, we investigated alternative pathways by which mycobacterial products could activate the inflammasome. To do so, we biochemically fractionated heat-killed H37Ra, successively extracting polar and nonpolar lipids, lipoglycans, mycolic acid methyl esters, and arabinogalactan, as described previously (22–25). We tested each of these soluble fractions as well as the insoluble fractions remaining after each step. We found that none of the soluble fractions triggered the inflammasome, whereas all of the insoluble fractions—including PGN alone—contained this activity (Fig. 2e, f). These data implicate PGN as the primary stimulus for inflammasome activation by heat-killed H37Ra.
Previous studies utilizing other bacteria (e.g. Salmonella) have shown that PGN can activate the inflammasome, and this activity has generally been ascribed to the ubiquitous PGN subunit MDP acting via the receptor NOD2 (28–30). However, we found that macrophages from Nod1 −/−Nod2 −/− mice displayed normal caspase 1 cleavage and IL-1β secretion in response to H37Ra PGN (Supplementary Fig. 3), indicating that mycobacterial PGN can activate the inflammasome independently of these NOD receptors.
The signaling adaptor CARD9 is required for pro-IL-1β production and Th17 differentiation in response to CFA
Having shown that CFA-induced Th17 differentiation involves the inflammasome, we next investigated how the pro-IL-1β substrate for the inflammasome was being produced in this model. Transcription of the Il1b gene is induced by signals such as TLRs that cause translocation of NFκB to the nucleus (17). To determine whether Il1b induction in response to CFA involves TLR signaling, we immunized WT and Myd88 −/− Trif −/− mice (which lack all TLR signaling) with CFA/OVA. We then harvested injection site skin from these mice at various time points and measured mRNA levels of Il1b and other pro-inflammatory cytokines. We found that although Il1b induction was slightly delayed in Myd88 −/− Trif −/− mice, total transcript levels were not reduced compared to WT mice (Fig. 3a), arguing that TLR signaling is largely dispensable for pro-IL-1β production in this system.
Figure 3.
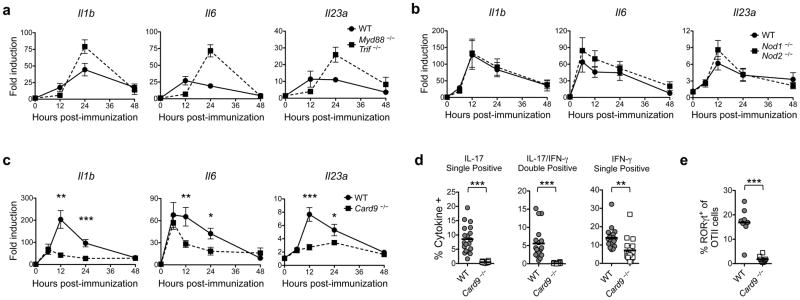
The signaling adaptor CARD9 is required for pro-IL-1β induction and Th17 differentiation in response to CFA. (a–c) The indicated mice were immunized with CFA/OVA and injection site skin was excised at the time points shown. RNA was isolated and transcript levels of the indicated genes were measured by quantitative PCR (qPCR). Fold induction over mRNA levels in unimmunized skin is shown. Data are mean ± SEM. (d) CD45.1 congenic Rag1 −/− OTII cells were transferred into CD45.2 congenic WT or Card9 −/− mice prior to immunization with CFA/OVA323-339. Draining LNs were harvested 10 days after immunization and restimulated with OVA323-339. IL-17 and IFN-γ production by the OTII cells was analyzed by ICS. (e) RORγt expression in the draining LN OTII cells described in d was analyzed by ICS. Data are representative of two independent experiments (a) or pooled from two to four experiments (b–e).
Bacterial peptidoglycan subunits can induce innate cytokine production via the cytosolic receptors NOD1 and NOD2 (31, 32), and mycobacteria have also been shown to trigger several C type lectins that signal via the adaptor CARD9 (9–12, 16, 33). To determine whether either of these pathways contribute to pro-IL-1β induction in response to CFA, we immunized Nod1 −/−Nod2 −/−or Card9 −/− mice and measured Il1b transcripts in the injection site skin. We found that while post-immunization Il1b mRNA levels were comparable in the skin of Nod1 −/− Nod2 −/− and WT mice (Fig. 3b), they were greatly reduced in Card9 −/− animals, particularly at later time points (Fig. 3c). Transcripts encoding the Th17-polarizing cytokines IL-6 and IL-23p19 were also diminished in the Card9 −/− mice (Fig. 3c). These data suggest that CFA provides “signal one” for IL-1β production largely via CARD9- rather than TLR- or NOD1/NOD2-dependent signaling.
To determine whether the defects in pro-inflammatory cytokine induction in the Card9 −/− mice affect CD4+ T cell responses, we adoptively transferred CD45.1 congenic Rag1 −/− OTII cells into CD45.2 congenic WT or Card9 −/− mice and then immunized the mice with CFA/OVA323-339. We found that the IL-17 and IL-17/IFN-γ DP OTII responses were nearly absent in the Card9 −/− recipients, as was the RORγt+ population (Fig. 3d, e), demonstrating that CFA-induced Th17 differentiation requires CARD9-dependent signaling.
Recognition of mycobacterial cord factor by mincle is a major CARD9-dependent signal for CFA-induced IL-1β production and Th17 polarization
Mycobacteria are known to trigger several CARD9-dependent C type lectin receptors, including dectin-1, dectin-2, and mincle (9–12). The latter is of particular interest since recent studies have identified trehalose dimycolate (TDM) as one of its ligands (9, 10). TDM, also known as “cord factor”, is a mycobacterial glycolipid with potent adjuvant activity that was discovered while investigating the cording phenomenon observed during infection with virulent mycobacteria (16, 34, 35). To determine whether TDM could account for the CARD9-dependent Il1b induction observed after CFA injection, we compared Il1b mRNA expression in the skin of mice immunized with incomplete Freund’s adjuvant (IFA; mineral oil and surfactant without mycobacteria), IFA supplemented with TDM, or CFA itself. IFA was a poor inducer of Il1b, but the addition of TDM resulted in robust Il1b upregulation comparable to that observed with CFA. Importantly, this response to TDM was ablated in Card9 −/− mice (Fig. 4a).
Figure 4.

Recognition of mycobacterial cord factor by mincle is upstream of the CARD9 requirement in CFA-induced pro-IL-1β production and Th17 polarization. (a) WT or Card9 −/−mice were immunized with OVA in PBS, IFA, IFA supplemented with cord factor (TDM), or CFA. Injection site skin was excised 12 hours post-immunization, RNA was isolated, and Il1b transcript levels were measured by qPCR. Fold induction over mRNA levels in unimmunized skin is shown. Data are mean ± SEM. (b) WT, Card9 −/−, or Mincle −/− (Clec4e −/−) mice were immunized with CFA/OVA and RNA was isolated from injection site skin 12 hours after immunization. Levels of indicated transcripts were measured by qPCR and fold induction over mRNA levels in unimmunized skin is shown. (c) CD45.1 congenic Rag1 −/− OTII cells were transferred into CD45.2 congenic WT, Card9 −/−, or Mincle −/− mice prior to immunization with CFA/OVA323-339. The draining LNs were harvested 10 days after immunization. IL-17 and IFN-γ production by OTII cells in the draining LNs was analyzed by ICS after restimulation with OVA323-339. (d) RORγt expression in the draining LN OTII cells described in c was analyzed by ICS. Data are pooled from three to six experiments (a–d).
Having shown that TDM in IFA stimulates Il1b expression, we asked whether recognition of TDM by mincle is required for CFA-induced pro-IL-1β production as well as Th17 polarization. We found that Clec4e −/− (Mincle −/−) mice exhibited reduced cutaneous Il1b induction after CFA immunization, although the response was less impaired than in the Card9 −/− mice (Fig. 4b). Moreover, OTII cells transferred into Mincle −/− mice were defective in their differentiation into IL-17-producing cells and in their expression of RORγt (Fig. 4c, d). Nevertheless, the Th17 response was not as greatly reduced in Mincle −/− as in Card9 −/− mice, thus implicating additional CARD9-dependent receptors in CFA-induced Th17 polarization.
IFA supplemented with TDM and PGN recapitulates the Th17-polarizing effects of CFA
The above experiments demonstrated major roles for mincle/CARD9-dependent recognition of TDM in driving pro-IL-1β production and for inflammasome-mediated recognition of PGN in triggering IL-1β maturation. Having found that IL-1β / IL-1R signaling as well as the CARD9 and inflammasome pathways are necessary for optimal Th17 responses after CFA immunization, we therefore asked whether the combination of TDM and PGN could recapitulate the Th17-polarizing capacity of the mycobacterial component of CFA by inducing production of IL-1β as well as other Th17-polarizing cytokines.
In contrast to CFA, IFA is incapable of inducing Th17 responses even though it promotes CD4+ T cell priming (Fig. 5a, b), thereby demonstrating the importance of the mycobacteria in CD4+ T cell polarization. To test the immunostimulatory effects of TDM and PGN, we added these components to IFA either individually or in combination. We then prepared emulsions with OVA323-339 and immunized mice that had received adoptively transferred CD45.1 Rag1 −/− OTII cells. IFA containing TDM or PGN alone induced little or no Th17 response, but IFA supplemented with TDM and PGN combined induced robust Th17 differentiation comparable to that induced by CFA itself (Fig. 5c and Supplementary Fig. 4). Importantly, as expected, the IL-17 response to the IFA+TDM+PGN adjuvant was dependent on both CARD9 and caspase 1 (Fig. 5d).
Figure 5.

Supplementation of IFA with cord factor and peptidoglycan recapitulates the Th17-inducing capacity of CFA. (a) CD45.1 congenic Rag1 −/− OTII cells were transferred into CD45.2 congenic WT mice prior to immunization with OVA323-339 in IFA or CFA. Draining LNs were harvested 10 days after immunization and the frequency of CD44hi CD45.1+ OTII cells was analyzed by flow cytometry. (b) Draining LN cells from the mice described in a were restimulated with OVA323-339 and IL-17 and IFN-γ production by the CD4+ CD45.1+ OTII cells was analyzed by ICS. (c) WT recipients of CD45.1 congenic Rag1 −/− OTII cells were immunized with OVA323-339 in IFA, IFA supplemented with TDM, IFA supplemented with PGN, or IFA with both TDM and PGN. Draining LNs were harvested 10 days after immunization. ICS was performed for IL-17 and IFN-γ production by the CD4+ CD45.1+ OTII cells after OVA323-339 restimulation. (d) CD45.1 congenic Rag1 −/− OTII cells were transferred into CD45.2 congenic WT, Card9 −/−, or Casp1 −/− mice prior to immunization with OVA323-339 in IFA or IFA supplemented with TDM and PGN. Draining LNs were harvested 10 days after immunization. IL-17 and IFN-γ production by the CD4+ CD45.1+ OTII cells was assayed by ICS after restimulation with OVA323-339. Data are representative of at least three independent experiments (a, b) or pooled from two to four experiments (c, d).
DISCUSSION
In this manuscript, we have deconstructed the key innate immune recognition events required for the induction of Th17 differentiation by the mycobacterial component of complete Freund’s adjuvant in vivo. We identify a two-step mechanism whereby dual recognition of mycobacterial cord factor and peptidoglycan synergistically drives production of IL-1β and other cytokines necessary for Th17 polarization. Our findings suggest a general strategy for the rational design of Th17-skewing adjuvants.
The role of MyD88 in CFA-induced CD4+ T cell responses was initially thought to reflect the involvement of TLRs in recognition of the mycobacterial component of the adjuvant (26). However, we found that deficiencies in TLRs implicated in the response to mycobacteria failed to have a major impact on Th17 differentiation following CFA immunization. Instead, the requirement for MyD88 in Th17 skewing could largely be explained by the role of IL-1β / IL-1R signaling, although a significant role for IL-1α was also observed. These findings are consistent with previous reports demonstrating a major function for IL-1R signaling in driving CFA-induced Th17 polarization, especially in the context of autoimmune disease models such as experimental autoimmune encephalomyelitis (EAE) and uveitis (EAU) (36–39).
Previous studies have shown that there is a T cell-intrinsic role for IL-1R / MyD88-dependent signaling in driving Th17 differentiation (36, 40). Interestingly, however, we found that MyD88-sufficient OTII cells transferred into Myd88 −/− recipients are unable to differentiate into Th17 cells after CFA immunization, suggesting that MyD88-dependent signaling in the non-T cell compartment also plays a critical role in inducing Th17 differentiation. Therefore, MyD88-dependent signaling may act in both the T cell and non-T cell compartments to drive Th17 differentiation.
After identifying IL-1β as a key mediator of the CFA-induced Th17 response, we investigated the innate immune pathways that drive production of this cytokine after immunization. We found that the Th17-skewing effects of CFA were partially dependent on the inflammasome. In agreement with previous studies, in vitro stimulation with heat-killed M.tb. induced caspase 1 cleavage and mature IL-1β production by macrophages in an NLRP3-dependent manner (41). However, Nlrp3 −/− mice displayed only a mild reduction in Th17 differentiation in response to CFA compared to Casp1 −/− and Asc −/− mice, suggesting the involvement of other inflammasome sensors in vivo. In addition, the Th17 defect in Casp1 −/− mice was not as profound as that in Il1b −/− mice, implicating inflammasome-independent mechanisms of IL-1β maturation acting in vivo. Multiple enzymes other than caspase 1 have been shown to be capable of cleaving pro-IL-1β, including several neutrophil serine proteases and caspase 8 (42, 43), and these alternative pathways for IL-1β maturation could explain the incomplete inflammasome requirement observed in vivo in our studies.
The results of our fractionation experiments implicate peptidoglycan as the primary trigger for the inflammasome in heat-killed M.tb. Interestingly, experiments performed decades ago identified the peptidoglycan subunit MDP as the minimal mycobacterial component that recapitulated the adjuvant activity of CFA when added to IFA (44). More recently, MDP has also been reported to activate the inflammasome in a NOD2-dependent manner (28–30). However, we found that, in contrast to MDP, polymeric peptidoglycan isolated from heat-killed M.tb. was able to induce inflammasome activation and IL-1β maturation in Nod1 −/− Nod2 −/− macrophages. While mycobacterial diaminopimelic acid (DAP)-containing peptidoglycan is distinct from Staphylococcus aureus lysine-type peptidoglycan, the latter has also recently been reported to activate the inflammasome independently of NOD2 (45). Therefore, in principle, peptidoglycan need not be processed into subunits recognized by NOD receptors in order to activate the inflammasome.
In addition to finding a role for the inflammasome in CFA-induced IL-1β cleavage and Th17 differentiation, we analyzed the signals involved in the induction of pro-IL-1β in response to CFA immunization. Although TLRs are thought to provide a major stimulus for NFκB-dependent pro-IL-1β induction (46), we found that upregulation of Il1b message in response to CFA was intact in Myd88 −/− Trif −/− mice. In contrast, we observed that CARD9-dependent signaling plays a major role in inducing pro-IL-1β production in CFA-immunized mice and that mincle is an important upstream receptor that triggers this pathway. However, while CFA-induced Th17 differentiation was strongly impaired in the Card9 −/− mice, it was only partially reduced in the Mincle −/− mice, suggesting the involvement of additional CARD9-dependent receptors in the recognition of CFA. Dectin-1 and dectin-2 have each been shown to recognize mycobacteria and are therefore logical candidates (11, 12). Moreover, the dectin-1 agonist curdlan was previously shown to augment Th17 differentiation in vivo via a CARD9-dependent pathway, although the role of IL-1 signaling was not investigated in this model (47).
Confirmation of the ability of mycobacterial cord factor and peptidoglycan to drive Th17 responses was obtained in experiments in which these components were admixed into IFA, and CARD9- and caspase 1-dependent Th17 polarization comparable to that observed with CFA was achieved. Importantly, either component alone did not trigger a substantial Th17 response, likely because cord factor and peptidoglycan act synergistically by providing the two signals necessary for mature IL-1β production. Recent studies investigating the basis of vaccine-induced protection to fungal and some bacterial infections have revealed a key role for the Th17 response (7, 48). For this reason, chemically defined agents that promote the differentiation of Th17 cells are likely to become important tools in the development of vaccines against these pathogens. Although we did not attempt to optimize our mycobacterial adjuvant (e.g. by changing the ratio of the two mycobacterial ligands or varying the nature of the oil component) to enhance the efficacy or safety of the emulsion, our findings establish the principle of combining a CARD9-dependent pro-IL-1β inducer with an inflammasome activator as a strategy for developing Th17-skewing immunostimulants. This rationale is not necessarily limited to mycobacterial products and could conceivably be applied to the formulation of adjuvants with components derived from other sources.
Supplementary Material
Acknowledgments
We are grateful to the NIAID Flow Cytometry Facility and Animal Care Branch for their major contributions to this work. We also thank Dr. Tod Merkel and Vanessa Kelly for generously providing Nod- and Il1-deficient mouse strains critical for our experiments, Dr. Hidekazu Shirota and Robin Winkler-Pickett for breeding and supplying Nlrp3 −/− and Nlrc4 −/− mice, and Drs. Stephanie Eisenbarth, Fayyaz Sutterwala, Pam Schwartzberg, Jason Hall, Jeffrey Actor, and Dan Cua for their invaluable advice during the course of our study.
This work was supported by the Intramural Research Program of the NIAID, the UK Medical Research Council, and Cancer Research UK (C399/A2291). K.S. was supported by a scholarship from the Rhodes Trust. G.S.B. acknowledges support in the form of a Personal Research Chair from Mr. James Bardrick, Royal Society Wolfson Research Merit Award, the Medical Research Council, and The Wellcome Trust (081569/Z/06/Z and 084923/B/08/Z).
Footnotes
AUTHOR CONTRIBUTIONS
K.S., V.C., and A.S. designed experiments and wrote the manuscript. K.S. performed experiments with assistance from S.O., S.H., and P.C. S.S.G. and G.S.B. performed biochemical fractionation experiments. D.L.B., K.D.M., D.J., and C.F. helped design experiments and interpret data. S.Y., X.L., G.T., and J.P.-Y.T. provided key reagents and advice on their use.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
References
- 1.Ott G, Van Nest G. Development of vaccine adjuvants: a historical perspective. In: Manmohan S, editor. Vaccine Adjuvants and delivery systems. Wiley-Interscience; Hoboken, N.J: 2007. pp. 1–32. [Google Scholar]
- 2.Luo Y, Henning J, O’Donnell MA. Th1 cytokine-secreting recombinant Mycobacterium bovis bacillus Calmette-Guerin and prospective use in immunotherapy of bladder cancer. Clin Dev Immunol. 2011;2011:728930. doi: 10.1155/2011/728930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Billiau A, Matthys P. Modes of action of Freund’s adjuvants in experimental models of autoimmune diseases. J Leukoc Biol. 2001;70:849–860. [PubMed] [Google Scholar]
- 4.Komiyama Y, Nakae S, Matsuki T, Nambu A, Ishigame H, Kakuta S, Sudo K, Iwakura Y. IL-17 plays an important role in the development of experimental autoimmune encephalomyelitis. J Immunol. 2006;177:566–573. doi: 10.4049/jimmunol.177.1.566. [DOI] [PubMed] [Google Scholar]
- 5.Peck A, Mellins ED. Precarious balance: Th17 cells in host defense. Infect Immun. 2010;78:32–38. doi: 10.1128/IAI.00929-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Milner JD, Brenchley JM, Laurence A, Freeman AF, Hill BJ, Elias KM, Kanno Y, Spalding C, Elloumi HZ, Paulson ML, Davis J, Hsu A, Asher AI, O’Shea J, Holland SM, Paul WE, Douek DC. Impaired T(H)17 cell differentiation in subjects with autosomal dominant hyper-IgE syndrome. Nature. 2008;452:773–776. doi: 10.1038/nature06764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Khader SA, Bell GK, Pearl JE, Fountain JJ, Rangel-Moreno J, Cilley GE, Shen F, Eaton SM, Gaffen SL, Swain SL, Locksley RM, Haynes L, Randall TD, Cooper AM. IL-23 and IL-17 in the establishment of protective pulmonary CD4+ T cell responses after vaccination and during Mycobacterium tuberculosis challenge. Nat Immunol. 2007;8:369–377. doi: 10.1038/ni1449. [DOI] [PubMed] [Google Scholar]
- 8.Lin Y, Ritchea S, Logar A, Slight S, Messmer M, Rangel-Moreno J, Guglani L, Alcorn JF, Strawbridge H, Park SM, Onishi R, Nyugen N, Walter MJ, Pociask D, Randall TD, Gaffen SL, Iwakura Y, Kolls JK, Khader SA. Interleukin-17 is required for T helper 1 cell immunity and host resistance to the intracellular pathogen Francisella tularensis. Immunity. 2009;31:799–810. doi: 10.1016/j.immuni.2009.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ishikawa E, Ishikawa T, Morita YS, Toyonaga K, Yamada H, Takeuchi O, Kinoshita T, Akira S, Yoshikai Y, Yamasaki S. Direct recognition of the mycobacterial glycolipid, trehalose dimycolate, by C-type lectin Mincle. J Exp Med. 2009;206:2879–2888. doi: 10.1084/jem.20091750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schoenen H, Bodendorfer B, Hitchens K, Manzanero S, Werninghaus K, Nimmerjahn F, Agger EM, Stenger S, Andersen P, Ruland J, Brown GD, Wells C, Lang R. Cutting edge: Mincle is essential for recognition and adjuvanticity of the mycobacterial cord factor and its synthetic analog trehalose-dibehenate. J Immunol. 2010;184:2756–2760. doi: 10.4049/jimmunol.0904013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McGreal EP, Rosas M, Brown GD, Zamze S, Wong SY, Gordon S, Martinez-Pomares L, Taylor PR. The carbohydrate-recognition domain of Dectin-2 is a C-type lectin with specificity for high mannose. Glycobiology. 2006;16:422–430. doi: 10.1093/glycob/cwj077. [DOI] [PubMed] [Google Scholar]
- 12.Rothfuchs AG, Bafica A, Feng CG, Egen JG, Williams DL, Brown GD, Sher A. Dectin-1 interaction with Mycobacterium tuberculosis leads to enhanced IL-12p40 production by splenic dendritic cells. J Immunol. 2007;179:3463–3471. doi: 10.4049/jimmunol.179.6.3463. [DOI] [PubMed] [Google Scholar]
- 13.Kleinnijenhuis J, Oosting M, Joosten LA, Netea MG, Van Crevel R. Innate immune recognition of Mycobacterium tuberculosis. Clin Dev Immunol. 2011;2011:405310. doi: 10.1155/2011/405310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fritz JH, Le Bourhis L, Sellge G, Magalhaes JG, Fsihi H, Kufer TA, Collins C, Viala J, Ferrero RL, Girardin SE, Philpott DJ. Nod1-mediated innate immune recognition of peptidoglycan contributes to the onset of adaptive immunity. Immunity. 2007;26:445–459. doi: 10.1016/j.immuni.2007.03.009. [DOI] [PubMed] [Google Scholar]
- 15.Magalhaes JG, Fritz JH, Le Bourhis L, Sellge G, Travassos LH, Selvanantham T, Girardin SE, Gommerman JL, Philpott DJ. Nod2-dependent Th2 polarization of antigen-specific immunity. J Immunol. 2008;181:7925–7935. doi: 10.4049/jimmunol.181.11.7925. [DOI] [PubMed] [Google Scholar]
- 16.Werninghaus K, Babiak A, Gross O, Holscher C, Dietrich H, Agger EM, Mages J, Mocsai A, Schoenen H, Finger K, Nimmerjahn F, Brown GD, Kirschning C, Heit A, Andersen P, Wagner H, Ruland J, Lang R. Adjuvanticity of a synthetic cord factor analogue for subunit Mycobacterium tuberculosis vaccination requires FcRgamma-Syk-Card9-dependent innate immune activation. J Exp Med. 2009;206:89–97. doi: 10.1084/jem.20081445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Davis BK, Wen H, Ting JP. The inflammasome NLRs in immunity, inflammation, and associated diseases. Annu Rev Immunol. 2011;29:707–735. doi: 10.1146/annurev-immunol-031210-101405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Park JH, Kim YG, McDonald C, Kanneganti TD, Hasegawa M, Body-Malapel M, Inohara N, Nunez G. RICK/RIP2 mediates innate immune responses induced through Nod1 and Nod2 but not TLRs. J Immunol. 2007;178:2380–2386. doi: 10.4049/jimmunol.178.4.2380. [DOI] [PubMed] [Google Scholar]
- 19.Hsu YM, Zhang Y, You Y, Wang D, Li H, Duramad O, Qin XF, Dong C, Lin X. The adaptor protein CARD9 is required for innate immune responses to intracellular pathogens. Nat Immunol. 2007;8:198–205. doi: 10.1038/ni1426. [DOI] [PubMed] [Google Scholar]
- 20.Yamasaki S, Matsumoto M, Takeuchi O, Matsuzawa T, Ishikawa E, Sakuma M, Tateno H, Uno J, Hirabayashi J, Mikami Y, Takeda K, Akira S, Saito T. C-type lectin Mincle is an activating receptor for pathogenic fungus, Malassezia. Proc Natl Acad Sci U S A. 2009;106:1897–1902. doi: 10.1073/pnas.0805177106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Goldszmid RS, Idoyaga J, Bravo AI, Steinman R, Mordoh J, Wainstok R. Dendritic cells charged with apoptotic tumor cells induce long-lived protective CD4+ and CD8+ T cell immunity against B16 melanoma. J Immunol. 2003;171:5940–5947. doi: 10.4049/jimmunol.171.11.5940. [DOI] [PubMed] [Google Scholar]
- 22.Dobson G, Minnikin DE, Minnikin SM, Parlett JH, Goodfellow M, Ridell M, Magnusson M. Systematic analysis of complex mycobacterial lipids. In: Goodfellow M, Minnikin DE, editors. Chemical methods in bacterial systematics. Academic Press; Orlando: 1985. pp. 237–265. [Google Scholar]
- 23.Mishra AK, Krumbach K, Rittmann D, Appelmelk B, Pathak V, Pathak AK, Nigou J, Geurtsen J, Eggeling L, Besra GS. Lipoarabinomannan biosynthesis in Corynebacterineae: the interplay of two alpha(1-->2)-mannopyranosyltransferases MptC and MptD in mannan branching. Mol Microbiol. 2011;80:1241–1259. doi: 10.1111/j.1365-2958.2011.07640.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Besra GS, Khoo KH, McNeil MR, Dell A, Morris HR, Brennan PJ. A new interpretation of the structure of the mycolyl-arabinogalactan complex of Mycobacterium tuberculosis as revealed through characterization of oligoglycosylalditol fragments by fast-atom bombardment mass spectrometry and 1H nuclear magnetic resonance spectroscopy. Biochemistry. 1995;34:4257–4266. doi: 10.1021/bi00013a015. [DOI] [PubMed] [Google Scholar]
- 25.Davidson LA, Draper P, Minnikin DE. Studies on the mycolic acids from the walls of Mycobacterium microti. J Gen Microbiol. 1982;128:823–828. doi: 10.1099/00221287-128-4-823. [DOI] [PubMed] [Google Scholar]
- 26.Schnare M, Barton GM, Holt AC, Takeda K, Akira S, Medzhitov R. Toll-like receptors control activation of adaptive immune responses. Nat Immunol. 2001;2:947–950. doi: 10.1038/ni712. [DOI] [PubMed] [Google Scholar]
- 27.Koo IC, Wang C, Raghavan S, Morisaki JH, Cox JS, Brown EJ. ESX-1-dependent cytolysis in lysosome secretion and inflammasome activation during mycobacterial infection. Cell Microbiol. 2008;10:1866–1878. doi: 10.1111/j.1462-5822.2008.01177.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Martinon F, Agostini L, Meylan E, Tschopp J. Identification of bacterial muramyl dipeptide as activator of the NALP3/cryopyrin inflammasome. Curr Biol. 2004;14:1929–1934. doi: 10.1016/j.cub.2004.10.027. [DOI] [PubMed] [Google Scholar]
- 29.Hsu LC, Ali SR, McGillivray S, Tseng PH, Mariathasan S, Humke EW, Eckmann L, Powell JJ, Nizet V, Dixit VM, Karin M. A NOD2-NALP1 complex mediates caspase-1-dependent IL-1beta secretion in response to Bacillus anthracis infection and muramyl dipeptide. Proc Natl Acad Sci U S A. 2008;105:7803–7808. doi: 10.1073/pnas.0802726105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pan Q, Mathison J, Fearns C, Kravchenko VV, Da Silva Correia J, Hoffman HM, Kobayashi KS, Bertin J, Grant EP, Coyle AJ, Sutterwala FS, Ogura Y, Flavell RA, Ulevitch RJ. MDP-induced interleukin-1beta processing requires Nod2 and CIAS1/NALP3. J Leukoc Biol. 2007;82:177–183. doi: 10.1189/jlb.1006627. [DOI] [PubMed] [Google Scholar]
- 31.Girardin SE, I, Boneca G, Carneiro LA, Antignac A, Jehanno M, Viala J, Tedin K, Taha MK, Labigne A, Zahringer U, Coyle AJ, DiStefano PS, Bertin J, Sansonetti PJ, Philpott DJ. Nod1 detects a unique muropeptide from gram-negative bacterial peptidoglycan. Science. 2003;300:1584–1587. doi: 10.1126/science.1084677. [DOI] [PubMed] [Google Scholar]
- 32.Inohara N, Ogura Y, Fontalba A, Gutierrez O, Pons F, Crespo J, Fukase K, Inamura S, Kusumoto S, Hashimoto M, Foster SJ, Moran AP, Fernandez-Luna JL, Nunez G. Host recognition of bacterial muramyl dipeptide mediated through NOD2. Implications for Crohn’s disease. J Biol Chem. 2003;278:5509–5512. doi: 10.1074/jbc.C200673200. [DOI] [PubMed] [Google Scholar]
- 33.Osorio F, Reis e Sousa C. Myeloid C-type lectin receptors in pathogen recognition and host defense. Immunity. 2011;34:651–664. doi: 10.1016/j.immuni.2011.05.001. [DOI] [PubMed] [Google Scholar]
- 34.Saito R, Tanaka A, Sugiyama K, Azuma I, Yamamura Y. Adjuvant effect of cord factor, a mycobacterial lipid. Infect Immun. 1976;13:776–781. doi: 10.1128/iai.13.3.776-781.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Davidsen J, Rosenkrands I, Christensen D, Vangala A, Kirby D, Perrie Y, Agger EM, Andersen P. Characterization of cationic liposomes based on dimethyldioctadecylammonium and synthetic cord factor from M. tuberculosis (trehalose 6,6′-dibehenate)-a novel adjuvant inducing both strong CMI and antibody responses. Biochim Biophys Acta. 2005;1718:22–31. doi: 10.1016/j.bbamem.2005.10.011. [DOI] [PubMed] [Google Scholar]
- 36.Chung Y, Chang SH, Martinez GJ, Yang XO, Nurieva R, Kang HS, Ma L, Watowich SS, Jetten AM, Tian Q, Dong C. Critical regulation of early Th17 cell differentiation by interleukin-1 signaling. Immunity. 2009;30:576–587. doi: 10.1016/j.immuni.2009.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sutton C, Brereton C, Keogh B, Mills KH, Lavelle EC. A crucial role for interleukin (IL)-1 in the induction of IL-17-producing T cells that mediate autoimmune encephalomyelitis. J Exp Med. 2006;203:1685–1691. doi: 10.1084/jem.20060285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Su SB, Silver PB, Grajewski RS, Agarwal RK, Tang J, Chan CC, Caspi RR. Essential role of the MyD88 pathway, but nonessential roles of TLRs 2, 4, and 9, in the adjuvant effect promoting Th1-mediated autoimmunity. J Immunol. 2005;175:6303–6310. doi: 10.4049/jimmunol.175.10.6303. [DOI] [PubMed] [Google Scholar]
- 39.Fang J, Fang D, Silver PB, Wen F, Li B, Ren X, Lin Q, Caspi RR, Su SB. The role of TLR2, TRL3, TRL4, and TRL9 signaling in the pathogenesis of autoimmune disease in a retinal autoimmunity model. Invest Ophthalmol Vis Sci. 2010;51:3092–3099. doi: 10.1167/iovs.09-4754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fukata M, Breglio K, Chen A, Vamadevan AS, Goo T, Hsu D, Conduah D, Xu R, Abreu MT. The myeloid differentiation factor 88 (MyD88) is required for CD4+ T cell effector function in a murine model of inflammatory bowel disease. J Immunol. 2008;180:1886–1894. doi: 10.4049/jimmunol.180.3.1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lalor SJ, Dungan LS, Sutton CE, Basdeo SA, Fletcher JM, Mills KH. Caspase-1-processed cytokines IL-1beta and IL-18 promote IL-17 production by gammadelta and CD4 T cells that mediate autoimmunity. J Immunol. 2011;186:5738–5748. doi: 10.4049/jimmunol.1003597. [DOI] [PubMed] [Google Scholar]
- 42.Stehlik C. Multiple interleukin-1beta-converting enzymes contribute to inflammatory arthritis. Arthritis Rheum. 2009;60:3524–3530. doi: 10.1002/art.24961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Maelfait J, Vercammen E, Janssens S, Schotte P, Haegman M, Magez S, Beyaert R. Stimulation of Toll-like receptor 3 and 4 induces interleukin-1beta maturation by caspase-8. J Exp Med. 2008;205:1967–1973. doi: 10.1084/jem.20071632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ellouz F, Adam A, Ciorbaru R, Lederer E. Minimal structural requirements for adjuvant activity of bacterial peptidoglycan derivatives. Biochem Biophys Res Commun. 1974;59:1317–1325. doi: 10.1016/0006-291x(74)90458-6. [DOI] [PubMed] [Google Scholar]
- 45.Shimada T, Park BG, Wolf AJ, Brikos C, Goodridge HS, Becker CA, Reyes CN, Miao EA, Aderem A, Gotz F, Liu GY, Underhill DM. Staphylococcus aureus evades lysozyme-based peptidoglycan digestion that links phagocytosis, inflammasome activation, and IL-1beta secretion. Cell Host Microbe. 2010;7:38–49. doi: 10.1016/j.chom.2009.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mariathasan S, Monack DM. Inflammasome adaptors and sensors: intracellular regulators of infection and inflammation. Nat Rev Immunol. 2007;7:31–40. doi: 10.1038/nri1997. [DOI] [PubMed] [Google Scholar]
- 47.LeibundGut-Landmann S, Gross O, Robinson MJ, Osorio F, Slack EC, Tsoni SV, Schweighoffer E, Tybulewicz V, Brown GD, Ruland J, Reis e Sousa C. Syk- and CARD9-dependent coupling of innate immunity to the induction of T helper cells that produce interleukin 17. Nat Immunol. 2007;8:630–638. doi: 10.1038/ni1460. [DOI] [PubMed] [Google Scholar]
- 48.Wuthrich M, Gern B, Hung CY, Ersland K, Rocco N, Pick-Jacobs J, Galles K, Filutowicz H, Warner T, Evans M, Cole G, Klein B. Vaccine-induced protection against 3 systemic mycoses endemic to North America requires Th17 cells in mice. J Clin Invest. 2011;121:554–568. doi: 10.1172/JCI43984. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.