

This review summarizes the molecular biology, clinical features, detection, and targeting of ROS1 and RET rearrangements in non-small cell lung cancer (NSCLC). Genetic alterations in ROS1 and RET were recently identified in NSCLC patients, and those affected appear to have unique clinical and pathologic features that may facilitate identification and enrichment strategies. Preclinical data and early clinical data suggest that both ROS1 and RET rearrangements may be viable therapeutic targets.
Keywords: Non-small cell lung cancer, ROS1, RET, Targeted therapy, Fusion oncogenes
Abstract
The discovery of chromosomal rearrangements involving the anaplastic lymphoma kinase (ALK) gene in non-small cell lung cancer (NSCLC) has stimulated renewed interest in oncogenic fusions as potential therapeutic targets. Recently, genetic alterations in ROS1 and RET were identified in patients with NSCLC. Like ALK, genetic alterations in ROS1 and RET involve chromosomal rearrangements that result in the formation of chimeric fusion kinases capable of oncogenic transformation. Notably, ROS1 and RET rearrangements are rarely found with other genetic alterations, such as EGFR, KRAS, or ALK. This finding suggests that both ROS1 and RET are independent oncogenic drivers that may be viable therapeutic targets. In initial screening studies, ROS1 and RET rearrangements were identified at similar frequencies (approximately 1%–2%), using a variety of genotyping techniques. Importantly, patients with either ROS1 or RET rearrangements appear to have unique clinical and pathologic features that may facilitate identification and enrichment strategies. These features may in turn expedite enrollment in clinical trials evaluating genotype-directed therapies in these rare patient populations. In this review, we summarize the molecular biology, clinical features, detection, and targeting of ROS1 and RET rearrangements in NSCLC.
Implications for Practice:
Treatment approaches for non-small cell lung cancer (NSCLC) have been advanced in recent years by the identification of distinct, molecularly-defined subsets of patients that derive benefit from targeted therapies. As a result, the field has placed greater focus on identifying new molecular targets. Recently, ROS1 and RET fusions were independently identified in 1%–2% of patients with NSCLC. Preclinical data and early clinical findings suggest that both may be viable therapeutic targets. In this review, we summarize this data as well as the clinical and pathologic features associated with these fusions. This may in turn inform screening strategies and the development of clinical trials evaluating genotype-driven therapies in these patient populations.
Introduction
The 2004 discovery that somatic mutations in the epidermal growth factor receptor (EGFR) confer sensitivity to EGFR tyrosine kinase inhibitors (TKIs) established a new treatment paradigm in non-small cell lung cancer (NSCLC) [1–3]. This discovery also placed greater focus on identifying new molecular subsets of NSCLC that may benefit from targeted therapies. In particular, emphasis has been placed on finding new “driver” oncogenes, so named because these genes are central to the growth and viability of cancer cells (e.g., mutant KRAS, BRAF, or HER2). Until recently, genetic alterations in NSCLC driver oncogenes were presumed to occur predominantly through point mutations or small insertions or deletions. Chromosomal translocations were thought to be infrequent events in solid tumors [4, 5]. In 2007, however, Soda and colleagues discovered a novel fusion gene generated by a chromosomal rearrangement involving the anaplastic lymphoma kinase (ALK) gene in NSCLC [6]. ALK rearrangements have since been identified in approximately 3%–5% of patients with lung cancer, defining a distinct molecular subset of NSCLC with unique clinicopathologic features and marked sensitivity to the ALK inhibitor crizotinib [7, 8].
The success of crizotinib in ALK-positive patients has prompted efforts to find new oncogenic fusions in NSCLC. Facilitated by advances in cytogenetic and molecular techniques, these efforts have identified novel oncogenic fusions involving ROS1 and RET. In this review, we summarize the molecular biology of these rearrangements, outline current detection methods, and introduce treatment approaches for these newly identified oncogenic drivers.
ROS1
Biology: Native ROS1
Initially identified in the early 1980s, ROS1 is located on chromosome 6, where it encodes an orphan receptor tyrosine kinase [9–11]. ROS1 consists of (a) a glycoprotein-rich extracellular domain, (b) a transmembrane domain, and (c) an intracellular tyrosine kinase [12]. To date, no ROS1 ligand has been identified, and insights into the normal function of native ROS1 in humans are limited. In murine models, ROS1 transcripts are temporally and spatially expressed in epithelial cells of the kidney, lung, heart, intestine, and testis [12–14]. It has been speculated that ROS1 may be involved in epithelial-mesenchymal transitions in these organs; however, ROS1 knockout mice are viable and appear healthy, with the exception of infertility among male knockout mice [15]. The latter has been attributed to abnormalities in epididymal differentiation causing defective sperm maturation.
ROS1 in Human Malignancy
ROS1 rearrangements were initially identified in a human glioblastoma cell line [16]. Further characterization of this cell line revealed that this rearrangement generates a novel fusion gene involving FIG (fused in glioblastoma; also known as GOPC) at the 5′ end and a portion of ROS1 at the 3′ end [17–19]. The entire ROS1 kinase domain is retained in this rearrangement. FIG-ROS1 arises through an intrachromosomal deletion, producing a constitutively active kinase [19, 20]. Recently, ROS1 rearrangements have been identified in several other malignancies, including cholangiocarcinoma [21], ovarian carcinoma [22], gastric carcinoma [23], and NSCLC [24–34].
ROS1 rearrangements in NSCLC were first identified in 2007 [24]. In this initial report, Rikova and colleagues characterized tyrosine kinase signaling in 41 NSCLC cell lines and 150 NSCLC tumors. Within one cell line, HCC78, the authors identified a novel ROS1 rearrangement involving SLC34A2 and ROS1. Like previously identified ROS1 rearrangements, SLC34A2-ROS1 retains the entire kinase domain of ROS1. The authors also identified a separate ROS1 rearrangement in a patient-derived NSCLC specimen [24]. In this sample, CD74, which encodes a type 2 transmembrane protein, was fused with ROS1, generating a novel CD74-ROS1 transcript. Subsequently, multiple ROS1 fusion partners have been identified in NSCLC: SDC4 [27, 33], EZR [29, 33, 34], KDELR2 [30], CCDC6 [31], TPM3 [33], LRIG3 [33], and FIG (Fig. 1) [28, 32]. In total, nine different ROS1 fusion partners have been identified, with CD74 being the most common. With the exception of FIG and EZR, ROS1 fusion partners are located on different chromosomes than the native ROS1 gene [32]. Despite a diversity of fusion partners, ROS1 rearrangements involve conserved ROS1 break points that preserve the tyrosine kinase domain [33].
Figure 1.

Schematic diagram of ROS1 fusions in non-small cell lung cancer. (A): ROS1 tyrosine kinase domain (dark green), ROS1 transmembrane domain (blue), and coiled-coil domains (pink) in ROS1 fusion partners; KDELR2-ROS1 is not shown. (B): Reported frequencies of different ROS1 fusion partners. Not all studies included reverse transcription polymerase chain reaction primers against all fusion partners listed.
Abbreviation: E, exon.
ROS1 fusions are potent oncogenic drivers. Indeed, expression of ROS1 fusions in vitro and in vivo leads to oncogenic transformation [20, 21, 33, 35, 36]. This is thought to occur through constitutive kinase activation [24, 37]. The exact mechanism by which ROS1 rearrangements lead to dysregulated kinase activity remains unclear. In the setting of ALK and RET rearrangements, coiled-coil domains in the 5′ fusion partners mediate ligand-independent homodimerization, leading to kinase activation [6, 38]. In contrast, a majority of ROS1 partner proteins lack dimerization domains (Fig. 1) [33, 39]. Nevertheless, ROS1 rearrangements are believed to promote signal transduction programs, leading to upregulation of SHP-1 and SHP-2 (also known as PTPN6 and PTPN11, respectively) and to activation of the phosphoinositide-3 kinase (PI3K)/AKT/mTOR, JAK/STAT (also known as SOAT1), and MAPK/ERK pathways [12]. Together, these downstream signals promote cell survival and proliferation.
Detection of ROS1 Fusions
ROS1 rearrangements have been identified in approximately 1%–2% of patients with NSCLC by using diverse genotyping techniques (Table 1) [24–34]. Several recent studies have used whole-genome and whole-transcriptome sequencing to identify ROS1 rearrangements, including several novel fusion partners [29, 30, 32]. Such approaches are not readily generalizable to widescale clinical testing at present. Consequently, ROS1 screening strategies have been largely informed by experiences with ALK testing, for which the three most commonly used methods of detection are fluorescent in situ hybridization (FISH), reverse transcription polymerase chain reaction (RT-PCR), and immunohistochemistry (IHC) [40].
Table 1.
Prevalence of ROS1 rearrangements in non-small cell lung cancer screening studies
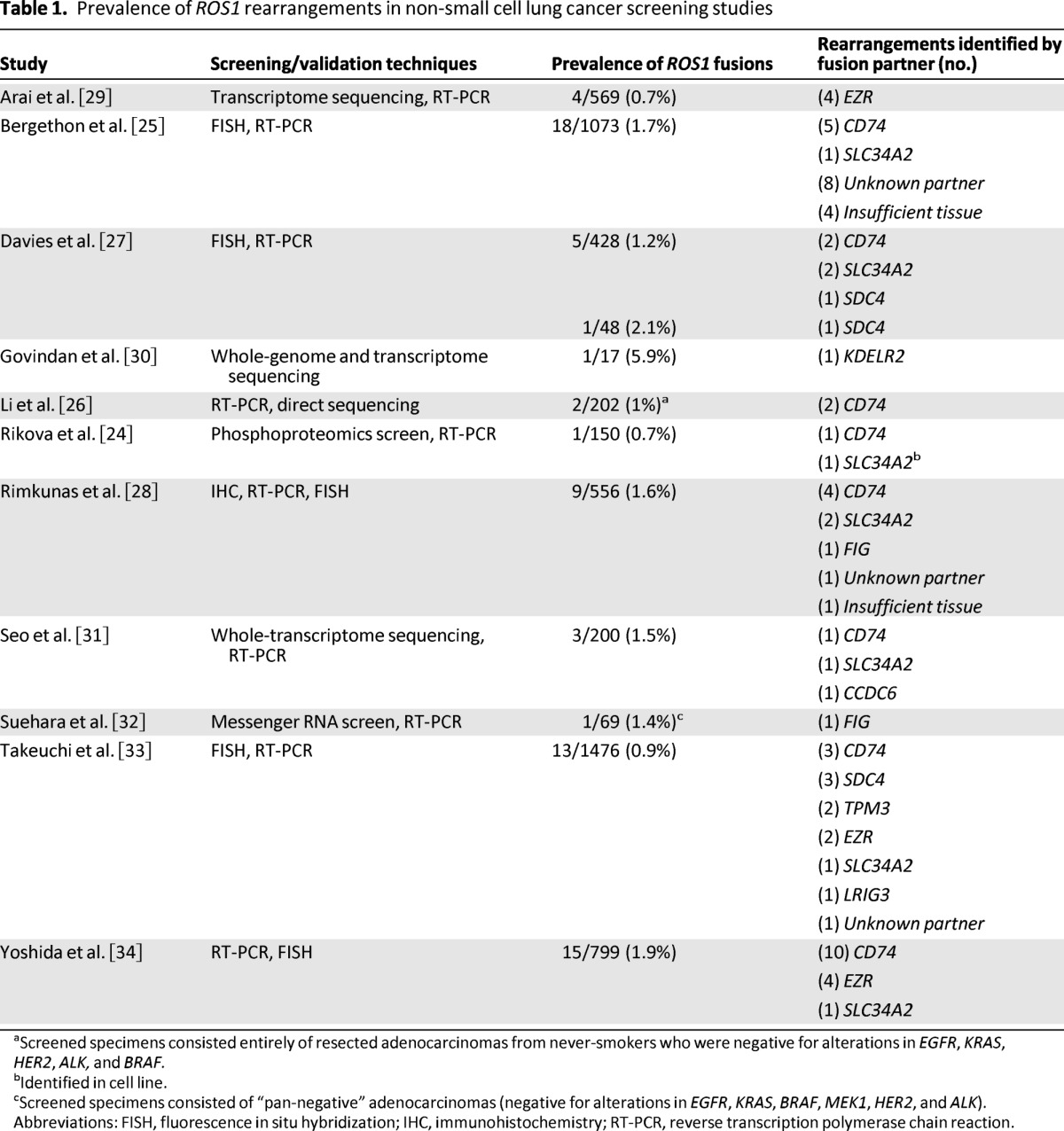
aScreened specimens consisted entirely of resected adenocarcinomas from never-smokers who were negative for alterations in EGFR, KRAS, HER2, ALK, and BRAF.
bIdentified in cell line.
cScreened specimens consisted of “pan-negative” adenocarcinomas (negative for alterations in EGFR, KRAS, BRAF, MEK1, HER2, and ALK).
Abbreviations: FISH, fluorescence in situ hybridization; IHC, immunohistochemistry; RT-PCR, reverse transcription polymerase chain reaction.
FISH is the gold standard assay for detection of ALK rearrangements [7, 40, 41]. Consequently, many early ROS1 screening studies used FISH as the predominant testing tool [25, 27, 33]. Several different ROS1 FISH assays have been developed. These assays generally use red and green fluorescent probes to hybridize with sequences adjacent to or including a portion of the ROS1 gene. In the absence of a ROS1 rearrangement, the overlapping probes produce a fused or yellow signal. When a ROS1 gene rearrangement is present, however, the two probes become separated, resulting in a “split” signal (Fig. 2). Isolated red 3′ signals can also be observed in the setting of ROS1 rearrangements [27, 33, 34]. Specimens are deemed positive if more than 15% of tumor cells demonstrate split or isolated 3′ signals.
Figure 2.

A ROS1 break-apart fluorescent in situ hybridization (FISH) assay. FISH reveals separation of the 5′ ROS1 probe (green) from the 3′ ROS1 probe (red), indicative of a ROS1 rearrangement in a patient with non-small cell lung cancer. Size bar = 10 μm. Reprinted from [25] with permission [Bergethon, K et al: J Clin Oncol 2012;30:863—870 © 2013 American Society of Clinical Oncology. All rights reserved].
Bergethon and colleagues recently screened 1,073 NSCLCs using ALK and ROS1 FISH and detected ROS1 rearrangements in 18 specimens (1.7%) [25]. Takeuchi et al. similarly examined 1,476 lung cancer specimens using FISH and identified 13 ROS1 rearrangements (0.9%) [33]. One advantage of FISH is that it can be performed on formalin-fixed, paraffin-embedded (FFPE) tissues. In contrast to RT-PCR, FISH is also capable of detecting rearrangements without prior knowledge of 5′ fusion partners [25]. Nevertheless, FISH has several limitations. This technically challenging technique is not uniformly available in all laboratories. Furthermore, certain ROS1 rearrangements may not be detected by currently available break-apart FISH assays. The native FIG gene, for example, is located only 134 kilobase pairs upstream of ROS1; therefore, intrachromosomal deletions generating FIG-ROS1 fusions may not create a sufficient change in FISH signal patterns to allow identification [32]. Lastly, FISH does not provide information on specific ROS1 fusion partners; however, it remains unclear whether this is biologically or clinically relevant.
Data on using RT-PCR alone in the detection of ROS1 rearrangements are limited at present. Potential advantages of RT-PCR as a screening tool include rapid turnaround time and limited tissue requirements. RT-PCR also permits identification of specific fusion partners, but standard RT-PCR requires that fusion-specific primers be included during testing; therefore, this technique is unable to identify rearrangements involving unknown fusion partners. Another drawback of RT-PCR is that results depend on the quality of RNA, which is often degraded in FFPE tissues.
In addition to FISH and RT-PCR, IHC has also been used to screen for ROS1 rearrangements. Rimkunas et al. recently developed a novel ROS1 IHC assay (Cell Signaling, Danvers, MA; clone D4D6) [28]. Screening 556 NSCLC specimens, they identified ROS1 expression in 9 cases (1.6%). In eight of these specimens, confirmatory ROS1 FISH was positive. The ninth sample could not be assessed because of high background signal intensity. Interestingly, several different ROS1 staining patterns were observed: diffuse cytoplasmic staining, membranous localization, vesicular localization, and perinuclear aggregates. These patterns did not correlate with specific ROS1 fusion partners. Although prior studies identified ROS1 mRNA expression in normal lung [12], Rimkunas et al. did not observe ROS1 staining in adjacent normal lung tissue using the D4D6 antibody [28]. However, non-neoplastic cells, such as macrophages and bronchial cells, exhibited positive staining in rare cases. No ROS1 staining was observed among 138 ROS1 FISH-negative cases.
Despite similar clinicopathologic features, ROS1 rearrangements do not overlap with mutations in other oncogenic drivers, such as EGFR or ALK. In one study of 1,073 tumor specimens, ROS1 and ALK rearrangements were mutually exclusive across the entire study population. In a separate study using ROS1 and ALK IHC, there was no evidence of coexpression among 556 tumors tested.
Additional studies are needed to validate ROS1 IHC. Implementation will also require development of standardized scoring criteria as well as additional assessments of antibody selectivity [42]. Nonetheless, these early findings by Rimkunas et al. suggest that ROS1 IHC may be a promising screening tool. In general, advantages of IHC include rapid turnaround time and the widespread availability of testing in most pathology laboratories. Finally, although our discussion has focused on IHC, RT-PCR, and FISH, future directions include use of chromogenic in situ hybridization and next-generation sequencing [39, 43].
ROS1 Fusions: Clinical and Pathologic Features
ROS1 rearrangements define a distinct molecular subtype of NSCLC with unique clinicopathologic features. Patients with ROS1 rearrangements share many features in common with ALK-positive patients. In one multi-institutional series, ROS1 rearrangements were associated with younger age, never-smoking history, Asian ethnicity, and advanced stage [25]. Furthermore, all 18 ROS1-positive patients in this series had adenocarcinoma histology. Additional studies have confirmed adenocarcinoma as the predominant histologic pattern, although ROS1 rearrangements have also been infrequently observed in large cell and squamous cell carcinomas [27, 28].
Despite similar clinicopathologic features, ROS1 rearrangements do not overlap with mutations in other oncogenic drivers, such as EGFR or ALK [24–26, 28]. In one study of 1,073 tumor specimens, ROS1 and ALK rearrangements were mutually exclusive across the entire study population [25]. In a separate study using ROS1 and ALK IHC, there was no evidence of coexpression among 556 tumors tested [28]. In this latter report, however, two ROS1-positive tumors also stained positive for EGFR mutations (EGFR L858R and E746-A750del) using mutation-specific IHC (and confirmed by sequencing). It is unknown whether both alterations were present in the same cells.
Targeting ROS1
In their initial report of ROS1 rearrangements in NSCLC, Rikova and colleagues demonstrated that cell lines containing ROS1 rearrangements undergo apoptosis on treatment with ROS1-specific small interfering RNAs, suggesting that ROS1-positive tumors are oncogene addicted [24]. Subsequently, it was recognized that ROS1 and ALK share a high degree of homology within their respective tyrosine kinase domains, prompting hypotheses that ALK TKIs may also inhibit ROS1 [39, 44]. McDermott et al. demonstrated that the HCC78 cell line, which contains SLC34A2-ROS1, is sensitive to treatment with the ALK inhibitor TAE684 [44]. More recently, crizotinib was shown to suppress ROS1 activity and downstream signaling in vitro, leading to growth inhibition [25, 27, 45].
Based on this preclinical data, a phase I trial of crizotinib (ClinicalTrials.gov identifier NCT00585195) was amended to include a ROS1-positive cohort. Preliminary results from this trial were presented at the 2012 American Society of Clinical Oncology annual meeting and updated at the 2012 European Society for Medical Oncology congress [46, 47]. Twenty ROS1-positive NSCLC patients were evaluable at the time of reporting. All ROS1 rearrangements were detected by FISH. In these patients, crizotinib was associated with an objective response rate (ORR) of 50%, including one complete response and nine partial responses. The disease control rate was 70% after 8 weeks of treatment. These results are similar to those seen with crizotinib in ALK-positive patients. Additional clinical trials evaluating first- and second-line crizotinib in ROS1-positive patients are being planned in Japan, China, Taiwan, and South Korea.
Several additional clinical trials are ongoing for ROS1-positive patients (Table 2). Given the homology between ALK and ROS1, many of these trials are evaluating structurally distinct, next-generation ALK TKIs, with separate cohorts for ALK- and ROS1-positive patients. Data from these ROS1-positive cohorts have not been reported to date. Several ongoing studies are also examining TKIs in combination with either chemotherapy or heat shock protein 90 (HSP90) inhibitors. The use of HSP90 inhibitors in this setting is based on preclinical models demonstrating synergy between crizotinib and HSP90 inhibition [48].
Table 2.
Current clinical trials for patients with ROS1 rearrangements
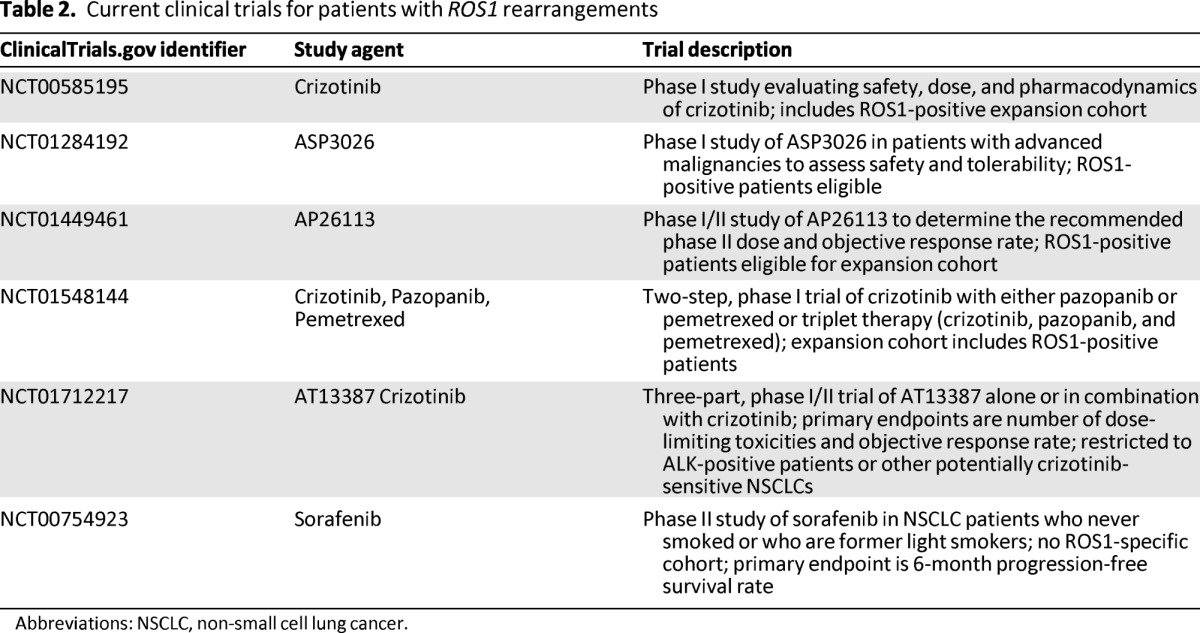
Abbreviations: NSCLC, non-small cell lung cancer.
At present, guidelines from the National Comprehensive Cancer Network (NCCN) note that clinicians may use crizotinib in patients known to harbor a ROS1 rearrangement (grade 2A recommendation) [49]. Although commercial testing is available, formal screening recommendations for ROS1 fusions have not been established, and ROS1 testing is not a universal standard. Testing should be considered in select patients, such as those with nonsquamous histology and negative EGFR and ALK testing. If ROS1 fusions are identified, such patients should be directed toward participation in clinical trials of ROS1-directed therapies.
RET
Biology: Native RET
The RET (rearranged during transfection) proto-oncogene was initially identified in 1985 through transfection of NIH3T3 cells with human lymphoma DNA [50]. RET was subsequently mapped to chromosome 10q11.2, where it encodes a receptor tyrosine kinase [51]. RET is normally expressed in neurons, sympathetic and parasympathetic ganglia, thyroid C cells, adrenal medullary cells, urogenital tract cells, and testis germ cells [52–54]. RET plays an important role in renal organogenesis and development of the enteric nervous system [55, 56]. RET consists of three domains: (a) an extracellular domain, (b) a transmembrane domain, and (c) an intracellular tyrosine kinase [57]. RET activation requires formation of a multimeric protein complex [58]. This involves binding of the glial cell line-derived neurotrophic factor (GDNF) family of ligands and a cell membrane-bound coreceptor, GFR-α [58–61]. On activation, RET undergoes autophosphorylation and engages downstream signaling pathways (RAS/MAPK/ERK, PI3K/AKT, and phospholipase C-γ), leading to cellular proliferation, migration, and differentiation [62]. Gain-of-function alterations in RET have been implicated in human malignancy. In contrast, loss-of-function RET mutations are associated with congenital absence of enteric ganglia, or Hirschsprung's disease [63, 64].
RET in Human Malignancy
In 1990, Grieco et al. reported chromosomal rearrangements involving the RET proto-oncogene in papillary thyroid cancer (PTC) [65]. These rearrangements generate fusion transcripts, pairing the 3′ end of RET, which encodes the tyrosine kinase domain, with the 5′ end of a separate gene (CCDC6, NCOA4, PRKAR1A, TRIM24, GOLGA5, TRIM33, KTN1, ERC1, MBD1, and TRIM27) [57, 66]. In general, RET fusions are identified in approximately 5%–40% of PTCs [52]. Notably, RET rearrangements in PTC are associated with ionizing radiation treatment or environmental radiation exposure, as has been observed among survivors of the atomic bombings in Japan and the Chernobyl nuclear power plant accident [67–69].
In addition to PTC, genetic alterations in RET are observed in medullary thyroid cancer (MTC). Instead of rearrangements, however, MTC is associated with gain-of-function point mutations in RET [70]. Specifically, germ-line mutations in RET are present in nearly all inherited forms of MTC, including multiple endocrine neoplasia 2 [71–73]. In sporadic MTC, RET mutations are identified in up to 50% of patients [70, 74, 75].
Chromosomal rearrangements involving RET were also recently found in NSCLC [31–33, 38, 76–83]. In late 2011 through early 2012, four independent groups of investigators used different screening strategies to discover RET rearrangements in approximately 1%–2% of NSCLCs [33, 38, 76, 77]. In one early report, Ju et al. performed massively parallel whole-genome and transcriptome sequencing of a lung adenocarcinoma from a 33-year-old never-smoker [38]. Prior genotyping of this patient's tumor revealed no ALK rearrangement or mutations in EGFR or KRAS. Sequencing of this specimen and paired normal tissue identified a novel fusion gene involving the 5′ end of KIF5B and the 3′ portion of RET within the tumor. In this study, two additional KIF5B-RET fusions were identified among 20 lung adenocarcinomas.
In a separate report, Kohno et al. identified a KIF5B-RET fusion using whole-transcriptome sequencing during a screening study for new oncogenic drivers in NSCLC [76]. The authors subsequently screened 433 lung adenocarcinoma specimens using RT-PCR and confirmatory FISH, identifying six additional KIF5B-RET fusions. In a contemporaneous report, Lipson et al. used targeted next-generation sequencing to screen for candidate oncogenic drivers and also identified KIF5B-RET fusions in a subset of tested specimens [77]. Takeuchi and colleagues relied on an alternative approach, using a FISH-based screen with confirmatory RT-PCR [33]. Among 1,529 specimens, 14 RET rearrangements (0.9%) were identified.
Importantly, preclinical models demonstrate that RET rearrangements are transforming in vitro and in vivo [33, 77]. To date, four different RET fusion partners have been identified in NSCLC: KIF5B, CCDC6, TRIM33, and NCOA4 [79, 84, 85]. Interestingly, CCDC6 (also known as PTC), NCOA4 (also known as PTC3) and TRIM33 (also known as PTC7) are also known RET fusion partners in PTC. With the exception of TRIM33, all of these genes are located on chromosome 10; therefore, RET fusions in NSCLC appear to arise predominantly through intrachromosomal rearrangements. KIF5B is the most common fusion partner in NSCLC. Seven different KIF5B-RET variants have been recognized; each differs with respect to KIF5B and RET break points (Fig. 3) [86]. Notably, the RET tyrosine kinase is preserved in all fusions. Moreover, in contrast to ROS1, each RET partner protein contains a coiled-coil domain, which is believed to promote ligand-independent dimerization and constitutive activation of RET (Fig. 4) [33, 38, 76, 77]. This is analogous to the mechanism of oncogenic activation in the setting of ALK rearrangements [6].
Figure 3.

Schematic diagram depicting RET fusions identified in non-small cell lung cancer. The RET tyrosine kinase domain (blue) is preserved in all fusions. Four different RET fusion partners are depicted: KIF5B, CCDC6, TRIM33, and NCOA4. KIF5B is the most common fusion partner, with seven different KIF5B-RET fusion variants described to date.
Abbreviation: TM, transmembrane.
Figure 4.

Models of RET rearrangements. (A): Schematic representation of the RET proto-oncogene (left). RET activation typically involves ligand binding, interactions with a coreceptor, and homodimerization leading to formation of a multiprotein complex (right). (B): Schematic representation of a KIF5B-RET fusion (left). The coiled-coil domain of KIF5B promotes ligand-independent homodimerization of RET, leading to constitutive activation of downstream growth signaling.
Abbreviations: CC, coiled-coil domain; GFL, glial cell line-derived neurotrophic factor family ligand; GFRα, GDNF family receptor α; KIF5B, kinesin family member 5B; P, phosphorylated tyrosine residue; RET, rearranged during transfection; TK, tyrosine kinase; TM, transmembrane.
Detection of RET Rearrangements
As noted, RET rearrangements in NSCLC were discovered using several different genomic screening strategies [33, 38, 76, 77]. Like ROS1 and ALK rearrangements, however, broad screening efforts have focused largely on three testing techniques: FISH, RT-PCR, and IHC. Each test has advantages and disadvantages, many of which have already been reviewed in our discussion of ROS1 testing. To date, no screening technique has emerged as a gold standard, and most screening studies have incorporated multiple testing techniques for initial detection and validation of RET fusions (Table 3).
Table 3.
Prevalence of RET rearrangements in non-small cell lung cancer screening studies
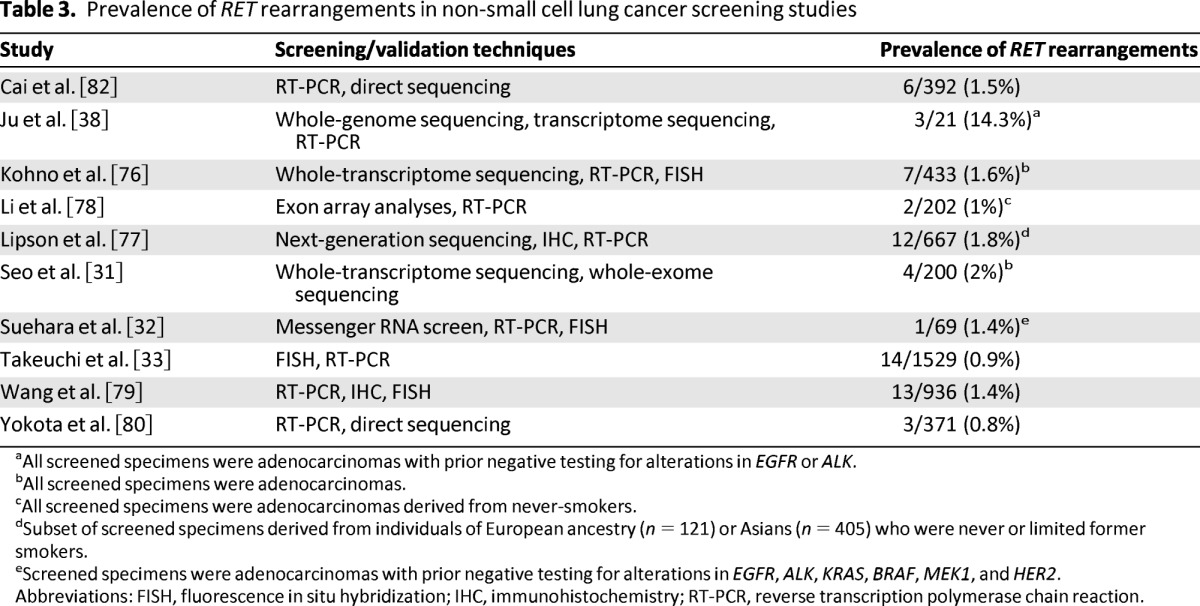
aAll screened specimens were adenocarcinomas with prior negative testing for alterations in EGFR or ALK.
bAll screened specimens were adenocarcinomas.
cAll screened specimens were adenocarcinomas derived from never-smokers.
dSubset of screened specimens derived from individuals of European ancestry (n = 121) or Asians (n = 405) who were never or limited former smokers.
eScreened specimens were adenocarcinomas with prior negative testing for alterations in EGFR, ALK, KRAS, BRAF, MEK1, and HER2.
Abbreviations: FISH, fluorescence in situ hybridization; IHC, immunohistochemistry; RT-PCR, reverse transcription polymerase chain reaction.
In general, FISH is an effective tool for detection of chromosomal rearrangements. In the largest screening study to date, FISH and RT-PCR were used in combination to identify 14 RET-positive cases among 1,529 specimens [33]. In more recent studies, FISH has been used as a confirmatory tool rather than a screening tool [32, 76, 79].
RT-PCR has been incorporated into a majority of RET screening studies. Because RT-PCR cannot identify rearrangements involving novel fusion partners, this technique has typically been combined with other testing modalities, such as IHC or FISH. In one study using RT-PCR alone, however, KIF5B-RET fusions were identified in 6 of 392 patients (1.5%) [82]. Notably, the multiplexed RT-PCR assay in this study did not assess for CCDC6-RET or NCOA4-RET; consequently, the prevalence of RET rearrangements may have been underestimated.
Despite low RET expression in normal lung tissue [87], RET IHC has been disappointing as a screening tool. Lipson et al., for example, identified moderate-intense RET staining in 22 of 117 NSCLC specimens (18.8%); however, only 1 of these specimens harbored a RET rearrangement using RT-PCR [77]. Similarly, Wang et al. found no significant differences between RET IHC staining patterns among RET-positive and RET-negative specimens identified by RT-PCR [79]. The experience of using RET IHC in PTC has been similarly disappointing because of variable staining patterns and weak immunoreactivity [88]. At present, RET IHC does not appear to be a reliable screening technique for detection of RET rearrangements.
RET Fusions: Clinical and Pathologic Features
RET rearrangements represent a distinct molecular subset of NSCLC, with specific clinicopathologic features and an estimated prevalence of 1%–2% (Table 3). Importantly, RET rearrangements are mutually exclusive with mutations in EGFR, KRAS, ALK, HER2, and BRAF, suggesting that RET fusions are independent oncogenic drivers in NSCLC [33, 38, 76, 77, 79].
Initial reports of RET rearrangements in NSCLC suggested that these alterations were more commonly found among never-smokers with adenocarcinoma histology [33, 38, 76, 77]. Additional insights into the clinicopathologic features of RET rearrangements were recently reported [79]. In this study, 13 RET-positive patients were identified among 936 surgically resected NSCLC patients using RT-PCR and quantitative real-time polymerase chain reaction. Confirmatory FISH testing was positive in all patients. Among RET-positive patients, adenocarcinoma histology was observed in 11 of 13 (85%). Two patients had adenosquamous histology. Patients with RET-positive adenocarcinomas were mostly never-smokers (81.8%), were relatively younger in age (72.7%, <60 years of age), and had roughly equal sex distribution, although none of these features reached statistical significance compared with RET-negative patients. Furthermore, patients with RET-positive adenocarcinomas tended to have small primary tumors (100%, ≤3 cm), solid-predominant histology (63.6%), N2 disease (54.4%), and more poorly differentiated tumors (63.6%). Interestingly, 36.4% of RET-positive adenocarcinomas had signet ring cells in ≥10% of tumor cells. No RET-positive patients reported prior radiation exposure.
Several preclinical studies suggest that RET fusions are viable targets in NSCLC. In cell line models expressing KIF5B-RET, for example, treatment with multitargeted TKIs with anti-RET activity (sunitinib, sorafenib, and vandetanib) leads to growth inhibition. Conversely, treatment of these cells with gefitinib or crizotinib, which do not possess anti-RET activity, is ineffective at suppressing growth.
Targeting RET
RET plays a central role in several thyroid malignancies, providing a basis for RET-directed therapies. Two agents, vandetanib and cabozantinib, recently received regulatory approval for the treatment of advanced MTC on the basis that each significantly prolongs progression-free survival (PFS) compared with placebo [89, 90]. Both agents possess activity against RET, but each also inhibits several other kinases believed to be important in MTC (e.g., vascular endothelial growth factor receptor). It remains unclear whether the antitumor effects of these agents result from RET inhibition alone [91].
Several preclinical studies suggest that RET fusions are viable targets in NSCLC [33, 76, 77]. In cell line models expressing KIF5B-RET, for example, treatment with multitargeted TKIs with anti-RET activity (sunitinib, sorafenib, and vandetanib) leads to growth inhibition [33, 77]. Conversely, treatment of these cells with gefitinib or crizotinib, which do not possess anti-RET activity, is ineffective at suppressing growth [33, 77]. More recently, a CCDC6-RET fusion was identified in the human lung cancer cell line LC-2/ad [92]. Consistent with findings from other in vitro models, these cells are also highly sensitive to treatment with vandetanib.
Several agents with anti-RET activity (e.g., sunitinib, sorafenib, and vandetanib) have been evaluated previously in NSCLC clinical trials. These agents were associated with relatively low response rates (0%–10%); however, all were evaluated in unselected patient populations [93–96]. Clinical trials enriching for RET rearrangements are clearly needed. Indeed, clinical trials using the RET inhibitors ponatinib (ClinicalTrials.gov identifier NCT01813734) and cabozantinib (ClinicalTrials.gov identifier NCT01639508) are currently being pursued in this patient population. Drilon and colleagues recently reported preliminary data from an ongoing trial using cabozantinib [85]. Among three RET-positive patients treated with cabozantinib, confirmed partial responses were observed in two patients, and a third has experienced prolonged stable disease (31 weeks). All patients were still on therapy at the time of reporting. Subsequently, Gautschi and colleagues described one patient with RET-positive NSCLC who responded to treatment with vandetanib [97]. Although the number of patients in these initial reports is small and follow-up is limited, such data offer early proof-of-principle that RET is a potentially targetable oncogenic driver in NSCLC.
Summary
The success of targeted therapy in ALK-positive NSCLC has prompted additional gene discovery efforts geared toward the identification of novel oncogenic fusions. In this setting, ROS1 and RET fusions have emerged as independent and potentially targetable oncogenic drivers in NSCLC. Although the prevalence of each alteration is low (approximately 1%–2%), this translates into a combined 4,000–8,000 cases in the United States annually. Preclinical and early clinical data suggest that ROS1 and RET fusions are promising therapeutic targets that may become more broadly applicable to practicing oncologists as additional data emerge from ongoing clinical trials in these molecularly defined populations. At present, NCCN guidelines specify that clinicians may use crizotinib in ROS1-positive patients [49], although formal recommendations regarding ROS1 screening have not been proposed. Similarly, screening recommendations for RET fusions have not been established.
It is our hope that knowledge of the unique clinicopathologic features associated with ROS1 and RET fusions will inform future screening strategies and enable clinicians to direct patients to clinical trials targeting such populations. Furthermore, as tumor genotyping practices evolve and are consolidated into multigene panels, clinicians will likely encounter patients with rare but clinically relevant genetic alterations such as ROS1 and RET fusions.
As targeted therapies are developed for such small, genetically defined subsets of patients, new challenges will emerge. Most ongoing trials involving ROS1- and RET-positive patients are single-arm studies with ORR or PFS as efficacy endpoints. This raises the question of whether the efficacy observed in single-arm studies will be viewed as sufficient by regulatory authorities or whether randomized trials of targeted therapies versus chemotherapy in these rare patient populations will be required for approval. Finally, as with other targeted therapy paradigms, resistance to ROS1- and RET-directed therapies is likely to be inevitable. Focus should be placed on identifying mechanisms of resistance and developing strategies to prevent or delay the emergence of resistant cancers.
Acknowledgments
This work was supported by a grant from the U.S. National Institutes of Health 5R01CA164273-02 and by a V Foundation Translational Research Grant.
Author Contributions
Conception/Design: Justin Gainor, Alice Shaw
Provision of study material or patients: None
Collection and/or assembly of data: Justin Gainor, Alice Shaw
Data of analysis and interpretation: None
Manuscript writing:Justin Gainor, Alice Shaw
Final approval of manuscript: Justin Gainor, Alice Shaw
Disclosures
Alice Shaw: Pfizer, Ariad, Novartis (C/A); Pfizer, Ariad, Novartis (RF). The other author indicated no financial relationship.
(C/A) Consulting/advisory relationship; (RF) Research funding; (E) Employment; (H) Honoraria received; (OI) Ownership interests; (IP) Intellectual property rights/inventor/patent holder; (SAB) Scientific advisory board
Reference
- 1.Lynch TJ, Bell DW, Sordella R, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350:2129–2139. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 2.Paez JG, Jänne PA, Lee JC, et al. EGFR mutations in lung cancer: Correlation with clinical response to gefitinib therapy. Science. 2004;304:1497–1500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- 3.Pao W, Miller V, Zakowski M, et al. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci U S A. 2004;101:13306–13311. doi: 10.1073/pnas.0405220101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mitelman F, Johansson B, Mertens F. The impact of translocations and gene fusions on cancer causation. Nat Rev Cancer. 2007;7:233–245. doi: 10.1038/nrc2091. [DOI] [PubMed] [Google Scholar]
- 5.Kaye FJ. Mutation-associated fusion cancer genes in solid tumors. Mol Cancer Ther. 2009;8:1399–1408. doi: 10.1158/1535-7163.MCT-09-0135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Soda M, Choi YL, Enomoto M, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. 2007;448:561–566. doi: 10.1038/nature05945. [DOI] [PubMed] [Google Scholar]
- 7.Shaw AT, Yeap BY, Mino-Kenudson M, et al. Clinical features and outcome of patients with non-small-cell lung cancer who harbor EML4-ALK. J Clin Oncol. 2009;27:4247–4253. doi: 10.1200/JCO.2009.22.6993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kwak EL, Bang YJ, Camidge DR, et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med. 2010;363:1693–1703. doi: 10.1056/NEJMoa1006448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Birchmeier C, Birnbaum D, Waitches G, et al. Characterization of an activated human ros gene. Mol Cell Biol. 1986;6:3109–3116. doi: 10.1128/mcb.6.9.3109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nagarajan L, Louie E, Tsujimoto Y, et al. The human c-ros gene (ROS) is located at chromosome region 6q16—-6q22. Proc Natl Acad Sci U S A. 1986;83:6568–6572. doi: 10.1073/pnas.83.17.6568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Matsushime H, Wang LH, Shibuya M. Human c-ros-1 gene homologous to the v-ros sequence of UR2 sarcoma virus encodes for a transmembrane receptorlike molecule. Mol Cell Biol. 1986;6:3000–3004. doi: 10.1128/mcb.6.8.3000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Acquaviva J, Wong R, Charest A. The multifaceted roles of the receptor tyrosine kinase ROS in development and cancer. Biochim Biophys Acta. 2009;1795:37–52. doi: 10.1016/j.bbcan.2008.07.006. [DOI] [PubMed] [Google Scholar]
- 13.Sonnenberg E, Gödecke A, Walter B, et al. Transient and locally restricted expression of the ros1 protooncogene during mouse development. EMBO J. 1991;10:3693–3702. doi: 10.1002/j.1460-2075.1991.tb04937.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tessarollo L, Nagarajan L, Parada LF. C-ros: The vertebrate homolog of the sevenless tyrosine kinase receptor is tightly regulated during organogenesis in mouse embryonic development. Development. 1992;115:11–20. doi: 10.1242/dev.115.1.11. [DOI] [PubMed] [Google Scholar]
- 15.Sonnenberg-Riethmacher E, Walter B, Riethmacher D, et al. The c-ros tyrosine kinase receptor controls regionalization and differentiation of epithelial cells in the epididymis. Genes Dev. 1996;10:1184–1193. doi: 10.1101/gad.10.10.1184. [DOI] [PubMed] [Google Scholar]
- 16.Birchmeier C, Sharma S, Wigler M. Expression and rearrangement of the ROS1 gene in human glioblastoma cells. Proc Natl Acad Sci U S A. 1987;84:9270–9274. doi: 10.1073/pnas.84.24.9270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sharma S, Birchmeier C, Nikawa J, et al. Characterization of the ros1-gene products expressed in human glioblastoma cell lines. Oncogene Res. 1989;5:91–100. [PubMed] [Google Scholar]
- 18.Charest A, Lane K, McMahon K, et al. Association of a novel PDZ domain-containing peripheral Golgi protein with the Q-SNARE (Q-soluble N-ethylmaleimide-sensitive fusion protein (NSF) attachment protein receptor) protein syntaxin 6. J Biol Chem. 2001;276:29456–29465. doi: 10.1074/jbc.M104137200. [DOI] [PubMed] [Google Scholar]
- 19.Charest A, Lane K, McMahon K, et al. Fusion of FIG to the receptor tyrosine kinase ROS in a glioblastoma with an interstitial del(6)(q21q21) Genes Chromosomes Cancer. 2003;37:58–71. doi: 10.1002/gcc.10207. [DOI] [PubMed] [Google Scholar]
- 20.Charest A, Kheifets V, Park J, et al. Oncogenic targeting of an activated tyrosine kinase to the Golgi apparatus in a glioblastoma. Proc Natl Acad Sci U S A. 2003;100:916–921. doi: 10.1073/pnas.242741799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gu TL, Deng X, Huang F, et al. Survey of tyrosine kinase signaling reveals ROS kinase fusions in human cholangiocarcinoma. PLoS One. 2011;6:e15640. doi: 10.1371/journal.pone.0015640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Birch AH, Arcand SL, Oros KK, et al. Chromosome 3 anomalies investigated by genome wide SNP analysis of benign, low malignant potential and low grade ovarian serous tumours. PLoS One. 2011;6:e28250. doi: 10.1371/journal.pone.0028250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee J, Lee SE, Kang SY, et al. Identification of ROS1 rearrangement in gastric adenocarcinoma. Cancer. 2013;119:1627–1635. doi: 10.1002/cncr.27967. [DOI] [PubMed] [Google Scholar]
- 24.Rikova K, Guo A, Zeng Q, et al. Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell. 2007;131:1190–1203. doi: 10.1016/j.cell.2007.11.025. [DOI] [PubMed] [Google Scholar]
- 25.Bergethon K, Shaw AT, Ou SH, et al. ROS1 rearrangements define a unique molecular class of lung cancers. J Clin Oncol. 2012;30:863–870. doi: 10.1200/JCO.2011.35.6345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li C, Fang R, Sun Y, et al. Spectrum of oncogenic driver mutations in lung adenocarcinomas from East Asian never smokers. PLoS One. 2011;6:e28204. doi: 10.1371/journal.pone.0028204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Davies KD, Le AT, Theodoro MF, et al. Identifying and targeting ROS1 gene fusions in non-small cell lung cancer. Clin Cancer Res. 2012;18:4570–4579. doi: 10.1158/1078-0432.CCR-12-0550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rimkunas VM, Crosby K, Kelly M, et al. Analysis of receptor tyrosine kinase ROS1-positive tumors in non-small cell lung cancer: Identification of a FIG-ROS1 fusion. Clin Cancer Res. 2012;18:4449–4457. doi: 10.1158/1078-0432.CCR-11-3351. [DOI] [PubMed] [Google Scholar]
- 29.Arai Y, Totoki Y, Takahashi H, et al. Mouse model for ROS1-rearranged lung cancer. PLoS One. 2013;8:e56010. doi: 10.1371/journal.pone.0056010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Govindan R, Ding L, Griffith M, et al. Genomic landscape of non-small cell lung cancer in smokers and never-smokers. Cell. 2012;150:1121–1134. doi: 10.1016/j.cell.2012.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Seo JS, Ju YS, Lee WC, et al. The transcriptional landscape and mutational profile of lung adenocarcinoma. Genome Res. 2012;22:2109–2119. doi: 10.1101/gr.145144.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Suehara Y, Arcila M, Wang L, et al. Identification of KIF5B-RET and GOPC-ROS1 fusions in lung adenocarcinomas through a comprehensive mRNA-based screen for tyrosine kinase fusions. Clin Cancer Res. 2012;18:6599–6608. doi: 10.1158/1078-0432.CCR-12-0838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Takeuchi K, Soda M, Togashi Y, et al. RET, ROS1 and ALK fusions in lung cancer. Nat Med. 2012;18:378–381. doi: 10.1038/nm.2658. [DOI] [PubMed] [Google Scholar]
- 34.Yoshida A, Kohno T, Tsuta K, et al. ROS1-rearranged lung cancer: A clinicopathologic and molecular study of 15 surgical cases. Am J Surg Pathol. 2013;37:554–562. doi: 10.1097/PAS.0b013e3182758fe6. [DOI] [PubMed] [Google Scholar]
- 35.Jun HJ, Johnson H, Bronson RT, et al. The oncogenic lung cancer fusion kinase CD74-ROS activates a novel invasiveness pathway through E-Syt1 phosphorylation. Cancer Res. 2012;72:3764–3774. doi: 10.1158/0008-5472.CAN-11-3990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Charest A, Wilker EW, McLaughlin ME, et al. ROS fusion tyrosine kinase activates a SH2 domain-containing phosphatase-2/phosphatidylinositol 3-kinase/mammalian target of rapamycin signaling axis to form glioblastoma in mice. Cancer Res. 2006;66:7473–7481. doi: 10.1158/0008-5472.CAN-06-1193. [DOI] [PubMed] [Google Scholar]
- 37.El-Deeb IM, Yoo KH, Lee SH. ROS receptor tyrosine kinase: A new potential target for anticancer drugs. Med Res Rev. 2011;31:794–818. doi: 10.1002/med.20206. [DOI] [PubMed] [Google Scholar]
- 38.Ju YS, Lee WC, Shin JY, et al. A transforming KIF5B and RET gene fusion in lung adenocarcinoma revealed from whole-genome and transcriptome sequencing. Genome Res. 2012;22:436–445. doi: 10.1101/gr.133645.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chin LP, Soo RA, Soong R, et al. Targeting ROS1 with anaplastic lymphoma kinase inhibitors: A promising therapeutic strategy for a newly defined molecular subset of non-small-cell lung cancer. J Thorac Oncol. 2012;7:1625–1630. doi: 10.1097/JTO.0b013e31826baf83. [DOI] [PubMed] [Google Scholar]
- 40.Shaw AT, Solomon B, Kenudson MM. Crizotinib and testing for ALK. J Natl Compr Canc Netw. 2011;9:1335–1341. doi: 10.6004/jnccn.2011.0115. [DOI] [PubMed] [Google Scholar]
- 41.Martelli MP, Sozzi G, Hernandez L, et al. EML4-ALK rearrangement in non-small cell lung cancer and non-tumor lung tissues. Am J Pathol. 2009;174:661–670. doi: 10.2353/ajpath.2009.080755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stumpfova M, Jänne PA. Zeroing in on ROS1 rearrangements in non-small cell lung cancer. Clin Cancer Res. 2012;18:4222–4224. doi: 10.1158/1078-0432.CCR-12-1812. [DOI] [PubMed] [Google Scholar]
- 43.Jänne PA, Meyerson M. ROS1 rearrangements in lung cancer: A new genomic subset of lung adenocarcinoma. J Clin Oncol. 2012;30:878–879. doi: 10.1200/JCO.2011.39.4197. [DOI] [PubMed] [Google Scholar]
- 44.McDermott U, Iafrate AJ, Gray NS, et al. Genomic alterations of anaplastic lymphoma kinase may sensitize tumors to anaplastic lymphoma kinase inhibitors. Cancer Res. 2008;68:3389–3395. doi: 10.1158/0008-5472.CAN-07-6186. [DOI] [PubMed] [Google Scholar]
- 45.Yasuda H, de Figueiredo-Pontes LL, Kobayashi S, et al. Preclinical rationale for use of the clinically available multitargeted tyrosine kinase inhibitor crizotinib in ROS1-translocated lung cancer. J Thorac Oncol. 2012;7:1086–1090. doi: 10.1097/JTO.0b013e3182570919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shaw A, Camidge D, Engelman J, et al. Clinical activity of crizotinib in advanced non-small cell lung cancer (NSCLC) harboring ROS1 gene rearrangement. J Clin Oncol. 2012;30(suppl):7508a. [Google Scholar]
- 47.Ou S, Camidge R, Engelman J, et al. Clinical activity of crizotinib in patients with advanced non-small cell lung cancer (NSCLC) harboring ROS1 gene rearrangement. Ann Oncol. 2012;23(suppl):2091a. [Google Scholar]
- 48.Proia D, Acquaviva J, Jiang Q, et al. Preclinical activity of the HSP90 inhibitor, ganetespib, in ALK- and ROS1-driven cancers. J Clin Oncol. 2012;30(suppl):3090a. [Google Scholar]
- 49.National Comprehensive Cancer Network. Non-small cell lung cancer (version 2.2013) [Accessed April 28, 2013]. Available at http://www.nccn.org/professionals/physician_gls/pdf/nscl.pdf.
- 50.Takahashi M, Ritz J, Cooper GM. Activation of a novel human transforming gene, ret, by DNA rearrangement. Cell. 1985;42:581–588. doi: 10.1016/0092-8674(85)90115-1. [DOI] [PubMed] [Google Scholar]
- 51.Ishizaka Y, Itoh F, Tahira T, et al. Human ret proto-oncogene mapped to chromosome 10q11.2. Oncogene. 1989;4:1519–1521. [PubMed] [Google Scholar]
- 52.Wells SA, Santoro M. Targeting the RET pathway in thyroid cancer. Clin Cancer Res. 2009;15:7119–7123. doi: 10.1158/1078-0432.CCR-08-2742. [DOI] [PubMed] [Google Scholar]
- 53.Nakamura T, Ishizaka Y, Nagao M, et al. Expression of the ret proto-oncogene product in human normal and neoplastic tissues of neural crest origin. J Pathol. 1994;172:255–260. doi: 10.1002/path.1711720305. [DOI] [PubMed] [Google Scholar]
- 54.Takahashi M, Buma Y, Iwamoto T, et al. Cloning and expression of the ret proto-oncogene encoding a tyrosine kinase with two potential transmembrane domains. Oncogene. 1988;3:571–578. [PubMed] [Google Scholar]
- 55.Schuchardt A, D'Agati V, Larsson-Blomberg L, et al. Defects in the kidney and enteric nervous system of mice lacking the tyrosine kinase receptor Ret. Nature. 1994;367:380–383. doi: 10.1038/367380a0. [DOI] [PubMed] [Google Scholar]
- 56.Pichel JG, Shen L, Sheng HZ, et al. Defects in enteric innervation and kidney development in mice lacking GDNF. Nature. 1996;382:73–76. doi: 10.1038/382073a0. [DOI] [PubMed] [Google Scholar]
- 57.Eng C. RET proto-oncogene in the development of human cancer. J Clin Oncol. 1999;17:380–393. doi: 10.1200/JCO.1999.17.1.380. [DOI] [PubMed] [Google Scholar]
- 58.Phay JE, Shah MH. Targeting RET receptor tyrosine kinase activation in cancer. Clin Cancer Res. 2010;16:5936–5941. doi: 10.1158/1078-0432.CCR-09-0786. [DOI] [PubMed] [Google Scholar]
- 59.Trupp M, Arenas E, Fainzilber M, et al. Functional receptor for GDNF encoded by the c-ret proto-oncogene. Nature. 1996;381:785–789. doi: 10.1038/381785a0. [DOI] [PubMed] [Google Scholar]
- 60.Durbec P, Marcos-Gutierrez CV, Kilkenny C, et al. GDNF signalling through the Ret receptor tyrosine kinase. Nature. 1996;381:789–793. doi: 10.1038/381789a0. [DOI] [PubMed] [Google Scholar]
- 61.Jing S, Wen D, Yu Y, et al. GDNF-induced activation of the ret protein tyrosine kinase is mediated by GDNFR-alpha, a novel receptor for GDNF. Cell. 1996;85:1113–1124. doi: 10.1016/s0092-8674(00)81311-2. [DOI] [PubMed] [Google Scholar]
- 62.Ibáñez CF. Structure and physiology of the RET receptor tyrosine kinase. Cold Spring Harb Perspect Biol. 2013;5:a009134. doi: 10.1101/cshperspect.a009134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Edery P, Lyonnet S, Mulligan LM, et al. Mutations of the RET proto-oncogene in Hirschsprung's disease. Nature. 1994;367:378–380. doi: 10.1038/367378a0. [DOI] [PubMed] [Google Scholar]
- 64.Romeo G, Ronchetto P, Luo Y, et al. Point mutations affecting the tyrosine kinase domain of the RET proto-oncogene in Hirschsprung's disease. Nature. 1994;367:377–378. doi: 10.1038/367377a0. [DOI] [PubMed] [Google Scholar]
- 65.Grieco M, Santoro M, Berlingieri MT, et al. PTC is a novel rearranged form of the ret proto-oncogene and is frequently detected in vivo in human thyroid papillary carcinomas. Cell. 1990;60:557–563. doi: 10.1016/0092-8674(90)90659-3. [DOI] [PubMed] [Google Scholar]
- 66.Santoro M, Melillo RM, Fusco A. RET/PTC activation in papillary thyroid carcinoma: European Journal of Endocrinology Prize Lecture. Eur J Endocrinol. 2006;155:645–653. doi: 10.1530/eje.1.02289. [DOI] [PubMed] [Google Scholar]
- 67.Bounacer A, Wicker R, Schlumberger M, et al. Oncogenic rearrangements of the ret proto-oncogene in thyroid tumors induced after exposure to ionizing radiation. Biochimie. 1997;79:619–623. doi: 10.1016/s0300-9084(97)82012-3. [DOI] [PubMed] [Google Scholar]
- 68.Ito T, Seyama T, Iwamoto KS, et al. Activated RET oncogene in thyroid cancers of children from areas contaminated by Chernobyl accident. Lancet. 1994;344:259. doi: 10.1016/s0140-6736(94)93024-4. [DOI] [PubMed] [Google Scholar]
- 69.Hamatani K, Eguchi H, Ito R, et al. RET/PTC rearrangements preferentially occurred in papillary thyroid cancer among atomic bomb survivors exposed to high radiation dose. Cancer Res. 2008;68:7176–7182. doi: 10.1158/0008-5472.CAN-08-0293. [DOI] [PubMed] [Google Scholar]
- 70.Drosten M, Pützer BM. Mechanisms of disease: Cancer targeting and the impact of oncogenic RET for medullary thyroid carcinoma therapy. Nat Clin Pract Oncol. 2006;3:564–574. doi: 10.1038/ncponc0610. [DOI] [PubMed] [Google Scholar]
- 71.Donis-Keller H, Dou S, Chi D, et al. Mutations in the RET proto-oncogene are associated with MEN 2A and FMTC. Hum Mol Genet. 1993;2:851–856. doi: 10.1093/hmg/2.7.851. [DOI] [PubMed] [Google Scholar]
- 72.Mulligan LM, Kwok JB, Healey CS, et al. Germ-line mutations of the RET proto-oncogene in multiple endocrine neoplasia type 2A. Nature. 1993;363:458–460. doi: 10.1038/363458a0. [DOI] [PubMed] [Google Scholar]
- 73.Carlson KM, Dou S, Chi D, et al. Single missense mutation in the tyrosine kinase catalytic domain of the RET protooncogene is associated with multiple endocrine neoplasia type 2B. Proc Natl Acad Sci U S A. 1994;91:1579–1583. doi: 10.1073/pnas.91.4.1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Romei C, Elisei R, Pinchera A, et al. Somatic mutations of the ret protooncogene in sporadic medullary thyroid carcinoma are not restricted to exon 16 and are associated with tumor recurrence. J Clin Endocrinol Metab. 1996;81:1619–1622. doi: 10.1210/jcem.81.4.8636377. [DOI] [PubMed] [Google Scholar]
- 75.Marsh DJ, Learoyd DL, Andrew SD, et al. Somatic mutations in the RET proto-oncogene in sporadic medullary thyroid carcinoma. Clin Endocrinol (Oxf) 1996;44:249–257. doi: 10.1046/j.1365-2265.1996.681503.x. [DOI] [PubMed] [Google Scholar]
- 76.Kohno T, Ichikawa H, Totoki Y, et al. KIF5B-RET fusions in lung adenocarcinoma. Nat Med. 2012;18:375–377. doi: 10.1038/nm.2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lipson D, Capelletti M, Yelensky R, et al. Identification of new ALK and RET gene fusions from colorectal and lung cancer biopsies. Nat Med. 2012;18:382–384. doi: 10.1038/nm.2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Li F, Feng Y, Fang R, et al. Identification of RET gene fusion by exon array analyses in “pan-negative” lung cancer from never smokers. Cell Res. 2012;22:928–931. doi: 10.1038/cr.2012.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wang R, Hu H, Pan Y, et al. RET fusions define a unique molecular and clinicopathologic subtype of non-small-cell lung cancer. J Clin Oncol. 2012;30:4352–4359. doi: 10.1200/JCO.2012.44.1477. [DOI] [PubMed] [Google Scholar]
- 80.Yokota K, Sasaki H, Okuda K, et al. KIF5B/RET fusion gene in surgically-treated adenocarcinoma of the lung. Oncol Rep. 2012;28:1187–1192. doi: 10.3892/or.2012.1908. [DOI] [PubMed] [Google Scholar]
- 81.Sasaki H, Shimizu S, Tani Y, et al. RET expression and detection of KIF5B/RET gene rearrangements in Japanese lung cancer. Cancer Med. 2012;1:68–75. doi: 10.1002/cam4.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cai W, Su C, Li X, et al. KIF5B-RET fusions in Chinese patients with non-small cell lung cancer. Cancer. 2013;119:1486–1494. doi: 10.1002/cncr.27940. [DOI] [PubMed] [Google Scholar]
- 83.Kim SC, Jung Y, Park J, et al. A high-dimensional, deep-sequencing study of lung adenocarcinoma in female never-smokers. PLoS One. 2013;8:e55596. doi: 10.1371/journal.pone.0055596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chao BH, Briesewitz R, Villalona-Calero MA. RET fusion genes in non-small-cell lung cancer. J Clin Oncol. 2012;30:4439–4441. doi: 10.1200/JCO.2012.45.8240. [DOI] [PubMed] [Google Scholar]
- 85.Drilon A, Wang L, Hasanovic A, et al. Response to cabozantinib in patients with RET fusion-positive lung adenocarcinomas. Cancer Discov. 2013 doi: 10.1158/2159-8290.CD-13-0035. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Pao W, Hutchinson KE. Chipping away at the lung cancer genome. Nat Med. 2012;18:349–351. doi: 10.1038/nm.2697. [DOI] [PubMed] [Google Scholar]
- 87.Su AI, Cooke MP, Ching KA, et al. Large-scale analysis of the human and mouse transcriptomes. Proc Natl Acad Sci U S A. 2002;99:4465–4470. doi: 10.1073/pnas.012025199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Marotta V, Guerra A, Sapio MR, et al. RET/PTC rearrangement in benign and malignant thyroid diseases: A clinical standpoint. Eur J Endocrinol. 2011;165:499–507. doi: 10.1530/EJE-11-0499. [DOI] [PubMed] [Google Scholar]
- 89.Wells SA, Robinson BG, Gagel RF, et al. Vandetanib in patients with locally advanced or metastatic medullary thyroid cancer: A randomized, double-blind phase III trial. J Clin Oncol. 2012;30:134–141. doi: 10.1200/JCO.2011.35.5040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Schoffski P, Ellisei R, Muller S, et al. An international, double-blind, randomized, placebo-controlled phase III trial (EXAM) of cabozantinib (XL184) in medullary thyroid carcinoma (MTC) patients (pts) with documented RECIST progression at baseline. J Clin Oncol. 2012;30(suppl):5508a. [Google Scholar]
- 91.Houvras Y. Completing the arc: Targeted inhibition of RET in medullary thyroid cancer. J Clin Oncol. 2012;30:200–202. doi: 10.1200/JCO.2011.38.7639. [DOI] [PubMed] [Google Scholar]
- 92.Matsubara D, Kanai Y, Ishikawa S, et al. Identification of CCDC6-RET fusion in the human lung adenocarcinoma cell line, LC-2/ad. J Thorac Oncol. 2012;7:1872–1876. doi: 10.1097/JTO.0b013e3182721ed1. [DOI] [PubMed] [Google Scholar]
- 93.Socinski MA, Novello S, Brahmer JR, et al. Multicenter, phase II trial of sunitinib in previously treated, advanced non-small-cell lung cancer. J Clin Oncol. 2008;26:650–656. doi: 10.1200/JCO.2007.13.9303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Novello S, Scagliotti GV, Rosell R, et al. Phase II study of continuous daily sunitinib dosing in patients with previously treated advanced non-small cell lung cancer. Br J Cancer. 2009;101:1543–1548. doi: 10.1038/sj.bjc.6605346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Blumenschein GR, Gatzemeier U, Fossella F, et al. Phase II, multicenter, uncontrolled trial of single-agent sorafenib in patients with relapsed or refractory, advanced non-small-cell lung cancer. J Clin Oncol. 2009;27:4274–4280. doi: 10.1200/JCO.2009.22.0541. [DOI] [PubMed] [Google Scholar]
- 96.Lee JS, Hirsh V, Park K, et al. Vandetanib versus placebo in patients with advanced non-small-cell lung cancer after prior therapy with an epidermal growth factor receptor tyrosine kinase inhibitor: A randomized, double-blind phase III trial (ZEPHYR) J Clin Oncol. 2012;30:1114–1121. doi: 10.1200/JCO.2011.36.1709. [DOI] [PubMed] [Google Scholar]
- 97.Gautschi O, Zander T, Keller FA, et al. A patient with lung adenocarcinoma and RET fusion treated with vandetanib. J Thorac Oncol. 2013;8:e43–e44. doi: 10.1097/JTO.0b013e31828a4d07. [DOI] [PubMed] [Google Scholar]