

Lymphoma is the most common malignancy arising in the ocular adnexa, which includes conjunctiva, lachrymal gland, lachrymal sac, eyelids, orbit soft tissue, and extraocular muscles. Ocular adnexal lymphoma (OAL) accounts for 1%–2% of non-Hodgkin lymphoma and 5%–15% of extranodal lymphoma. The most common and well-studied histotype is represented by marginal zone B-cell lymphoma. This review covers the prevalence, clinical presentation, behavior, and histological and molecular features of uncommon forms of primary OAL and provides practical recommendations for therapeutic management.
Keywords: Ocular adnexal lymphoma, Diffuse large B-cell lymphoma, Follicular lymphoma, Mantle cell lymphoma, T-cell lymphoma
Abstract
Lymphoma is the most common malignancy arising in the ocular adnexa, which includes conjunctiva, lachrymal gland, lachrymal sac, eyelids, orbit soft tissue, and extraocular muscles. Ocular adnexal lymphoma (OAL) accounts for 1%–2% of non-Hodgkin lymphoma and 5%–15% of extranodal lymphoma. Histology, stage, and primary localizations are the most important variables influencing the natural history and therapeutic outcome of these malignancies. Among the various lymphoma variants that could arise in the ocular adnexa, marginal zone B-cell lymphoma (OA-MZL) is the most common one. Other types of lymphoma arise much more rarely in these anatomical sites; follicular lymphoma is the second most frequent histology, followed by diffuse large B-cell lymphoma and mantle cell lymphoma. Additional lymphoma entities, like T-cell/natural killer cell lymphomas and Burkitt lymphoma, only occasionally involve orbital structures. Because they are so rare, related literature mostly consists of anecdotal cases included within series focused on OA-MZL and sporadic case reports. This bias hampers a global approach to clinical and molecular properties of these types of lymphoma, with a low level of evidence supporting therapeutic options. This review covers the prevalence, clinical presentation, behavior, and histological and molecular features of uncommon forms of primary OAL and provides practical recommendations for therapeutic management.
Implications for Practice:
The vast majority of literature's data on ocular adnexa lymphomas focus on the most frequent herein encountered histotype, i.e., marginal zone B-cell lymphoma. However, when a physician encounters less common histologies in this anatomical site, difficulties in their recognition and management may arise, because these tumors are usually reported as minor groups within large series describing marginal zone B-cell lymphomas or under case report format. The present review aims to offer a comprehensive, easily accessible review on these uncommon histologies in order to collect and present available pathologic, clinical, and therapeutic data other than providing own authors' experience in individual considered entities.
Introduction
Orbital lymphomas can be divided into three main groups: (a) the primary ocular adnexal lymphomas (POALs), which includes those forms arising in the conjunctiva, lachrymal gland, lachrymal sac, eyelids, orbit soft tissue, and extraocular muscles, accounting for 1%–2% of all non-Hodgkin lymphomas (NHLs) and 5%–15% of extranodal lymphomas [1–3]; (b) the secondary ocular adnexal lymphomas (SOALs), including disseminated lymphomas affecting the ocular adnexa at diagnosis or relapse, which are diagnosed and treated like their systemic counterparts; and (c) intraocular lymphoma, which is usually a diffuse large B-cell lymphoma (DLBCL) arising in the eyeball, classified within the aggressive primary central nervous system (CNS) lymphoma category. The most frequent and better studied POAL is ocular adnexal marginal zone lymphoma (OA-MZL) of mucosa-associated lymphoid tissue (MALT) type. Although there is a large amount of data covering both the clinical and biological properties of OA-MZL, the available literature on POALs of other histology is represented by sporadic case reports and a few cases included within the series, mostly OA-MZL cases.
This review summarizes the available clinical, pathological, molecular, and therapeutic data on rare forms of POAL other than OA-MZL, aiming to better characterize their clinical and molecular features and to provide practical guidelines for their correct recognition, which is critical to avoid confusion with OA-MZL and, most importantly, to drive the best therapeutic management. To allow reliable comparison among reported series, only the articles published after the introduction of the Revised European American Lymphoma Classification in 1994 and the worldwide acceptance of some lymphoma entities, such as MALT and mantle cell lymphomas, were considered.
Clinical Presentation and Imaging
Clinical and radiological features of POAL, although highly indicative, do not usually differentiate lymphomas from benign hyperplasia or between indolent and aggressive lymphomas. Presentation varies according to involved structures. Usually, the lymphomatous lesion appears as an enlarging mass, causing proptosis and swelling, with or without minimal pain and inflammation, but other common presentations include conjunctival salmon patches, ptosis, exophtalm, diplopia, pain, loss of vision, epiphora, dacriocystitis, and/or gritty sensation [3–6]. Site-specific manifestations include tearing (lachrymal sac involvement), limited eye excursions (extraocular muscle involvement), visual function impairment (rapidly growing forms determining choroidal folds), and defects of visual acuity and visual field. Acute, inflammatory-like signs and symptoms mimicking dacryocystitis, dacryoadenitis, and orbital pseudotumor can also be observed. Indolent lymphomas mainly remain localized to orbital tissues, whereas the erosion of the orbital osseous walls is more common in aggressive lymphomas, sometimes resulting in the direct infiltration of intracranial structures and meningeal dissemination.
Orbit ultrasonography and magnetic resonance imaging (MRI) play an important role when suspecting OAL. Orbit ultrasonography could be useful to differentiate lymphomas from other tumors and tumor-like lesions, but MRI is the gold standard in terms of imaging techniques in the evaluation of ocular adnexal malignancies [7]. When MRI is not available, contrasted CT scan may also be useful to assess lymphomatous lesions.
Diagnosis, Histology, and Sites of Involvement
Clinical and imaging findings are unable to define lymphoma histology; accordingly, surgical biopsy is mandatory to obtain tissue samples for morphologic, immunophenotypical, and molecular diagnosis of orbital lesions [8, 9]. In all, 55%–60% of orbital lymphomas are OA-MZL, 19% are follicular lymphomas (FL), and 15%–20% are aggressive lymphomas (DLBCL or mantle cell lymphoma [MCL]). The orbit is involved in 46%–74% of POAL cases, conjunctiva in 20%–33%, lachrymal gland in 15%–20%, and the eyelid in 5%–20% of cases [10]. Conjunctiva is usually infiltrated by OA-MZL; in the lachrymal gland, OA-MZL and DLBCL are equally prevalent [10]. OAL occurring in the eyelid—a rare phenomenon—has a more aggressive course than orbital or conjunctival disease. Bilateral involvement of ocular adnexal structures was reported in 10%–20% of cases [3, 4, 11, 12], with lower figures for non-OA-MZL histologies [13].
Staging
OAL staging is critical because the presence of extraorbital disease usually drives therapeutic decisions and negatively influences prognosis [3, 14, 15]. Basically, staging should include complete blood count, bone marrow biopsy, and total body computed tomography (CT) scan [5, 9]. Further investigations vary according to histology and/or accompanying symptoms. For instance, whole brain MRI and cerebrospinal fluid evaluation are suggested in patients with OA-DLBCL [16]. Positron emission tomography (PET) shows higher sensitivity in the detection of systemic disease when compared with traditional imaging techniques, sometimes with a subsequent major change in therapeutic decision [17] and mostly in patients with aggressive lymphomas, because their tumor cells display higher 18-FDG avidity compared with indolent lymphomas. Conversely, PET provides lower sensitivity in detecting orbital lesions in comparison to traditional imaging (27% vs. 73%), mostly due to the interference of preferential uptake of 18-FDG by extraocular muscles and brain parenchyma and to the usually small size of OAL lesions. PET/CT, used according to consensus guidelines for systemic lymphomas, may overcome this limitation (Fig. 1) [17, 18].
Figure 1.

Computed tomography scan (A) and corresponding positron emission tomography imaging (B) of a male patient presenting with right lachrymal sac diffuse large B-cell lymphoma (arrows).
In patients with OA-MCL, gastrointestinal and peripheral blood involvement should be assessed by endoscopy and flow cytometry, respectively, because occult disseminated disease is detected in three-fourths of patients with MCL who are asymptomatic at presentation [19–21].
Correlation With Infectious Agents and Other Pathogenetic Hypotheses
The interest in investigating the role of pathogens with lymphoma genic capability in OALs has increased after the characterization of their association with Chlamydophila psittaci infection by means of polymerase chain reaction, immunohistochemistry, immunofluorescence, electron microscopy, and in vitro cultures [22–24]. With geographical variations [25], C. psittaci is strongly associated with OA-MZL. There are little data on other OAL histologys, mostly showing the association between C. psittaci and de novo OA-DLBCL [22, 25] and a few cases of OA-FL and OA-MCL [23, 26]; the assessment of this infection by polymerase chain reaction in OA-MZL should be performed in daily clinical practice, whereas the assessment in other OAL categories should only be performed for investigational purposes. This lymphoma-bacteria association plays a relevant therapeutic role because nearly half of patients with C. psittaci-positive OA-MZL experience lymphoma regression following antibiotic treatment with doxycycline [27], and more than 80% of these patients went into lymphoma regression when C. psittaci was successfully eradicated [28]. To date, this antibiotic therapy has only been successfully used in a single case of DLBCL developing outside ocular adnexal site (bronchus) [29]. Thus, C. psittaci-eradicating antibiotic therapy should only be used in patients with OA-MZL, whereas patients with OAL of other histologies should only be considered in clinical trials. Helicobacter pylori, herpes simplex viruses 1 and 2, adenovirus 8, and hepatitis C virus have been investigated in OA-MZL, but not in other OAL histologies. A single exception is represented by the occurrence of Epstein-Barr virus (EBV) infection in six out of eight patients with OA natural killer (NK)/T-cell lymphomas [30].
Interestingly, a few DLBCL cases transformed from indolent OAL were reported in the setting of patients with immunoglobulin (Ig)G4-related chronic sclerosing dacryoadenitis [31, 32]. OAL have consistently been associated with several autoimmune conditions, such as rheumatoid arthritis, Sjögren syndrome, and systemic lupus erythematous. The exact mechanism of lymphomagenesis in the context of autoimmunity remains largely unexplained, but it may be related to chronic antigenic stimulation, chronic inflammatory response, and deficiency in immune surveillance, promoting a multistep process of genetic instability resulting in accumulation of genetic alterations. In addition, immunosuppressive medications (e.g., methotrexate) may concur to alter patients' immune status.
Prognostic Factors and Therapeutic Principles
Histology and stage are the most influential prognostic variables in OAL [33–35]. In unselected series, the 4-year overall survival (OS) rate stood at 95% in OA-MZL, 65% in OA-FL, 50% in OA-DLBCL, and 0% in patients with OA-PTCL/NK lymphomas and OA-MCL [6]. The prognostic role of different involved ocular adnexa structures is controversial because multiple histologies are considered as a whole group. In general terms, conjunctival lymphomas present lower systemic dissemination rates and better outcome compared with eyelid- and orbital-infiltrating lymphomas [6]. The deepest seated OAL are usually monolateral (91%), with uncommon dissemination (13%) and frequent local dissemination (13%), resulting in paranasal sinus infiltration in 7% of cases [33]. A few unconfirmed studies suggested a prognostic role for lachrymal gland localization. Therapeutic choice in OAL strongly depends on histology, stage, comorbidities, age, performance status, and the impact of the treatment of visual function [36]. Conventional treatments for OAL include surgery, watch and wait, radiotherapy, chemotherapy and immunotherapy.
The prognostic role of different involved ocular adnexa structures is controversial because multiple histologies are considered as a whole group. In general terms, conjunctival lymphomas present lower systemic dissemination rates and better outcome compared with eyelid- and orbital-infiltrating lymphomas.
Surgical excision is limited to lesions of the conjunctiva and orbit in patients with indolent lymphomas who cannot be treated with alternative strategies [4, 37]. Exclusive radiation therapy only retains its role for patients with contraindications to chemotherapy. CNS prophylaxis should be performed in patients with high-risk aggressive OAL. The best regimen or combination remains to be defined, but a combination of intrathecal drug delivery and intravenous high-dose methotrexate seems to be adequate [38].
Peculiar Features and Treatment of Individual Lymphoma Entities
Follicular Lymphoma
Follicular lymphoma (FL) is the second most frequent POAL [39, 40]. It typically occurs in elderly patients (median age: 60 years), shows previous recurrent conjunctivitis, and often involves orbital soft tissue and conjunctiva [40]. Stage at presentation varies largely among studies, which may reflect heterogeneous selection criteria and sample sizes (Table 1). Histologic features of OA-FL recapitulate nodal counterpart, also in terms of frequency of morphological grading and immunophenotype (CD20+, CD10+, bcl-6+, bcl-2+, cytoplasmic Ig light, mainly κ and heavy chains+) [40, 41]. According to WHO classification, grades 1–2 range grades 1–2 range between 20% and 50% [3].
Table 1.
Distribution of the types of ocular adnexal lymphoproliferative lesions in different series (%)

Abbreviation: NK, natural killer.
T(14;18)(q32;q21) is the most common cytogenetic abnormality (70%–95% of cases). Interestingly, aberrant nuclear/cytoplasmic bcl-10 expression was observed in 30%–40% of OA-FL, a much higher prevalence rate in comparison with 10% of corresponding nodal cases [42, 43]. Bcl-10, which is encoded by the BCL10 gene located on chromosome 1p, is a regulatory molecule of apoptosis involved in the activation of nuclear factor-kappa B (NF-κB) pathway, the action for which is critical for cell survival, proliferation, and apoptosis [44]. This aberrant nuclear/cytoplasmic bcl-10 expression does not seem to play a prognostic role [43].
OA-FL shows an indolent course and, despite a high systemic relapse rate, occurs mostly in bone marrow and lymph nodes [13, 39, 40, 45]. It has a good prognosis, with a 5-year OS rate of 72%. Better OS rates have been reported in patients with stage IE disease. Most patients with stage-I/II OA-FL are treated with radiotherapy (Table 2). For FL grade 1 and 2, orbital radiotherapy results in a >85% local disease control rate. Local recurrence rates are between 0% and 15% and distant relapse occurs in 6%–50% of patients, at a median follow-up of >7 years and a 10-year OS rate of 83% [12, 46–52].
Table 2.
Therapeutic management of non-Hodgkin lymphomas involving the ocular adnexa
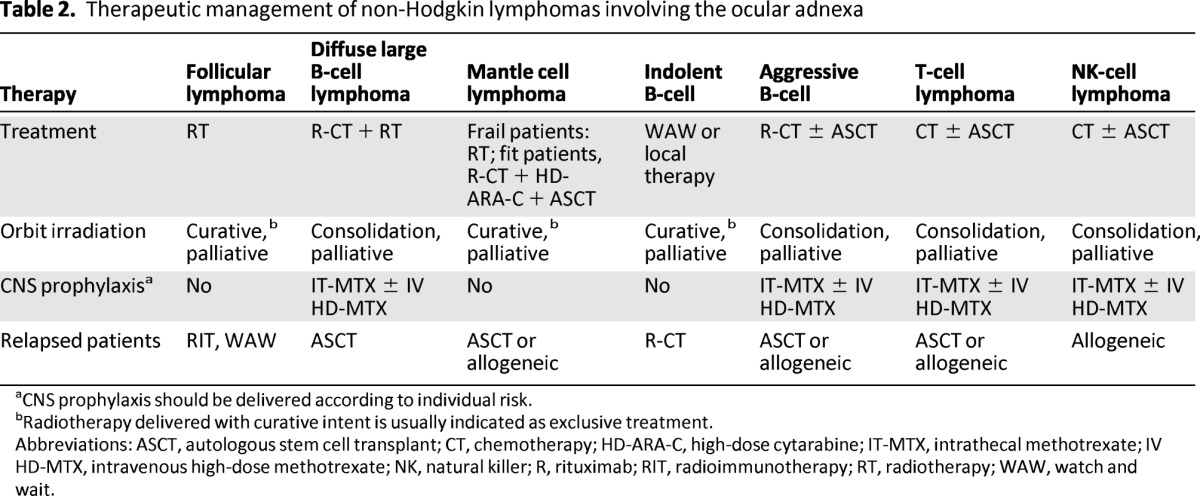
aCNS prophylaxis should be delivered according to individual risk.
bRadiotherapy delivered with curative intent is usually indicated as exclusive treatment.
Abbreviations: ASCT, autologous stem cell transplant; CT, chemotherapy; HD-ARA-C, high-dose cytarabine; IT-MTX, intrathecal methotrexate; IV HD-MTX, intravenous high-dose methotrexate; NK, natural killer; R, rituximab; RIT, radioimmunotherapy; RT, radiotherapy; WAW, watch and wait.
Recommended doses of electron or photon beams for OA-FL oscillate between 28 and 30 Gy [53]. A randomized trial (recently reported as a meeting abstract) demonstrated that a dose of 4 Gy in 2 fractions was as effective as palliative treatment, whereas it was significantly inferior to 24 Gy in 12 fractions in 473 patients with nodal FL [54]. Although this radiation dose was associated with excellent response rates, local disease control, and tolerability, results should be confirmed in OA-FL. In all, 38% of randomized patients had disseminated disease, only 40% of the whole series was irradiated with curative intent, several patients received prior treatments (25% radiotherapy; 34% chemotherapy), and median follow-up was only 23 months. Minor toxicities (i.e., moderate cutaneous or conjunctival reactions, keratoconjunctivitis, dryness or excessive tearing, cataract, or xerophthalmia) may occur, whereas more adverse reactions (i.e., ischemic retinopathy, glaucoma, or corneal ulceration) may arise much less often [47, 55, 56]. For grade 3 OA-FL, systemic chemotherapy is indicated, following standard regimens currently used for nodal FL [57]. Anthracycline-based regimens combined with rituximab are suitable for treating these patients.
In the last few years, anti-CD20 monoclonal antibodies have been added to the treatment of OAL. Anecdotal cases have been reported of patients who were successfully treated with intravenous infusions of rituximab at conventional doses, with excellent tolerability.
In the last few years, anti-CD20 monoclonal antibodies have been added to the treatment of OAL [58]. Anecdotal cases have been reported of patients who were successfully treated with intravenous infusions of rituximab at conventional doses, with excellent tolerability [59]. Intralesional administration of rituximab, consisting of four weekly injections followed by six monthly injections of undiluted rituximab together with xylocaine 2%, was recently reported in indolent, relapsed OAL [60]. This is a well-tolerated and active strategy, even for patients refractory to systemic rituximab treatment. Interestingly, one patient with stage IE FL of the eyelid who did not achieve lymphoma regression after intravenous rituximab received further intralesional injections of rituximab enhanced with autologous serum, thus achieving long-lasting lymphoma remission. Systemic rituximab inefficacy in this patient was probably related to low bioavailability of effectors in the tumor tissue, which was likely improved by the addition of autologous serum [60].
90Yttrium-labeled ibritumomab tiuxetan was highly active in a small series of patients with grade 1–2 OA-FL [61]. This radioimmunoconjugate merges the advantages of radiotherapy to a remarkably lower dose delivery in the surrounding uninvolved tissues. In such instances, the estimated orbital radiation dose with 90Y-ibritumomab tiuxetan (<3 Gy) corresponds to about a tenth of the dose with external radiotherapy [61].
Diffuse Large B-Cell Lymphoma
DLBCL is the third most common type of POAL (7%–21% of cases) [3, 6, 13, 33, 34, 40, 53, 62–66]. Its usual onset is stage I disease [6], with frequent involvement of the retrobulbar compartment [67], followed by lachrymal gland, eyelid, and palpebral conjunctiva [64, 68]. Intraconal lesions are more likely associated with systemic disease and poor prognosis [69]. DLBCL may evolve either de novo or secondarily during the course of a less aggressive lymphoma, most commonly represented by FL [8]. The tumor often directly infiltrates surrounding tissues, including the orbital bone component.
From a histological standpoint, OA-DLBCL is characterized by a diffuse infiltration of medium to large B-lymphocytes exhibiting anaplastic, centroblastic, or immunoblastic features [40], often accompanied by atypical mitotic figures. CD20 is almost invariably expressed by neoplastic lymphocytes, whose proliferation rate ranges between 50% and 80%. The highest proliferation rates have been associated with diffuse immunoreactivity for p53 protein (mean value: 35%) [3]. Seventy-five percent of OA-DLBCL cases correspond to germinal-center B-cell-like lymphomas according to the Choi algorithm [70], but the distinction from activated B-cell-like DLBCL does not play a prognostic role [69]. It is likely that this lack of difference may be due to the small numbers of the evaluated series.
Treatment of OA-DLBCL (Table 2) should follow the widely accepted therapeutic guidelines for nodal DLBCL [57, 71]. In particular, patients with stage I/II OA-DLBCL should be treated with upfront anthracycline-based chemotherapy in combination with rituximab (R-CHOP regimen or derivatives), followed by consolidation radiotherapy [65]. This modern strategy is associated with excellent results in patients with stage I disease, with a 5-year progression-free survival (PFS) and OS rates of 100% and 94%, respectively [53, 65]. Eyelid involvement seems to be associated with worse prognosis [65].
Radiotherapy is currently used as consolidation therapy after primary chemotherapy with or without immunotherapy in patients with limited-stage OA-DLBCL [72, 73], with doses of 30–40 Gy [53], whereas its use as exclusive therapy, restricted to patients with absolute chemotherapy contraindications, has been associated with more widespread relapse rates [46, 73, 74].
OA-DLBCL is associated with an increased risk of CNS dissemination [16, 75], either by hematogenous route or by direct infiltration through the skull bones (Fig. 2). Once CNS relapse occurs, the median OS time is 2–5 months, with only a very few long-term survivors [16]. CNS prophylaxis is therefore advisable in patients with OA-DLBCL, but it should be used with caution because of its relevant toxicity. National Comprehensive Cancer Network guidelines recommend prophylaxis with four to eight doses of intrathecal methotrexate and/or cytarabine given during primary chemotherapy. Intrathecal drug delivery by lumbar injection produces short-lived drug levels and an erratic drug bioavailability, whereas the intraventricular drug administration via Ommaya reservoir enables more efficient cerebrospinal-fluid distribution. Liposomal cytarabine delivered by intrathecal injection appears promising and allows dosing once every 14 days [76]. However, intrathecal chemotherapy does not prevent brain parenchymal relapses in systemic DLBCL. Accordingly, systematic release, during or after conventional chemotherapy, of high doses of methotrexate and/or other drugs with good CNS bioavailability (i.e., cytarabine, ifosfamide, or nitrosoureas), could effectively prevent both meningeal and parenchymal relapses [16].
Figure 2.

T1-weighted magnetic resonance imaging from a woman with diffuse large B-cell lymphoma arising in the left orbit (asterisk) and associated with direct infiltration of left frontal lobe (arrows).
Mantle Cell Lymphoma
OA-MCL represents 1%–5% of POALs [37, 40, 77] and does not differ from systemic MCL in terms of clinical and biologic profiles [78]. Elderly men (median age: 74 years) are more frequently affected [77, 78]. At diagnosis, patients most commonly have disseminated disease with bilateral involvement in 71% of cases [13, 77]. Almost all OA-MCL patients show involvement of multiple ocular adnexa, but orbital soft tissue is the most commonly involved structure (71% of POA-MCL) [77]. Orbital involvement seems to be associated with shorter survival in comparison to other disseminated presentations [77, 79].
Histopathological features of OA-MCL recall cytological features of their nodal counterpart [43]. Tumor cells may be immunoreactive for p53 [3]. Importantly, some cases with monocytoid-like features may occur as well, thus generating important differential diagnosis problems with OA-MZL [80] (M. Ponzoni, personal observation); in such instances, cyclin D1 immunoreactivity allows for the correct identification of MCL [80]. It is therefore an intriguing hypothesis that, before the availability of cyclin D1 immunostaining, some OA-MZLs with adverse prognosis were actually unrecognized cases of MCL. Cyclin D1 is particularly helpful in cases of CD5-negative MCL, which are not uncommon in the orbit [78] (M. Ponzoni, personal observation). Rare OA-MCL cases may actually be negative for cyclin D1; in such instances, SOX11 immunoreactivity is a reliable diagnostic marker [81]. T(11;14)(q32;q32) is a useful diagnostic tool. It is detected by fluorescence in situ hybridization analysis in 95% of OA-MCL cases and by cytogenetic analysis in 80% of cases [53, 78, 82].
Chemotherapy is the cornerstone of the management of OA-MCL because this lymphoma is mostly diagnosed at advanced stage [13, 77]. A few cases of limited-stage OA-MCL treated with radiotherapy alone displayed almost invariably early distant dissemination, sometimes with brain involvement [77]. As a rule, upfront treatment should include rituximab, high-dose cytarabine, and high-dose chemotherapy (HDC) supported by autologous stem cell transplantation (ASCT), with a median OS of 43 months [77, 79]. Notably, the addition of rituximab significantly improved response and survival rates, with 5-year OS rates of 83% and 8% for patients treated with or without this antibody, respectively [77].
Other B-Cell Lymphomas
Burkitt lymphoma rarely involves the orbit. The majority of Burkitt lymphomas occur sporadically in children, although extremely rare cases have been reported in immunocompetent adults [8]. Morphological variants, including those with plasmacytoid appearance, often encountered in nonendemic cases or admixed centroblasts/immunoblasts, are frequently observed in immunedeficient patients [8, 83]. A single case of a 16-year-old patient with stage-IE Burkitt lymphoma of the conjunctiva who was successfully treated with combined chemotherapy has been reported [84]. Treatment consists of intensified short-term chemoimmunotherapy regimens used for other patients with Burkitt lymphoma.
A case of intravascular large B-cell lymphoma presenting as an orbital mass lesion in a 77-year-old woman was recently reported [85]. The patient complained of fatigue and diplopia; MRI revealed both a frontal lobe lesion and right orbital disease. First-line treatment for this rare and extremely aggressive lymphoma is anthracycline- and rituximab-based chemoimmunotherapy in association with CNS prophylaxis with high doses of drugs with good CNS bioavailability [86, 87]. Small lymphocytic lymphoma/chronic lymphocytic leukemia usually reflects the ocular adnexal infiltration of a systemic disease; in fact, no cases of stage-I OA-SLL have been reported [40].
T-Cell Lymphomas
Only 1%–3% of all OALs display T-cell immunophenotype [3]. POA-T-cell lymphomas usually originate from natural killer or cytotoxic T-lymphocytes [88–90]. Ocular adnexal involvement could be the result of direct invasion by T/NK lymphomas originating in the nasal cavity or paranasal sinuses. T/NK lymphomas, in part represented by “nasal-type” entities, both present necrosis and infiltration/destruction of the vascular wall made by a population of atypical lymphocytes, usually immunoreactive for CD2, CD56, cytoplasmic CD3 (surface CD3 is often lacking), tumor intracellular antigen 1 (TIA-1), perforin, and granzyme B. EBV is almost always present. The proliferation rate is usually high (60%–70%), as well as the proportion of p53-positive neoplastic cells [3].
The median age of affected patients is 45 years (range: 26–65 years); nearly half of this population has limited-stage lymphoma, 62% of which have concurrent sinus-nasal involvement, whereas 37% present with disease limited to the orbit [91]. Although the available literature is anecdotal and fragmented, reported experiences suggest that nearly 90% of these patients die early due to lymphoma progression, with a median OS time of approximately 6 months. Only exceptional patients are alive at 5 years—a feature that even exceeds the well-known adverse mortality rates reported for NK/T-cell lymphomas involving other sites [30, 91].
The best treatment for extranodal NK/T-cell lymphoma, nasal type, remains to be defined. Surgical resection of the lesions has been proposed, essentially for diagnostic purposes, but also to promote drainage of necrotic cavities. External beam radiotherapy with a dose of ∼52 Gy delivered according to classical fractionation is recommended for localized stages (stages I and II), which is associated with a 40%–80% complete response rate (CRR) and a 5-year OS rate of 40%–59% [92, 93]. Aggressive chemotherapy is the only available treatment option for advanced forms (stage III and IV), but CRR is around 15%, with the combination of CHOP (cyclophosphamide, hydroxydaunorubicin, vincristine, and prednisone) chemotherapy and radiotherapy being effective only in patients with limited-stage disease [94].
In the largest reported series [30], eight patients with OA-NK/T-cell lymphoma were treated with radiotherapy and/or chemotherapy, often including cyclophosphamide, doxorubicin, vincristine, and prednisone. Seven patients died early (1–13 months). A single patient was alive and disease-free at 5-year follow-up. Two patients developed CNS involvement. Taken together, this data strongly suggests that more intensive chemotherapy, possibly followed by autologous bone marrow transplantation and CNS prophylaxis, should be indicated for these patients [5, 30]. Overall, data from some recent reports seem to suggest that patients with nasal NK/T-cell lymphoma of stage IE or contiguous stage IIE with cervical node involvement should be managed with concurrent chemoradiotherapy, with reduced-dose DeVIC (dexamethasone, etoposide, ifosfamide, and carboplatin) chemotherapy as the first choice [95]. Intensified l-asparaginase-containing chemotherapies, such as the dexamethasone+methotrexate+ifosfamide+l-asparaginase+etoposide regimen, are recommended as an induction therapy in young patients, whereas other less intensive l-asparaginase- and/or etoposide-based combinations are good options for elderly or frail patients [95]. EBV-DNA monitoring in peripheral blood is useful both for prognosis definition and disease surveillance in follow-up.
Rare cases of primary and secondary involvement of the ocular adnexa by T/NK cell lymphoma and adult T-cell leukemia associated with human T-cell lymphotropic virus type I infection have been reported [30, 96, 97]. A few cases of primary involvement of the ocular adnexa by anaplastic lymphoma kinase-positive, anaplastic large T-cell lymphomas have been described [36, 96]. Treatment follows worldwide guidelines applied to systemic anaplastic lymphoma, with chemotherapy followed by consolidation radiotherapy if clinically indicated [98].
Peripheral T-cell lymphoma not otherwise specified can present both intraocular and extra ocular involvement as well. Ectropion is the most common presentation; additional ocular manifestations may include edema of eyelid, tight skin, blepharitis, and corneal abnormalities [99]. A CHOP-like regimen is the standard option, but also other agents potentially active for T-cell lymphomas, like methotrexate, l-asparaginase, cytarabine, interferon-alpha, etoposide, and bleomycin, have been used [100]. Upfront HDC/ASCT has been evaluated in clinical trials, with encouraging results in young patients; allogeneic transplant plays a role in relapsed cases (Table 2). Radiotherapy may be used as a palliative or consolidative approach in relapsed patients [101].
Other Lymphoproliferative Disorders
Ocular adnexa infiltration by Hodgkin lymphoma is usually a manifestation of advanced or relapsing disease, followed by intracranial extension in some cases [8, 73]. These patients should be treated following well-known guidelines for advanced-stage or failed Hodgkin lymphoma [102]. Lymphomatoid granulomatosis is an EBV-related lymphoproliferative disease, where B-cells progressively overcome the background of accompanying T-cells, become monoclonal, and eventually progress to aggressive large B-cell lymphoma. Less than 20% of 21 reported cases, with age onset ranging from 7 to 75 years, present with ocular involvement, hepatosplenomegaly, adenopathy, and arthralgia [103]. Diagnosis is usually established by lung biopsy; the histological details of rare cases reported in sclera and eyebrow are almost lacking. The conventional treatment based on immunosuppression with prednisolone and cyclophosphamide was accomplished by antiviral therapy with interferon-α [99]. In grade I–II diseases, interferon-α was associated with 60% CRR, with 90% CRR in patients with CNS involvement, and a 5-year PFS rate of 56% [104]. Nearly 20% of patients progress to interferon with grade III disease, and many of them have been successfully treated with immunochemotherapy [104]. Interestingly, patients with low-grade lesions can relapse with high-grade disease and vice versa; there may also be simultaneous discordant disease at different sites; thus, rebiopsy is critical to drive treatment strategy. Intensified chemoimmunotherapy is associated with 66% CRR and a 2-year PFS of 40% in patients with grade III disease [104]. Radiotherapy has been used in localized disease. Overall, nearly a third of patients die of lymphoma. Median OS time is approximately 6 months [105]. Encouraging results have been reported with rituximab and HDC/ASCT [106].
Conclusions
POALs (other than MZL) are a relatively heterogeneous group of rare disorders. This heterogeneity is reflected in poorly known molecular and biological properties and a lack of therapeutic recommendations for individual entities. The effort of future studies will require the collection of a larger number of cases of these entities, with the aim to better highlight the potential differences compared with their systemic counterparts and to evaluate the efficacy of modern therapeutic approaches. Association with infectious agents and the role of antimicrobial therapies deserve further investigation in view of the recently reported encouraging results with this conservative strategy [28]. Because POALs are so rare, clinical trials addressing novel drugs and therapeutic modalities in series constituted exclusively by patients with POALs will be challenging. Future therapeutic progress will more likely result from studies focused on related systemic lymphoma counterparts. However, the peculiar physiologic and anatomic properties of ocular adnexa should always be considered before defining overall strategies for treating these intriguing malignancies.
Author Contributions
Conception/Design: Maurilio Ponzoni, Silvia Govi, Andrés J. M. Ferreri
Collection and/or assembly of data: Maurilio Ponzoni, Giada Licata, Antonio Giordano Resti, Letterio S. Politi, Eliana Di Cairano, Andrés J. M. Ferreri
Data analysis and interpretation: Maurilio Ponzoni, Silvia Govi, Giada Licata, Silvia Mappa, Antonio Giordano Resti, Letterio S. Politi, Lorenzo Spagnuolo, Claudio Doglioni, Andrés J. M. Ferreri
Manuscript writing: Maurilio Ponzoni, Silvia Govi, Giada Licata, Silvia Mappa, Andrés J. M. Ferreri
Final approval of manuscript: Maurilio Ponzoni, Silvia Govi, Silvia Mappa, Antonio Giordano Resti, Letterio S. Politi, Lorenzo Spagnuolo, Eliana Di Cairano, Claudio Doglioni, Andrés J. M. Ferreri
Disclosures
The authors indicated no financial relationships.
Reference
- 1.Spraul CW, Mattfeldt T, Lang GK. [Conjunctival pseudolymphoma in infectious mononucleosis] Klin Monatsbl Augenheilkd. 1999;215:68–69. doi: 10.1055/s-2008-1034673. [DOI] [PubMed] [Google Scholar]
- 2.Tovilla-Canales JL, Tovilla y Pomar JL, Ceron JR. Lymphoproliferative disorders of the ocular adnexa. Curr Opin Ophthalmol. 2004;15:401–405. doi: 10.1097/01.icu.0000139302.16648.80. [DOI] [PubMed] [Google Scholar]
- 3.Coupland SE, Krause L, Delecluse HJ, et al. Lymphoproliferative lesions of the ocular adnexa. Analysis of 112 cases. Ophthalmology. 1998;105:1430–1441. doi: 10.1016/S0161-6420(98)98024-1. [DOI] [PubMed] [Google Scholar]
- 4.Shields CL, Shields JA, Carvalho C, et al. Conjunctival lymphoid tumors: Clinical analysis of 117 cases and relationship to systemic lymphoma. Ophthalmology. 2001;108:979–984. doi: 10.1016/s0161-6420(01)00547-4. [DOI] [PubMed] [Google Scholar]
- 5.Sullivan TJ, Grimes D, Bunce I. Monoclonal antibody treatment of orbital lymphoma. Ophthal Plast Reconstr Surg. 2004;20:103–106. doi: 10.1097/01.iop.0000115594.98470.ac. [DOI] [PubMed] [Google Scholar]
- 6.McKelvie PA, McNab A, Francis IC, et al. Ocular adnexal lymphoproliferative disease: A series of 73 cases. Clin Experiment Ophthalmol. 2001;29:387–393. doi: 10.1046/j.1442-9071.2001.d01-18.x. [DOI] [PubMed] [Google Scholar]
- 7.Polito E, Galieni P, Leccisotti A. Clinical and radiological presentation of 95 orbital lymphoid tumors. Graefes Arch Clin Exp Ophthalmol. 1996;234:504–509. doi: 10.1007/BF00184859. [DOI] [PubMed] [Google Scholar]
- 8.Coupland SE, Foss HD, Hidayat AA, et al. Extranodal marginal zone B cell lymphomas of the uvea: An analysis of 13 cases. J Pathol. 2002;197:333–340. doi: 10.1002/path.1130. [DOI] [PubMed] [Google Scholar]
- 9.Bernardini G, Braconi D, Lusini P, et al. Helicobacter pylori: Immunoproteomics related to different pathologies. Expert Rev Proteomics. 2007;4:679–689. doi: 10.1586/14789450.4.5.679. [DOI] [PubMed] [Google Scholar]
- 10.Sjo LD, Juhl BR, Buchwald C, et al. Epstein-Barr positive T-cell lymphoma in the ocular region. Eur J Ophthalmol. 2006;16:181–185. doi: 10.1177/112067210601600132. [DOI] [PubMed] [Google Scholar]
- 11.Nakata M, Matsuno Y, Katsumata N, et al. Histology according to the Revised European-American Lymphoma Classification significantly predicts the prognosis of ocular adnexal lymphoma. Leuk Lymphoma. 1999;32:533–543. doi: 10.3109/10428199909058411. [DOI] [PubMed] [Google Scholar]
- 12.Stafford SL, Kozelsky TF, Garrity JA, et al. Orbital lymphoma: Radiotherapy outcome and complications. Radiother Oncol. 2001;59:139–144. doi: 10.1016/s0167-8140(00)00328-5. [DOI] [PubMed] [Google Scholar]
- 13.Hatef E, Roberts D, McLaughlin P, et al. Prevalence and nature of systemic involvement and stage at initial examination in patients with orbital and ocular adnexal lymphoma. Arch Ophthalmol. 2007;125:1663–1667. doi: 10.1001/archopht.125.12.1663. [DOI] [PubMed] [Google Scholar]
- 14.Raderer M, Vorbeck F, Formanek M, et al. Importance of extensive staging in patients with mucosa-associated lymphoid tissue (MALT)-type lymphoma. Br J Cancer. 2000;83:454–457. doi: 10.1054/bjoc.2000.1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cahill M, Barnes C, Moriarty P, et al. Ocular adnexal lymphoma-comparison of MALT lymphoma with other histological types. Br J Ophthalmol. 1999;83:742–747. doi: 10.1136/bjo.83.6.742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cheung CW, Burton C, Smith P, et al. Central nervous system chemoprophylaxis in non-Hodgkin lymphoma: Current practice in the UK. Br J Haematol. 2005;131:193–200. doi: 10.1111/j.1365-2141.2005.05756.x. [DOI] [PubMed] [Google Scholar]
- 17.Valenzuela AA, Allen C, Grimes D, et al. Positron emission tomography in the detection and staging of ocular adnexal lymphoproliferative disease. Ophthalmology. 2006;113:2331–2337. doi: 10.1016/j.ophtha.2006.05.059. [DOI] [PubMed] [Google Scholar]
- 18.Roe RH, Finger PT, Kurli M, et al. Whole-body positron emission tomography/computed tomography imaging and staging of orbital lymphoma. Ophthalmology. 2006;113:1854–1858. doi: 10.1016/j.ophtha.2006.04.029. [DOI] [PubMed] [Google Scholar]
- 19.Romaguera JE, Medeiros LJ, Hagemeister FB, et al. Frequency of gastrointestinal involvement and its clinical significance in mantle cell lymphoma. Cancer. 2003;97:586–591. doi: 10.1002/cncr.11096. [DOI] [PubMed] [Google Scholar]
- 20.Salar A, Juanpere N, Bellosillo B, et al. Gastrointestinal involvement in mantle cell lymphoma: A prospective clinic, endoscopic, and pathologic study. Am J Surg Pathol. 2006;30:1274–1280. doi: 10.1097/01.pas.0000208899.15859.cb. [DOI] [PubMed] [Google Scholar]
- 21.Ferrer A, Salaverria I, Bosch F, et al. Leukemic involvement is a common feature in mantle cell lymphoma. Cancer. 2007;109:2473–2480. doi: 10.1002/cncr.22715. [DOI] [PubMed] [Google Scholar]
- 22.Ferreri AJ, Guidoboni M, Ponzoni M, et al. Evidence for an association between Chlamydia psittaci and ocular adnexal lymphomas. J Natl Cancer Inst. 2004;96:586–594. doi: 10.1093/jnci/djh102. [DOI] [PubMed] [Google Scholar]
- 23.Ponzoni M, Ferreri AJ, Guidoboni M, et al. Chlamydia infection and lymphomas: Association beyond ocular adnexal lymphomas highlighted by multiple detection methods. Clin Cancer Res. 2008;14:5794–5800. doi: 10.1158/1078-0432.CCR-08-0676. [DOI] [PubMed] [Google Scholar]
- 24.Ferreri AJ, Dolcetti R, Magnino S, et al. Chlamydial infection: The link with ocular adnexal lymphomas. Nat Rev Clin Oncol. 2009;6:658–669. doi: 10.1038/nrclinonc.2009.147. [DOI] [PubMed] [Google Scholar]
- 25.Chanudet E, Zhou Y, Bacon CM, et al. Chlamydia psittaci is variably associated with ocular adnexal MALT lymphoma in different geographical regions. J Pathol. 2006;209:344–351. doi: 10.1002/path.1984. [DOI] [PubMed] [Google Scholar]
- 26.De Cremoux P, Subtil A, Ferreri AJ, et al. Low prevalence of Chlamydia psittaci infection in French patients with ocular adnexal lymphomas. J Natl Cancer Inst. 2006;98:365–366. doi: 10.1093/jnci/djj079. [DOI] [PubMed] [Google Scholar]
- 27.Ferreri AJ, Ponzoni M, Guidoboni M, et al. Bacteria-eradicating therapy with doxycycline in ocular adnexal MALT lymphoma: A multicenter prospective trial. J Natl Cancer Inst. 2006;98:1375–1382. doi: 10.1093/jnci/djj373. [DOI] [PubMed] [Google Scholar]
- 28.Ferreri AJ, Govi S, Pasini E, et al. Chlamydophila psittaci eradication with doxycycline as first-line targeted therapy for ocular adnexae lymphoma: Final results of an international phase II trial. J Clin Oncol. 2012;30:2988–2994. doi: 10.1200/JCO.2011.41.4466. [DOI] [PubMed] [Google Scholar]
- 29.Ferreri AJ, Dolcetti R, Magnino S, et al. A woman and her canary: A tale of chlamydiae and lymphomas. J Natl Cancer Inst. 2007;99:1418–1419. doi: 10.1093/jnci/djm118. [DOI] [PubMed] [Google Scholar]
- 30.Woog JJ, Kim YD, Yeatts RP, et al. Natural killer/T-cell lymphoma with ocular and adnexal involvement. Ophthalmology. 2006;113:140–147. doi: 10.1016/j.ophtha.2005.09.036. [DOI] [PubMed] [Google Scholar]
- 31.Cheuk W, Yuen HK, Chan AC, et al. Ocular adnexal lymphoma associated with IgG4+ chronic sclerosing dacryoadenitis: A previously undescribed complication of IgG4-related sclerosing disease. Am J Surg Pathol. 2008;32:1159–1167. doi: 10.1097/PAS.0b013e31816148ad. [DOI] [PubMed] [Google Scholar]
- 32.Sato Y, Omura K, Harada H, et al. Lymphoepithelial cyst arising in the parotid gland: A report of 3 cases. Kokubyo Gakkai Zasshi. 2008;75:162–167. doi: 10.5357/koubyou.75.162. [DOI] [PubMed] [Google Scholar]
- 33.Jenkins C, Rose GE, Bunce C, et al. Clinical features associated with survival of patients with lymphoma of the ocular adnexa. Eye. 2003;17:809–820. doi: 10.1038/sj.eye.6700379. [DOI] [PubMed] [Google Scholar]
- 34.Auw-Haedrich C, Coupland SE, Kapp A, et al. Long term outcome of ocular adnexal lymphoma subtyped according to the REAL classification. Revised European and American Lymphoma. Br J Ophthalmol. 2001;85:63–69. doi: 10.1136/bjo.85.1.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Plaisier MB, Sie-Go DM, Berendschot TT, et al. Ocular adnexal lymphoma classified using the WHO classification: Not only histology and stage, but also gender is a predictor of outcome. Orbit. 2007;26:83–88. doi: 10.1080/01676830601169148. [DOI] [PubMed] [Google Scholar]
- 36.Ferreri AJ, Ernberg I, Copie-Bergman C. Infectious agents and lymphoma development: Molecular and clinical aspects. J Intern Med. 2009;265:421–438. doi: 10.1111/j.1365-2796.2009.02083.x. [DOI] [PubMed] [Google Scholar]
- 37.Coupland SE, Heimann H, Bechrakis NE. Primary intraocular lymphoma: A review of the clinical, histopathological and molecular biological features. Graefes Arch Clin Exp Ophthalmol. 2004;242:901–913. doi: 10.1007/s00417-004-0973-0. [DOI] [PubMed] [Google Scholar]
- 38.Abramson JS, Hochberg EP. Intravenous methotrexate as central nervous system (CNS) prophylaxis is associated with a low risk of CNS recurrence in high-risk patients with diffuse large B-cell lymphoma. Cancer. 2010;116:4283–4290. doi: 10.1002/cncr.25278. [DOI] [PubMed] [Google Scholar]
- 39.Jakobiec FA. Ocular adnexal lymphoid tumors: Progress in need of clarification. Am J Ophthalmol. 2008;145:941–950. doi: 10.1016/j.ajo.2008.03.013. [DOI] [PubMed] [Google Scholar]
- 40.Ferry JA, Fung CY, Zukerberg L, et al. Lymphoma of the ocular adnexa: A study of 353 cases. Am J Surg Pathol. 2007;31:170–184. doi: 10.1097/01.pas.0000213350.49767.46. [DOI] [PubMed] [Google Scholar]
- 41.Harris NL, Jaffe ES, Diebold J, et al. World Health Organization classification of neoplastic diseases of the hematopoietic and lymphoid tissues: Report of the Clinical Advisory Committee meeting-Airlie House, Virginia, November 1997. J Clin Oncol. 1999;17:3835–3849. doi: 10.1200/JCO.1999.17.12.3835. [DOI] [PubMed] [Google Scholar]
- 42.Ye H, Dogan A, Karran L, et al. BCL10 expression in normal and neoplastic lymphoid tissue. Nuclear localization in MALT lymphoma. Am J Pathol. 2000;157:1147–1154. doi: 10.1016/S0002-9440(10)64630-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vejabhuti C, Harris GJ, Erickson BA, et al. BCL10 expression in ocular adnexal lymphomas. Am J Ophthalmol. 2005;140:836–843. doi: 10.1016/j.ajo.2005.05.038. [DOI] [PubMed] [Google Scholar]
- 44.Thome M. CARMA1, BCL-10 and MALT1 in lymphocyte development and activation. Nat Rev Immunol. 2004;4:348–359. doi: 10.1038/nri1352. [DOI] [PubMed] [Google Scholar]
- 45.Norton AJ. Monoclonal antibodies in the diagnosis of lymphoproliferative diseases of the orbit and orbital adnexa. Eye (Lond) 2006;20:1186–1188. doi: 10.1038/sj.eye.6702478. [DOI] [PubMed] [Google Scholar]
- 46.Fung CY, Tarbell NJ, Lucarelli MJ, et al. Ocular adnexal lymphoma: Clinical behavior of distinct World Health Organization classification subtypes. Int J Radiat Oncol Biol Phys. 2003;57:1382–1391. doi: 10.1016/s0360-3016(03)00767-3. [DOI] [PubMed] [Google Scholar]
- 47.Bolek TW, Moyses HM, Marcus RB, et al. Radiotherapy in the management of orbital lymphoma. Int J Radiat Oncol Biol Phys. 1999;44:31–36. doi: 10.1016/s0360-3016(98)00535-5. [DOI] [PubMed] [Google Scholar]
- 48.Pelloski CE, Wilder RB, Ha CS, et al. Clinical stage IEA-IIEA orbital lymphomas: outcomes in the era of modern staging and treatment. Radiother Oncol. 2001;59:145–151. doi: 10.1016/s0167-8140(01)00338-3. [DOI] [PubMed] [Google Scholar]
- 49.Bhatia S, Paulino AC, Buatti JM, et al. Curative radiotherapy for primary orbital lymphoma. Int J Radiat Oncol Biol Phys. 2002;54:818–823. doi: 10.1016/s0360-3016(02)02966-8. [DOI] [PubMed] [Google Scholar]
- 50.Martinet S, Ozsahin M, Belkacemi Y, et al. Outcome and prognostic factors in orbital lymphoma: A Rare Cancer Network study on 90 consecutive patients treated with radiotherapy. Int J Radiat Oncol Biol Phys. 2003;55:892–898. doi: 10.1016/s0360-3016(02)04159-7. [DOI] [PubMed] [Google Scholar]
- 51.Ejima Y, Sasaki R, Okamoto Y, et al. Ocular adnexal mucosa-associated lymphoid tissue lymphoma treated with radiotherapy. Radiother Oncol. 2006;78:6–9. doi: 10.1016/j.radonc.2005.11.005. [DOI] [PubMed] [Google Scholar]
- 52.Baldini L, Blini M, Guffanti A, et al. Treatment and prognosis in a series of primary extra nodal lymphomas of the ocular adnexa. Ann Oncol. 1998;9:779–781. doi: 10.1023/a:1008327301372. [DOI] [PubMed] [Google Scholar]
- 53.Sullivan TJ, Whitehead K, Williamson R, et al. Lymphoproliferative disease of the ocular adnexa: A clinical and pathologic study with statistical analysis of 69 patients. Ophthal Plast Reconstr Surg. 2005;21:177–188. doi: 10.1097/01.iop.0000159173.42243.ad. [DOI] [PubMed] [Google Scholar]
- 54.Hoskin P, Kirkwood A, Popova B, et al. A phase 3 multi-center peospective randomized trial of low dose radiation therapy for follicular and marginal zone lymphoma. Rad Oncol Int J Biol Phys. 2012;84(suppl 3) Abstract LAB2. [Google Scholar]
- 55.Liao SL, Kao SC, Hou PK, et al. Results of radiotherapy for orbital and adnexal lymphoma. Orbit. 2002;21:117–123. doi: 10.1076/orbi.21.2.117.7192. [DOI] [PubMed] [Google Scholar]
- 56.Uno T, Isobe K, Shikama N, et al. Radiotherapy for extranodal, marginal zone, B-cell lymphoma of mucosa-associated lymphoid tissue originating in the ocular adnexa: A multiinstitutional, retrospective review of 50 patients. Cancer. 2003;98:865–871. doi: 10.1002/cncr.11539. [DOI] [PubMed] [Google Scholar]
- 57.Bardenstein DS. Ocular adnexal lymphoma: classification, clinical disease, and molecular biology. Ophthalmol Clin North Am. 2005;18:187–197. doi: 10.1016/j.ohc.2004.11.001. [DOI] [PubMed] [Google Scholar]
- 58.Esmaeli B, Murray JL, Ahmadi MA, et al. Immunotherapy for low-grade non-hodgkin secondary lymphoma of the orbit. Arch Ophthalmol. 2002;120:1225–1227. [PubMed] [Google Scholar]
- 59.Zinzani PL, Alinari L, Stefoni V, et al. Rituximab in primary conjunctiva lymphoma. Leuk Res. 2005;29:107–108. doi: 10.1016/j.leukres.2004.05.011. [DOI] [PubMed] [Google Scholar]
- 60.Ferreri AJ, Govi S, Colucci A, et al. Intralesional rituximab: A new therapeutic approach for patients with conjunctival lymphomas. Ophthalmology. 2011;118:24–28. doi: 10.1016/j.ophtha.2010.04.036. [DOI] [PubMed] [Google Scholar]
- 61.Esmaeli B, McLaughlin P, Pro B, et al. Prospective trial of targeted radioimmunotherapy with Y-90 ibritumomab tiuxetan (Zevalin) for front-line treatment of early-stage extra nodal indolent ocular adnexal lymphoma. Ann Oncol. 2009;20:709–714. doi: 10.1093/annonc/mdn692. [DOI] [PubMed] [Google Scholar]
- 62.Meunier J, Lumbroso-Le Rouic L, Vincent-Salomon A, et al. Ophthalmologic and intraocular non-Hodgkin's lymphoma: A large single centre study of initial characteristics, natural history, and prognostic factors. Hematol Oncol. 2004;22:143–158. doi: 10.1002/hon.741. [DOI] [PubMed] [Google Scholar]
- 63.Coupland SE, Hellmich M, Auw-Haedrich C, et al. Prognostic value of cell-cycle markers in ocular adnexal lymphoma: An assessment of 230 cases. Graefes Arch Clin Exp Ophthalmol. 2004;242:130–145. doi: 10.1007/s00417-003-0831-5. [DOI] [PubMed] [Google Scholar]
- 64.Coupland SE, Damato B. Lymphomas involving the eye and the ocular adnexa. Curr Opin Ophthalmol. 2006;17:523–531. doi: 10.1097/ICU.0b013e328010948d. [DOI] [PubMed] [Google Scholar]
- 65.Madge SN, McCormick A, Patel I, et al. Ocular adnexal diffuse large B-cell lymphoma: Local disease correlates with better outcomes. Eye (Lond) 2010;24:954–961. doi: 10.1038/eye.2009.283. [DOI] [PubMed] [Google Scholar]
- 66.Rootman DB, Mavrikakis I, Connors JM, et al. Primary, unilateral ocular adnexal lymphoma: Disease progression and long-term survival. Ophthal Plast Reconstr Surg. 2011;27:405–409. doi: 10.1097/IOP.0b013e31821cc4bb. [DOI] [PubMed] [Google Scholar]
- 67.Sasai K, Yamabe H, Dodo Y, et al. Non-Hodgkin's lymphoma of the ocular adnexa. Acta Oncol. 2001;40:485–490. doi: 10.1080/028418601750288217. [DOI] [PubMed] [Google Scholar]
- 68.Huerva V, Canto LM, Marti M. Primary diffuse large B-cell lymphoma of the lower eyelid. Ophthal Plast Reconstr Surg. 2003;19:160–161. doi: 10.1097/01.IOP.0000056160.54804.DB. [DOI] [PubMed] [Google Scholar]
- 69.Stacy RC, Jakobiec FA, Herwig MC, et al. Diffuse large B-cell lymphoma of the orbit: Clinicopathologic, immunohistochemical, and prognostic features of 20 cases. Am J Ophthalmol. 2012;154:87–98. doi: 10.1016/j.ajo.2012.01.021. [DOI] [PubMed] [Google Scholar]
- 70.Choi WW, Weisenburger DD, Greiner TC, et al. A new immunostain algorithm classifies diffuse large B-cell lymphoma into molecular subtypes with high accuracy. Clin Cancer Res. 2009;15:5494–5502. doi: 10.1158/1078-0432.CCR-09-0113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Armitage JO. How I treat patients with diffuse large B-cell lymphoma. Blood. 2007;110:29–36. doi: 10.1182/blood-2007-01-041871. [DOI] [PubMed] [Google Scholar]
- 72.Yadav BS, Sharma SC. Orbital lymphoma: Role of radiation. Indian J Ophthalmol. 2009;57:91–97. doi: 10.4103/0301-4738.44516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Farmer JP, Lamba M, Lamba WR, et al. Lymphoproliferative lesions of the lacrimal gland: Clinicopathological, immunohistochemical and molecular genetic analysis. Can J Ophthalmol. 2005;40:151–160. doi: 10.1016/S0008-4182(05)80026-2. [DOI] [PubMed] [Google Scholar]
- 74.Huerva V, Canto LM. Eyelid primary diffuse large B-cell lymphoma. Can J Ophthalmol. 2008;43:240–241. doi: 10.3129/i08-013. [DOI] [PubMed] [Google Scholar]
- 75.Bollen EL, Brouwer RE, Hamers S, et al. Central nervous system relapse in non-Hodgkin lymphoma. A single-center study of 532 patients. Arch Neurol. 1997;54:854–859. doi: 10.1001/archneur.1997.00550190044013. [DOI] [PubMed] [Google Scholar]
- 76.McClune B, Buadi FK, Aslam N, et al. Intrathecal liposomal cytarabine for prevention of meningeal disease in patients with acute lymphocytic leukemia and high-grade lymphoma. Leuk Lymphoma. 2007;48:1849–1851. doi: 10.1080/10428190701573232. [DOI] [PubMed] [Google Scholar]
- 77.Rasmussen P, Sjo LD, Prause JU, et al. Mantle cell lymphoma in the orbital and adnexal region. Br J Ophthalmol. 2009;93:1047–1051. doi: 10.1136/bjo.2008.146910. [DOI] [PubMed] [Google Scholar]
- 78.Looi A, Gascoyne RD, Chhanabhai M, et al. Mantle cell lymphoma in the ocular adnexal region. Ophthalmology. 2005;112:114–119. doi: 10.1016/j.ophtha.2004.07.025. [DOI] [PubMed] [Google Scholar]
- 79.Dreyling M, Hiddemann W. Dose-intense treatment of mantle cell lymphoma: Can durable remission be achieved? Curr Opin Oncol. 2008;20:487–494. doi: 10.1097/CCO.0b013e32830b61c2. [DOI] [PubMed] [Google Scholar]
- 80.Yang WI, Zukerberg LR, Motokura T, et al. Cyclin D1 (Bcl-1, PRAD1) protein expression in low-grade B-cell lymphomas and reactive hyperplasia. Am J Pathol. 1994;145:86–96. [PMC free article] [PubMed] [Google Scholar]
- 81.Mozos A, Roue G, Lopez-Guillermo A, et al. The expression of the endoplasmic reticulum stress sensor BiP/GRP78 predicts response to chemotherapy and determines the efficacy of proteasome inhibitors in diffuse large B-cell lymphoma. Am J Pathol. 2011;179:2601–2610. doi: 10.1016/j.ajpath.2011.07.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Panayiotidis P, Kotsi P. Genetics of small lymphocyte disorders. Semin Hematol. 1999;36:171–177. [PubMed] [Google Scholar]
- 83.Jaffe ES. Lymphoid lesions of the head and neck: A model of lymphocyte homing and lymphomagenesis. Mod Pathol. 2002;15:255–263. doi: 10.1038/modpathol.3880521. [DOI] [PubMed] [Google Scholar]
- 84.Weisenthal RW, Streeten BW, Dubansky AS, et al. Burkitt lymphoma presenting as a conjunctival mass. Ophthalmology. 1995;102:129–134. doi: 10.1016/s0161-6420(95)31069-x. [DOI] [PubMed] [Google Scholar]
- 85.Berbos ZJ, Lee MS, Zaldivar RA, et al. Intravascular lymphoma presenting as an orbital mass lesion: A case report. Orbit. 2010;29:91–93. doi: 10.3109/01676830903336288. [DOI] [PubMed] [Google Scholar]
- 86.Ferreri AJ, Dognini GP, Bairey O, et al. The addition of rituximab to anthracycline-based chemotherapy significantly improves outcome in “Western” patients with intravascular large B-cell lymphoma. Br J Haematol. 2008;143:253–257. doi: 10.1111/j.1365-2141.2008.07338.x. [DOI] [PubMed] [Google Scholar]
- 87.Shimada K, Murase T, Matsue K, et al. Central nervous system involvement in intravascular large B-cell lymphoma: A retrospective analysis of 109 patients. Cancer Sci. 2010;101:1480–1486. doi: 10.1111/j.1349-7006.2010.01555.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Coupland SE, Foss HD, Anagnostopoulos I, et al. Immunoglobulin VH gene expression among extranodal marginal zone B-cell lymphomas of the ocular adnexa. Invest Ophthalmol Vis Sci. 1999;40:555–562. [PubMed] [Google Scholar]
- 89.Shields CL, Shields JA, Eagle RC. Clinicopathologic reports, case reports, and small case series: Rapidly progressive T-cell lymphoma of the conjunctiva. Arch Ophthalmol. 2002;120:508–509. [PubMed] [Google Scholar]
- 90.Kirn TJ, Levy NB, Gosselin JJ, et al. Peripheral T-cell lymphoma presenting as sclerouveitis. Cornea. 2007;26:1147–1149. doi: 10.1097/ICO.0b013e31812dfa88. [DOI] [PubMed] [Google Scholar]
- 91.Yousuf SJ, Kumar N, Kidwell ED, Jr, et al. Rapidly fatal nasal natural killer/T-cell lymphoma: Orbital and ocular adnexal presentations. Orbit. 2011;30:120–121. doi: 10.3109/01676830.2010.546933. [DOI] [PubMed] [Google Scholar]
- 92.Al-Hakeem DA, Fedele S, Carlos R, et al. Extranodal NK/T-cell lymphoma, nasal type. Oral Oncol. 2007;43:4–14. doi: 10.1016/j.oraloncology.2006.03.011. [DOI] [PubMed] [Google Scholar]
- 93.You JY, Chi KH, Yang MH, et al. Radiation therapy versus chemotherapy as initial treatment for localized nasal natural killer (NK)/T-cell lymphoma: A single institute survey in Taiwan. Ann Oncol. 2004;15:618–625. doi: 10.1093/annonc/mdh143. [DOI] [PubMed] [Google Scholar]
- 94.Kwong YL. Natural killer-cell malignancies: Diagnosis and treatment. Leukemia. 2005;19:2186–2194. doi: 10.1038/sj.leu.2403955. [DOI] [PubMed] [Google Scholar]
- 95.Yamaguchi M. Current and future management of NK/T-cell lymphoma based on clinical trials. Int J Hematol. 2012;96:562–571. doi: 10.1007/s12185-012-1189-4. [DOI] [PubMed] [Google Scholar]
- 96.Coupland SE, Foss HD, Assaf C, et al. T-cell and T/natural killer-cell lymphomas involving ocular and ocular adnexal tissues: A clinicopathologic, immunohistochemical, and molecular study of seven cases. Ophthalmology. 1999;106:2109–2120. doi: 10.1016/S0161-6420(99)90492-X. [DOI] [PubMed] [Google Scholar]
- 97.Davis JL. Diagnosis of intraocular lymphoma. Ocul Immunol Inflamm. 2004;12:7–16. doi: 10.1076/ocii.12.1.7.28072. [DOI] [PubMed] [Google Scholar]
- 98.Mencia-Gutierrez E, Gutierrez-Diaz E, Salamanca J, et al. Cutaneous presentation on the eyelid of primary, systemic, CD30+, anaplastic lymphoma kinase (ALK)-negative, anaplastic large-cell lymphoma (ALCL) Int J Dermatol. 2006;45:766–769. doi: 10.1111/j.1365-4632.2004.02412.x. [DOI] [PubMed] [Google Scholar]
- 99.Cook BE, Jr, Bartley GB, Pittelkow MR. Ophthalmic abnormalities in patients with cutaneous T-cell lymphoma. Ophthalmology. 1999;106:1339–1344. doi: 10.1016/S0161-6420(99)00721-6. [DOI] [PubMed] [Google Scholar]
- 100.Kwong YL, Chan AC, Liang RH. Natural killer cell lymphoma/leukemia: Pathology and treatment. Hematol Oncol. 1997;15:71–79. doi: 10.1002/(sici)1099-1069(199705)15:2<71::aid-hon601>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 101.Ishikawa M, Watabe H, Hayakawa M, et al. Peripheral T-cell lymphoma of the eyelid. Clin Ophthalmol. 2009;3:527–529. doi: 10.2147/opth.s7717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Gobbi PG, Ferreri AJ, Ponzoni M, et al. Hodgkin lymphoma. Crit Rev Oncol Hematol. 2013;85:216–237. doi: 10.1016/j.critrevonc.2012.07.002. [DOI] [PubMed] [Google Scholar]
- 103.Jaffe ES, Wilson WH. Lymphomatoid granulomatosis: Pathogenesis, pathology and clinical implications. Cancer Surv. 1997;30:233–248. [PubMed] [Google Scholar]
- 104.Dunleavy K, Roschewski M, Wilson WH. Lymphomatoid granulomatosis and other Epstein-Barr virus associated lymphoproliferative processes. Curr Hematol Malig Rep. 2012;7:208–215. doi: 10.1007/s11899-012-0132-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Pradeep TG, Cannon P, Dodd T, et al. Lacrimal gland involvement in lymphomatoid granulomatosis and review of the literature. J Ophthalmol. 2010;2010:358121. doi: 10.1155/2010/358121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kampalath B, Peltier WL, Vesole DH. Successful treatment of systemic and central nervous system lymphomatoid granulomatosis with rituximab. Leuk Lymphoma. 2004;45:777–780. doi: 10.1080/10428190310001625854. [DOI] [PubMed] [Google Scholar]
- 107.Sharara N, Holden JT, Wojno TH, et al. Ocular adnexal lymphoid proliferations: Clinical, histologic, flow cytometric, and molecular analysis of forty-three cases. Ophthalmology. 2003;110:1245–1254. doi: 10.1016/S0161-6420(03)00330-0. [DOI] [PubMed] [Google Scholar]
- 108.Mannami T, Yoshino T, Oshima K, et al. Clinical, histopathological, and immunogenetic analysis of ocular adnexal lymphoproliferative disorders: Characterization of malt lymphoma and reactive lymphoid hyperplasia. Mod Pathol. 2001;14:641–649. doi: 10.1038/modpathol.3880366. [DOI] [PubMed] [Google Scholar]
- 109.Cho EY, Han JJ, Ree HJ, et al. Clinicopathologic analysis of ocular adnexal lymphomas: Extra nodal marginal zone B-cell lymphoma constitutes the vast majority of ocular lymphomas among Koreans and affect younger patients. Am J Hematol. 2003;73:87–96. doi: 10.1002/ajh.10332. [DOI] [PubMed] [Google Scholar]
- 110.White WL, Ferry JA, Harris NL, et al. Ocular adnexal lymphoma. A clinicopathologic study with identification of lymphomas of mucosa-associated lymphoid tissue type. Ophthalmology. 1995;102:1994–2006. doi: 10.1016/s0161-6420(95)30764-6. [DOI] [PubMed] [Google Scholar]