

Targeted therapy against epidermal growth factor receptor (EGFR) is one of the most promising therapeutics for head and neck squamous cell carcinoma, and EGFR is overexpressed in a wide range of malignancies. An improved understanding of the resistance to EGFR inhibitors may provide new treatment options. This review summarizes some mechanisms and decribes strategies to overcome this resistance.
Keywords: Head and neck squamous cell carcinoma, Anti-EGFR therapy, Cetuximab, Intrinsic and acquired resistance
Abstract
Targeted therapy against the epidermal growth factor receptor (EGFR) is one of the most promising molecular therapeutics for head and neck squamous cell carcinoma (HNSCC). EGFR is overexpressed in a wide range of malignancies, including HNSCC, and initiates important signal transduction pathways in HNSCC carcinogenesis. However, primary and acquired resistance are serious problems and are responsible for low single-agent response rate and tumor recurrence. Therefore, an improved understanding of the molecular mechanisms of resistance to EGFR inhibitors may provide valuable indications to identify biomarkers that can be used clinically to predict response to EGFR blockade and to establish new treatment options to overcome resistance. To date, no predictive biomarker for HNSCC is available in the clinic. Therapeutic resistance to anti-EGFR therapy may arise from mechanisms that can compensate for reduced EGFR signaling and/or mechanisms that can modulate EGFR-dependent signaling. In this review, we will summarize some of these molecular mechanisms and describe strategies to overcome that resistance.
Implications for Practice:
The introduction of epidermal growth factor receptor (EGFR) targeted therapeutic agents in the treatment of head and neck squamous cell carcinoma (HNSCC) has led to a new therapeutic challenge, namely resistance. Resistance can either be intrinsic or acquired during treatment. The knowledge we have gained from the underlying molecular mechanisms of resistance in HNSCC as well as other cancer types will be helpful in learning both how to predict resistance, and, if possible, how to overcome this resistance. Eventually, this will lead to personalized therapy for cancer patients where the right drug will be selected for the right patient.
Introduction
Most cancers originating from the squamous epithelium of the upper aerodigestive tract, including lip, oral cavity, pharynx (oropharynx, hypopharynx, and nasopharynx), larynx, and paranasal sinuses, are grouped as head and neck squamous cell carcinoma (HNSCC). Overall, HNSCC comprise 90% of all head and neck cancers and represent the sixth most common form of cancer worldwide [1]. In only 50% of HNSCC patients, the current conventional treatment strategies, including surgery, chemotherapy, and radiation, are effective, underscoring the need for new approaches to treat this malignancy [2, 3]. The existing cytotoxic therapies are nonselective and associated with considerable toxicity in HNSCC patients. Therefore, the need for additional treatment options that improve clinical outcome and have a better toxicity profile is pressing. As our understanding of the molecular biology of HNSCC continues to improve, this may provide the opportunity to develop targeted therapy for HNSCC treatment. Ideally, targeted agents are directed against unique molecular features of cancer cells, which cause, promote, or maintain the malignant behavior of these cells. To maximally exploit these features, characterization of the tumor at the molecular level, understanding the biological heterogeneity of human cancer, and insight into the inter-individual variation in the human genome are essential [4–6]. Thus, identifying biological markers, or biomarkers, that allow prediction of response to therapy has become increasingly important [7–9]. Eventually, this will lead to a personalized therapy for cancer patients where the right drug will be selected for the right patient.
Currently, HNSCCs are classified according to the TNM system, based on morphology and anatomic distribution. However, it is obvious that this classification lacks biological and molecular markers [10], leading to the same treatment for malignancies with a different biology [9].
The epidermal growth factor receptor (EGFR) and its ligands play an essential role in proliferation, differentiation, antiapoptotic signaling, and the processes of angiogenesis and metastasis, thereby driving the malignant behavior of the tumor [11, 12]. The oncoprotein EGFR belongs to the ErbB family of cell surface receptors and is also known as ErbB1 or HER1 [11]. Expression of EGFR is found in up to 90% of HNSCC cases, and it is an independent prognostic marker, as high expression is associated with increased tumor size, decreased radiation sensitivity, and increased risk of recurrence [2, 13]. As a result, EGFR overexpression is related to decreased overall survival [14].
Because EGFR is overexpressed in a wide range of malignancies, and initiates important signal transduction pathways in carcinogenesis, it has emerged as a promising therapeutic target (Fig. 1). Considering EGFR targeting therapies, two main categories of molecules are of key importance: monoclonal antibodies (mABs, such as cetuximab and panitumumab) and tyrosine kinase inhibitors (TKIs, such as gefitinib and erlotinib). The therapeutic effect of the mABs is exerted by binding to the extracellular domain of EGFR, thereby preventing ligands to activate EGFR, while promoting EGFR internalization and some by antibody-dependent cell-mediated cytotoxicity (ADCC) [15, 16]. In contrast, the quinazoline-derived TKIs directly inhibit the kinase function by blocking ATP binding to the intracellular tyrosine kinase domain of EGFR, thereby preventing downstream signaling [17]. Currently, the EGFR monoclonal antibody cetuximab (Erbitux) is the only FDA-approved EGFR targeting strategy for HNSCC, in three specific settings: either as a single agent for metastatic/recurrent disease (after failure of platinum-based chemotherapy), in combination with radiation for locally or regionally advanced HNSCC, or in combination with platinum/5-FU in the first-line metastatic/recurrent disease setting [18–20].
Figure 1.
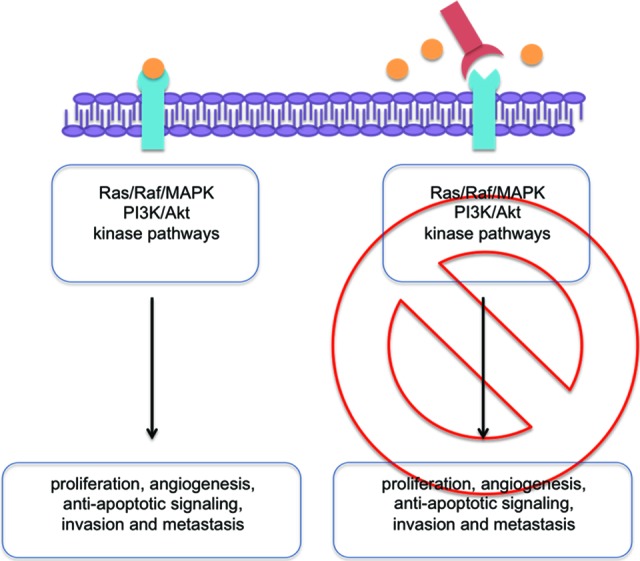
EGFR signaling and anti-EGFR mAB. Left: Activation of EGFR by binding of a natural ligand to the receptor and subsequent activation of signaling pathways. Right: Blockade of the EGFR receptor by an anti-EGFR mAB preventing the subsequent activation of the signaling pathway.
Abbreviation: EGFR, epidermal growth factor receptor.
Nevertheless, one main challenge in the targeted therapy of HNSCC remains, namely (intrinsic and acquired) drug resistance. Many HNSCC tumors remain nonresponsive to EGFR targeting agents, as the response rate with such agents, as for instance cetuximab as a single agent, is consistently lower than 15% [21]. Nevertheless, EGFR inhibition has shown to be promising also in the clinical setting, when combined with conventional cytotoxic approaches [18, 22]. Therefore, an improved understanding of the molecular mechanisms of resistance to EGFR inhibitors might allow identification of biomarkers that can be used clinically to predict response to EGFR blockade and/or to establish new treatment options to overcome resistance [7–9].
This applies not only to HNSCC, but also to other forms of cancers where anti-EGFR targeting agents are used, such as non-small cell lung cancer (NSCLC) and colorectal cancer (CRC). In fact, in these types of cancer, biomarkers of response have already been identified (EGFR tyrosine kinase and K-Ras mutations). However, as not all unresponsive CRC and NSCLC cases could be clarified by these mutations, other genes must be involved too.
Because cetuximab has been most successful in improving clinical outcomes in HNSCC and is approved by the FDA and EMEA for the treatment of HNSCC, this review focuses on mechanisms of resistance to monoclonal-based anti-EGFR therapy, mainly cetuximab.
Potential Predictive Markers for Anti-EGFR Therapy in HNSCC
Until now, the only clinical marker for response to cetuximab therapy is the severity of skin rash, which is correlated with outcome in HNSCC patients [22]. However, in the literature, several possible causes for altered responses to anti-EGFR therapy in HNSCC have been described, and will be discussed below. Therapeutic resistance to anti-EGFR therapy may arise from mechanisms that either compensate for reduced EGFR signaling and/or modulate EGFR-dependent signaling (Fig. 2).
Figure 2.

Despite mAB-mediated anti-EGFR treatment, the signaling cascades induced by EGFR activation may still be active because of molecular resistance mechanisms at different levels, leading to proliferation, angiogenesis, antiapoptotic signaling, invasion, and metastasis.
Abbreviation: EGFR, epidermal growth factor receptor.
The genes and proteins discussed below are involved in altered response to anti-EGFR therapy in HNSCC patients, and can be considered potential predictive biomarkers for anti-EGFR therapy. However, their role has not been crystalized yet and more studies are warranted to identify new reliable predictive biomarkers and effective therapeutic combinations that overcome treatment resistance and improve clinical outcome in HNSCC patients.
Altered Response Elicited at the Level of EGFR
Sustained EGFR signaling can be elicited at the level of the target itself by ligand or receptor overexpression, amplification, or mutation. Moreover, EGFR can escape lysosomal degradation routes, and subsequently functions as a transcription factor in the nucleus, thereby inducing prolonged EGFR signaling [23, 24].
Ligand Overexpression
Binding of ligands to EGFR drives homodimerization or heterodimerization with ErbB family members, resulting in the initiation of downstream signaling pathways. Therefore, overexpression of its ligands may contribute to cetuximab resistance.
Hatakeyama et al. showed that cetuximab-sensitive HNSCC cell lines become resistant to cetuximab when stimulated with the ligand heparin binding EGF (HB-EGF), whereas knockdown of HB-EGF reverses resistance to cetuximab in the resistant HNSCC cell lines [25]. Additionally, activated EGFR was evoked by three ligands, amphiregulin, HB-EGF, and TGF-α even in the presence of cetuximab [25]. Transactivation of EGFR and ERK signaling can be blocked by neutralization of TGF-α [26]. Furthermore, an in vivo study showed that HNSCC xenografts grown in the presence of cetuximab resulted in the development of resistant tumor cells that expressed relatively higher levels of TGF-β compared with untreated tumor-bearing mice [27]. Combination therapy with cetuximab and a TGF-β blocking antibody prevented the development of such resistant tumor cells and induced complete regression [27].
A correlation with enhanced response to cetuximab therapy and overexpression of the EGFR ligands amphiregulin and epiregulin in K-Ras wild-type metastatic colorectal tumors has been reported [28].
In HNSCC patients receiving cetuximab-docetaxel treatment, high amphiregulin levels were detected in 45% of the patients. A significant correlation was found between high amphiregulin levels and shortened overall survival and progression-free survival compared with patients with low amphiregulin expression [29].
Activating Mutations in the EGFR Gene
Until now, neither the expression level of the EGFR protein nor the amplification status of the EGFR gene could be linked to therapeutic response [30, 31].
Activating mutations have been observed in the tyrosine kinase domain or in the extracellular ligand-binding domain of EGFR [32]. The most common tyrosine kinase EGFR mutations include deletion of four conserved amino acids residues (leucine-arginine-glutamic acid-alanine) in exon 19 and a point mutation, L858R, in exon 21, which account for 90% of all EGFR tyrosine kinase mutations in NSCLC [33–35]. These EGFR tyrosine kinase mutations are associated with an improved clinical response to TKIs (gefitinib or erlotinib) in NSCLC patients but they are rarely found in HNSCC. Literature data suggest that the incidence of such activating mutations in HNSCC patients range from 0 to 15.7% (Table 1) [33–48]. In these studies, a total of 889 HNSCC samples were screened for EGFR tyrosine kinase mutations, of which 34 (3.8%) contained a mutation.
Table 1.
Frequency of EGFR tyrosine kinase mutations in HNSCC patients
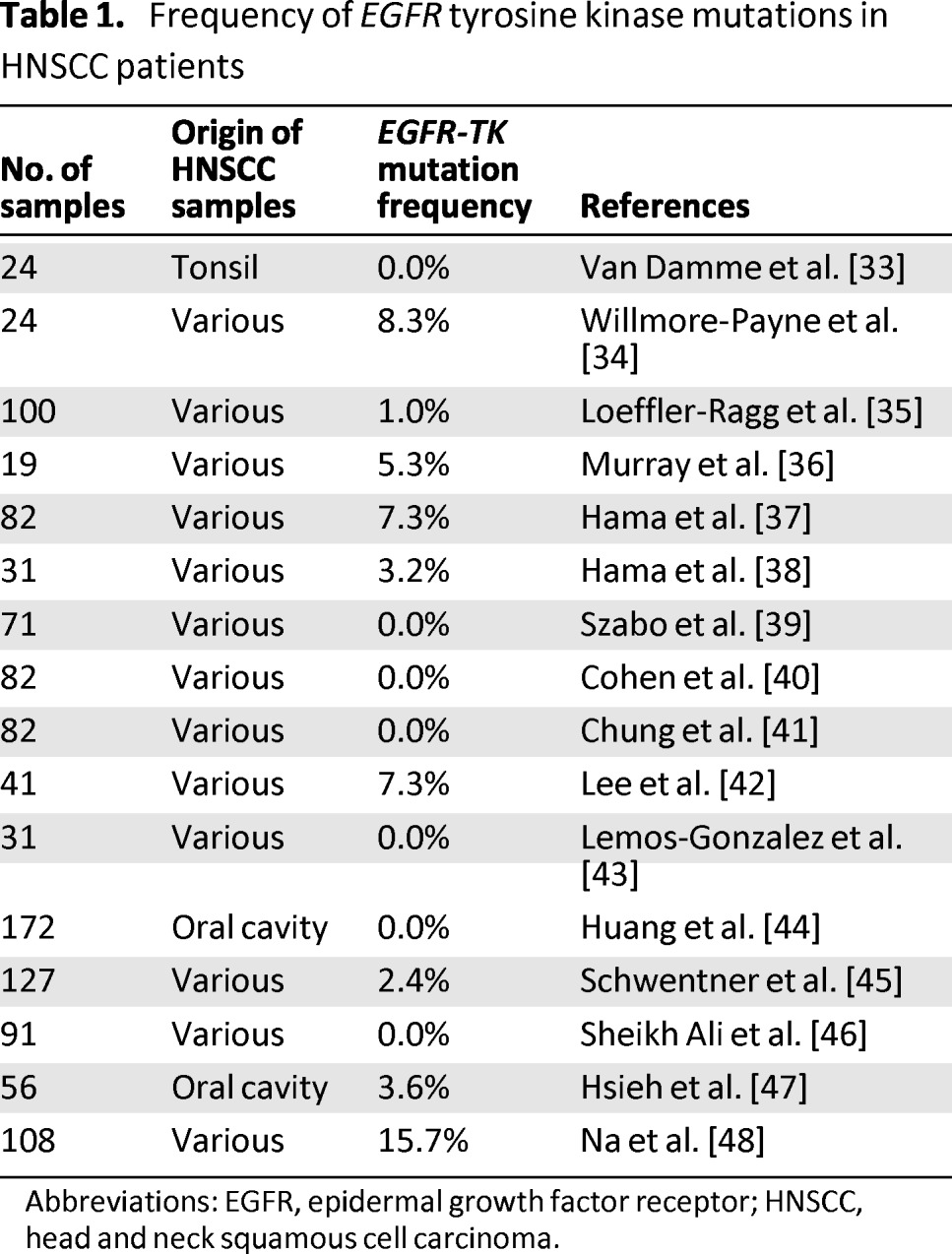
Abbreviations: EGFR, epidermal growth factor receptor; HNSCC, head and neck squamous cell carcinoma.
The EGFRvIII Mutation
Next to the above-discussed activating mutation, the EGFR variant III (EGFRvIII) is a truncated form of EGFR. The causing mutation consists of an in frame deletion of 801 base pairs (exon 2–7) in the coding sequence of the extracellular domain, resulting in ligand-independent tyrosine kinase activity [49, 50]. Interestingly, the deletion creates a novel glycine epitope, which might be used as a target for specific antibody-based vaccines in the future [51]. Moreover, in contrast to wild-type EGFR, EGFRvIII seems to preferentially activate the phosphatidylinositol-3 kinase (PI3K) pathway. The mutant EGFRvIII form is associated with increased proliferation, tumor growth, cell motility and invasion in vitro, and resistance to anti-EGFR therapy [50, 51]. Studies in glioma cells have shown that cetuximab binds to EGFRvIII, attenuating its expression and reducing its phosphorylation [52]. Furthermore, cetuximab induces ADCC against EGFRvIII expressing glioma cells [53]. However, treatment with cetuximab does not inhibit the activation of EGFRvIII expressing cells, nor its downstream Akt and MAPK signaling pathways [53, 54].
The mutation frequency of EGFRvIII in HNSCC ranges from 0 to 48%. However, this mutation frequency was reported in only seven studies, of which five studies reported an EGFRvIII mutation frequency ranging from 17% to 48% [29, 39, 51, 55, 56]. In contrast, the two other studies detected a lower frequency of EGFRvIII in HNSCC patients [37, 57]. In these latter studies, the EGFRvIII could not be found at all [37] or only in 2% of cases of oral squamous cell carcinoma [57]. It has been suggested that the EGFRvIII might be more available in the recurrent/metastatic disease setting and be responsible for the lack of response to EGFR-targeted therapies [57]. However, most of the time only archival formalin-fixed paraffin-embedded primary tumor biopsy material is available, as was the case in the study reported by Tinhofer et al. Tinhofer et al. analyzed tumor biopsies from 47 recurrent or metastatic HNSCC patients relapsing after platinum-containing chemoradiotherapy or after platinum-containing first-line chemotherapy who were treated in a single-arm phase II multicenter study with cetuximab and docetaxel. High expression of EGFRvIII was found in eight patients (17%) and was significantly associated with reduced disease control rate and shortened progression-free survival (HR: 3.3, p = .005), but not with overall survival [29]. Consequently, more studies involving the mutation frequency of EGFRvIII in HNSCC and its association with response to anti-EGFR therapy are warranted. The clinical implications of the presence of EGFRvIII in HNSCC patients have not been studied in prospective clinical trials.
The presence of nuclear EGFR is not only associated with poor prognosis, but also with treatment resistance. For example, it has been reported that nuclear EGFR expression plays a role in the therapeutic response to cisplatin and radiation by modulation of DNA repair kinetics and may have implications for EGFR-targeted combination therapies. Moreover, cells with acquired resistance to cetuximab have upregulated HER family ligands and this enhances the translocation of EGFR to the nucleus.
Nuclear EGFR
Occasionally, a part of the EGFR receptor escapes the internalization and lysosomal degradation route and translocates to the nucleus [23, 24]. Nuclear EGFR functions either as a transcription factor of cyclin D1, iNOS, b-myb, and COX-2, or as a tyrosine kinase phosphorylating and stabilizing proliferating cell nuclear antigen (PCNA), resulting in an activation of the nitric oxide pathway and increased G1/S progression of the cell cycle [58–62]. Consequently, the proliferative potential of the cancer cells is thereby enhanced. The presence of nuclear EGFR is not only associated with poor prognosis, but also with treatment resistance [13, 63, 64]. For example, it has been reported that nuclear EGFR expression plays a role in the therapeutic response to cisplatin and radiation by modulation of DNA repair kinetics and may have implications for EGFR-targeted combination therapies [24]. Moreover, cells with acquired resistance to cetuximab have upregulated HER family ligands and this enhances the translocation of EGFR to the nucleus [65]. Perinuclear and nuclear EGFR have been found in gefitinib-resistant cancer cells [66, 67]. Furthermore, A341 epidermal carcinoma cells with acquired gefitinib resistance also show increased levels of nuclear EGFR [68].
Besides its potential involvement in resistance mechanisms, nuclear EGFR is also associated with local recurrence [13]. In oral squamous cancers, nuclear EGFR was observed in 24.3% of patients [63].
The Protein Tyrosine Phosphatase Receptor S
The protein tyrosine phosphatase receptor S (PTPRS) directly interacts with EGFR and phosphorylates and inactivates EGFR. Loss of PTPRS has been reported to enhance EGFR-induced transformation [69, 70]. Therefore, inactivating mutations or other mechanisms responsible for loss of function could contribute to anti-EGFR therapy resistance. One study detected intragenic PTPRS deletion in 26% of HNSCC tumors, resulting in loss of mRNA expression and promoting EGFR/PI3K pathway activation [71]. Moreover, in vitro results showed that PTPRS expression could predict response to cetuximab in HNSCC cell lines [71]. Evidently, more studies are warranted to define the relationship between loss of PTPRS expression and resistance to anti-EGFR therapy.
Molecular Alterations in Effectors Downstream of EGFR
Stimulation of EGFR leads to activation of different signaling pathways, which are probably among the best-studied pathways in cancer biology. Aberrant EGFR signaling can be provoked by molecular changes in downstream effectors of EGFR; particularly the K-Ras, PIK3CA, PTEN, and signal transducer and activator of transcription (STAT) proteins have been shown to contribute to resistance to EGFR-targeted therapies in other malignancies.
K-Ras
K-Ras is a protein located downstream of EGFR in the Ras-MAPK pathway. Somatic point mutations in K-Ras occur in a variety of human malignancies, most frequently in pancreatic cancer, NSCLC, and colon cancers [72, 73]. A mutation in codon 12 or 13 in this gene leads to constitutive activation of the protein, regardless of upstream activating signals. In colorectal tumors, these mutations confer resistance to therapy with the EGFR targeting monoclonal antibodies cetuximab and/or panitumumab [74–77]. Approximately 30%–40% of colorectal tumors harbor a K-Ras mutation [78, 79].
In contrast, in HNSCC, these K-Ras mutations are infrequent; in different reports, the frequency of K-Ras mutations in HNSCC is ranging from 0 to 9.1% (Table 2) [33, 39, 80, 82–89]. In areas with a betel quid chewing habit, a higher K-Ras mutation incidence (±20%) has been noticed [81]. Overall, these mutations are rare, and therefore little is known about the predictive value of K-Ras mutations in HNSCC patients.
Table 2.
Studies involving K-Ras mutations in HNSCC patients
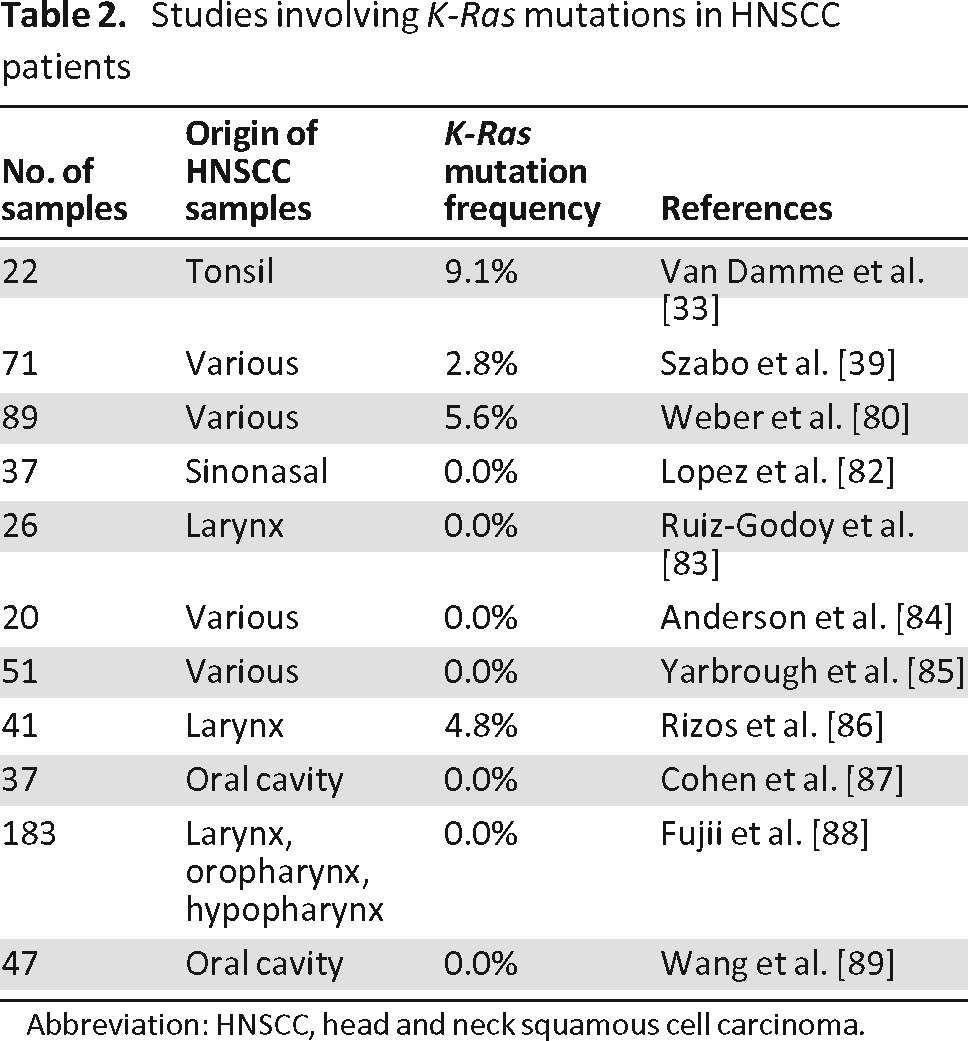
Abbreviation: HNSCC, head and neck squamous cell carcinoma.
K-Ras expression could also be regulated by alterations in binding of microRNAs (miRNAs). These are short, noncoding RNAs, which bind the evolutionarily highly conserved 3′ untranslated regions (3′-UTR) of mRNAs, thereby preventing translation of the mRNA. The family of let-7 miRNAs downregulates the Ras gene family, including K-Ras, after binding to specific sites in the 3′-UTR of Ras mRNA. Recently, a functional single nucleotide polymorphism (T > G) has been identified in the let-7 complementary site (LCS6) in the K-Ras 3′-UTR, which alters let-7 binding, resulting in increased K-Ras expression and decreased let-7 exposure. Several studies have investigated the role of this variant as a prognostic or predictive biomarker for anti-EGFR therapy. However, the results were contradictory. In NSCLC, Chin et al. reported an association between the G variant and increased risk for NSCLC among moderate smokers [90], whereas Nelson et al. could not find any association between the G variant and survival [91]. In HNSCC, the variant is associated with poor prognosis and the prognosis was worst among cases of oral cancer [92]. Moreover, in HNSCC patients, the G allele variant may be associated with tumor progression rather than initiation [92]. Furthermore, knockdown of let-7d promotes epithelial-to-mesenchymal transition (EMT) traits and migratory/invasive capabilities in oral SCC cells, whereas lentiviral-mediated let-7a overexpression significantly inhibited the stemness signature and the chemoresistant abilities of head and neck cancer cells [93, 94]. In CRC, LCS6 G-allele variant and response to cetuximab have been reported in three studies. In 2010, it was shown that G allele carriers had a worse overall survival and a shorter progression-free survival [95]. Strikingly, early stage CRC patients with the LCS6 variant had a better prognosis, whereas the opposite was observed in advanced disease [96]. Additionally, LCS6 allele G carriers seemed to have a better response rate than wild-type carriers regardless of stage [97].
H-Ras
Another family member of the Ras proto-oncogenes is H-Ras. Mutations in H-Ras have been reported in the literature and vary between 0 and 22% [85, 98–101]. Rampias et al. showed that silencing of H-Ras in H-Ras mutant HNSCC cell lines restored sensitivity to cetuximab and caused a direct downregulation of pERK1/2 levels [101]. Furthermore, treatment of a H-Ras mutated BB49 cell line with cetuximab and PI3K inhibitor LY294002 led to a marked reduction of their viability, whereas these same HNSCC cells were found to be resistant to cetuximab [100]. Collectively, these data suggest that H-Ras mutations might play a role in cetuximab resistance and therefore further studies are warranted.
Dual-Specificity Phosphatase (DUSP)
Further downstream of K-Ras in the MAPK signaling pathway, a member of the dual-specificity phosphatase (DUSP) family is located. DUSP proteins are involved in a negative feedback mechanism of the MAPK signaling pathway by dephosphorylation of the threonine-glutamic acid-tyrosine motif on MAP kinases [102]. Therefore, DUSP proteins can be seen as tumor suppressor proteins, and loss of their expression may promote constitutive activation of ERK and uncontrolled cell growth. Moreover, inhibition of the MAPK pathway can be compensated by suppression of the DUSP enzymes [103]. Both the cytoplasmic DUSP5 and the nuclear DUSP6 can dephosphorylate ERK1/2, thereby blocking the MAPK signal transduction cascade [104].
Human carcinomas frequently express one or more members of the ErbB family, and therefore, cross-activation of EGFR downstream signaling might be present. Agents that block other members of the ErbB family simultaneously or that bind multiple ErbB receptors irreversibly are promising and are currently investigated in clinical trials for HNSCC patients.
A recent study investigated DUSP6 expression in esophageal squamous cell carcinoma (ESCC) and nasopharyngeal carcinoma (NPC) tumor tissue. Reduced expression was observed in 40% and 75% of ESCC and NPC tumor tissue, respectively [105]. Reduced expression of DUSP6 can be accomplished either by loss of heterozygosity of the DUSP6 locus or by promoter methylation [106]. Moreover, regulation of DUSP6 is mediated at the promoter level by Ets1, a nuclear target of activated ERK [107]. Furthermore, suppressive effects of DUSP6 in tumor formation and cancer cell mobility were seen in vitro and in vivo, and DUSP6 overexpression impairs EMT-associated properties [105]. Finally, using microarray analysis, Oliveras-Ferraros et al. reported that molecular functioning of cetuximab in A431 epidermoid cancer cells was dependent on EGFR ligands, reduced expression of DUSP6, and EMT-associated proteins [108].
Taken together, the exact function of the DUSP family proteins in relation to cetuximab resistance in HNSCC needs to be further elucidated.
The PI3K/Akt Pathway
Besides activation of the Ras/Raf/MAPK signaling pathway, EGFR can also mediate activation of the PI3K/Akt pathway. Consequently, alterations in proteins involved in the PI3K/Akt pathway might also play a role in resistance to anti-EGFR therapy.
Mutations in the PIK3CA protein occur in 6%–11% of HNSCC patients [71, 109, 110] and are associated with activation of the Akt signaling pathway [111]. Treatment of cetuximab-resistant HNSCC cells with cetuximab did not result in the decreased levels of Akt phosphorylation that were seen in cetuximab-sensitive HNSCC cells. A mutation in exon 20 of the PIK3CA gene was identified in a HNSCC cell line, leading to persistent Akt activation [112]. Furthermore, persistent activation of either MAPK or Akt, or both, was observed in HNSCC and colon cell lines showing limited efficacy of cetuximab therapy [113].
In addition, loss of the tumor suppressor protein PTEN also resulted in persistent activation of the PI3K/Akt pathway. Reintroducing PTEN in PTEN null prostate cancer cells was associated with restoration of cetuximab-induced cell growth inhibition and apoptosis induction. Strikingly, in HNSCC cells, it was reported that treatment of PTEN-silenced Cal27 cells with cetuximab led to decreased pAkt and pERK1/2 levels [114]. Although screening of 16 HNSCC cell lines for mutations in PIK3CA and PTEN identified two mutations for each gene (12.5%), no correlation was found between these mutations and the response to cetuximab therapy [115].
To define the exact role of mutations in the PI3K/Akt pathway, regarding response to anti-EGFR therapy, more in-depth studies are needed.
Src Kinases
Src kinases are upstream as well as downstream activators of EGFR and other receptor tyrosine kinases. Upon EGFR stimulation, Src kinases are activated and associate with EGFR. As such, they can affect cellular proliferation and survival by activation of STAT family of transcription factors, especially STAT3 and STAT5 [116, 117].
In vitro studies showed reduced activity of Src kinases following EGFR inhibition [118]. Elevated Src levels and/or kinase activity have been shown in HNSCC and other malignancies [117, 119]. Therefore, activation of Src kinases by EGFR upstream or downstream signaling might result in resistance to anti-EGFR therapy.
Src-specific inhibitors resulted in decreased activation of STAT3 and STAT5 and reduced growth rates in vitro [117]. However, sustained Src inhibition resulted in only a transient inhibition, because of a compensatory mechanism leading to altered JAK-STAT3 binding and JAK kinase activity [116]. Furthermore, Koppikar et al. reported that mutual inhibition of Src by AZD0530 and EGFR by gefitinib resulted in an increased inhibition of invasion and growth, compared with nonmutual blockade of either tyrosine kinase alone [120]. The effect of saracatinib, a Src inhibitor, was examined in vitro as well as in vivo. These results showed inhibition of growth, cell cycle progression, and transwell Matrigel invasion using HNSCC cell lines. However, this drug had no significant growth inhibitory effect in an orthotopic mouse model [121].
As mentioned earlier, nuclear translocation of EGFR is a possible mechanism of resistance to therapy and this has been observed in patients treated with cetuximab and radiotherapy. Phosphorylation of EGFR on tyrosine 845 by the Src kinases enhanced EGFR-mediated mitogenesis by binding and phosphorylating the STAT5b transcription factor, and this has been described as the underlying mechanism responsible for nuclear translocation of EGFR [122, 123]. Indeed, dasatinib, an Src inhibitor, blocked EGFR translocation to the nucleus in HNSCC cell lines and, therefore, might be a potential way to evade resistance to anti-EGFR therapy [124]. Moreover, dasatinib enhanced radiosensitivity of the HN5 HNSCC cell line by interfering with nuclear localization of EGFR and by blocking DNA repair pathways [125]. In cetuximab-resistant NSCLC cells, EGFR was shown to be responsible for activation of Src kinases, and the cells were highly dependent on this activity for proliferation and survival. Accordingly, dasatinib decreased HER3 and PI3K/Akt activity and resensitized these cells to cetuximab therapy [126].
Collectively, these results indicate that Src inhibitors may be useful in overcoming anti-EGFR resistance by decreasing activated STAT3 and STAT5.
Signal Transducer and Activator of Transcription (STAT) Proteins
When considering resistance to anti-EGFR therapy, the family of STAT proteins are also important downstream EGFR effectors. This family plays an important role in transmitting survival signals and antiapoptotic signals that are initiated through activation of EGFR; especially activation of STAT3 and STAT5 has been linked to phosphorylation of EGFR [117, 127, 128]. Therefore, dysregulation of the STAT signaling pathway has been proposed to be implicated in malignant transformation.
Activation of STAT3 leads to the activation of several survival proteins, including bcl-xl, bcl-2, and survivin [129]. In HNSCC, STAT3 activation can be mediated by JAK and Src signaling, and partially by EGFR signaling [124, 130]. Consequently, STAT3 can be inhibited via EGFR blocking in vitro as well as in vivo [131]. However, recent work reported that the mutant EGFRvIII increased STAT3 activation in vitro [50].
It has been shown that the antiproliferative effects of cetuximab, as well as cetuximab-induced apoptosis, are more pronounced in STAT3 knockdown cells compared with control cells [129]. These antitumor effects were also seen in HNSCC cells in vitro and in vivo using erlotinib in combination with a STAT3 transcription factor decoy [3]. Likewise, upon addition of erlotinib, less growth inhibition induced by erlotinib was detected in HNSCC cells expressing constitutive STAT5 compared with empty vector-transfected control cells [132]. Moreover, the natural STAT3 inhibitor guggulsterone enhanced the efficacy of erlotinib, cetuximab, and cisplatin treatment in HNSCC cell lines by inducing apoptosis, cell cycle arrest, and inhibition of invasion [133]. Additionally, similar results were found in vivo [133]. However, guggulsterone failed to confer protection against oral-induced carcinogenesis in a murine model, whereas erlotinib was able to decrease the incidence of preneoplastic and neoplastic lesions by 69% [134]. On the basis of these results, targeting STAT3 and EGFR together seems promising in HNSCC carcinogenesis.
STAT1 is activated by interferon γ (INF-γ), independent of STAT3 overexpression [135]. The INF-γ-phospho STAT1 signaling pathway is able to downregulate components of the antigen-processing machinery, involved in tumor antigen presentation [135]. This pathway has been identified in chronically adapted cetuximab-resistant vulvar squamous carcinoma cell clones, inducing prosurvival signals instead of apoptosis [136]. In addition, another study showed that STAT1 is required to promote the tumor-killing effects of STAT3 inhibition when cells were treated with JAK inhibitors [137].
Mechanism of Resistance Through Crosstalk with Other Receptor Tyrosine Kinases
Selective stress of anti-EGFR therapy may lead to activation of alternative parallel signaling pathways to compensate for the reduced EGFR signaling, thereby promoting cell survival. Other receptor pathways involving other ErbB family members, insulin growth factor type 1 receptor, MET, and so forth can activate common downstream EGFR effectors and therefore might also contribute to anti-EGFR resistance in HNSCC.
The ErbB Receptor Family
As mentioned earlier, EGFR is a family member of the ErbB receptor family, and activation of other members of this family might result in resistance to anti-EGFR therapy. In the literature, activation of HER2 signaling has been associated with cetuximab resistance because its signaling occurs through many of the same downstream effectors of EGFR. With use of an in vitro model of acquired cetuximab resistance, a marked increase in the phosphorylation status of the C-terminal fragment of HER2, 611-CTF, was observed. Combination therapy of afatinib, an irreversible dual EGFR/HER2 inhibitor, and cetuximab resulted in a dramatic reduction in cetuximab-resistant tumor volumes compared with either agent alone in monotherapy [138]. Therefore, it was suggested that dual inhibition of EGFR and HER2 could be an effective approach to enhance the efficacy of cetuximab, to prevent and/or overcome cetuximab resistance. Likewise, a study by Yonesaka et al. has shown that cetuximab resistance could be induced by activation of ErbB2 signaling [139]. The underlying mechanism involved amplification of ErbB2 or upregulation of heregulin, both leading to persistent ERK1/2 activation. Moreover, restoring cetuximab sensitivity was accomplished by inhibition of ErbB2 or by disruption of ErbB2/ErbB3 heterodimerization in vitro as well as in vivo.
Only one study investigated and reported the link between HER2 activation and response to TKI in HNSCC. Of four evaluable cases of TKI-responsive HNSCC patients, one harbored a heterozygous V773A mutation in HER2 and this mutation was not present in surrounding normal stromal tissue [40]. Given the very small sample population, caution is needed with regard to the association between HER2 mutations and EGFR-TKI sensitivity. More studies are warranted to determine the frequency of HER2 mutations in HNSCC and their role in the response to TKIs.
The Insulin Growth Factor Type 1 Receptor
Activation of the insulin growth factor type 1 receptor (IGF-1R) leads to downstream activation of the Ras/Raf/MAPK and PI3K/Akt pathway and enhances survivin expression, all contributing to cell proliferation, altered cell adhesion, enhanced motility properties, and impaired apoptosis [140, 141]. Furthermore, IGF-1R/EGFR heterodimerization has been reported in multiple epithelial cancers as well as in several HNSCC cell lines after stimulation of these cells with IGF or EGF [142].
In nasopharyngeal carcinoma cell lines, cetuximab resistance was associated with gene amplification and overexpression of H-Ras, which was probably associated with increased activity of the IGF-1R signaling pathway [143]. Moreover, treatment with the anti-IGF-1R antibody A12 in combination with cetuximab was more effective at reducing cell proliferation and migration of HNSCC cell lines than either agent alone [142]. In addition, complete regression of tongue cancer cell xenografts was more frequent in the combination group than in the monotherapy group [142]. Similar results were observed in cutaneous squamous cell carcinoma cell lines and tumor xenografts [144]. Concerning radiation therapy, this anti-IGF-1R antibody was reported to enhance the radiosensitivity of HNSCC cell lines and the radioresponse of FaDu xenografts [145]. The IGF-1R pathway has also been proposed to be involved in resistance to gefitinib and radiation [146, 147]. In vitro results have shown that dual inhibition of EGFR and IGF-1R by tyrosine kinase inhibitors is more efficacious compared with single agents [148].
MET
The MET proto-oncogene encodes a transmembrane receptor tyrosine kinase MET, also known as c-MET or hepatocyte growth factor receptor (HGFR). The MET pathway can be deregulated in two different ways: on the one hand, by mutation and/or amplification of MET, and on the other hand, by increased ligand expression and/or activity, both resulting in persistent activation of the PI3K/Akt signaling pathway [149]. Approximately 80% of primary HNSCC tumors express the ligand hepatocyte growth factor (HGF), MET, or both, thus activating important downstream signals, which overlap with EGFR signaling [55, 150]. Moreover, MET mutations or amplifications have been observed in 13.5% and 13% of HNSCC tumors, respectively [151]. As high MET expression could be observed in 58% of patients with recurrent/metastatic HNSCC [55], the role of MET in resistance to anti-EGFR therapy has been investigated in a number of studies. Chau et al. did not detect any association between response to erlotinib and time to progression or overall survival in recurrent/metastatic HNSCC patients with high MET expression [55]. In contrast, in metastatic colorectal cancer, a significant correlation has been reported between high MET expression and both shorter median progression-free survival and median overall survival in patients treated with cetuximab [152]. Likewise, MET activation has been associated with cetuximab resistance in gastric cell lines [153]. Interestingly, the MET mutation frequency increases from 4% to 7% in untreated lung tumors to 20% in tumors with acquired resistance to EGFR inhibition [154–159]. Moreover, in vitro results demonstrated an additive effect of cetuximab plus a MET inhibitor in cetuximab-resistant lung cancer cell lines [160]. Similar results were observed in colorectal cancer cells [161]. Furthermore, the inhibitory effect of cetuximab could be compensated by overexpression of the MET ligand HGF in colorectal cancer cells [161]. In a HNSCC xenograft model, a delay in tumor growth was observed after administration of crizotinib, a MET TKI [150]. Collectively, these data suggest that high MET expression might play a role in cetuximab resistance.
Other Potential Mechanisms of Resistance
Not only alterations in proteins linked to the EGFR signaling pathway but also proteins involved in more general cancer characteristics, such as proliferation, apoptosis, invasion, and metastasis, might confer resistance to anti-EGFR therapeutics. Moreover, alterations in these genes/proteins may also lead to resistance to other anticancer agents.
Aurora Kinase
The Aurora kinases A and B are highly conserved serine/threonine kinases that play an essential and distinct role in mitosis [162, 163]. Overexpression of both kinases is frequently present in many types of malignant tumors, and in the case of HNSCC, overexpression of Aurora kinase A is found in up to 90% of tumors [163–165]. Overexpression of Aurora kinase A is correlated with tumor progression, a metastatic phenotype, and shortened survival, and is therefore regarded as a negative prognostic marker [162, 164, 165]. High expression levels of Aurora kinase B are found in glioblastoma, ovarian carcinoma, and hepatocellular carcinoma and are associated with poor prognosis [166].
EGFR can elicit overexpression of Aurora kinase A at two different levels, by increasing the translational efficiency of Aurora kinase A, or by binding to the Aurora kinase A promoter and thereby increasing its transcription, both resulting in chromosome instability and tumorigenesis [61, 167].
Next to its role as a prognostic factor, recent studies indicated evidence for a role of Aurora kinase A in the response to therapy. Overexpression of Aurora kinase A triggered the activation of two important molecules involved in the regulation of drug resistance, Akt and NF-κB [168]. Interestingly, knockdown of Aurora kinase A in HeLa cells resulted in sensitization to cisplatin, and Aurora kinase A overexpression could overcome cell death induced by paclitaxel [168]. Furthermore, treatment of HNSCC cells with cetuximab and a pan-Aurora kinase inhibitor R763 resulted in a rapid and efficient decrease in the level of the Aurora kinase substrate S10HH3. These results could not be confirmed by using a specific Aurora kinase A inhibitor, and therefore it was concluded that the effects of the pan-Aurora kinase inhibitor were most likely mediated by its blockage of Aurora kinase B activity [162].
Collectively, these results indicate that the Aurora kinases may be an interesting target for HNSCC tumors resistant to anti-EGFR therapy.
Cyclin D1
The G1/S-specific cyclin D1 forms a complex with CDK4 and CDK6 and functions as a regulatory subunit of CDK4 and CDK6, the activity of which is required for cell cycle G1/S transition. As previously mentioned, nuclear EGFR functions as a transcription factor for cyclin D1. Moreover, constitutive activation of STAT3 is required for EGFR-mediated cell growth and results in elevated levels of STAT3 target genes, including cyclin D1 [128, 169]. These observations make cyclin D1 an interesting potential marker for predicting anti-EGFR therapy.
The cyclin D1 A870G polymorphism was evaluated in 58 advanced colorectal cancer patients treated with a combination therapy of cetuximab and irinotecan. This study identified the G allele as being associated with a shorter time to progression in wild-type K-Ras patients and a shorter overall survival in all patients [170]. On the basis of these results, the authors concluded that the cyclin D1 A870G polymorphism might be used as an additional marker for predicting cetuximab efficacy. In addition, cotargeting EGFR and cyclin D1 seemed promising in lung cancer [171, 172], as combination therapy with erlotinib and the cyclin-dependent kinase inhibitor seliciclib resulted in a synergistic effect in both in vitro and in vivo studies [173]. With use of HNSCC cell lines, gefitinib resistance was shown to be associated with deregulated cyclin D1 overexpression [174]. However, more studies are warranted to reveal the exact role of cyclin D1 in anti-EGFR therapy resistance.
p53
The tumor suppressor protein p53 has a critical role in controlling cell cycle progression, and consequently, loss of its function is linked to the carcinogenic process. In response to a variety of cellular stimuli, p53 can induce cell cycle arrest, apoptosis, or senescence.
A study investigating the difference between cetuximab-resistant and their sensitive parental lung cancer cells identified p53 as the most downregulated and pERK1/2 as the most upregulated cellular signaling protein. Downregulation of p53 was also observed in erlotinib-resistant cells. Furthermore, silencing of p53 in cetuximab-sensitive cells resulted in reduced sensitivity to the drug, whereas restoring p53 function in resistant cells resulted in enhanced cetuximab sensitivity [175]. In vivo experiments, using a stable cetuximab-resistant clone with tetracycline-inducible p53, showed that repair of p53 restored cetuximab sensitivity in tumor xenografts resistant to cetuximab [175]. Another study, investigating the role of p53 in the response to gefitinib, suggested that, in human lung cancer A549 cells, gefitinib-induced apoptosis was at least partly mediated by phosphorylation of p53 at Ser15, resulting in activation of p53 [176]. A study investigating the p53 mutation status of 31 colorectal cancer patients observed that loss of functional p53 limited the response to gefitinib and chemotherapy, particularly in tumors with intact p21 [177]. In addition, cetuximab was able to inhibit cell growth in p53 wild-type cells, but not in p53-mutated cells [178]. In general, there is insufficient experimental evidence to unequivocally state that loss of functional p53 can be predictive of resistance to anti-EGFR therapy.
Epithelial-to-Mesenchymal Transition
Epithelial-to-mesenchymal transition (EMT) is characterized by loss of epithelial cell characteristics and acquisition of mesenchymal phenotypic traits, causing tumor cells to detach from neighboring cells and to migrate into adjacent tissue [179–181].
TGF-β is, together with the Ras pathway, a potent inducer of EMT [180]. Haddad et al. showed that erlotinib-resistant cell lines exhibited a greater migratory capacity and invasive potential compared with erlotinib-sensitive cell lines, and response to erlotinib was correlated with an “epithelial” molecular phenotype [182]. Furthermore, Skvortsova et al. suggested that c-myc, E-cadherin, and vimentin might be considered to be predictive biomarkers for HNSCC patients treated with cetuximab in combination with radiotherapy [183]. Moreover, upregulation of vimentin and downregulation of the EMT markers E-cadherin, claudin 4, and claudin 7 were associated with gefitinib resistance in HNSCC and NSCLC cell lines [184]. Similar results were found in other studies investigating the effect of erlotinib in NSCLC [185, 186]. In addition, pancreatic and colorectal tumor cell lines, resistant to EGFR inhibition, showed features of EMT [187]. HNSCC cells with a mesenchymal-like morphology and elevated migratory potential were found to be less sensitive to irradiation and cetuximab [188]. However, when these treatment modalities were combined, the authors observed an increased sensitivity [188].
Hypoxia and Hypoxia-Inducible Factors
Regions within solid tumors often experience mild to severe oxygen deprivation (hypoxia) and it has been well-documented that poor oxygenation is a pathophysiological property of the majority of human solid tumors, including HNSCC [189]. Importantly, oxygen deficiency has a major impact on clinical responses to cancer treatment, and it was shown that hypoxic tumor regions often contain viable cells that are intrinsically more resistant to treatment with radiotherapy and/or chemotherapy [190, 191]. Interestingly, preclinical and clinical studies support an important link between hypoxia and upregulation of EGFR in cancers that do not display genetic alteration of the receptor [192]. For example, in a fraction of aggressive human HNSCC, immunohistochemistry data showed that hypoxia induced EGFR activation [193]. These results were supported by a recent study, reporting colocalization of EGFR and the hypoxia marker pimonidazole in patients with HNSCC, predominantly at increasing distances from blood vessels [194]. Subsequent EGFR signaling stimulates hypoxia-inducible factor (HIF) signaling. As the HIF transcription factors play a pivotal role in the cellular adaptation to hypoxic stress, EGFR-induced HIF signaling thus augments the induction of proteins that promote cellular survival in a hostile microenvironment. Consequently, the presence of tumor hypoxia may contribute to resistance to EGFR inhibitors. HNSCC patients with high levels of hypoxia-associated factors indeed were more likely to relapse, following induction therapy that included cetuximab [195].
Interestingly, recent data suggest that the lack of clinical responses to EGFR-directed therapy may be circumvented by supplementation of the anti-EGFR therapy with additional approaches targeting HIF. For example, downregulation of HIF-1 by siRNA or a small molecule inhibitor enhanced responses of cetuximab-resistant HNSCC cells to cetuximab plus radiation [196]. Findings from such studies may provide important guidance for designing novel therapeutic strategies in which EGFR inhibitors are combined with approaches targeting tumor hypoxia to enhance the tumor response.
Clinical Studies Targeting EGFR Resistance Networks in HNSCC
Overcoming mechanisms of intrinsic and acquired resistance to current ErbB-targeted therapies is a critical area of investigation. In general, more preclinical in vitro and in vivo studies, evaluating the role of the above-described genes/proteins, are needed to associate one or more of these mechanisms to sensitivity or resistance to anti-EGFR therapy. As such, specific inhibitors of these signaling cascades, showing promising results in preclinical studies, can be evaluated in clinical trials with HNSCC patients.
First, both monoclonal antibodies and tyrosine kinase inhibitors, targeting IGF-1R, have entered the clinic. The efficacy and toxicity of the anti-IGF-1R antibody figitumumab was evaluated in 17 palliative HNSCC patients. No significant clinical activity was detected using figitumumab as a single agent in unselected palliative HNSCC patients. In contrast, even an acceleration in progression seemed to occur. Moreover, main relevant grade 3–4 toxicities (hyperglycemia, asthenia, anorexia, infection, anemia, and gastrointestinal bleeding) were observed. Contrary to a downregulation of IGF-1R, the EGFR pathway was found activated, leading to upregulation of pEGFR and an increase in the plasma level of TGF-α [197]. Similarly, no antitumor activity of the anti-IGF-1R antibody A12 as single agent was observed in colorectal cancer patients. Additionally, the combination of cetuximab and A12 was investigated in a randomized phase II study and did not show a significant benefit in colorectal cancer patients refractory to EGFR inhibitors [198]. Currently, this combination is evaluated in a phase II clinical trial in HNSCC patients (ClinicalTrials.gov identifier: NCT00957853). In contrast, treatment of cetuximab-refractory HNSCC patients with the IGF-1R inhibitor AMG-479 did result in conversion of a gene-expression profile associated with cetuximab resistance to a profile associated with cetuximab sensitivity [199].
A recently started phase II study will assess the progression-free survival of HNSCC patients, treated with cetuximab plus the dual kinase inhibitor of both IGF-1R and insulin receptor, OSI-906, and this treatment schedule will be compared with cetuximab plus placebo (ClinicalTrials.gov identifier: NCT01427205).
Second, another promising molecular target in HNSCC treatment is the family of Src kinases. The Src inhibitor dasatinib was evaluated in a phase II trial in advanced HNSCC patients. Despite Src inhibition, no significant clinical activity was seen in single-agent therapy [200]. Similar results were obtained with single-agent saracatinib, another Src inhibitor, in a phase II study of patients with recurrent or metastatic HNSCC [201]. At the moment, clinical trials are ongoing with a combination therapy of dasatinib and EGFR inhibitors. A phase II study will evaluate the objective response rate with this combination in recurrent HNSCC patients who have recurred after cetuximab-containing therapy (ClinicalTrials.gov identifier: NCT01488318). Furthermore, activity and toxicity of dasatinib will be assessed when given in combination with cetuximab and radiotherapy or in combination with cetuximab and cisplatin and radiotherapy (ClinicalTrials.gov identifier: NCT00882583).
Third, human carcinomas frequently express one or more members of the ErbB family, and therefore, cross-activation of EGFR downstream signaling might be present. Agents that block other members of the ErbB family simultaneously or that bind multiple ErbB receptors irreversibly are promising and are currently investigated in clinical trials for HNSCC patients. The efficacy and safety of afatinib, an aniline-quinazoline compound binding irreversibly to EGFR and HER2, is being assessed in different phase II and phase III trials. The objective of one of these trials is to explore different molecular pathways to identify tumor response and resistance mechanisms (ClinicalTrials.gov identifier: NCT01538381). Additionally, a phase II trial (ClinicalTrials.gov identifier: NCT00514943) has explored the efficacy of afatinib versus cetuximab in patients with metastatic or recurrent HNSCC and concluded that afatinib has at least comparable antitumor activity to cetuximab in HNSCC that failed on platinum-based therapy. Moreover, disease control rates were particularly notable for afatinib in stage 2 (crossover between treatment arms after disease progression or toxicity), 38.9% (afatinib as second treatment) versus 18.8% (cetuximab second) by investigator review, and 33.3% (afatinib as second treatment) versus 18.8% (cetuximab second) by independent central review. Patients on afatinib had more diarrhea and less rash/acne than those on the cetuximab arm.
Lapatinib, another dual inhibitor of EGFR and HER2, has been investigated in HNSCC also. In a phase II study, lapatinib in monotherapy did not result in complete or partial responses, but only in stable disease [202]. However, in a randomized phase II study, the activity and safety of concurrent chemoradiotherapy and lapatinib followed by maintenance treatment in locally advanced unresected advanced HNSCC was assessed. This combination proved to be well-tolerated with a numeric increase in complete response at 6 months post-CRT and median progression-free survival in p16-negative disease [203]. However, very recently, the combination of lapatinib with full dose docetaxel, cisplatin, and 5-fluorouracil showed unexpected renal toxicity and needs further evaluation [204]. The combination of lapatinib with cetuximab is under evaluation in an ongoing phase I trial to determine the maximum dosages patients can tolerate when these two drugs are given at the same time (ClinicalTrials.gov identifier: NCT01184482).
Finally, although the STAT family of proteins has attracted interest as therapeutic targets in HNSCC, no STAT inhibitors have been tested in the clinic yet. Especially the combination of STAT3 inhibitors with EGFR inhibitors might be an attractive therapeutic strategy. Currently, four phase I clinical trials are ongoing in patients with advanced cancers to evaluate the safety profile and recommended dose for use in subsequent studies and to investigate the pharmacokinetics and antitumor effect of the compound (ClinicalTrials.gov identifier: NCT01184807, NCT00657176, NCT01423903, and NCT01563302).
Conclusion
Most HNSCC tumors rely on activation of the EGFR pathway for their proliferation, differentiation, antiapoptotic signaling, angiogenesis, and metastasis. Therefore, targeted therapies inhibiting this pathway are promising. However, intrinsic and acquired resistance to anti-EGFR therapies is a serious problem in the treatment of HNSCC. Although in other cancer types certain mechanisms of resistance have already been identified, it is becoming clear that what is relevant in one cancer type may not necessarily apply to other forms of cancer. In HNSCC cancer, mechanisms of resistance at the level of EGFR or its downstream effectors are potential causes, but also activation of alternative parallel signaling pathways might result in resistance to anti-EGFR therapeutics. Figure 3 gives an overview of all described mechanisms of resistance in this review.
Figure 3.

Possible mechanisms of resistance to EGFR-targeted therapy in HNSCC described in this review: (1) overexpression of ligands, (2) activating mutations in EGFR, (3) translocation of EGFR to the nucleus, (4) loss of PTPRS, (5) activating Ras mutations, (6) downregulation of DUSP, (7) activating mutations in PIK3CA or inactivating mutations in PTEN, (8) activation of Src kinases, (9) dysregulation of the STAT pathway, (10) heterodimerization with other ErbB family members, (11) parallel signaling of other receptors, IGF-1R or MET, (12) overexpression of the Aurora kinase A and B, (13) deregulation of cyclin D1 or the A870G polymorphism, (14) loss of functional p53, and (15) activation of HIF-1 signaling.
Abbreviations: EGFR, epidermal growth factor receptor; HNSCC, head and neck squamous cell carcinoma; STAT, signal transducer and activator of transcription.
In addition, inhibiting the EGFR pathway at multiple steps or in parallel signaling pathways might overcome intrinsic and/or acquired resistance. The findings discussed illustrate the complexity of the EGFR cascade. Further unraveling of this cascade will undoubtedly reveal markers for anti-EGFR therapy resistance and combinations of targeted therapies able to overcome this resistance.
Acknowledgments
A.W. holds a postdoctoral fellowship from the FWO (Fonds voor Wetenschappelijk Onderzoek), Vlaanderen.
Author Contributions
Conception and design: Carolien Boeckx, Marc Baay, Jan B. Vermorken
Manuscript Writing: Carolien Boeckx, An Wouters
Final Approval of Manuscript: Marc Baay, Pol Specenier, Jan B. Vermorken, Marc Peeters, Filip Lardon
Disclosures
Jan B. Vermorken: Merck-Serono (C/A); Merck-Serono, Bristol-Myers Squibb (H); Merck-Serono, Eli Lilly, BMS (travel compensation and honorarium for lecturing); Marc Peeters: Amgen/Merck-Serono (C/A); Amgen/Merck-Serono (RF); Amgen/Merck-Serono (H). The other authors indicated no financial relationships.
(C/A) Consulting/advisory relationship; (RF) Research funding; (E) Employment; (H) Honoraria received; (OI) Ownership interests; (IP) Intellectual property rights/inventor/patent holder; (SAB) Scientific advisory board
Reference
- 1.Leemans CR, Braakhuis BJ, Brakenhoff RH. The molecular biology of head and neck cancer. Nat Rev Cancer. 2011;11:9–22. doi: 10.1038/nrc2982. [DOI] [PubMed] [Google Scholar]
- 2.Molinolo AA, Amornphimoltham P, Squarize CH, et al. Dysregulated molecular networks in head and neck carcinogenesis. Oral Oncol. 2009;45:324–334. doi: 10.1016/j.oraloncology.2008.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boehm AL, Sen M, Seethala R, et al. Combined targeting of epidermal growth factor receptor, signal transducer and activator of transcription-3, and bcl-X (L) enhances antitumor effects in squamous cell carcinoma of the head and neck. Mol Pharmacol. 2008;73:1632–1642. doi: 10.1124/mol.107.044636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schilsky RL. Personalized medicine in oncology: The future is now. Nat Rev Drug Discov. 2010;9:363–366. doi: 10.1038/nrd3181. [DOI] [PubMed] [Google Scholar]
- 5.Kelloff GJ, Sigman CC. Cancer biomarkers: Selecting the right drug for the right patient. Nat Rev Drug Discov. 2012;11:201–214. doi: 10.1038/nrd3651. [DOI] [PubMed] [Google Scholar]
- 6.Sawyers CL. The cancer biomarker problem. Nature. 2008;452:548–552. doi: 10.1038/nature06913. [DOI] [PubMed] [Google Scholar]
- 7.Schaaij-Visser TB, Brakenhoff RH, Leemans CR, et al. Protein biomarker discovery for head and neck cancer. J Proteomics. 2010;73:1790–1803. doi: 10.1016/j.jprot.2010.01.013. [DOI] [PubMed] [Google Scholar]
- 8.Chang SS, Califano J. Current status of biomarkers in head and neck cancer. J Surg Oncol. 2008;97:640–643. doi: 10.1002/jso.21023. [DOI] [PubMed] [Google Scholar]
- 9.Ferreira MB, De Souza JA, Cohen EE. Role of molecular markers in the management of head and neck cancers. Curr Opin Oncol. 2011;23:259–264. doi: 10.1097/CCO.0b013e328344f53a. [DOI] [PubMed] [Google Scholar]
- 10.van der Schroeff MP, Baatenburg de Jong RJ. Staging and prognosis in head and neck cancer. Oral Oncol. 2009;45:356–360. doi: 10.1016/j.oraloncology.2008.05.022. [DOI] [PubMed] [Google Scholar]
- 11.Glazer CA, Chang SS, Ha PK, et al. Applying the molecular biology and epigenetics of head and neck cancer in everyday clinical practice. Oral Oncol. 2009;45:440–446. doi: 10.1016/j.oraloncology.2008.05.013. [DOI] [PubMed] [Google Scholar]
- 12.Normanno N, De Luca A, Bianco C, et al. Epidermal growth factor receptor (EGFR) signaling in cancer. Gene. 2006;366:2–16. doi: 10.1016/j.gene.2005.10.018. [DOI] [PubMed] [Google Scholar]
- 13.Psyrri A, Yu Z, Weinberger PM, et al. Quantitative determination of nuclear and cytoplasmic epidermal growth factor receptor expression in oropharyngeal squamous cell cancer by using automated quantitative analysis. Clin Cancer Res. 2005;11:5856–5862. doi: 10.1158/1078-0432.CCR-05-0420. [DOI] [PubMed] [Google Scholar]
- 14.Ang KK, Berkey BA, Tu X, et al. Impact of epidermal growth factor receptor expression on survival and pattern of relapse in patients with advanced head and neck carcinoma. Cancer Res. 2002;62:7350–7356. [PubMed] [Google Scholar]
- 15.Schneider-Merck T, Lammerts van Bueren JJ, Berger S, et al. Human IgG2 antibodies against epidermal growth factor receptor effectively trigger antibody-dependent cellular cytotoxicity but, in contrast to IgG1, only by cells of myeloid lineage. J Immunol. 2010;184:512–520. doi: 10.4049/jimmunol.0900847. [DOI] [PubMed] [Google Scholar]
- 16.Lee SC, Lopez-Albaitero A, Ferris RL. Immunotherapy of head and neck cancer using tumor antigen-specific monoclonal antibodies. Curr Oncol Rep. 2009;11:156–162. doi: 10.1007/s11912-009-0023-5. [DOI] [PubMed] [Google Scholar]
- 17.Lurje G, Lenz HJ. Egfr signaling and drug discovery. Oncology. 2009;77:400–410. doi: 10.1159/000279388. [DOI] [PubMed] [Google Scholar]
- 18.Bonner JA, Harari PM, Giralt J, et al. Radiotherapy plus cetuximab for squamous-cell carcinoma of the head and neck. N Engl J Med. 2006;354:567–578. doi: 10.1056/NEJMoa053422. [DOI] [PubMed] [Google Scholar]
- 19.Vermorken JB, Trigo J, Hitt R, et al. Open-label, uncontrolled, multicenter phase II study to evaluate the efficacy and toxicity of cetuximab as a single agent in patients with recurrent and/or metastatic squamous cell carcinoma of the head and neck who failed to respond to platinum-based therapy. J Clin Oncol. 2007;25:2171–2177. doi: 10.1200/JCO.2006.06.7447. [DOI] [PubMed] [Google Scholar]
- 20.Vermorken JB, Mesia R, Rivera F, et al. Platinum-based chemotherapy plus cetuximab in head and neck cancer. N Engl J Med. 2008;359:1116–1127. doi: 10.1056/NEJMoa0802656. [DOI] [PubMed] [Google Scholar]
- 21.Vermorken JB, Herbst RS, Leon X, et al. Overview of the efficacy of cetuximab in recurrent and/or metastatic squamous cell carcinoma of the head and neck in patients who previously failed platinum-based therapies. Cancer. 2008;112:2710–2719. doi: 10.1002/cncr.23442. [DOI] [PubMed] [Google Scholar]
- 22.Burtness B, Goldwasser MA, Flood W, et al. Phase III randomized trial of cisplatin plus placebo compared with cisplatin plus cetuximab in metastatic/recurrent head and neck cancer: An eastern cooperative oncology group study. J Clin Oncol. 2005;23:8646–8654. doi: 10.1200/JCO.2005.02.4646. [DOI] [PubMed] [Google Scholar]
- 23.Dittmann K, Mayer C, Rodemann HP. Nuclear EGFR as novel therapeutic target: Insights into nuclear translocation and function. Strahlenther Onkol. 2010;186:1–6. doi: 10.1007/s00066-009-2026-4. [DOI] [PubMed] [Google Scholar]
- 24.Liccardi G, Hartley JA, Hochhauser D. EGFR nuclear translocation modulates DNA repair following cisplatin and ionizing radiation treatment. Cancer Res. 2011;71:1103–1114. doi: 10.1158/0008-5472.CAN-10-2384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hatakeyama H, Cheng H, Wirth P, et al. Regulation of heparin-binding EGF-like growth factor by miR-212 and acquired cetuximab-resistance in head and neck squamous cell carcinoma. PLoS ONE. 2010;5:e12702. doi: 10.1371/journal.pone.0012702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jijon HB, Buret A, Hirota CL, et al. The EGF receptor and HER2 participate in TNF-α-dependent mapk activation and IL-8 secretion in intestinal epithelial cells. Mediat Inflamm. 2012;2012:207398. doi: 10.1155/2012/207398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bedi A, Chang X, Noonan K, et al. Inhibition of TGF-β enhances the in vivo antitumor efficacy of EGF receptor-targeted therapy. Mol Cancer Ther. 2012;11:2429–2439. doi: 10.1158/1535-7163.MCT-12-0101-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Khambata-Ford S, Garrett CR, Meropol NJ, et al. Expression of epiregulin and amphiregulin and K-ras mutation status predict disease control in metastatic colorectal cancer patients treated with cetuximab. J Clin Oncol. 2007;25:3230–3237. doi: 10.1200/JCO.2006.10.5437. [DOI] [PubMed] [Google Scholar]
- 29.Tinhofer I, Klinghammer K, Weichert W, et al. Expression of amphiregulin and EGFRvIII affect outcome of patients with squamous cell carcinoma of the head and neck receiving cetuximab-docetaxel treatment. Clin Cancer Res. 2011;17:5197–5204. doi: 10.1158/1078-0432.CCR-10-3338. [DOI] [PubMed] [Google Scholar]
- 30.Licitra L, Mesia R, Rivera F, et al. Evaluation of EGFR gene copy number as a predictive biomarker for the efficacy of cetuximab in combination with chemotherapy in the first-line treatment of recurrent and/or metastatic squamous cell carcinoma of the head and neck: Extreme study. Ann Oncol. 2011;22:1078–1087. doi: 10.1093/annonc/mdq588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rivera F, Garcia-Castano A, Vega N, et al. Cetuximab in metastatic or recurrent head and neck cancer: The extreme trial. Expert Rev Anticancer Ther. 2009;9:1421–1428. doi: 10.1586/era.09.113. [DOI] [PubMed] [Google Scholar]
- 32.Laurent-Puig P, Lievre A, Blons H. Mutations and response to epidermal growth factor receptor inhibitors. Clin Cancer Res. 2009;15:1133–1139. doi: 10.1158/1078-0432.CCR-08-0905. [DOI] [PubMed] [Google Scholar]
- 33.Van Damme N, Deron P, Van Roy N, et al. Epidermal growth factor receptor and K-ras status in two cohorts of squamous cell carcinomas. BMC Cancer. 2010;10:189. doi: 10.1186/1471-2407-10-189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Willmore-Payne C, Holden JA, Layfield LJ. Detection of EGFR- and HER2-activating mutations in squamous cell carcinoma involving the head and neck. Mod Pathol. 2006;19:634–640. doi: 10.1038/modpathol.3800552. [DOI] [PubMed] [Google Scholar]
- 35.Loeffler-Ragg J, Witsch-Baumgartner M, Tzankov A, et al. Low incidence of mutations in EGFR kinase domain in caucasian patients with head and neck squamous cell carcinoma. Eur J Cancer. 2006;42:109–111. doi: 10.1016/j.ejca.2005.08.034. [DOI] [PubMed] [Google Scholar]
- 36.Murray S, Bobos M, Angouridakis N, et al. Screening for EGFR mutations in patients with head and neck cancer treated with gefitinib on a compassionate-use program: A hellenic cooperative oncology group study. J Oncol. 2010;2010:709678. doi: 10.1155/2010/709678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hama T, Yuza Y, Saito Y, et al. Prognostic significance of epidermal growth factor receptor phosphorylation and mutation in head and neck squamous cell carcinoma. The Oncologist. 2009;14:900–908. doi: 10.1634/theoncologist.2009-0058. [DOI] [PubMed] [Google Scholar]
- 38.Hama T, Yuza Y, Suda T, et al. Functional mutation analysis of EGFR family genes and corresponding lymph node metastases in head and neck squamous cell carcinoma. Clin Exp Metastasis. 2012;29:19–25. doi: 10.1007/s10585-011-9425-5. [DOI] [PubMed] [Google Scholar]
- 39.Szabo B, Nelhubel GA, Karpati A, et al. Clinical significance of genetic alterations and expression of epidermal growth factor receptor (EGFR) in head and neck squamous cell carcinomas. Oral Oncol. 2011;47:487–496. doi: 10.1016/j.oraloncology.2011.03.020. [DOI] [PubMed] [Google Scholar]
- 40.Cohen EE, Lingen MW, Martin LE, et al. Response of some head and neck cancers to epidermal growth factor receptor tyrosine kinase inhibitors may be linked to mutation of ERBB2 rather than EGFR. Clin Cancer Res. 2005;11:8105–8108. doi: 10.1158/1078-0432.CCR-05-0926. [DOI] [PubMed] [Google Scholar]
- 41.Chung CH, Ely K, McGavran L, et al. Increased epidermal growth factor receptor gene copy number is associated with poor prognosis in head and neck squamous cell carcinomas. J Clin Oncol. 2006;24:4170–4176. doi: 10.1200/JCO.2006.07.2587. [DOI] [PubMed] [Google Scholar]
- 42.Lee JW, Soung YH, Kim SY, et al. Somatic mutations of EGFR gene in squamous cell carcinoma of the head and neck. Clin Cancer Res. 2005;11:2879–2882. doi: 10.1158/1078-0432.CCR-04-2029. [DOI] [PubMed] [Google Scholar]
- 43.Lemos-Gonzalez Y, Paez de la Cadena M, Rodriguez-Berrocal FJ, et al. Absence of activating mutations in the EGFR kinase domain in spanish head and neck cancer patients. Tumour Biol. 2007;28:273–279. doi: 10.1159/000110425. [DOI] [PubMed] [Google Scholar]
- 44.Huang SF, Chuang WY, Chen IH, et al. EGFR protein overexpression and mutation in areca quid-associated oral cavity squamous cell carcinoma in taiwan. Head Neck. 2009;31:1068–1077. doi: 10.1002/hed.21067. [DOI] [PubMed] [Google Scholar]
- 45.Schwentner I, Witsch-Baumgartner M, Sprinzl GM, et al. Identification of the rare EGFR mutation p.G796S as somatic and germline mutation in white patients with squamous cell carcinoma of the head and neck. Head Neck. 2008;30:1040–1044. doi: 10.1002/hed.20831. [DOI] [PubMed] [Google Scholar]
- 46.Sheikh Ali MA, Gunduz M, Nagatsuka H, et al. Expression and mutation analysis of epidermal growth factor receptor in head and neck squamous cell carcinoma. Cancer Sci. 2008;99:1589–1594. doi: 10.1111/j.1349-7006.2008.00861.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hsieh CH, Chang JW, Hsieh JJ, et al. Epidermal growth factor receptor mutations in patients with oral cavity cancer in a betel nut chewing-prevalent area. Head Neck. 2011;33:1758–1764. doi: 10.1002/hed.21665. [DOI] [PubMed] [Google Scholar]
- 48.Na II, Kang HJ, Cho SY, et al. EGFR mutations and human papillomavirus in squamous cell carcinoma of tongue and tonsil. Eur J Cancer. 2007;43:520–526. doi: 10.1016/j.ejca.2006.09.025. [DOI] [PubMed] [Google Scholar]
- 49.Uribe P, Gonzalez S. Epidermal growth factor receptor (EGFR) and squamous cell carcinoma of the skin: Molecular bases for EGFR-targeted therapy. Pathol Res Pract. 2011;207:337–342. doi: 10.1016/j.prp.2011.03.002. [DOI] [PubMed] [Google Scholar]
- 50.Wheeler SE, Suzuki S, Thomas SM, et al. Epidermal growth factor receptor variant III mediates head and neck cancer cell invasion via STAT3 activation. Oncogene. 2010;29:5135–5145. doi: 10.1038/onc.2009.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sok JC, Coppelli FM, Thomas SM, et al. Mutant epidermal growth factor receptor (EGFRvIII) contributes to head and neck cancer growth and resistance to EGFR targeting. Clin Cancer Res. 2006;12:5064–5073. doi: 10.1158/1078-0432.CCR-06-0913. [DOI] [PubMed] [Google Scholar]
- 52.Patel D, Lahiji A, Patel S, et al. Monoclonal antibody cetuximab binds to and down-regulates constitutively activated epidermal growth factor receptor vIII on the cell surface. Anticancer Res. 2007;27:3355–3366. [PubMed] [Google Scholar]
- 53.Fukai J, Nishio K, Itakura T, et al. Antitumor activity of cetuximab against malignant glioma cells overexpressing EGFR deletion mutant variant III. Cancer Sci. 2008;99:2062–2069. doi: 10.1111/j.1349-7006.2008.00945.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dreier A, Barth S, Goswami A, et al. Cetuximab induces mitochondrial translocalization of EGFRvIII, but not EGFR: Involvement of mitochondria in tumor drug resistance? Tumour Biol. 2012;33:85–94. doi: 10.1007/s13277-011-0248-4. [DOI] [PubMed] [Google Scholar]
- 55.Chau NG, Perez-Ordonez B, Zhang K, et al. The association between EGFR variant III, HPV, p16, c-MET, EGFR gene copy number and response to EGFR inhibitors in patients with recurrent or metastatic squamous cell carcinoma of the head and neck. Head Neck Oncol. 2011;3:11. doi: 10.1186/1758-3284-3-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pectasides E, Fountzilas G, Kountourakis P, et al. Evalutaion of the incidence and prognostic value of mutant epidermal growth factor receptor (EGFRvIII) protein expression in head and neck squamous cell carcinomas (HNSCC) using AQUA. Paper presented at: ASCO Annual Meeting; June 4–8, 2010; Chicago. [Google Scholar]
- 57.McIntyre JB, Bose P, Klimowicz AC, et al. Specific and sensitive hydrolysis probe-based real-time PCR detection of epidermal growth factor receptor variant III in oral squamous cell carcinoma. PLoS ONE. 2012;7:e31723. doi: 10.1371/journal.pone.0031723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang SC, Nakajima Y, Yu YL, et al. Tyrosine phosphorylation controls PCNA function through protein stability. Nat Cell Biol. 2006;8:1359–1368. doi: 10.1038/ncb1501. [DOI] [PubMed] [Google Scholar]
- 59.Lin SY, Makino K, Xia W, et al. Nuclear localization of EGF receptor and its potential new role as a transcription factor. Nat Cell Biol. 2001;3:802–808. doi: 10.1038/ncb0901-802. [DOI] [PubMed] [Google Scholar]
- 60.Hanada N, Lo HW, Day CP, et al. Co-regulation of B-Myb expression by E2F1 and EGF receptor. Mol Carcinog. 2006;45:10–17. doi: 10.1002/mc.20147. [DOI] [PubMed] [Google Scholar]
- 61.Hung LY, Tseng JT, Lee YC, et al. Nuclear epidermal growth factor receptor (EGFR) interacts with signal transducer and activator of transcription 5 (STAT5) in activating Aurora-A gene expression. Nucleic Acids Res. 2008;36:4337–4351. doi: 10.1093/nar/gkn417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lo HW, Hsu SC, Ali-Seyed M, et al. Nuclear interaction of EGFR and STAT3 in the activation of the iNOS/NO pathway. Cancer Cell. 2005;7:575–589. doi: 10.1016/j.ccr.2005.05.007. [DOI] [PubMed] [Google Scholar]
- 63.Lo HW, Xia W, Wei Y, et al. Novel prognostic value of nuclear epidermal growth factor receptor in breast cancer. Cancer Res. 2005;65:338–348. [PubMed] [Google Scholar]
- 64.Hoshino M, Fukui H, Ono Y, et al. Nuclear expression of phosphorylated EGFR is associated with poor prognosis of patients with esophageal squamous cell carcinoma. Pathobiology. 2007;74:15–21. doi: 10.1159/000101047. [DOI] [PubMed] [Google Scholar]
- 65.Li C, Iida M, Dunn EF, et al. Nuclear EGFR contributes to acquired resistance to cetuximab. Oncogene. 2009;28:3801–3813. doi: 10.1038/onc.2009.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kwak EL, Sordella R, Bell DW, et al. Irreversible inhibitors of the EGF receptor may circumvent acquired resistance to gefitinib. Proc Natl Acad Sci USA. 2005;102:7665–7670. doi: 10.1073/pnas.0502860102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nishimura Y, Yoshioka K, Bereczky B, et al. Evidence for efficient phosphorylation of EGFR and rapid endocytosis of phosphorylated EGFR via the early/late endocytic pathway in a gefitinib-sensitive non-small cell lung cancer cell line. Mol Cancer. 2008;7:42. doi: 10.1186/1476-4598-7-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Huang WC, Chen YJ, Li LY, et al. Nuclear translocation of epidermal growth factor receptor by Akt-dependent phosphorylation enhances breast cancer-resistant protein expression in gefitinib-resistant cells. J Biol Chem. 2011;286:20558–20568. doi: 10.1074/jbc.M111.240796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Suarez Pestana E, Tenev T, Gross S, et al. The transmembrane protein tyrosine phosphatase RPTPσ modulates signaling of the epidermal growth factor receptor in A431 cells. Oncogene. 1999;18:4069–4079. doi: 10.1038/sj.onc.1202794. [DOI] [PubMed] [Google Scholar]
- 70.Vijayvargia R, Kaur S, Krishnasastry MV. α-Hemolysin-induced dephosphorylation of EGF receptor of A431 cells is carried out by RPTPσ. Biochem Biophys Res Commun. 2004;325:344–352. doi: 10.1016/j.bbrc.2004.10.038. [DOI] [PubMed] [Google Scholar]
- 71.Morris LG, Taylor BS, Bivona TG, et al. Genomic dissection of the epidermal growth factor receptor (EGFR)/PI3K pathway reveals frequent deletion of the EGFR phosphatase PTPRS in head and neck cancers. Proc Natl Acad Sci USA. 2011;108:19024–19029. doi: 10.1073/pnas.1111963108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–954. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- 73.Bos JL. ras Oncogenes in human cancer: A review. Cancer Res. 1989;49:4682–4689. [PubMed] [Google Scholar]
- 74.Lievre A, Bachet JB, Le Corre D, et al. KRAS mutation status is predictive of response to cetuximab therapy in colorectal cancer. Cancer Res. 2006;66:3992–3995. doi: 10.1158/0008-5472.CAN-06-0191. [DOI] [PubMed] [Google Scholar]
- 75.Lievre A, Bachet JB, Boige V, et al. KRAS mutations as an independent prognostic factor in patients with advanced colorectal cancer treated with cetuximab. J Clin Oncol. 2008;26:374–379. doi: 10.1200/JCO.2007.12.5906. [DOI] [PubMed] [Google Scholar]
- 76.Amado RG, Wolf M, Peeters M, et al. Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J Clin Oncol. 2008;26:1626–1634. doi: 10.1200/JCO.2007.14.7116. [DOI] [PubMed] [Google Scholar]
- 77.De Roock W, Piessevaux H, De Schutter J, et al. KRAS wild-type state predicts survival and is associated to early radiological response in metastatic colorectal cancer treated with cetuximab. Ann Oncol. 2008;19:508–515. doi: 10.1093/annonc/mdm496. [DOI] [PubMed] [Google Scholar]
- 78.Deschoolmeester V, Boeckx C, Baay M, et al. KRAS mutation detection and prognostic potential in sporadic colorectal cancer using high-resolution melting analysis. Br J Cancer. 2010;103:1627–1636. doi: 10.1038/sj.bjc.6605959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Andreyev HJ, Norman AR, Cunningham D, et al. Kirsten ras mutations in patients with colorectal cancer: The multicenter “RASCAL” study. J Natl Cancer Inst. 1998;90:675–684. doi: 10.1093/jnci/90.9.675. [DOI] [PubMed] [Google Scholar]
- 80.Weber A, Langhanki L, Sommerer F, et al. Mutations of the BRAF gene in squamous cell carcinoma of the head and neck. Oncogene. 2003;22:4757–4759. doi: 10.1038/sj.onc.1206705. [DOI] [PubMed] [Google Scholar]
- 81.Kuo MY, Jeng JH, Chiang CP, et al. Mutations of Ki-ras oncogene codon 12 in betel quid chewing-related human oral squamous cell carcinoma in taiwan. J Oral Pathol Med. 1994;23:70–74. doi: 10.1111/j.1600-0714.1994.tb00259.x. [DOI] [PubMed] [Google Scholar]
- 82.Lopez F, Llorente JL, Oviedo CM, et al. Gene amplification and protein overexpression of EGFR and ERBB2 in sinonasal squamous cell carcinoma. Cancer. 2012;118:1818–1826. doi: 10.1002/cncr.26451. [DOI] [PubMed] [Google Scholar]
- 83.Ruiz-Godoy RL, Garcia-Cuellar CM, Herrera Gonzalez NE, et al. Mutational analysis of K-ras and Ras protein expression in larynx squamous cell carcinoma. J Exp Clin Cancer Res. 2006;25:73–78. [PubMed] [Google Scholar]
- 84.Anderson JA, Irish JC, Ngan BY. Prevalence of RAS oncogene mutation in head and neck carcinomas. J Otolaryngol. 1992;21:321–326. [PubMed] [Google Scholar]
- 85.Yarbrough WG, Shores C, Witsell DL, et al. ras Mutations and expression in head and neck squamous cell carcinomas. Laryngoscope. 1994;104:1337–1347. doi: 10.1288/00005537-199411000-00005. [DOI] [PubMed] [Google Scholar]
- 86.Rizos E, Sourvinos G, Arvanitis DA, et al. Low incidence of H-, K- and N-ras oncogene mutations in cytological specimens of laryngeal tumours. Oral Oncol. 1999;35:561–563. doi: 10.1016/s1368-8375(99)00032-9. [DOI] [PubMed] [Google Scholar]
- 87.Cohen Y, Goldenberg-Cohen N, Shalmon B, et al. Mutational analysis of PTEN/PIK3CA/AKT pathway in oral squamous cell carcinoma. Oral Oncol. 2011;47:946–950. doi: 10.1016/j.oraloncology.2011.07.013. [DOI] [PubMed] [Google Scholar]
- 88.Fujii S, Uryu H, Akashi K, et al. Clinical significance of KRAS gene mutation and epidermal growth factor receptor expression in japanese patients with squamous cell carcinoma of the larynx, oropharynx and hypopharynx. Int J Clin Oncol. 2012 doi: 10.1007/s10147-012-0402-z. doi: 10.1007/s10147-012-0402-z. [DOI] [PubMed] [Google Scholar]
- 89.Wang WY, Chien YC, Wong YK, et al. Effects of KRAS mutation and polymorphism on the risk and prognosis of oral squamous cell carcinoma. Head Neck. 2012;34:663–666. doi: 10.1002/hed.21792. [DOI] [PubMed] [Google Scholar]
- 90.Chin LJ, Ratner E, Leng S, et al. A SNP in a let-7 microRNA complementary site in the KRAS 3′ untranslated region increases non-small cell lung cancer risk. Cancer Res. 2008;68:8535–8540. doi: 10.1158/0008-5472.CAN-08-2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Nelson HH, Christensen BC, Plaza SL, et al. KRAS mutation, KRAS-LCS6 polymorphism, and non-small cell lung cancer. Lung Cancer. 2010;69:51–53. doi: 10.1016/j.lungcan.2009.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Christensen BC, Moyer BJ, Avissar M, et al. A let-7 microRNA-binding site polymorphism in the KRAS 3′UTR is associated with reduced survival in oral cancers. Carcinogenesis. 2009;30:1003–1007. doi: 10.1093/carcin/bgp099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Chang CJ, Hsu CC, Chang CH, et al. Let-7d functions as novel regulator of epithelial–mesenchymal transition and chemoresistant property in oral cancer. Oncol Rep. 2011;26:1003–1010. doi: 10.3892/or.2011.1360. [DOI] [PubMed] [Google Scholar]
- 94.Yu CC, Chen YW, Chiou GY, et al. MicroRNA let-7a represses chemoresistance and tumourigenicity in head and neck cancer via stem-like properties ablation. Oral Oncol. 2011;47:202–210. doi: 10.1016/j.oraloncology.2010.12.001. [DOI] [PubMed] [Google Scholar]
- 95.Graziano F, Canestrari E, Loupakis F, et al. Genetic modulation of the Let-7 microRNA binding to KRAS 3′-untranslated region and survival of metastatic colorectal cancer patients treated with salvage cetuximab-irinotecan. Pharmacogenomics J. 2010;10:458–464. doi: 10.1038/tpj.2010.9. [DOI] [PubMed] [Google Scholar]
- 96.Smits KM, Paranjape T, Nallur S, et al. A let-7 microRNA SNP in the KRAS 3′UTR is prognostic in early-stage colorectal cancer. Clin Cancer Res. 2011;17:7723–7731. doi: 10.1158/1078-0432.CCR-11-0990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kjersem JB, Ikdahl T, Guren T, et al. Let-7 miRNA-binding site polymorphism in the KRAS 3′UTR; colorectal cancer screening population prevalence and influence on clinical outcome in patients with metastatic colorectal cancer treated with 5-fluorouracil and oxaliplatin +/− cetuximab. BMC Cancer. 2012;12:534. doi: 10.1186/1471-2407-12-534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Sheng ZM, Barrois M, Klijanienko J, et al. Analysis of the c-Ha-ras-1 gene for deletion, mutation, amplification and expression in lymph node metastases of human head and neck carcinomas. Br J Cancer. 1990;62:398–404. doi: 10.1038/bjc.1990.306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Anderson JA, Irish JC, McLachlin CM, et al. H-ras oncogene mutation and human papillomavirus infection in oral carcinomas. Arch Otolaryngol Head Neck Surg. 1994;120:755–760. doi: 10.1001/archotol.1994.01880310059011. [DOI] [PubMed] [Google Scholar]
- 100.Rampias T, Giagini A, Florou K, et al. H-RAS and PIK3CA mutations and response to cetuximab in head and neck squamous cell carcinoma (HNSCC). Paper presented at: ASCO Annual Meeting; June 3–7, 2011; Chicago. [Google Scholar]
- 101.Rampias T, Giagini A, Matsuzaki H, et al. Genetic alterations in HRAS gene in relation to outcoume and response to cetuximab in head and neck squamous cell carcinoma. Paper presented at: ASCO Annual Meeting; June 1–5, 2012; Chicago. [Google Scholar]
- 102.Keyse SM. Protein phosphatases and the regulation of mitogen-activated protein kinase signalling. Curr Opin Cell Biol. 2000;12:186–192. doi: 10.1016/s0955-0674(99)00075-7. [DOI] [PubMed] [Google Scholar]
- 103.Gazel A, Nijhawan RI, Walsh R, et al. Transcriptional profiling defines the roles of ERK and p38 kinases in epidermal keratinocytes. J Cell Physiol. 2008;215:292–308. doi: 10.1002/jcp.21394. [DOI] [PubMed] [Google Scholar]
- 104.Arkell RS, Dickinson RJ, Squires M, et al. DUSP6/MKP-3 inactivates ERK1/2 but fails to bind and inactivate ERK5. Cell Signal. 2008;20:836–843. doi: 10.1016/j.cellsig.2007.12.014. [DOI] [PubMed] [Google Scholar]
- 105.Wong VC, Chen H, Ko JM, et al. Tumor suppressor dual-specificity phosphatase 6 (DUSP6) impairs cell invasion and epithelial-mesenchymal transition (EMT)-associated phenotype. Int J Cancer. 2012;130:83–95. doi: 10.1002/ijc.25970. [DOI] [PubMed] [Google Scholar]
- 106.Okudela K, Yazawa T, Woo T, et al. Down-regulation of DUSP6 expression in lung cancer: Its mechanism and potential role in carcinogenesis. Am J Pathol. 2009;175:867–881. doi: 10.2353/ajpath.2009.080489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Zhang Z, Kobayashi S, Borczuk AC, et al. Dual specificity phosphatase 6 (DUSP6) is an ETS-regulated negative feedback mediator of oncogenic ERK signaling in lung cancer cells. Carcinogenesis. 2010;31:577–586. doi: 10.1093/carcin/bgq020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Oliveras-Ferraros C, Vazquez-Martin A, Cufi S, et al. Stem cell property epithelial-to-mesenchymal transition is a core transcriptional network for predicting cetuximab (Erbitux) efficacy in KRAS wild-type tumor cells. J Cell Biochem. 2011;112:10–29. doi: 10.1002/jcb.22952. [DOI] [PubMed] [Google Scholar]
- 109.Ligresti G, Militello L, Steelman LS, et al. PIK3CA mutations in human solid tumors: Role in sensitivity to various therapeutic approaches. Cell Cycle. 2009;8:1352–1358. doi: 10.4161/cc.8.9.8255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Qiu W, Schonleben F, Li X, et al. Pik3ca mutations in head and neck squamous cell carcinoma. Clin Cancer Res. 2006;12:1441–1446. doi: 10.1158/1078-0432.CCR-05-2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Pedrero JM, Carracedo DG, Pinto CM, et al. Frequent genetic and biochemical alterations of the PI 3-K/AKT/PTEN pathway in head and neck squamous cell carcinoma. Int J Cancer. 2005;114:242–248. doi: 10.1002/ijc.20711. [DOI] [PubMed] [Google Scholar]
- 112.Rebucci M, Peixoto P, Dewitte A, et al. Mechanisms underlying resistance to cetuximab in the HNSCC cell line: Role of AKT inhibition in bypassing this resistance. Int J Oncol. 2011;38:189–200. [PubMed] [Google Scholar]
- 113.Yamatodani T, Ekblad L, Kjellen E, et al. Epidermal growth factor receptor status and persistent activation of Akt and p44/42 MAPK pathways correlate with the effect of cetuximab in head and neck and colon cancer cell lines. J Cancer Res Clin Oncol. 2009;135:395–402. doi: 10.1007/s00432-008-0475-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Mriouah J, Boura C, Pinel S, et al. Cellular response to cetuximab in PTEN-silenced head and neck squamous cell carcinoma cell line. Int J Oncol. 2010;37:1555–1563. doi: 10.3892/ijo_00000809. [DOI] [PubMed] [Google Scholar]
- 115.Kondo N, Tsukuda M, Taguchi T, et al. Gene status of head and neck squamous cell carcinoma cell lines and cetuximab-mediated biological activities. Cancer Sci. 2011;102:1717–1723. doi: 10.1111/j.1349-7006.2011.01999.x. [DOI] [PubMed] [Google Scholar]
- 116.Sen B, Saigal B, Parikh N, et al. Sustained Src inhibition results in signal transducer and activator of transcription 3 (STAT3) activation and cancer cell survival via altered Janus-activated kinase-STAT3 binding. Cancer Res. 2009;69:1958–1965. doi: 10.1158/0008-5472.CAN-08-2944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Xi S, Zhang Q, Dyer KF, et al. Src kinases mediate stat growth pathways in squamous cell carcinoma of the head and neck. J Biol Chem. 2003;278:31574–31583. doi: 10.1074/jbc.M303499200. [DOI] [PubMed] [Google Scholar]
- 118.Yang Z, Bagheri-Yarmand R, Wang RA, et al. The epidermal growth factor receptor tyrosine kinase inhibitor ZD1839 (Iressa) suppresses c-Src and Pak1 pathways and invasiveness of human cancer cells. Clin Cancer Res. 2004;10:658–667. doi: 10.1158/1078-0432.ccr-0382-03. [DOI] [PubMed] [Google Scholar]
- 119.van Oijen MG, Rijksen G, ten Broek FW, et al. Overexpression of c-Src in areas of hyperproliferation in head and neck cancer, premalignant lesions and benign mucosal disorders. J Oral Pathol Med. 1998;27:147–152. doi: 10.1111/j.1600-0714.1998.tb01931.x. [DOI] [PubMed] [Google Scholar]
- 120.Koppikar P, Choi SH, Egloff AM, et al. Combined inhibition of c-Src and epidermal growth factor receptor abrogates growth and invasion of head and neck squamous cell carcinoma. Clin Cancer Res. 2008;14:4284–4291. doi: 10.1158/1078-0432.CCR-07-5226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Ammer AG, Kelley LC, Hayes KE, et al. Saracatinib impairs head and neck squamous cell carcinoma invasion by disrupting invadopodia function. J Cancer Sci Ther. 2009;1:52–61. doi: 10.4172/1948-5956.1000009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Dittmann K, Mayer C, Kehlbach R, et al. Radiation-induced caveolin-1 associated EGFR internalization is linked with nuclear EGFR transport and activation of DNA-PK. Mol Cancer. 2008;7:69. doi: 10.1186/1476-4598-7-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Kloth MT, Laughlin KK, Biscardi JS, et al. STAT5b, a mediator of synergism between c-Src and the epidermal growth factor receptor. J Biol Chem. 2003;278:1671–1679. doi: 10.1074/jbc.M207289200. [DOI] [PubMed] [Google Scholar]
- 124.Li C, Iida M, Dunn EF, et al. Dasatinib blocks cetuximab- and radiation-induced nuclear translocation of the epidermal growth factor receptor in head and neck squamous cell carcinoma. Radiother Oncol. 2010;97:330–337. doi: 10.1016/j.radonc.2010.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Raju U, Riesterer O, Wang ZQ, et al. Dasatinib, a multi-kinase inhibitor increased radiation sensitivity by interfering with nuclear localization of epidermal growth factor receptor and by blocking DNA repair pathways. Radiother Oncol. 2012;105:241–249. doi: 10.1016/j.radonc.2012.08.010. [DOI] [PubMed] [Google Scholar]
- 126.Wheeler DL, Iida M, Kruser TJ, et al. Epidermal growth factor receptor cooperates with Src family kinases in acquired resistance to cetuximab. Cancer Biol Ther. 2009;8:696–703. doi: 10.4161/cbt.8.8.7903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Lee TL, Yeh J, Van Waes C, et al. Epigenetic modification of SOCS-1 differentially regulates STAT3 activation in response to interleukin-6 receptor and epidermal growth factor receptor signaling through JAK and/or MEK in head and neck squamous cell carcinomas. Mol Cancer Ther. 2006;5:8–19. doi: 10.1158/1535-7163.MCT-05-0069. [DOI] [PubMed] [Google Scholar]
- 128.Kijima T, Niwa H, Steinman RA, et al. STAT3 activation abrogates growth factor dependence and contributes to head and neck squamous cell carcinoma tumor growth in vivo. Cell Growth Differ. 2002;13:355–362. [PubMed] [Google Scholar]
- 129.Bonner JA, Yang ES, Trummell HQ, et al. Inhibition of STAT-3 results in greater cetuximab sensitivity in head and neck squamous cell carcinoma. Radiother Oncol. 2011;99:339–343. doi: 10.1016/j.radonc.2011.05.070. [DOI] [PubMed] [Google Scholar]
- 130.Onishi A, Chen Q, Humtsoe JO, et al. STAT3 signaling is induced by intercellular adhesion in squamous cell carcinoma cells. Exp Cell Res. 2008;314:377–386. doi: 10.1016/j.yexcr.2007.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Hambek M, Baghi M, Strebhardt K, et al. STAT 3 activation in head and neck squamous cell carcinomas is controlled by the EGFR. Anticancer Res. 2004;24:3881–3886. [PubMed] [Google Scholar]
- 132.Koppikar P, Lui VW, Man D, et al. Constitutive activation of signal transducer and activator of transcription 5 contributes to tumor growth, epithelial-mesenchymal transition, and resistance to epidermal growth factor receptor targeting. Clin Cancer Res. 2008;14:7682–7690. doi: 10.1158/1078-0432.CCR-08-1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Leeman-Neill RJ, Wheeler SE, Singh SV, et al. Guggulsterone enhances head and neck cancer therapies via inhibition of signal transducer and activator of transcription-3. Carcinogenesis. 2009;30:1848–1856. doi: 10.1093/carcin/bgp211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Leeman-Neill RJ, Seethala RR, Singh SV, et al. Inhibition of EGFR-STAT3 signaling with erlotinib prevents carcinogenesis in a chemically induced mouse model of oral squamous cell carcinoma. Cancer Prev Res (Phila) 2011;4:230–237. doi: 10.1158/1940-6207.CAPR-10-0249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Leibowitz MS, Andrade Filho PA, Ferrone S, et al. Deficiency of activated STAT1 in head and neck cancer cells mediates TAP1-dependent escape from cytotoxic T lymphocytes. Cancer Immunol Immunother. 2011;60:525–535. doi: 10.1007/s00262-010-0961-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Oliveras-Ferraros C, Vazquez-Martin A, Queralt B, et al. Interferon/STAT1 and neuregulin signaling pathways are exploratory biomarkers of cetuximab (Erbitux) efficacy in KRAS wild-type squamous carcinomas: A pathway-based analysis of whole human-genome microarray data from cetuximab-adapted tumor cell-line models. Int J Oncol. 2011;39:1455–1479. doi: 10.3892/ijo.2011.1155. [DOI] [PubMed] [Google Scholar]
- 137.Shim SH, Sung MW, Park SW, et al. Absence of STAT1 disturbs the anticancer effect induced by STAT3 inhibition in head and neck carcinoma cell lines. Int J Mol Med. 2009;23:805–810. doi: 10.3892/ijmm_00000196. [DOI] [PubMed] [Google Scholar]
- 138.Quesnelle KM, Grandis JR. Dual kinase inhibition of EGFR and HER2 overcomes resistance to cetuximab in a novel in vivo model of acquired cetuximab resistance. Clin Cancer Res. 2011;17:5935–5944. doi: 10.1158/1078-0432.CCR-11-0370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Yonesaka K, Zejnullahu K, Okamoto I, et al. Activation of ERBB2 signaling causes resistance to the EGFR-directed therapeutic antibody cetuximab. Sci Transl Med. 2011;3:99ra86. doi: 10.1126/scitranslmed.3002442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Pollak M. Insulin and insulin-like growth factor signalling in neoplasia. Nat Rev Cancer. 2008;8:915–928. doi: 10.1038/nrc2536. [DOI] [PubMed] [Google Scholar]
- 141.van der Veeken J, Oliveira S, Schiffelers RM, et al. Crosstalk between epidermal growth factor receptor- and insulin-like growth factor-1 receptor signaling: Implications for cancer therapy. Curr Cancer Drug Targets. 2009;9:748–760. doi: 10.2174/156800909789271495. [DOI] [PubMed] [Google Scholar]
- 142.Barnes CJ, Ohshiro K, Rayala SK, et al. Insulin-like growth factor receptor as a therapeutic target in head and neck cancer. Clin Cancer Res. 2007;13:4291–4299. doi: 10.1158/1078-0432.CCR-06-2040. [DOI] [PubMed] [Google Scholar]
- 143.Zuo Q, Shi M, Li L, et al. Development of cetuximab-resistant human nasopharyngeal carcinoma cell lines and mechanisms of drug resistance. Biomed Pharmacother. 2010;64:550–558. doi: 10.1016/j.biopha.2010.03.003. [DOI] [PubMed] [Google Scholar]
- 144.Galer CE, Corey CL, Wang Z, et al. Dual inhibition of epidermal growth factor receptor and insulin-like growth factor receptor I: Reduction of angiogenesis and tumor growth in cutaneous squamous cell carcinoma. Head Neck. 2011;33:189–198. doi: 10.1002/hed.21419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Riesterer O, Yang Q, Raju U, et al. Combination of anti-IGF-1R antibody A12 and ionizing radiation in upper respiratory tract cancers. Int J Radiat Oncol Biol Phys. 2011;79:1179–1187. doi: 10.1016/j.ijrobp.2010.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Hurbin A, Wislez M, Busser B, et al. Insulin-like growth factor-1 receptor inhibition overcomes gefitinib resistance in mucinous lung adenocarcinoma. J Pathol. 2011;225:83–95. doi: 10.1002/path.2897. [DOI] [PubMed] [Google Scholar]
- 147.Yang L, Li J, Ran L, et al. Phosphorylated insulin-like growth factor 1 receptor is implicated in resistance to the cytostatic effect of gefitinib in colorectal cancer cells. J Gastrointest Surg. 2011;15:942–957. doi: 10.1007/s11605-011-1504-z. [DOI] [PubMed] [Google Scholar]
- 148.Tandon R, Kapoor S, Vali S, et al. Dual epidermal growth factor receptor (EGFR)/insulin-like growth factor-1 receptor (IGF-1R) inhibitor: A novel approach for overcoming resistance in anticancer treatment. Eur J Pharmacol. 2011;667:56–65. doi: 10.1016/j.ejphar.2011.04.066. [DOI] [PubMed] [Google Scholar]
- 149.Sierra JR, Tsao MS. c-MET as a potential therapeutic target and biomarker in cancer. Ther Adv Med Oncol. 2011;3:S21–S35. doi: 10.1177/1758834011422557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Knowles LM, Stabile LP, Egloff AM, et al. HGF and c-Met participate in paracrine tumorigenic pathways in head and neck squamous cell cancer. Clin Cancer Res. 2009;15:3740–3750. doi: 10.1158/1078-0432.CCR-08-3252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Seiwert TY, Jagadeeswaran R, Faoro L, et al. The met receptor tyrosine kinase is a potential novel therapeutic target for head and neck squamous cell carcinoma. Cancer Res. 2009;69:3021–3031. doi: 10.1158/0008-5472.CAN-08-2881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Inno A, Di Salvatore M, Cenci T, et al. Is there a role for IGF1R and c-MET pathways in resistance to cetuximab in metastatic colorectal cancer? Clin Colorectal Cancer. 2011;10:325–332. doi: 10.1016/j.clcc.2011.03.028. [DOI] [PubMed] [Google Scholar]
- 153.Heindl S, Eggenstein E, Keller S, et al. Relevance of MET activation and genetic alterations of KRAS and E-cadherin for cetuximab sensitivity of gastric cancer cell lines. J Cancer Res Clin Oncol. 2012;138:843–858. doi: 10.1007/s00432-011-1128-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Turke AB, Zejnullahu K, Wu YL, et al. Preexistence and clonal selection of MET amplification in EGFR mutant NSCLC. Cancer Cell. 2010;17:77–88. doi: 10.1016/j.ccr.2009.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Cappuzzo F, Janne PA, Skokan M, et al. Met increased gene copy number and primary resistance to gefitinib therapy in non-small-cell lung cancer patients. Ann Oncol. 2009;20:298–304. doi: 10.1093/annonc/mdn635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Chen HJ, Mok TS, Chen ZH, et al. Clinicopathologic and molecular features of epidermal growth factor receptor T790M mutation and c-MET amplification in tyrosine kinase inhibitor-resistant Chinese non-small cell lung cancer. Pathol Oncol Res. 2009;15:651–658. doi: 10.1007/s12253-009-9167-8. [DOI] [PubMed] [Google Scholar]
- 157.Bean J, Brennan C, Shih JY, et al. MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc Natl Acad Sci USA. 2007;104:20932–20937. doi: 10.1073/pnas.0710370104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Beau-Faller M, Ruppert AM, Voegeli AC, et al. MET gene copy number in non-small cell lung cancer: Molecular analysis in a targeted tyrosine kinase inhibitor naive cohort. J Thorac Oncol. 2008;3:331–339. doi: 10.1097/JTO.0b013e318168d9d4. [DOI] [PubMed] [Google Scholar]
- 159.Onitsuka T, Uramoto H, Ono K, et al. Comprehensive molecular analyses of lung adenocarcinoma with regard to the epidermal growth factor receptor, K-ras, MET, and hepatocyte growth factor status. J Thorac Oncol. 2010;5:591–596. doi: 10.1097/JTO.0b013e3181d0a4db. [DOI] [PubMed] [Google Scholar]
- 160.Krumbach R, Schuler J, Hofmann M, et al. Primary resistance to cetuximab in a panel of patient-derived tumour xenograft models: Activation of MET as one mechanism for drug resistance. Eur J Cancer. 2011;47:1231–1243. doi: 10.1016/j.ejca.2010.12.019. [DOI] [PubMed] [Google Scholar]
- 161.Liska D, Chen CT, Bachleitner-Hofmann T, et al. HGF rescues colorectal cancer cells from EGFR inhibition via MET activation. Clin Cancer Res. 2011;17:472–482. doi: 10.1158/1078-0432.CCR-10-0568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Hoellein A, Pickhard A, von Keitz F, et al. Aurora kinase inhibition overcomes cetuximab resistance in squamous cell cancer of the head and neck. Oncotarget. 2011;2:599–609. doi: 10.18632/oncotarget.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Kelly KR, Ecsedy J, Mahalingam D, et al. Targeting aurora kinases in cancer treatment. Curr Drug Targets. 2011;12:2067–2078. doi: 10.2174/138945011798829410. [DOI] [PubMed] [Google Scholar]
- 164.McLaughlin J, Markovtsov V, Li H, et al. Preclinical characterization of Aurora kinase inhibitor R763/AS703569 identified through an image-based phenotypic screen. J Cancer Res Clin Oncol. 2010;136:99–113. doi: 10.1007/s00432-009-0641-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Reiter R, Gais P, Jutting U, et al. Aurora kinase a messenger RNA overexpression is correlated with tumor progression and shortened survival in head and neck squamous cell carcinoma. Clin Cancer Res. 2006;12:5136–5141. doi: 10.1158/1078-0432.CCR-05-1650. [DOI] [PubMed] [Google Scholar]
- 166.Lens SM, Voest EE, Medema RH. Shared and separate functions of polo-like kinases and aurora kinases in cancer. Nat Rev Cancer. 2010;10:825–841. doi: 10.1038/nrc2964. [DOI] [PubMed] [Google Scholar]
- 167.Lai CH, Tseng JT, Lee YC, et al. Translational up-regulation of Aurora-A in EGFR-overexpressed cancer. J Cell Mol Med. 2010;14:1520–1531. doi: 10.1111/j.1582-4934.2009.00919.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Wu CC, Yu CT, Chang GC, et al. Aurora-A promotes gefitinib resistance via a NF-κB signaling pathway in p53 knockdown lung cancer cells. Biochem Biophys Res Commun. 2011;405:168–172. doi: 10.1016/j.bbrc.2011.01.001. [DOI] [PubMed] [Google Scholar]
- 169.Arany I, Chen SH, Megyesi JK, et al. Differentiation-dependent expression of signal transducers and activators of transcription (STATs) might modify responses to growth factors in the cancers of the head and neck. Cancer Lett. 2003;199:83–89. doi: 10.1016/s0304-3835(03)00345-8. [DOI] [PubMed] [Google Scholar]
- 170.Dahan L, Norguet E, Etienne-Grimaldi MC, et al. Pharmacogenetic profiling and cetuximab outcome in patients with advanced colorectal cancer. BMC Cancer. 2011;11:496. doi: 10.1186/1471-2407-11-496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Kim ES, Lee JJ, Wistuba II. Cotargeting cyclin D1 starts a new chapter in lung cancer prevention and therapy. Cancer Prev Res (Phila) 2011;4:779–782. doi: 10.1158/1940-6207.CAPR-11-0143. [DOI] [PubMed] [Google Scholar]
- 172.Freemantle SJ, Dmitrovsky E. Cyclin E transgenic mice: Discovery tools for lung cancer biology, therapy, and prevention. Cancer Prev Res (Phila) 2010;3:1513–1518. doi: 10.1158/1940-6207.CAPR-10-0297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Fleming IN, Hogben M, Frame S, et al. Synergistic inhibition of ErbB signaling by combined treatment with seliciclib and ErbB-targeting agents. Clin Cancer Res. 2008;14:4326–4335. doi: 10.1158/1078-0432.CCR-07-4633. [DOI] [PubMed] [Google Scholar]
- 174.Kalish LH, Kwong RA, Cole IE, et al. Deregulated cyclin D1 expression is associated with decreased efficacy of the selective epidermal growth factor receptor tyrosine kinase inhibitor gefitinib in head and neck squamous cell carcinoma cell lines. Clin Cancer Res. 2004;10:7764–7774. doi: 10.1158/1078-0432.CCR-04-0012. [DOI] [PubMed] [Google Scholar]
- 175.Huang S, Benavente S, Armstrong EA, et al. P53 modulates acquired resistance to egfr inhibitors and radiation. Cancer Res. 2011;71:7071–7079. doi: 10.1158/0008-5472.CAN-11-0128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Chang GC, Yu CT, Tsai CH, et al. An epidermal growth factor inhibitor, Gefitinib, induces apoptosis through a p53-dependent upregulation of pro-apoptotic molecules and downregulation of anti-apoptotic molecules in human lung adenocarcinoma A549 cells. Eur J Pharmacol. 2008;600:37–44. doi: 10.1016/j.ejphar.2008.10.024. [DOI] [PubMed] [Google Scholar]
- 177.Ogino S, Meyerhardt JA, Cantor M, et al. Molecular alterations in tumors and response to combination chemotherapy with gefitinib for advanced colorectal cancer. Clin Cancer Res. 2005;11:6650–6656. doi: 10.1158/1078-0432.CCR-05-0738. [DOI] [PubMed] [Google Scholar]
- 178.Huether A, Hopfner M, Baradari V, et al. EGFR blockade by cetuximab alone or as combination therapy for growth control of hepatocellular cancer. Biochem Pharmacol. 2005;70:1568–1578. doi: 10.1016/j.bcp.2005.09.007. [DOI] [PubMed] [Google Scholar]
- 179.Cowling VH, Cole MD. E-cadherin repression contributes to c-Myc-induced epithelial cell transformation. Oncogene. 2007;26:3582–3586. doi: 10.1038/sj.onc.1210132. [DOI] [PubMed] [Google Scholar]
- 180.Thiery JP. Epithelial–mesenchymal transitions in development and pathologies. Curr Opin Cell Biol. 2003;15:740–746. doi: 10.1016/j.ceb.2003.10.006. [DOI] [PubMed] [Google Scholar]
- 181.Guarino M. Epithelial–mesenchymal transition and tumour invasion. Int J Biochem Cell Biol. 2007;39:2153–2160. doi: 10.1016/j.biocel.2007.07.011. [DOI] [PubMed] [Google Scholar]
- 182.Haddad Y, Choi W, McConkey DJ. Delta-crystallin enhancer binding factor 1 controls the epithelial to mesenchymal transition phenotype and resistance to the epidermal growth factor receptor inhibitor erlotinib in human head and neck squamous cell carcinoma lines. Clin Cancer Res. 2009;15:532–542. doi: 10.1158/1078-0432.CCR-08-1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Skvortsova I, Skvortsov S, Raju U, et al. Epithelial-to-mesenchymal transition and c-myc expression are the determinants of cetuximab-induced enhancement of squamous cell carcinoma radioresponse. Radiother Oncol. 2010;96:108–115. doi: 10.1016/j.radonc.2010.04.017. [DOI] [PubMed] [Google Scholar]
- 184.Frederick BA, Helfrich BA, Coldren CD, et al. Epithelial to mesenchymal transition predicts gefitinib resistance in cell lines of head and neck squamous cell carcinoma and non-small cell lung carcinoma. Mol Cancer Ther. 2007;6:1683–1691. doi: 10.1158/1535-7163.MCT-07-0138. [DOI] [PubMed] [Google Scholar]
- 185.Thomson S, Buck E, Petti F, et al. Epithelial to mesenchymal transition is a determinant of sensitivity of non-small-cell lung carcinoma cell lines and xenografts to epidermal growth factor receptor inhibition. Cancer Res. 2005;65:9455–9462. doi: 10.1158/0008-5472.CAN-05-1058. [DOI] [PubMed] [Google Scholar]
- 186.Yauch RL, Januario T, Eberhard DA, et al. Epithelial versus mesenchymal phenotype determines in vitro sensitivity and predicts clinical activity of erlotinib in lung cancer patients. Clin Cancer Res. 2005;11:8686–8698. doi: 10.1158/1078-0432.CCR-05-1492. [DOI] [PubMed] [Google Scholar]
- 187.Buck E, Eyzaguirre A, Barr S, et al. Loss of homotypic cell adhesion by epithelial-mesenchymal transition or mutation limits sensitivity to epidermal growth factor receptor inhibition. Mol Cancer Ther. 2007;6:532–541. doi: 10.1158/1535-7163.MCT-06-0462. [DOI] [PubMed] [Google Scholar]
- 188.Holz C, Niehr F, Boyko M, et al. Epithelial-mesenchymal-transition induced by EGFR activation interferes with cell migration and response to irradiation and cetuximab in head and neck cancer cells. Radiother Oncol. 2011;101:158–164. doi: 10.1016/j.radonc.2011.05.042. [DOI] [PubMed] [Google Scholar]
- 189.Vaupel P, Mayer A. Hypoxia in cancer: Significance and impact on clinical outcome. Cancer Metastasis Rev. 2007;26:225–239. doi: 10.1007/s10555-007-9055-1. [DOI] [PubMed] [Google Scholar]
- 190.Bertout JA, Patel SA, Simon MC. The impact of O2 availability on human cancer. Nat Rev Cancer. 2008;8:967–975. doi: 10.1038/nrc2540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Wouters A, Pauwels B, Lardon F, et al. Review: Implications of in vitro research on the effect of radiotherapy and chemotherapy under hypoxic conditions. Oncologist. 2007;12:690–712. doi: 10.1634/theoncologist.12-6-690. [DOI] [PubMed] [Google Scholar]
- 192.Wouters A, Boeckx C, Vermorken JB, et al. The intriguing interplay between therapies targeting the epidermal growth factor receptor, the hypoxic microenvironment and hypoxia-inducible factors. Curr Pharm Des. 2012;19:907–917. [PubMed] [Google Scholar]
- 193.Wang X, Schneider A. HIF-2α-mediated activation of the epidermal growth factor receptor potentiates head and neck cancer cell migration in response to hypoxia. Carcinogenesis. 2010;31:1202–1210. doi: 10.1093/carcin/bgq078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Hoogsteen IJ, Marres HA, van den Hoogen FJ, et al. Expression of EGFR under tumor hypoxia: Identification of a subpopulation of tumor cells responsible for aggressiveness and treatment resistance. Int J Radiat Oncol Biol Phys. 2012;84:807–814. doi: 10.1016/j.ijrobp.2012.01.002. [DOI] [PubMed] [Google Scholar]
- 195.Byers LA, Holsinger FC, Kies MS, et al. Serum signature of hypoxia-regulated factors is associated with progression after induction therapy in head and neck squamous cell cancer. Mol Cancer Ther. 2010;9:1755–1763. doi: 10.1158/1535-7163.MCT-09-1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Lu H, Liang K, Lu Y, et al. The anti-EGFR antibody cetuximab sensitizes human head and neck squamous cell carcinoma cells to radiation in part through inhibiting radiation-induced upregulation of HIF-1α. Cancer Lett. 2012;322:78–85. doi: 10.1016/j.canlet.2012.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Schmitz S, Kaminsky-Forrett MC, Henry S, et al. Phase II study of figitumumab in patients with recurrent and/or metastatic squamous cell carcinoma of the head and neck: Clinical activity and molecular response (GORTEC 2008-02) Ann Oncol. 2012;23:2153–2161. doi: 10.1093/annonc/mdr574. [DOI] [PubMed] [Google Scholar]
- 198.Reidy DL, Vakiani E, Fakih MG, et al. Randomized, phase II study of the insulin-like growth factor-1 receptor inhibitor IMC-A12, with or without cetuximab, in patients with cetuximab- or panitumumab-refractory metastatic colorectal cancer. J Clin Oncol. 2010;28:4240–4246. doi: 10.1200/JCO.2010.30.4154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Chung CH, Pohlmann PR, Rothenberg ML, et al. Insulin-like growth factor-1 receptor inhibitor, AMG-479, in cetuximab-refractory head and neck squamous cell carcinoma. Head Neck. 2011;33:1804–1808. doi: 10.1002/hed.21478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Brooks HD, Glisson BS, Bekele BN, et al. Phase 2 study of dasatinib in the treatment of head and neck squamous cell carcinoma. Cancer. 2011;117:2112–2119. doi: 10.1002/cncr.25769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Fury MG, Baxi S, Shen R, et al. Phase II study of saracatinib (AZD0530) for patients with recurrent or metastatic head and neck squamous cell carcinoma (HNSCC) Anticancer Res. 2011;31:249–253. [PMC free article] [PubMed] [Google Scholar]
- 202.de Souza JA, Davis DW, Zhang Y, et al. A phase II study of lapatinib in recurrent/metastatic squamous cell carcinoma of the head and neck. Clin Cancer Res. 2012;18:2336–2343. doi: 10.1158/1078-0432.CCR-11-2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Harrington KJ, Berrier A, Robinson M, et al. Randomised phase II study of oral lapatinib combined with chemoradiotherapy in patients with advanced squamous cell carcinoma of the head and neck: Rationale for future randomised trials in human papillomavirus-negative disease. Eur J Cancer. 2012;49:1609–1618. doi: 10.1016/j.ejca.2012.11.023. [DOI] [PubMed] [Google Scholar]
- 204.Lalami Y, Specenier PM, Awada A, et al. EORTC 24051: Unexpected side effects in a phase I study of TPF induction chemotherapy followed by chemoradiation with lapatinib, a dual EGFR/ErbB2 inhibitor, in patients with locally advanced resectable larynx and hypopharynx squamous cell carcinoma. Radiother Oncol. 2012;105:238–240. doi: 10.1016/j.radonc.2012.08.006. [DOI] [PubMed] [Google Scholar]