

Abstract
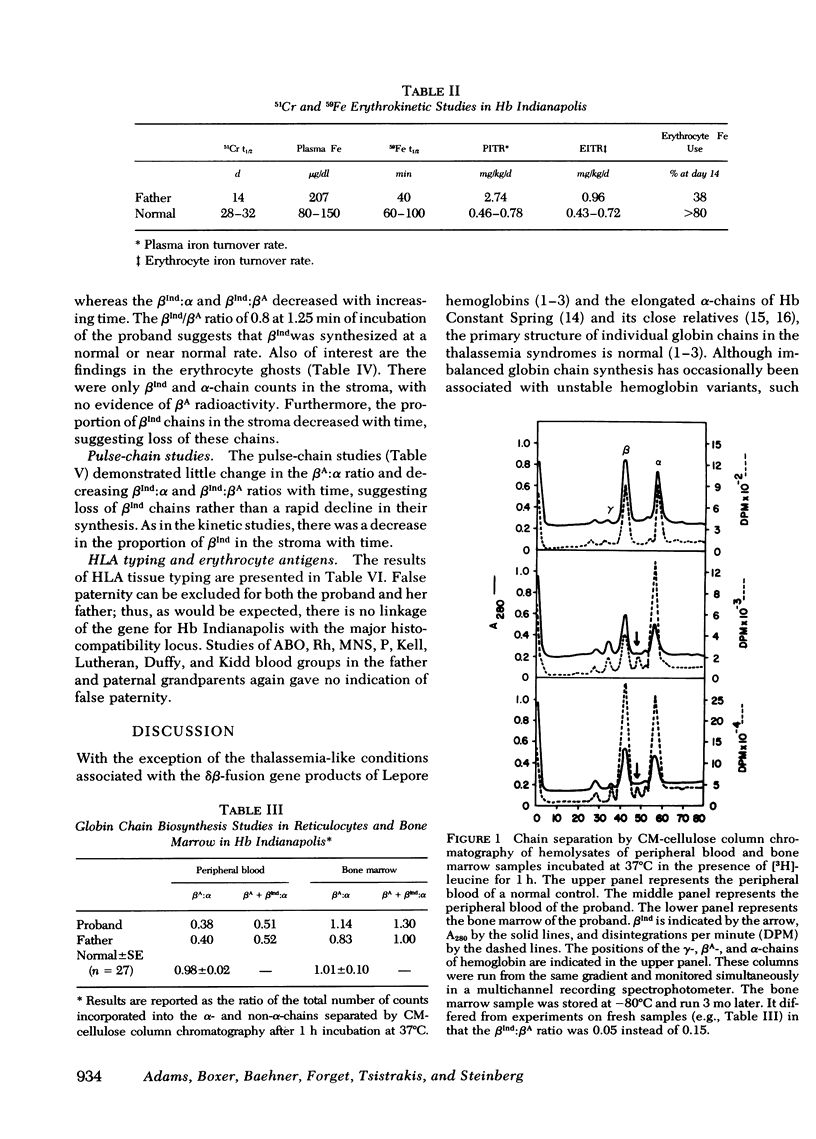
Hemoglobin (Hb) Indianapolis is an extremely labile beta-chain variant, present in such small amounts that it was undetectable by usual techniques. Clinically, it produces the phenotype of severe beta-thalassemia. Biosynthetic studies showed a beta:alpha ratio of 0.5 in reticulocytes and about 1.0 in marrow after a 1-h incubation. These results, similar to those seen in typical heterozygous beta-thalassemia, suggested that betaIndianapolis was destroyed so rapidly that its net synthesis was essentially zero. To examine the kinetics of globin synthesis, reticulocyte incubations of 1.25--20 min were performed with [3H]leucine. The betaIndianapolis:beta A ratio at 1.25 min was 0.80 suggesting that beta Indianapolis was synthesized at a near normal rate. At 20 min, this ratio was 0.46 reflecting rapid turnover of beta Indianapolis. The erythrocyte ghosts from these incubations contained only betaIndianapolis and alpha-chains, and the proportion of betaIndianapolis decreased with time, indicating loss of betaIndianapolis. Pulse-chase studies showed little change in beta A:alpha ratio and decreasing betaIndianapolis:alpha and betaIndianapolis:beta A with time. The half-life of betaIndianapolis in the soluble hemoglobin was approximately equal to 7 min. There was also rapid loss of beta Indianapolis from the erythrocyte membrane. From these results, it may be inferred that betaIndianapolis is rapidly precipitated from the soluble cell phase to the membrane, where it is catabolized. Heterozygotes for beta 0-thalassemia usually have minimal hematologic abnormalities, whereas heterozygotes with betaIndianapolis, having a similar net content of beta-chain, have severe disease. The extremely rapid precipitation and catabolism of betaIndianapolis and the resulting excess of alpha-chains, both causing membrane damage, may be responsible for the severe clinical manifestations associated with this variant. It seems likely that other, similar disturbances in the primary sequence of globin polypeptide chains may produce clinical findings similar to those seen with hemoglobin Indianapolis and thus produce the phenotype of severe beta-thalassemia.
Full text
PDF





Selected References
These references are in PubMed. This may not be the complete list of references from this article.
- Bank A., O'Donnell J. V., Braverman A. S. Globin chain synthesis in heterozygotes for beta chain mutations. J Lab Clin Med. 1970 Oct;76(4):616–621. [PubMed] [Google Scholar]
- Bank A. The thalassemia syndromes. Blood. 1978 Mar;51(3):369–384. [PubMed] [Google Scholar]
- Boyer S. H., Crosby E. F., Noyes A. N. Hemoglobin switching in non-anemic sheep. I. Mediation by plasma from anemic animals. Johns Hopkins Med J. 1968 Aug;123(2):85–91. [PubMed] [Google Scholar]
- Cavill I., Ricketts C., Jacobs A., Letsky E. Erythropoiesis and the effect of transfusion in homozygous beta-thalassemia. N Engl J Med. 1978 Apr 6;298(14):776–778. doi: 10.1056/NEJM197804062981406. [DOI] [PubMed] [Google Scholar]
- Clegg J. B., Naughton M. A., Weatherball D. J. Abnormal human haemoglobins. Separation and characterization of the alpha and beta chains by chromatography, and the determination of two new variants, hb Chesapeak and hb J (Bangkok). J Mol Biol. 1966 Aug;19(1):91–108. doi: 10.1016/s0022-2836(66)80052-9. [DOI] [PubMed] [Google Scholar]
- Clegg J. B., Weatherall D. J., Contopolou-Griva I., Caroutsos K., Poungouras P., Tsevrenis H. Haemoglobin Icaria, a new chain-termination mutant with causes alpha thalassaemia. Nature. 1974 Sep 20;251(5472):245–247. doi: 10.1038/251245a0. [DOI] [PubMed] [Google Scholar]
- Clegg J. B., Weatherall D. J. Haemoglobin synthesis during erythroid maturation in -thalassaemia. Nat New Biol. 1972 Dec 6;240(101):190–192. doi: 10.1038/newbio240190a0. [DOI] [PubMed] [Google Scholar]
- Clegg J. B., Weatherall D. J., Milner P. F. Haemoglobin Constant Spring--a chain termination mutant? Nature. 1971 Dec 10;234(5328):337–340. doi: 10.1038/234337a0. [DOI] [PubMed] [Google Scholar]
- DODGE J. T., MITCHELL C., HANAHAN D. J. The preparation and chemical characteristics of hemoglobin-free ghosts of human erythrocytes. Arch Biochem Biophys. 1963 Jan;100:119–130. doi: 10.1016/0003-9861(63)90042-0. [DOI] [PubMed] [Google Scholar]
- DeSimone J., Adams J. G., 3rd, Shaeffer J. Evidence for rapid loss of newly synthesized haemoglobin S molecules in sickle cell anaemia and sickle cell trait. Br J Haematol. 1977 Mar;35(3):373–385. doi: 10.1111/j.1365-2141.1977.tb00597.x. [DOI] [PubMed] [Google Scholar]
- Drysdale J. W., Righetti P., Bunn H. F. The separation of human and animal hemoglobins by isoelectric focusing in polyacrylamide gel. Biochim Biophys Acta. 1971 Jan 19;229(1):42–50. doi: 10.1016/0005-2795(71)90315-1. [DOI] [PubMed] [Google Scholar]
- ERLANDSON M. E., SCHULMAN I., STERN G., SMITH C. H. Studies of congenital hemolytic syndromes. I. Rates of destruction and production of erythrocytes in thalassemia. Pediatrics. 1958 Nov;22(5):910–922. [PubMed] [Google Scholar]
- Finch C. A., Deubelbeiss K., Cook J. D., Eschbach J. W., Harker L. A., Funk D. D., Marsaglia G., Hillman R. S., Slichter S., Adamson J. W. Ferrokinetics in man. Medicine (Baltimore) 1970 Jan;49(1):17–53. doi: 10.1097/00005792-197001000-00002. [DOI] [PubMed] [Google Scholar]
- Forget B. G., Benz E. J., Jr, Skoultchi A., Baglioni C., Housman D. Absence of messenger RNA for beta globin chain in beta(0) thalassaemia. Nature. 1974 Feb 8;247(5440):379–381. doi: 10.1038/247379a0. [DOI] [PubMed] [Google Scholar]
- Friedman S., Ozsoylu S., Luddy R., Schwartz E. Heterozygous beta thalassaemia of unusual severity. Br J Haematol. 1976 Jan;32(1):65–77. doi: 10.1111/j.1365-2141.1976.tb01876.x. [DOI] [PubMed] [Google Scholar]
- Honig G. R., Mason R. G., Vida L. N., Shamsuddin M. Synthesis of hemoglobin Abraham Lincoln (beta32leu leads to pro). Blood. 1974 May;43(5):657–664. [PubMed] [Google Scholar]
- Huehns E. R. The unstable haemoglobins. Bull Soc Chim Biol (Paris) 1970;52(11):1131–1146. [PubMed] [Google Scholar]
- Kacian D. L., Gambino R., Dow L. W., Grossbard E., Natta C., Ramirez F., Spiegelman S., Marks P. A., Bank A. Decreased globin messenger RNA in thalassemia detected by molecular hybridization. Proc Natl Acad Sci U S A. 1973 Jun;70(6):1886–1890. doi: 10.1073/pnas.70.6.1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kan Y. W., Dozy A. M., Trecartin R., Todd D. Identification of a nondeletion defect in alpha-thalassemia. N Engl J Med. 1977 Nov 17;297(20):1081–1084. doi: 10.1056/NEJM197711172972002. [DOI] [PubMed] [Google Scholar]
- Kan Y. W., Dozy A. M., Varmus H. E., Taylor J. M., Holland J. P., Lie-Injo L. E., Ganesan J., Todd D. Deletion of alpha-globin genes in haemoglobin-H disease demonstrates multiple alpha-globin structural loci. Nature. 1975 May 15;255(5505):255–256. doi: 10.1038/255255a0. [DOI] [PubMed] [Google Scholar]
- Kazazian H. H., Jr, Ginder G. D., Snyder P. G., Van Beneden R. J., Woodhead A. P. Further evidence of a quantitative deficiency of chain-specific globin mRNA in the thalassemia syndromes. Proc Natl Acad Sci U S A. 1975 Feb;72(2):567–571. doi: 10.1073/pnas.72.2.567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nronha P. A., Honig G. R. beta-Thalassemia arising as a new mutation in an American child. Am J Hematol. 1978;4(2):187–192. doi: 10.1002/ajh.2830040211. [DOI] [PubMed] [Google Scholar]
- Ottolenghi S., Lanyon W. G., Paul J., Williamson R., Weatherall D. J., Clegg J. B., Pritchard J., Pootrakul S., Boon W. H. The severe form of alpha thalassaemia is caused by a haemoglobin gene deletion. Nature. 1974 Oct 4;251(5474):389–392. doi: 10.1038/251389a0. [DOI] [PubMed] [Google Scholar]
- Ramirez F., Natta C., O'Donnell J. V., Canale V., Bailey G., Sanguensermsri T., Maniatis G. M., Marks P. A., Bank A. Relative numbers of human globin genes assayed with purified alpha and beta complementary human DNA. Proc Natl Acad Sci U S A. 1975 Apr;72(4):1550–1554. doi: 10.1073/pnas.72.4.1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez F., O'Donnell J. V., Marks P. A., Bank A., Musumeci S., Schilirò G., Pizzarelli G., Russo G., Luppis B., Gambino R. Abnormal or absent beta mRNA in betao Ferrara and gene deletion in delta beta thalassaemia. Nature. 1976 Oct 7;263(5577):471–475. doi: 10.1038/263471a0. [DOI] [PubMed] [Google Scholar]
- Rieder R. F. Human hemoglobin stability and instability: molecular mechanisms and some clinical correlations. Semin Hematol. 1974 Oct;11(4):423–440. [PubMed] [Google Scholar]
- Rieder R. F., James G. W., 3rd Imbalance in alpha and beta globin synthesis associated with a hemoglobinopathy. J Clin Invest. 1974 Oct;54(4):948–956. doi: 10.1172/JCI107835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz E. Heterozygous beta thalassemia: balanced globin synthesis in bone marrow cells. Science. 1970 Mar 13;167(3924):1512–1514. [PubMed] [Google Scholar]
- Schwartz E. The silent carrier of beta thalassemia. N Engl J Med. 1969 Dec 11;281(24):1327–1333. doi: 10.1056/NEJM196912112812403. [DOI] [PubMed] [Google Scholar]
- Stamatoyannopoulos G., Woodson R., Papayannopoulou T., Heywood D., Kurachi S. Inclusion-body beta-thalassemia trait. A form of beta thalassemia producing clinical manifestations in simple heterozygotes. N Engl J Med. 1974 Apr 25;290(17):939–943. doi: 10.1056/NEJM197404252901705. [DOI] [PubMed] [Google Scholar]
- Steadman J. H., Yates A., Huehns E. R. Idiopathic Heinz body anaemia: Hb-Bristol (beta67 (E11) Val to Asp). Br J Haematol. 1970 Apr;18(4):435–446. doi: 10.1111/j.1365-2141.1970.tb01457.x. [DOI] [PubMed] [Google Scholar]
- TERASAKI P. I., MCCLELLAND J. D. MICRODROPLET ASSAY OF HUMAN SERUM CYTOTOXINS. Nature. 1964 Dec 5;204:998–1000. doi: 10.1038/204998b0. [DOI] [PubMed] [Google Scholar]
- Taylor J. M., Dozy A., Kan Y. W., Varmus H. E., Lie-Injo L. E., Ganesan J., Todd D. Genetic lesion in homozygous alpha thalassaemia (hydrops fetalis). Nature. 1974 Oct 4;251(5474):392–393. doi: 10.1038/251392a0. [DOI] [PubMed] [Google Scholar]
- Tolstoshev P., Mitchell J., Lanyon G., Williamson R., Ottolenghi S., Comi P., Giglioni B., Masera G., Modell B., Weatherall D. J. Presence of gene for beta globin in homozygous beta0 thalassaemia. Nature. 1976 Jan 15;259(5539):95–98. doi: 10.1038/259095a0. [DOI] [PubMed] [Google Scholar]
- Tönz O., Winterhalter K. H., Glatthaar B. E. New mutation leading to -thalassaemia minor. Nat New Biol. 1973 Jan 24;241(108):127–128. doi: 10.1038/newbio241127a0. [DOI] [PubMed] [Google Scholar]
- White J. M., Brain M. C. Defective synthesis of an unstable haemoglobin: haemoglobin Koln (beta 98 Val-Met). Br J Haematol. 1970 Feb;18(2):195–209. doi: 10.1111/j.1365-2141.1970.tb01434.x. [DOI] [PubMed] [Google Scholar]
- White J. M., Dacie J. V. In vitro synthesis of Hb Hammersmith (CDI phe-ser). Nature. 1970 Feb 28;225(5235):860–861. doi: 10.1038/225860a0. [DOI] [PubMed] [Google Scholar]