

Abstract
Oxidative stress and mitochondrial impairment are the main pathogenic mechanisms of Amyotrophic Lateral Sclerosis (ALS), a severe neurodegenerative disease still lacking of effective therapy. Recently, the coenzyme-Q (CoQ) complex, a key component of mitochondrial function and redox-state modulator, has raised interest for ALS treatment. However, while the oxidized form ubiquinone10 was ineffective in ALS patients and modestly effective in mouse models of ALS, no evidence was reported on the effect of the reduced form ubiquinol10, which has better bioavailability and antioxidant properties. In this study we compared the effects of ubiquinone10 and a new stabilized formulation of ubiquinol10 on the disease course of SOD1G93A transgenic mice, an experimental model of fALS. Chronic treatments (800 mg/kg/day orally) started from the onset of disease until death, to mimic the clinical trials that only include patients with definite ALS symptoms. Although the plasma levels of CoQ10 were significantly increased by both treatments (from <0.20 to 3.0–3.4 µg/mL), no effect was found on the disease progression and survival of SOD1G93A mice. The levels of CoQ10 in the brain and spinal cord of ubiquinone10- or ubiquinol10-treated mice were only slightly higher (≤10%) than the endogenous levels in vehicle-treated mice, indicating poor CNS availability after oral dosing and possibly explaining the lack of pharmacological effects. To further examine this issue, we measured the oxidized and reduced forms of CoQ9/10 in the plasma, brain and spinal cord of symptomatic SOD1G93A mice, in comparison with age-matched SOD1WT. Levels of ubiquinol9/10, but not ubiquinone9/10, were significantly higher in the CNS, but not in plasma, of SOD1G93A mice, suggesting that CoQ redox system might participate in the mechanisms trying to counteract the pathology progression. Therefore, the very low increases of CoQ10 induced by oral treatments in CNS might be not sufficient to provide significant neuroprotection in SOD1G93A mice.
Introduction
Coenzyme Q (CoQ) is an amphipathic molecule structurally composed of a quinone ring synthesized from p-hydroxybenzoate and of a polyisoprene chain synthesized from acetil-CoA [1]. There are different forms of CoQ, related to the length of polyisoprene chain: the predominant form in humans is CoQ10 which contains 10 isoprenoid units, whereas CoQ9 is the predominant form in rodents [2]. CoQ exists in three redox states: a fully oxidized form (ubiquinone), a fully reduced form (ubiquinol) and an intermediate semiquinone radical (CoQ.−). The redox state is tissue-specific: for example, in mice ubiquinol is the predominant form in liver and skeletal muscle while ubiquinone is predominant in the brain [3]. Several factors, such as drugs, age, diet and pathologies affect total levels and redox state of CoQ [4], [5], [6], [7] and their alterations may be regarded as markers of oxidative stress and mitochondrial dysfunctions [8].
Amyotrophic Lateral Sclerosis (ALS) is a severe neurodegenerative disorder in which the loss of motor neurons induces a progressive motor paralysis associated with dysphagia, dysarthria, respiratory failure and death within 3–5 years from the diagnosis. Analysis of plasma [9] and cerebrospinal fluid (CSF) [10] of sporadic ALS (sALS) patients have shown an increase of ubiquinone10 with a shift in the redox state of the coenzyme, interpreted as an index of oxidative stress.
Because of the substantial involvement of oxidative stress and mitochondrial impairment in this disease [11] different therapeutic approaches have been aimed towards counteracting these mechanisms. Among them, the use of CoQ10 has raised some interest for the treatment of ALS. In fact, on the basis of a modest but significant increase of survival observed in a mouse model of familial ALS (fALS) after chronic treatment with ubiquinone10 [12], a phase II trial was designed to investigate the effects of a chronic treatment with the same molecule (2700 mg daily) in patients with sALS. Unfortunately, the data collected after 9 months of treatment did not provide sufficient evidence of a benefit to justify a phase III trial [13].
The use of the reduced form ubiquinol, characterized by potent antioxidant properties, was difficult because it is readily oxidized in air. Recently, however, a new stabilized formulation of ubiquinol10 has been produced (Kaneka QH™) which overcomes this problem [14]. Notably, pharmacokinetic studies showed that ubiquinol10 has a better oral bioavailability than ubiquinone10, in humans [14], dogs and rats [15]. It may be expected that chronic treatment with this formulation results in higher CNS levels of CoQ10, with superior neuroprotective properties. Consistently, ubiquinol10 was more active than ubiquinone10 in protecting mice from striatal neurodegeneration induced by acute treatment with 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), a model for Parkinson’s disease [16].
On this basis, in the present study we evaluated: 1) the effects of a chronic oral administration with ubiquinol10 or ubiquinone10 on disease progression of transgenic SOD1G93A mice, a model of fALS [11], 2) the endogenous levels of total, reduced and oxidized CoQ10 and CoQ9 in plasma, brain and lumbar spinal cord of transgenic SOD1G93A mice at symptomatic stage.
Our data show that exogenous administration of ubiquinol10 or ubiquinone10 to SOD1G93A mice, starting at the onset of symptoms, had no effect on the disease course. Since the basal levels of reduced CoQ9/10 are increased in the CNS of symptomatic SOD1G93A mice, suggesting an attempt of equilibrium toward a protective antioxidant state, we think that the small increase of ubiquinol after the oral treatment is insufficient to exacerbate this effect.
Materials and Methods
Animals and Treatments
Transgenic SOD1G93A and SOD1WT mice on 129S2/SvHsd genetic background, and non-transgenic littermates were used in this study. The SOD1G93A mouse line derives from the line originally obtained from Jackson Laboratories (B6SJL-TgNSOD-1-SOD1G93A-1Gur) expressing approximately 20 copies of mutant human SOD1 with a Gly93Ala substitution (SOD1G93A) or wild-type human SOD1 (SOD1WT) and were maintained on 129S2/SvHsd background for more than 15 generations at Harlan Italy S.R.L., Bresso (MI), Italy [17]. We observed no differences between the males and females as regards the disease progression and survival, as reported in supporting figure 1 (Fig. S1). Therefore, to reduce at minimum the number of transgenic mice used, chronic treatments were carried out in female whereas the biochemical profile was examined in males.
Figure 1. Ubiquinol10 chronic treatment has no effect on disease course in SOD1G93A mice.

Effect of oral treatment with 800 mg/kg/day ubiquinone10, ubiquinol10 or vehicle, on motor dysfunction and disease progression of 129Sv SOD1G93A mice (n = 15 mice per group). Treatment started at the age of 91 days (arrows) until the sacrifice (at the end-stage of the disease, when mice were unable to right themselves within 10 seconds after being placed on both sides). Treatment with ubiquinone10 or ubiquinol10 had no significant effects on body weight (A), on the latency of rotarod (B) and PaGE test (C) (Two-way ANOVA), or on disease onset (D) and survival length (E). Each point represents the mean; for sake of clarity standard deviations are not indicated but they were always less than 15% of the value. Table reports the mean and standard deviations of symptoms onset and life-span for each group.
All mice were maintained at a temperature of 21±1°C with a relative humidity 55±10% and 12 h of light. Food (standard pellets) and water were supplied ad libitum.
Ubiquinone10 (Ubidecarenone from Kaneka) and ubiquinol10 (Kaneka QH™ from Kaneka) were administered by gavage after solubilization in sunflower seed oil (vehicle); preparation procedures were carried out at room temperature protecting from light. All the solutions were administered within four hours from preparation.
Ethics statement
Procedures involving animals and their care were conducted in conformity with the institutional guidelines of “Mario Negri” institute (IRFMN), that are in compliance with national (D.L. no. 116, G.U. suppl. 40, Feb. 18, 1992, Circular No.8, G.U., 14 luglio 1994) and international laws and policies (EEC Council Directive 86/609, OJ L 358, 1 Dec.12, 1987; NIH Guide for the Care and use of Laboratory Animals, U.S. National Research Council, 1996). All the experiments and the protocol proposed in the projects were reviewed and approved by the IRFMN Animal Care and Use Committee (IACUC), that includes members “ad hoc” for ethical issues. The mice were bred and maintained in a SPF environment. Animals with substantial motor impairment had food on the cage bottom and water bottles with long drinking spouts. The survival time was defined as the time when the animals were unable to right themselves within 10 s after being placed on either side. At this point, the animals were deeply anesthetized with Equithesin and then sacrificed by decapitation before proceeding to the dissection of tissues for biochemical analyses.
Preliminary acute treatment in 129Sv mice
Two preliminary pharmacokinetic studies were carried out in non-transgenic 129Sv female mice aging 9-11 weeks. For the time-course study, mice were treated orally with 200 mg/kg of ubiquinone10 or ubiquinol10 and killed at various times thereafter (30, 60, 180, 360 min and 24 hours after dosing, four mice/group). In a separate single-dose study, ubiquinone10 and ubiquinol10 were administered orally at various doses (200, 400, 800 and 1600 mg/kg) and the animals (4 mice/dose) were killed 3 hours after dosing. Four mice were treated with vehicle alone.
Chronic treatment in 129Sv SOD1G93A mice and analysis of their motor dysfunction and survival
Female 129Sv SOD1G93A mice were treated by oral gavage with ubiquinone10 or ubiquinol10 (800mg/kg), or vehicle (n = 15 each group). The treatment started when the mice were 13 weeks old, which is the mean age that the growth of their body weight starts to differentiate from that of non-transgenic littermates and they show first alterations of hindlimbs abduction when raised by tail. This occurs just before the appearance of motor impairments (onset of motor deficit). Mice were treated daily until the end-stage of the disease, when they were sacrificed (see below).
Disease progression was assessed two times a week from the start of the experiment by an operator blinded to the treatment. The parameters included body weight, latency on rotating bar (rotarod) and paw grip endurance (PaGE) and were evaluated as previously described [17]. The motor symptoms onset was determined by the first impairment in grip strength or on rotarod performance for two consecutive time points. The mice were sacrificed when they were unable to right themselves within 10 seconds after being placed on both sides. This time point was considered as the end-stage of the disease and was used to estimate the survival time.
Statistical analysis of the data of body weight, grip strength and rotarod performance was carried out by Two-way ANOVA for repeated measures (time) and different groups (treatments), followed by post-hoc Bonferroni’s test to compare the effect of treatments in respect to vehicle at each time point. The analysis was applied at the time points when all animals for each group were still alive in order to maintain the number per group balanced. The age at onset and the survival length were statistically evaluated by the Log-rank test to compare probabilities.
HPLC Analysis of CoQ10 and CoQ9 Levels
Animals were sacrificed by decapitation. Blood was collected from the heart in heparinized tubes. After centrifugation at 5000 g for 10 min, plasma was collected and stored at −80°C. CNS tissues were removed immediately, blotted with paper to remove surface blood, quickly frozen in dry ice and stored at −80°C.
All solvents and reagents used were HPLC grade (Carlo Erba, Milan, Italy). Ubiquinone10, ubiquinone9, FeCl3, 1,4-benzoquinone and sodium perchlorate were purchased from Sigma-Aldrich (St. Louis, MO, USA).
Plasma levels of total CoQ10 and CoQ9 in treated SOD1G93A mice
The method described by Mosca et al. [18] was used. Fifty µL of plasma were pre-incubated for 10 min at room temperature with 15 µL of 1,4-benzoquinone (2 mg/mL in ethanol), in order to allow a complete oxidation of ubiquinol9/10. The samples were then added to 235 µL of 1-propanol, vortexed and then centrifuged at 10000 g for 2 minutes. Two hundred µL of supernatant were then injected into the HPLC system with UV detection at 275 nm (Waters 1525 Binary Pump; Waters 2487 Dual λ Absorbance Detector; Waters mod. 717-plus autosampler). Separation was carried out using a Supelcosil LC 18-DB column (15×0.46 cm, i.d. 5 µm, Supelco Inc., Bellefonte, PA, USA), with a Supelcosil LC 18-DB Supelguard Cartridge (2×0.4 cm, i.d. 5 µm, Supelco Inc., Bellefonte, PA, USA) by isocratic elution with ethanol/methanol (65∶35 v/v) at flow rate of 1 mL/min. The total run time was 19 min and the retention times for CoQ9 and CoQ10 were 7.1 and 8.9 min, respectively.
Total levels of CoQ10 and CoQ9 were quantified by reference to calibration curves with ubiquinone10 or ubiquinone9, run in parallel with each analytical session. Calibration curves were linear in the range 0.2–3.2 µg/ml for both coenzymes, with r2 values >0.99.
CNS levels of total CoQ10 and CoQ9 in treated SOD1G93A mice
CoQs were extracted from CNS tissues and measured by HPLC with UV detection at 275 nm, based on the method described by Kitano et al. [14], with slight modifications.
CNS tissues were homogenized in ultrapure water (1 g in 20 mL), aliquots of 0.15 mL were added to 1 mL of acetonitrile and the samples were centrifuged at 2500 rpm for 5 min; the supernatants were collected and 1 ml aliquots were added to 0.1 mL of 2% FeCl3 to allow the oxidation of ubiquinols to ubiquinones. After 20 minutes incubation at room temperature the extraction procedure started by adding 2 ml methanol, 0.5 mL purified water and 5 ml hexane, followed by 15 minutes shaking, centrifugation (1200 g, 5 min) and collection of the organic phase. This procedure was repeated twice and the combined extracts were evaporated to dryness, the residue was dissolved in 150 µL of water/methanol (20∶80 v/v), transferred in Eppendorf tubes, centrifuged (2500 rpm, 30 sec) and 100 µl were injected into the chromatographic system (Waters 1525 Binary Pump; Waters 2487 Dual λ absorbance detector; Waters mod. 717-plus autosampler). Separation was carried out on a Kinetex 2.6 μ PFP 100 Å column (100×4.6 mm, Phenomenex Inc., Torrance, CA, USA), with a HPLC KrudKatcher 5 μ ultra column in-line filter (Phenomenex Inc., Torrance, CA, USA), using gradient elution with water/methanol (20∶80 v/v; solvent A) and acetonitrile/methanol (4∶96 v/v; solvent B). During HPLC analysis the solvent gradient was programmed to operate at a flow rate of 1 mL/min for 25 minutes at the following gradient conditions: step1 - from the initial condition of 100% solvent A to 100% solvent B over 16 min; step 2 - from 100% solvent B to 100% solvent A over 1 min. Retention times of CoQ9 and CoQ10 were 17.8 and 18.3 minutes, respectively.
Total levels of CoQ10 and CoQ9 were quantified by reference to calibration curves with ubiquinone10 or ubiquinone9, run in parallel with each analytical session. Calibration curves were linear in the range 4.0–16.0 µg/ml (CoQ10) and 10.0–60.0 µg/ml (CoQ9), with r2 values >0.99.
CNS levels of ubiquinol9/10 and ubiquinone9/10
The method described by Takada et al. [19] was used with slight modifications. Tissues were homogenized (1 g in 10 mL of ultrapure water) and divided in two aliquots, one to measure total CoQ9/10 levels (aliquot A) and the other to measure ubiquinone9/10 only (aliquot B). 100 µL of aliquot A were added to 10 µL of ultrapure water and 60 µL of 1,4-benzoquinone (2 mg/mL in ethanol) to allow a complete oxidation of ubiquinol. In parallel, 100 µL of aliquot B were added to 10 µL of 10% Na2EDTA (to protect ubiquinol from oxidation), and 60 µL of ethanol. After 10 min, both aliquots were added to 750 µL of ethanol/hexane (2∶5, v/v), shaken, and centrifuged at 4000g for 3 min (4°C). The supernatants were dried under nitrogen flow, reconstituted with 100 µL of ethanol and 50 µL were injected into a HPLC system (Waters 1525 Binary pump; Waters mod. 717-plus autosampler) coupled to a coulometric detector (ESA Couluchem 5100A with ESA analytical cell mod. 5011). The electrochemical analytical cell consisted of a series of two coulometric electrodes: the first one (−650mV) was for ubiquinone9/10 reduction and the second one (+400mV) was for detection of CoQ9/10 in the reduced form. The chromatographic column was a reverse phase Supelcosil LC-18 DB (15×0.46 cm, i.d. 3 µm; Supelco Inc., Bellefonte, PA, USA). The separation was carried out in isocratic operational mode with ethanol/methanol/HClO4 (700∶300:1, v/v/v) +7% sodium perchlorate at 0.8 mL/min for 13 minutes. Retention times were 5.3 min (ubiquinol9), 6.3 min (ubiquinone9), 7.1 min (ubiquinol10) and 8.8 min (ubiquinone10).
Total levels of CoQ9/10 (aliquot A), and ubiquinone9/10 (aliquot B) were quantified by reference to calibration curves with ubiquinone9/10, run in parallel with each analytical session. Calibration curves were linear in the range 5–50 µg/g tissue (ubiquinone10) and 20–100 µg/g tissue (ubiquinone9), with r2 values >0.99. Endogenous levels of ubiquinol9/10 were obtained as difference between total CoQ9/10 levels (determined in fully oxidized aliquot A) and ubiquinone9/10 levels (determined in aliquot B).
All the procedures were carried out rapidly, on ice, with protection from light, and in the presence of Na2EDTA, to limit the possibility that endogenous ubiquinol oxidizes to ubiquinone during the sample preparation. In these conditions, we verified that the storage of CNS homogenate at 4°C, for up to one hour, did not affect ubiquinol/total CoQ ratios (data not shown). We also note that the ubiquinol/total CoQ ratios detected in our study are very similar to published data [3], [19], [20]. Most importantly, we operated to ensure reliable comparisons between the different groups (SOD1G93A, SOD1WT and non-transgenic), always including matched samples in the same analytical session, with random processing.
Plasma levels of ubiquinol9 and ubiquinone9
The method described by Takada et al. [21] was used with slight modifications. Plasma samples were divided in two aliquots, one to measure total CoQ9 levels (aliquot A) and the other to measure ubiquinone9 only (aliquot B). 50 µL of aliquot A were added to 50 µL of ultrapure water and 25 µL of 1,4-benzoquinone (2 mg/mL ethanol), to allow a complete oxidation of ubiquinol9. In parallel, 50 µL of aliquot B were added to 50 µL of 10% Na2EDTA (to protect ubiquinol from oxidation), and 25 µL of ethanol. After 10 min, both aliquots were added to 750 µL of ethanol/hexane (2∶5, v/v), shaken, and centrifuged at 4000 g for 3 min (4°C). The supernatant was dried under nitrogen flow, reconstituted with 200 µL of H2O/2-propanol (1∶5, v/v) and 150 µL were injected in a HPLC system as reported for CNS samples. The chromatographic separation was carried out in isocratic operational mode with ethanol/methanol/H2O/HClO4 (700∶300:10∶1, v/v/v/v) +7% sodium perchlorate at 1 mL/min for 15 minutes. Retention times for ubiquinol9 and ubiquinone9 were 6.2 and 8.6 minutes, respectively.
Total levels of CoQ9 (aliquot A), and ubiquinone9 (aliquot B) were quantified by reference to calibration curves with ubiquinone9, run in parallel with each analytical session. Calibration curve was linear in the range 0.05–1.20 µg/mL, with r2 values >0.99. Endogenous levels of ubiquinol9 were obtained as difference between total CoQ9 levels (determined in fully oxidized aliquot A) and ubiquinone9 levels (determined in aliquot B).
All the procedures were carried out rapidly, on ice, with protection from light, and in the presence of Na2EDTA. The ubiquinol/total CoQ ratios detected in our study are very similar to published data [3], [19], [20]. Most importantly, we operated to ensure reliable comparisons between the different groups (SOD1G93A, SOD1WT and NTg), always including matched samples in the same analytical session, with random processing.
Results
Mice Treatment with Ubiquinone10 and Ubiquinol10
Since available data indicated that the stabilized formulation of ubiquinol10 has a better oral bioavailability than ubiquinone10 in different animal species [14], [15], preliminary studies were carried out to verify if this was true also in 129Sv mice. With this aim, plasmatic CoQ10 levels were measured in wild-type mice treated acutely with both ubiquinol10 and ubiquinone10. Fig. S2A shows that, after oral treatment with 200 mg/kg, the area under the plasma concentration-time curve over the dosing interval (AUC0−24h), was 1.5-fold higher for ubiquinol10 (14.2 µg.h/mL) than ubiquinone10 (9.8 µg.h/mL). Fig. S2B shows the plasma CoQ10 levels in mice treated with different doses of the two compounds, three hours before sacrifice, confirming higher CoQ10 levels in the plasma of ubiquinol10–treated mice than in the plasma of ubiquinone10-treated mice, except at the highest dose tested (1600 mg/kg). In particular, treatment with 800 mg/kg ubiquinol10 resulted in CoQ10 levels double than those found after treatment with the same dose of ubiquinone10 (1.5±0.2 and 0.8±0.4 µg/mL, respectively, p<0.05 Student’s t test).
Brain CoQ10 levels (13 µg/g tissue on average) were not affected by acute treatment with either ubiquinone10 or ubiquinol10, at any time-point or any concentration (data not shown).
Chronic Treatment with Ubiquinone10 and Ubiquinol10 in SOD1G93A Mice
Effect on motor dysfunction, body weight and survival of SOD1G93A mice
Fig. 1 shows that chronic oral treatment of SOD1G93A mice with 800 mg/kg/day ubiquinone10 or ubiquinol10, started at the onset of disease symptoms, did not modify the motor impairment progression assessed by rotarod performance (fig. 1B) and grip strength (fig. 1C), in respect to the vehicle-treated mice. The progressive loss of body weight (fig. 1A) was also unaffected by both treatments. The lack of effect was clearly evidenced by the overlapping Kaplan-Meyer curves and the similar mean for motor symptom onset (fig. 1D) and survival length (fig. 1E).
Total CoQ10 and CoQ9 plasma levels
Table 1 reports the plasma concentrations of total CoQ10 and CoQ9 after chronic administration of ubiquinone10 and ubiquinol10 in the same SOD1G93A mice used for behavioral tests. The last dose of both formulations was administered two hours before sacrifice.
Table 1. Levels of CoQ9/10 in the plasma of SOD1G93A mice treated chronically with ubiquinone10 or ubiquinol10.
| Chronic treatment | Total CoQ10 (µg/mL) | Total CoQ9 (µg/mL) |
| Vehicle | <0.20 (5) | 1.63±0.40 (5) |
| Ubiquinol10 | 3.40±0.22 (13) | 1.25±0.12 (13) |
| Ubiquinone10 | 3.03±0.26 (9) | 1.19±0.20 (9) |
Female 129Sv SOD1G93A mice were treated orally with 800 mg/kg of ubiquinol10 or ubiquinone10, or vehicle (sunflower seed oil), once a day, starting from age of 91 days until the last stage of the disease (age of 125 days, on average) when they were sacrificed, two hours after the last treatment. Values are means±SEM of (n) mice.
As expected, no detectable levels of CoQ10 were found in mice chronically treated with vehicle (<0.20 µg/mL), whereas levels of 3.0 and 3.4 µg/mL were measured in the mice treated with ubiquinone10 and ubiquinol10, respectively (difference not statistically significant). Plasma levels of CoQ9 were not significantly affected by treatment with either ubiquinone10 or ubiquinol10 (table 1).
Total CoQ10 and CoQ9 CNS tissues levels
Figure 2 shows the levels of CoQ10 and CoQ9 measured in the brain and spinal cord (cervicothoracic, CT-SpC and lumbar, L-SpC) of SOD1G93A mice after chronic treatment with ubiquinone10 or ubiquinol10, or vehicle.
Figure 2. CoQ9/10 levels in brain and spinal cord of SOD1G93A chronically treated with ubiquinol10 or ubiquinone10.
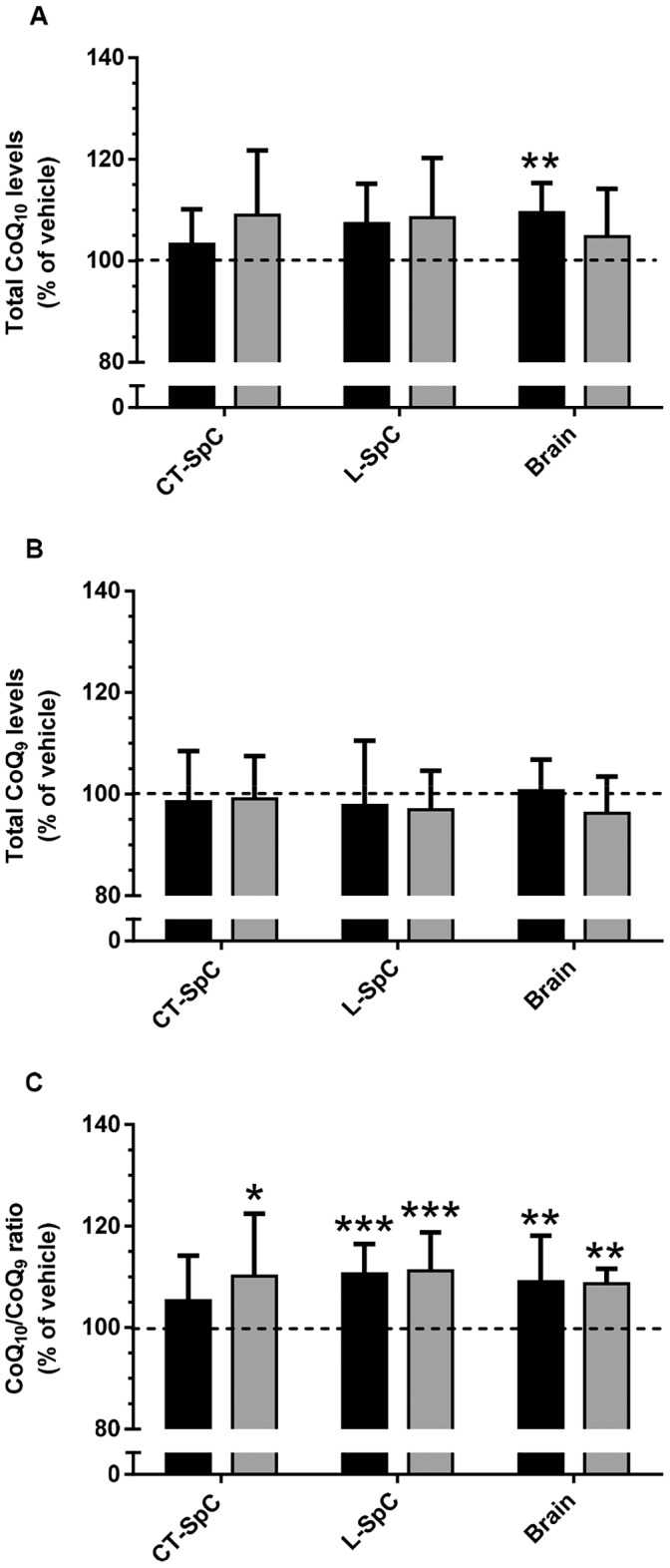
Total levels of CoQ10 (A) and CoQ9 (B), and CoQ10/CoQ9 ratio (C), in brain, and in cervicothoracic (CT-SpC) and lumbar (L-SpC) spinal cord of mice chronically treated with 800 mg/kg ubiquinol10 (black) or ubiquinone10 (grey). Each value is the mean±SEM of 10–12 mice and is indicated as percentage of levels measured in mice treated with vehicle. * P<0.05, ** P<0.01 compared to vehicle (Dunnet’s multiple comparison tests following One-way ANOVA).
CoQ10 levels in brain, CT-SpC and L-SpC of vehicle-treated mice were 17.6±0.3, 10.8±0.3 and 11.0±0.3 µg/g tissue (mean±SEM, n = 5), respectively; levels were slightly (4–11%) higher in all CNS tissues obtained from both ubiquinone10- or ubiquinol10-treated mice, with statistically significant differences detected in the brain of mice treated with ubiquinol10 only (Fig. 2A). CoQ9 levels were not affected by treatment with ubiquinone10 or ubiquinol10 (Fig. 2B). This finding prompted us to normalize, for each mouse, the CoQ10 level on the corresponding CoQ9 content, in order to minimize variability between mice. After this normalization, the levels of CoQ10 (expressed as CoQ10/CoQ9 ratio) appeared to be slightly but significantly increased (10%) by treatment with both ubiquinone10 or ubiquinol10 in all tissues, with the only exception for the levels in CT-SpC after treatment with ubiquinol10 (Fig. 2C).
Levels of Reduced and Oxidized Forms of CoQ10 and CoQ9 in SOD1G93A Mice
Basal levels of reduced and oxidized forms of both coenzymes were measured in male 129Sv SOD1G93A mice at the symptomatic stage of the disease (16 weeks), and in age-matched 129Sv SOD1WT and non-transgenic 129Sv mice. In figures 3, 4, and 5 , the levels in transgenic mice are shown as a percentage of levels in non-transgenic mice (absolute values in table S1), in order to highlight the effect of the over-expression of the human SOD1 transgene, and any difference related to the G93A mutation.
Figure 3. Increased levels of ubiquinol9/10 in the brain of symptomatic SOD1G93A mice.

Endogenous brain levels of CoQ9 (A–C) and CoQ10 (D–F) in 16 week-old male 129Sv SOD1WT and SOD1G93A mice are shown as percentage of the levels measured in age-matched 129Sv non-transgenic mice (absolute values in table S1). Panels A, D show the total CoQs levels, whereas panels B, E and panels C, F show the reduced and oxidized forms, respectively. Each value is the mean±SEM of 4–7 mice. * P<0.05, ** P<0.01 compared to SOD1WT mice (Student’s T-test).
Figure 4. Increased levels of ubiquinol9/10 in the lumbar spinal cord of symptomatic SOD1G93A mice.

Endogenous levels of CoQ9 (A–C) and CoQ10 (D–F) in the lumbar spinal cord of male 16 week-old 129Sv SOD1WT and SOD1G93A mice are shown as percentage of the levels measured in age-matched 129Sv non-transgenic mice (absolute values in table S1). Panels A, D show the total CoQs levels, whereas panels B, E and panels C, F show the reduced and oxidized forms, respectively. Each value is mean±SEM of 4–7 mice. * P<0.05, ** P<0.01 compared to SOD1WT mice; ° P<0.05 compared to 129Sv NTg mice (Student’s T-test).
Figure 5. Unchanged plasma levels of ubiquinol9 in symptomatic SOD1G93A mice.
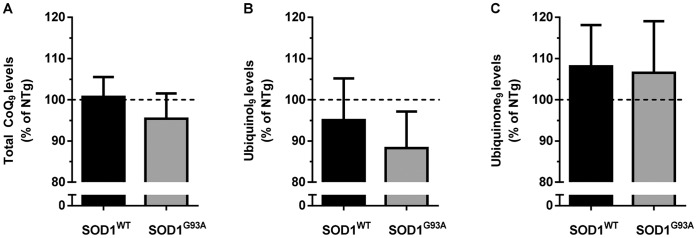
Levels of CoQ9 (A), ubiquinol9 (B) and ubiquinone9 (C) in plasma of male 16 week-old 129Sv SOD1WT and SOD1G93A mice are shown as percentage of the levels measured in age-matched 129Sv non-transgenic mice (table S1). Each value is mean±SEM of 4–7 mice.
Brain
Figure 3 shows that over-expression of human SOD1WT enzyme does not alter the endogenous levels of oxidized and reduced CoQ9/10, in comparison with non-transgenic mice. Over-expression of the mutated protein (SOD1G93A) was instead associated with a significant increase (10–25%) of endogenous total levels of both CoQ9 and CoQ10, in comparison with age-matched SOD1WT mice (Fig. 3A,D). This effect was associated with a significant increase (30–40%) of the reduced forms only (Fig. 3B,E), whereas no significant changes were observed for the oxidized forms (Fig. 3C,F).
Lumbar spinal cord
Endogenous levels of CoQ9 and CoQ10 in lumbar spinal cord of 16 weeks-old SOD1WT mice were significantly lower (−20%), than in age-matched non-transgenic mice, a difference due to a decrease of the oxidized forms. In line with the results found in the brain, basal levels of CoQ9 and CoQ10 in SOD1G93A were significantly higher (16–20%) than corresponding levels in age-matched SOD1WT mice (Fig. 4A,D) and this increase was associated with a significant increase (36–60%) of the reduced forms only (Fig. 4B,E), whereas no or much lower changes where observed for the oxidized forms (Fig. 4C,F).
Plasma
Figure 5 shows that over-expression of human SOD1WT enzyme does not alter the endogenous plasma levels of total CoQ9, ubiquinol9 and ubiquinone9, in comparison with non-transgenic mice. Moreover, no significant differences were observed between SOD1WT and SOD1G93A mice. CoQ10 levels were under the limit of detection.
Discussion
Oxidative stress and mitochondrial impairment are the most prominent pathogenic mechanisms of ALS. The CoQ complex is a key component of the mitochondrial function, and its reduced form ubiquinol is fundamental for the antioxidant response, by direct interaction with radical species or by regenerating antioxidant agents [22].
It had been previously reported that chronic treatment with ubiquinone10 had a significant, although slight, effect on the life-span of a mouse model of familial ALS [12]. Regrettably, this finding was not confirmed in a phase II trial in humans [13]. One of the potential reasons underlying these results regarded the poor pharmacokinetic profile of ubiquinone10. In fact, a stabilized formulation of ubiquinol10, with a better bioavailability in respect to the oxidized form [14], [15] showed a better neuroprotective activity on MPTP-induced degeneration in mice [16].
On these basis we chronically treated a mouse model of familial ALS (SOD1G93A) with ubiquinol10, and ubiquinone10 in parallel, to increase CNS CoQ10 levels with the aim to obtain beneficial effect on the disease course.
Chronic treatment with both formulation resulted in a significant increase of plasmatic CoQ10 levels (from <0.2 to ≥3 µg/mL) that was associated to a very mild increase (10% on average) of the CoQ10 levels in CNS tissues (brain and spinal cord). Despite the better pharmacokinetic profile of the new formulation of ubiquinol10 after acute treatment [14,15 and present data], we found no differences in the CoQ10 levels after chronic treatment with both formulations.
No effect of the chronic treatments with either ubiquinone10or ubiquinol10 (both at 800 mg/kg/day) was observed on the disease progression or the life-span of SOD1G93A mice.
This finding is at variance with previous data showing a 8% increase of survival after chronic treatment with a lower dose (200 mg/kg) of ubiquinone10 [12]. Unfortunately, that study did not report the levels of CoQ10 reached in the brain of SOD1G93A mice after the chronic treatment. Two possible reasons may account for the discrepancy: 1) the strain of SOD1G93A mice is different (C57BL6/SJL in [12] and 129Sv in our study), although the disease onset and progression is very similar in the two strains; and 2) in our study, the treatment started at the appearance of the first symptoms of disease (13 weeks of age), while Matthews et al. [12] started the treatment when mice were 7-weeks old, well before the onset of symptoms. According to the recent guidelines for preclinical animal research in ALS [23], the poor impact of preclinical studies on clinical practice is in part due to the fact that most of the pharmacological interventions are intended to be disease-preventive rather than being disease-modifying once the symptoms are evident. In this respect, since we start treatment when the mice exhibited the first disease symptoms, our data are in line with the lack of effect found in phase II clinical trials with ubiquinone10 [13].
The observation that the chronic treatments with ubiquinone10 and ubiquinol10 resulted in a very small increase of CoQ10 levels in the CNS of SOD1G93A mice (≤10%) may provide an explanation for the lack of effect on disease progression and life span. In fact, it was previously shown that the neuroprotection observed after 1-week pretreatment of non-transgenic rats with 200 mg/kg ubiquinone10, i.e. prevention of the neurotoxicity induced by 3-nitropropionic acid, was associated with a ∼30% increase of brain CoQ10 [12]. All these data may suggest that an high enough increase of CNS concentrations of CoQ10 over basal levels is required to support neuroprotection and that the increase associated with our treatments in 129Sv SOD1G93A mice, started at the time of the onset of symptoms, was too low to provide an efficient neuroprotection.
To better understand this point, in the second part of the study we measured the endogenous levels of CoQ10 and CoQ9 in CNS tissues and plasma of symptomatic SOD1G93A mice, in comparison with age- and sex-matched SOD1WT and non-transgenic mice.
Our results show for the first time that the CNS tissues of SOD1G93A mice have CoQ9/10 levels 10–25% higher than those of age-matched SOD1WT mice, while no differences were found in the plasma between the two strains. The increase was due to the reduced form only, ubiquinol, indicating a shift in the redox equilibrium toward a protective antioxidant state, and suggesting an attempt of the CNS to maintain an antioxidant environment to counteract the pathological processes of the disease. According with the antioxidant function of the reduced form of CoQ, it has been shown that ubiquinol10 is involved in the control of the mitochondrial levels of nitric oxide [24] and that the increase of ubiquinol pool in mitochondria (by antimycin) results in a decrease of protein nitration induced by ONOO− [25]. Notably, increased protein tyrosine nitration has been observed in spinal cord of ALS transgenic mice (C57BL/6 SOD1G93A), from the pre-symptomatic till late stages of the disease, suggesting a role of nitrative stress in ALS pathogenesis and progression [26], [27]. Therefore, the increased levels of ubiquinol in the spinal cord of SOD1G93A mice are likely not sufficient to counteract the intense nitrative stress occuring in these mice, with consequent lack of effect on their disease progression and life span.
An increase of ubiquinone10 with shift in the redox state of the coenzyme has been reported in the plasma [9] and CSF [10] of sALS patients and was suggested as a marker of the underlying oxidative stress. Our data, showing that the increase of ubiquinol9/10 in the CNS of SOD1G93A mice was not paralleled by changes in plasma levels, indicate that the peripheral and central compartments react differently to the oxidative stress induced by mutant SOD1, and suggest that plasmatic CoQ levels do not represent sensitive markers for the changes associated to oxidative stress in the CNS. This is in line with the observations of Kontush et al. [28] who could not detect changes of ubiquinol10 in the plasma of patients with Alzheimer’s disease (AD), another neurodegenerative condition associated with increased free radical production in the CNS, even if higher levels of total CoQ were reported in post mortem brain tissues from AD patients [29].
In summary, our data show that chronic treatment of SOD1G93A mice with ubiquinone10 or ubiquinol10 had no effect on the disease progression. This lack of efficacy may be due to the negligible changes of CoQ9/10 levels in the brain and spinal cord of treated mice, which in turn may be the consequence of a poor CNS availability of the compounds and/or the fact that endogenous CoQ9/10 levels are already significantly increased in SOD1G93A mice.
Supporting Information
No gender effect on disease progression in SOD1G93A mice. Disease progression and survival length in males (gray) and females (black) SOD1G93A mice on 129Sv background. Each point represent the mean of n = 10 male and n = 13 female. Table reports the mean and standard deviations of symptoms onset, life-span and disease duration for each group.
(TIF)
Ubiquinol10 shows a better oral pharmacokinetic profile than ubiquinone10. Plasma CoQ10 levels in female 129Sv non-transgenic mice treated orally with ubiquinone10 (grey) or ubiquinol10 (black). (A) Time-course after acute treatment with 200 mg/kg. (B) Dose-dependency after acute treatment with 200, 400, 800 and 1600 mg/kg and sacrifice 3 hours later. Each value is the mean±SEM of 4 mice. Plasma basal levels of CoQ10 were under the limit of quantification (<0.2 µg/mL).
(TIF)
Endogenous levels of CoQ9/10 in 129Sv NTg mice. Endogenous levels (µg/g tissue or ng/mL) of total CoQ9/10, ubiquinol9/10 and ubiquinone9/10 in brain, lumbar spinal cord (L-SpC) and plasma of 16 week-old male 129Sv non-transgenic mice. Values are expressed as means±SEM of (n) mice.
(DOC)
Acknowledgments
We thank Elisa Battaglia and Laura Pasetto for help in mice behavioral tests and treatment.
Funding Statement
The work was partly funded by Sintofarm S.p.A., Guastalla (RE), Italy. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. No additional external funding was received for this study.
References
- 1. Tran UC, Clarke CF (2007) Endogenous synthesis of coenzyme Q in eukaryotes. Mitochondrion 7 Suppl: S62–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lass A, Agarwal S, Sohal RS (1997) Mitochondrial ubiquinone homologues, superoxide radical generation, and longevity in different mammalian species. J Biol Chem 272: 19199–19204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Tang PH, Miles MV, Miles L, Quinlan J, Wong B, et al. (2004) Measurement of reduced and oxidized coenzyme Q9 and coenzyme Q10 levels in mouse tissues by HPLC with coulometric detection. Clin Chim Acta 341: 173–184. [DOI] [PubMed] [Google Scholar]
- 4. Littarru GP, Langsjoen P (2007) Coenzyme Q10 and statins: biochemical and clinical implications. Mitochondrion 7 Suppl: S168–174 [DOI] [PubMed] [Google Scholar]
- 5. Pravst I, Zmitek K, Zmitek J (2010) Coenzyme Q10 contents in foods and fortification strategies. Crit Rev Food Sci Nutr 50: 269–280. [DOI] [PubMed] [Google Scholar]
- 6. Huertas JR, Battino M, Lenaz G, Mataix FJ (1991) Changes in mitochondrial and microsomal rat liver coenzyme Q9 and Q10 content induced by dietary fat and endogenous lipid peroxidation. FEBS Lett 287: 89–92. [DOI] [PubMed] [Google Scholar]
- 7. Edlund C, Soderberg M, Kristensson K (1994) Isoprenoids in aging and neurodegeneration. Neurochem Int 25: 35–38. [DOI] [PubMed] [Google Scholar]
- 8. Lagendijk J, Ubbink JB, Vermaak WJ (1996) Measurement of the ratio between the reduced and oxidized forms of coenzyme Q10 in human plasma as a possible marker of oxidative stress. J Lipid Res 37: 67–75. [PubMed] [Google Scholar]
- 9. Sohmiya M, Tanaka M, Suzuki Y, Tanino Y, Okamoto K, et al. (2005) An increase of oxidized coenzyme Q-10 occurs in the plasma of sporadic ALS patients. J Neurol Sci 228: 49–53. [DOI] [PubMed] [Google Scholar]
- 10. Murata T, Ohtsuka C, Terayama Y (2008) Increased mitochondrial oxidative damage in patients with sporadic amyotrophic lateral sclerosis. J Neurol Sci 267: 66–69. [DOI] [PubMed] [Google Scholar]
- 11. Bendotti C, Carri MT (2004) Lessons from models of SOD1-linked familial ALS. Trends Mol Med 10: 393–400. [DOI] [PubMed] [Google Scholar]
- 12. Matthews RT, Yang L, Browne S, Baik M, Beal MF (1998) Coenzyme Q10 administration increases brain mitochondrial concentrations and exerts neuroprotective effects. Proc Natl Acad Sci U S A 95: 8892–8897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kaufmann P, Thompson JL, Levy G, Buchsbaum R, Shefner J, et al. (2009) Phase II trial of CoQ10 for ALS finds insufficient evidence to justify phase III. Ann Neurol 66: 235–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hosoe K, Kitano M, Kishida H, Kubo H, Fujii K, et al. (2007) Study on safety and bioavailability of ubiquinol (Kaneka QH) after single and 4-week multiple oral administration to healthy volunteers. Regul Toxicol Pharmacol 47: 19–28. [DOI] [PubMed] [Google Scholar]
- 15. Kitano M, Watanabe D, Oda S, Kubo H, Kishida H, et al. (2008) Subchronic oral toxicity of ubiquinol in rats and dogs. Int J Toxicol 27: 189–215. [DOI] [PubMed] [Google Scholar]
- 16. Cleren C, Yang L, Lorenzo B, Calingasan NY, Schomer A, et al. (2008) Therapeutic effects of coenzyme Q10 (CoQ10) and reduced CoQ10 in the MPTP model of Parkinsonism. J Neurochem 104: 1613–1621. [DOI] [PubMed] [Google Scholar]
- 17. Pizzasegola C, Caron I, Daleno C, Ronchi A, Minoia C, et al. (2009) Treatment with lithium carbonate does not improve disease progression in two different strains of SOD1 mutant mice. Amyotroph Lateral Scler 10: 221–228. [DOI] [PubMed] [Google Scholar]
- 18. Mosca F, Fattorini D, Bompadre S, Littarru GP (2002) Assay of coenzyme Q(10) in plasma by a single dilution step. Anal Biochem 305: 49–54. [DOI] [PubMed] [Google Scholar]
- 19. Takada M, Ikenoya S, Yuzuriha T, Katayama K (1984) Simultaneous determination of reduced and oxidized ubiquinones. Methods Enzymol 105: 147–155. [DOI] [PubMed] [Google Scholar]
- 20. Podda M, Weber C, Traber MG, Packer L (1996) Simultaneous determination of tissue tocopherols, tocotrienols, ubiquinols, and ubiquinones. J Lipid Res 37: 893–901. [PubMed] [Google Scholar]
- 21. Takada M, Ikenoya S, Yuzuriha T, Katayama K (1982) Studies on reduced and oxidized coenzyme Q (ubiquinones). II. The determination of oxidation-reduction levels of coenzyme Q in mitochondria, microsomes and plasma by high-performance liquid chromatography. Biochim Biophys Acta 679: 308–314. [DOI] [PubMed] [Google Scholar]
- 22. Bentinger M, Brismar K, Dallner G (2007) The antioxidant role of coenzyme Q. Mitochondrion. 7 Suppl: S41–50 [DOI] [PubMed] [Google Scholar]
- 23. Ludolph AC, Bendotti C, Blaugrund E, Chio A, Greensmith L, et al. (2010) Guidelines for preclinical animal research in ALS/MND: A consensus meeting. Amyotroph Lateral Scler 11: 38–45. [DOI] [PubMed] [Google Scholar]
- 24. Poderoso JJ (2009) The formation of peroxynitrite in the applied physiology of mitochondrial nitric oxide. Arch Biochem Biophys 484: 214–220. [DOI] [PubMed] [Google Scholar]
- 25. Schopfer F, Riobo N, Carreras MC, Alvarez B, Radi R, et al. (2000) Oxidation of ubiquinol by peroxynitrite: implications for protection of mitochondria against nitrosative damage. Biochem J 349: 35–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Casoni F, Basso M, Massignan T, Gianazza E, Cheroni C, et al. (2005) Protein nitration in a mouse model of familial amyotrophic lateral sclerosis: possible multifunctional role in the pathogenesis. J Biol Chem 280: 16295–16304. [DOI] [PubMed] [Google Scholar]
- 27. Basso M, Samengo G, Nardo G, Massignan T, D’Alessandro G, et al. (2009) Characterization of detergent-insoluble proteins in ALS indicates a causal link between nitrative stress and aggregation in pathogenesis. PLoS One 4: e8130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kontush A, Schippling S, Spranger T, Beisiegel U (1999) Plasma ubiquinol-10 as a marker for disease: is the assay worthwhile? Biofactors 9: 225–229. [DOI] [PubMed] [Google Scholar]
- 29. Edlund C, Soderberg M, Kristensson K, Dallner G (1992) Ubiquinone, dolichol, and cholesterol metabolism in aging and Alzheimer’s disease. Biochem Cell Biol 70: 422–428. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
No gender effect on disease progression in SOD1G93A mice. Disease progression and survival length in males (gray) and females (black) SOD1G93A mice on 129Sv background. Each point represent the mean of n = 10 male and n = 13 female. Table reports the mean and standard deviations of symptoms onset, life-span and disease duration for each group.
(TIF)
Ubiquinol10 shows a better oral pharmacokinetic profile than ubiquinone10. Plasma CoQ10 levels in female 129Sv non-transgenic mice treated orally with ubiquinone10 (grey) or ubiquinol10 (black). (A) Time-course after acute treatment with 200 mg/kg. (B) Dose-dependency after acute treatment with 200, 400, 800 and 1600 mg/kg and sacrifice 3 hours later. Each value is the mean±SEM of 4 mice. Plasma basal levels of CoQ10 were under the limit of quantification (<0.2 µg/mL).
(TIF)
Endogenous levels of CoQ9/10 in 129Sv NTg mice. Endogenous levels (µg/g tissue or ng/mL) of total CoQ9/10, ubiquinol9/10 and ubiquinone9/10 in brain, lumbar spinal cord (L-SpC) and plasma of 16 week-old male 129Sv non-transgenic mice. Values are expressed as means±SEM of (n) mice.
(DOC)