

Abstract
In almost 30 years since the introduction of HMG-CoA reductase inhibitors (statins), no other class of lipid modulators has entered the market. Elevation of high-density lipoprotein-cholesterol (HDL-C) via inhibiting cholesteryl ester transfer protein (CETP) is an attractive strategy for reducing the risk of cardiovascular events in high-risk patients. Triglyceride and cholesteryl ester (CE) transfer between lipoproteins is mediated by CETP; thus inhibition of this pathway increases the concentration of HDL-C. Torcetrapib was the first CETP inhibitor evaluated in Phase 3 clinical trials. Because of off-target effects, torcetrapib raised blood pressure and increased the concentration of serum aldosterone leading to higher cardiovascular events and mortality. Torcetrapib showed positive effects on the cardiovascular risk especially in patients with a greater increase in HDL-C and Apolipoprotein A-1 (apoA-1) levels.
The Phase 3 clinical trial of dalcetrapib, the second CETP inhibitor that has entered clinical development, was terminated because of ineffectiveness. Dalcetrapib is a CETP modulator that elevated HDL-C level but did not reduce the concentration of low-density lipoprotein cholesterol (LDL-C). Both heterotypic and homotypic CE transfer between lipoproteins are mediated by some CETP inhibitors including torcetrapib, anacetrapib and evacetrapib while dalcetrapib only affect the heterotypic CE transfer. Dalcetrapib has a chemical structure that is distinct from other CETP inhibitors with smaller molecular weight and lack of trifluoride moieties. Dalcetrapib is a pro-drug that must be hydrolyzed to a pharmacologically active thiol form.
Two other CETP inhibitors, anacetrapib and evacetrapib, are currently undergoing evaluation in Phase 3 clinical trial. Both molecules have shown beneficial effects by increasing HDL-C and decreasing LDL-C concentration. The success of anacetrapib and evacetrapib will remain to be confirmed upon the completion of Phase 3 clinical trials in 2017 and 2015, respectively.
Generally, the concentration of HDL-C has been considered as biomarker for the activity of CETP inhibitors. However, it is not clear whether a fundamental relationship exist between HDL-C and the risk of coronary artery diseases (CAD). The most crucial role for HDL-C is cholesterol efflux capacity in which HDL can reverse transport cholesterol from foam cells in atherosclerotic plaques. In view of the heterogeneity in HDL-C particle size, charge, and composition, the mere concentration of HDL-C may not be a good surrogate marker for HDL functionality. Recent clinical studies reported that increased HDL-C functionality inversely correlate with the development of atherosclerotic plaque. Future development of CETP inhibitors may therefore benefit from the use of biomarkers that better predict HDL functionality.
1. Introduction
1.1 Development of cholesterol ester transfer protein (CETP) inhibitors
Numerous studies have identified a significant association between the concentration of high-density lipoprotein cholesterol (HDL-C) and the risk of coronary artery diseases (CAD)[1–4]. Boosting the concentration of HDL-C thus appears to be an attractive strategy for atherosclerosis risk reduction[5]. Several other drugs, including fibrates and niacin, also increase HDL-C levels without a definitive effect on cardiovascular risk [6].
Fibrates including gemfibrozil, bezafibrate, fenofibrate and clofibrate, is a class of amphipathic carboxylic acids with lipid modulating properties that include raising HDL-C levels [7]. The results of Veterans Affairs HDL-C Intervention Trial (VA-HIT) showed that treatment with gemfibrozil reduced cardiovascular events by 24% over a median follow up of 5.1 years [8]. In contrast, the results of clinical studies with bezafibrate and fenofibrate were negative and unfortunately clofibrate was associated with higher cardiovascular events[5]. In addition, the combination of HMG-CoA reductase inhibitors (statins) with fibrates can increase the risk of rhabdomyolysis and subsequent acute renal failure [9]. Based on the variable results of clinical outcome studies and their safety concerns, the exact treatment position of fibrates remains uncertain.[5]
Niacin is the most effective current drug for elevation of HDL-C levels [10]. Coronary Drug Project (CDP), one of the most influential niacin trials, evaluated niacin monotherapy in 8341 patients with previous cardiovascular events. The result of this study showed that niacin decreased the incidence of nonfatal myocardial infarction by 27% at 6 years and total mortality was decreased by 11% at 15 years. Another clinical study named High density lipoprotein Atherosclerosis Treatment Study (HATS) [82], the combination of simvastatin and niacin was evaluated in CAD patients over a 3 years period. The results showed that the treatment group was associated with significant improvement in coronary artery atherosclerosis based on angiography and clinical markers. In general, niacin as monotherapy or in combination with other antihyperlipidemic drugs has beneficial clinical effect on atherosclerotic. Unfortunately, at pharmacological doses, niacin exhibit numerous side effects that preclude the widespread use of niacin for increasing HDL-C levels [11].
Cholesteryl ester transfer protein (CETP) is a plasma protein that naturally transfers cholesterol between lipoproteins, more importantly from HDL-C to very low density or low density lipoproteins (VLDL or LDL)[12]. Thus, inhibition of the CETP would raise the concentration of HDL-C and may reduce the risk of CAD[13].
To date, four CETP inhibitors including torcetrapib[14], dalcetrapib[15] anacetrapib[16] and evacetrapib[17] has entered clinical development (Table I), and a few other agents are also in the development pipeline (BAY 60–5521[18] and JNJ-28545595[19]). Figure 1 and Table I present the chemical structure and physiochemical properties of CETP inhibitors, respectively.
Table I.
Physicochemical properties of Cholesterol Ester Transfer Protein (CETP) inhibitors
| Drug | Pharmaceutical company | Molecular formula | XLogP | Topological Polar Surface Area (Angstrom squared) | CETP IC50 (nM)[67] |
|---|---|---|---|---|---|
| Torcetrapib | Pfizer | C26H25F9N2O4 | 7.0 | 59.1 | 39 |
| Dalcetrapib | Hoffmann La-Roche | C23H35NO2S | 7.1 | 71.5 | NA |
| Anacetrapib | Merck & Co. | C30H25F10NO3 | 8.8 | 38.8 | 46 |
| Evacetrapib | Eli Lilly & Company | C31H36F6N6O2 | 7.7 | 87.4 | 26 |
NA: not available
IC50 concentration of compound causing a 50% inhibition of CETP activity
Unless otherwise stated, all information were obtained from Pubchem compound (http://pubchem.ncbi.nlm.nih.gov/)
Fig. 1.

Chemical structure of CETP inhibitors with corresponding molecular weight
In 2006, torcetrapib development was stopped prematurely due to increased mortality[20]. In May of 2012, Hoffmann–La Roche terminated a Phase 3 trial of dalcetrapib upon recommendation of Data and Safety Monitoring Board (DSMB) because of lack of clinically meaningful efficacy [21]. Two other CETP inhibitors, anacetrapib and evacetrapib, are still undergoing development. This article analyses available pharmacological data on CETP inhibitors, with an emphasis on pharmacokinetic and pharmacodynamic properties of these agents and will correlate these with the expected efficacy outcomes, e.g. HDL-C increase through CETP inhibition.
1.2 Physiologic actions of CETP
Triglyceride and cholesteryl ester (CE) transfer between lipoproteins is mediated by CETP[12]. A Schematic diagram of cholesterol transfer by different mechanisms is shown in Figure 2. In the heterotypic CE transfer pathway, CE and triglyceride are moved between apolipoprotein B (apoB)-containing lipoproteins, including particles of VLDL or LDL and HDL. In the homotypic pathway, CE is transferred between sub-particles of HDL including HDL3, HDL2 and pre-β HDL[22]. Both heterotypic and homotypic CE transfer between lipoproteins are affected by some CETP inhibitors including torcetrapib, anacetrapib and evacetrapib while dalcetrapib affects the heterotypic transfer only. Shuttle and tunnel mechanisms are two main processes for neutral lipid transport between lipoproteins by CETP[23]. In the shuttle mechanism, CETP gathers CE molecules from one lipoprotein and transport these through the aqueous phase. In the tunnel mechanism, CETP mediates lipoprotein transport by forming a ternary complex[24].
Fig. 2.
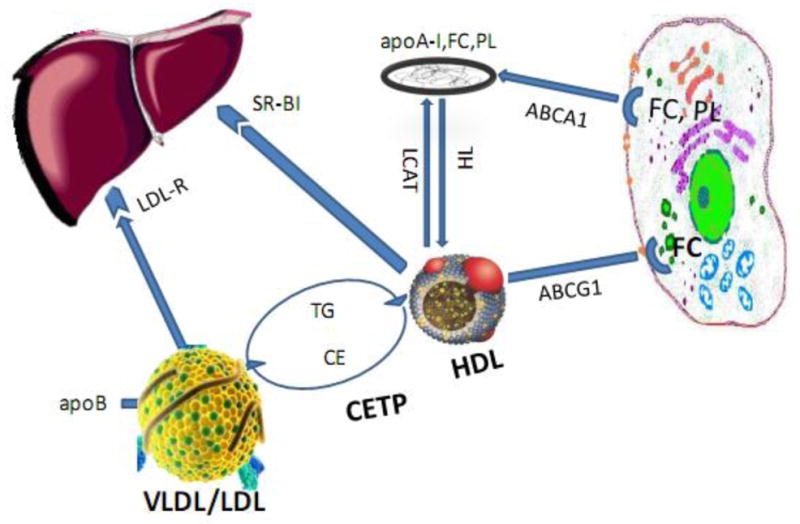
Schematic diagram showing inhibition of various pathways by CETP inhibitors
ABCA1: adenosine triphosphate (ATP)-binding cassette transporter A1; ABCG1: ATP-binding cassette transporter G1; apo B: Apolipoprotein B; apoA-I: Apolipoprotein A-I; CE: cholesteryl esters; CETP: Cholesteryl ester transfer protein; FC: Free Cholesterol; HDL: High density lipoprotein; HL: Hepatic lipase; LCAT: Lecithin:cholesterol acyltransferase; LDL: Low density lipoprotein; LDL-R: low-density lipoprotein receptors; PL: Phospholipids; SR-B1: scavenger receptor class B member 1; TG: Triglyceride; VLDL: Very low density lipoprotein
Moreover, CETP inhibitors may also influence HDL-mediated reverse cholesterol transport that mediates cholesterol removal from macrophage foam cells to the liver followed by biliary excretion. The main reverse cholesterol transport pathways consist of adenosine triphosphate (ATP)-binding cassette transporter G1 (ABCG1) that mediates cholesterol efflux to large HDL particles and ATP-binding cassette transporter A1 (ABCA1) that mediates cholesterol efflux to lipid-poor apoA-1[20].
1.3 Epidemiological evidence supporting the role of CETP in the development of coronary artery diseases
Several epidemiological studies suggest a link between genetic variants of CETP deficiency and the risk of CAD. It is generally believed that gene mutations resulting in CETP deficiency are associated with higher concentration of HDL-C[25–28]. As reviewed in Boekholdt and Thompson[29], controversy exists regarding the association between single-nucleotide polymorphisms (SNPs) within the CETP gene and the risk of CAD. A meta-analysis consisting of 92 epidemiological studies and 113,833 participants showed that CETP polymorphisms that resulted in reduced CETP activity were associated with a decreased risk of cardiovascular events[26]. Similarly, a prospective genome wide association study in a cohort of 18,245 healthy women showed that several SNPs in the CETP gene were associated with an increase in HDL-C levels and reduced risk of future myocardial infarction[30]. Based on this evidence, the CETP pathway appears to be a good target for reducing the risk of cardiovascular events.
2. Pharmacological properties of CETP inhibitors
2.1 Torcetrapib
Torcetrapib was originally developed by Pfizer and tested in Phase 3 clinical trials until its development was halted. Pfizer also investigated another CETP inhibitor, CP-800,569, but its development was also discontinued in 2008 for strategic reasons[31]. Torcetrapib binds to CETP with 1:1 stoichiometry and induces a non-effective complex between CETP and HDL-C. It inhibits both heterotypic and homotypic CE transfer pathways resulting in the complete inhibition of CETP. Clinical studies also showed that torcetrapib could slightly increase reverse cholesterol transport pathways[22].
2.1.1 Pharmacokinetics
A summary of pharmacokinetic properties of torcetrapib and other CETP inhibitors is presented in Table II. Dalvie and colleagues[32] described the result of a 21-day-long, mass balance study of orally administered of [14C]torcetrapib at a dose of 120 mg. This article also contains information on pre-clinical and Phase 1 studies of torcetrapib[32]. In healthy volunteers, the time to maximum concentration (tmax) of torcetrapib was approximately 6 hour[32]. Average elimination half-lives were 373 and 211 hour, for total and unchanged torcetrapib radioactivity, respectively[32]. Torcetrapib was biotransformed to numerous metabolites mainly via oxidative biotransformation[32]. The initial pathway for torcetrapib metabolism was decarbamoylation mediated by cytochrome P450 (CYP) 3A. Two metabolites, M1 (bistrifluoromethylbenzoic acid) and M4 (quinaldic acid), were identified as major metabolites in urine and plasma. In plasma, the concentration of M4 was 3-fold higher than torcetrapib concentration and ~40% of the dose was excreted as M4 and its glucuronide and urea conjugates in the urine. Only 7% of the dose was excreted in urine as the M1 metabolite[32]. Moreover, in Phase 1 studies, torcetrapib given to healthy subjects showed non-linear increase in Cmax and area under the concentration-time curve (AUC) with an increase in doses ranging from 10 to 1000 mg[32]. Torcetrapib exposure was higher in fed than fasted healthy volunteers[32]. Pre-clinical pharmacokinetic studies in rat and monkey indicate that torcetrapib has an oral bioavailability of 33 to 45% and volume of distribution of 1.1–2.5 L/kg[32].
Table II.
Summary of pharmacokinetic and pharmacodynamic properties of Cholesterol Ester Transfer Protein (CETP) inhibitors
| Parameter | Torcetrapib | Dalcetrapib | Anacetrapib | Evacetrapib |
|---|---|---|---|---|
| Dose | 60 mg/day | 600 mg/day | 100 mg/day | 130 mg/day |
| Pharmacokinetics | ||||
| Bioavailability | 33 to 45% in rat and monkey | NA | 38% in rats, 13% in monkeys | NA |
| Effect of food on bioavailability | Exposure is higher in fed than fasted state | Exposure is higher (~65%) in fed versus fasted state | Exposure is 2–3 fold higher after low fat meal and 6–8 fold higher after high fat meal | NA |
| tmax (hour) | ~6 | ~3 | ~4 | NA |
| Main route of elimination | Hepatic metabolism | Hepatic metabolism | Hepatic metabolism | NA |
| Metabolism pathways | Oxidation (CYP3A4/5) and glucuronidation | Hydrolysis, glucuronidation, oxidation and methylation | Oxidation (CYP3A4 major) and glucuronidation | NA |
| Metabolites | To M2, then M1 and M4 | To dalcetrapib-thiol (active metabolite), then to dalcetrapib-S-Glu and dalcetrapib-S-methyl | To M1, then M2 and M3 | NA |
| Elimination half life (hour) | 373 total 211 unchanged |
25.5 ± 3.9 as dalcetrapib-thiol | 9 to 62 fasted 42–83 fed |
NA |
| Urine | 63% as conjugated metabolites | NA | < 2% | NA |
| Bile | 13% as metabolites | NA | Major route as oxidative metabolites | NA |
| Pharmacodynamics | ||||
| HDL-C | ↑ 72% | ↑ 31% | ↑ 138% | ↑ 129% |
| LDL-C | ↓ 25% | No change | ↓ 40% | ↓ 36% |
| Effect on CETP | Complete inhibition | Modulation | Complete inhibition | Complete inhibition |
| Heterotypic CE transfer | Inhibition | Inhibition | Inhibition | NA |
| Homotypic CE transfer | Inhibition | No effect | Inhibition | NA |
NA: not available, tmax: Time to maximum concentration, CE: Cholesteryl Ester, HDL-C:High-density lipoprotein cholesterol, LDL-C: Low-density lipoprotein cholesterol
2.1.2 Clinical trials
The effect of torcetrapib on atherosclerosis was evaluated in three clinical studies, ILLUSTRATE[34], RADIANCE 1[35] and 2[36]. The ILLUSTRATE trial used intravascular ultrasound to evaluate coronary atheroma burden in patients with existing coronary atheroma[34], whereas, RADIANCE 1 and 2 trials used B-mode ultrasound to evaluate the intima-media thickness in patients with dyslipidemia[35, 36]. The results of these studies showed torcetrapib increased HDL-C level by ~60% and decreased low-density lipoprotein cholesterol (LDL-C) level by ~20%. Torcetrapib had no beneficial effect on either atheroma burden in the coronary arteries[34] or carotid intima-media thickness[35, 36].
The ILLUMINATE study was a Phase 3 trial aiming to investigate the effects of torcetrapib on cardiovascular outcomes[37]. Patients were randomized to take atorvastatin plus torcetrapib (60 mg) or atorvastatin plus placebo. Patients in the torcetrapib arm showed a 72% increase in HDL-C concentration and moderate decrease in LDL-C and triglyceride levels. However, the primary endpoint of the trial, a composite of first major cardiovascular and cerebrovascular events, defined as death because of coronary heart disease, nonfatal myocardial infarction, stroke, or hospitalization for unstable angina was 25% higher in the torcetrapib group. Total mortality due to cardiovascular events or cancer and infections also increased in the torcetrapib arm. Torcetrapib adverse effects bears a resemblance to mineralocorticoid excess syndrome that included higher systolic blood pressure, serum aldosterone, sodium and bicarbonate levels but lower serum potassium level[37].
The underlying mechanisms of increased cardiovascular mortality in ILLUMINATE trial are still not entirely determined but the effect of torcetrapib on aldosterone level is the most notable “off-target” effects of this drug. Expression of several renin-angiotensin-aldosterone system genes, in arteries and adrenal glands, can be up-regulated and the production of mineralocorticoid hormones can be induced by torcetrapib. The pressor effect of torcetrapib may be mediated by several actions of adrenal glands through the calcium pathway. Several studies showed that torcetrapib increased synthesis of aldosterone and cortisol in vitro. Aldosterone can induce hypertension and directly generate endothelial dysfunction, increased vascular smooth-muscle migration, myocardial fibrosis, and increased inflammation in the cardiovascular system. The possibility that elevation of aldosterone level may be related to the torcetrapib adverse effects was supported by the observation of a higher mortality from cardiovascular events in patients with greater changes in serum potassium and bicarbonate levels[38].
There is also evidence for beneficial effects of torcetrapib. The result of post hoc analysis in ILLUMINATE trial showed that lower cardiac events were seen in the patient group with higher increase in HDL-C and apoA-1[37]. Torcetrapib with 60 mg daily did not alter cholesterol efflux capacity through ABCG1 pathway and with 120 mg daily increased HDL-C functionality via previously mentioned pathway[20, 39]. Post hoc analysis of the ILLUSTRATE trial revealed a regression of coronary atheroma in torcetrapib group patients with HDL-C concentrations in the upper quartile[39].
2.2 Dalcetrapib
Dalcetrapib is a thioester prodrug (Figure 1) with a structure that is distinct from other CETP inhibitors (smaller molecular weight and lack of trifluoride moiety). It is hydrolyzed by non-specific esterases and lipases in the biological media to generate a pharmacologically active thiol form[40]. It modulates the activity of CETP by the formation of a disulfide bond at cytosine residue and induction of a conformational change in this protein[41]. Dalcetrapib inhibits heterotypic rather than homotypic CE transfer resulting in the partial inhibition of CETP. Through this mechanism, the homotypic CE transfer that produces larger HDL2 and smaller pre-β HDL from HDL3 will not be affected by dalcetrapib[22].
2.2.1 Pharmacokinetics
Dalcetrapib is well tolerated and exhibit dose-proportional pharmacokinetics up to a dose of 4500 mg/day[42]. Dalcetrapib is rapidly hydrolyzed to generate dalcetrapib-thiol that is pharmacologically active. Dalcetrapib-thiol covalently binds to CETP and to other plasma proteins but the compound is cleared with a relatively short half-life (t1/2 25.5 hour) thus generating a relatively transient change in CETP activity [43]. The hydrolysis of dalcetrapib to dalcetrapib-thiol is mediated by non-specific esterases and lipases[40]. Dalcetrapib-thiol is further biotransformed to two major metabolites, dalcetrapib-S-methyl and dalcetrapib-S-glucuronide metabolites[44]. Dalcetrapib exposure was significantly higher (~65%) in fed versus fasted state[45]. In comparison with a standard meal, exposure to dalcetrapib was ~15% lower after a light meal but was ~35% higher after a high fat meal[45]. Co-administration of anti-obesity agent orlistat (doses 10–120 mg) with dalcetrapib (600 mg) reduced dalcetrapib exposure by more than 50% in all dose levels except for 10 mg of orlistat[40]. The activity of CETP, measured ex vivo, was also pronouncedly reduced upon co-administration with orlistat[40]. Orlistat is a potent inhibitor of carboxylesterase-2, an enzyme expressed abundantly in the gastrointestinal tract and liver and is subjected to genetic polymorphism[46].
Drug interactions with dalcetrapib have been studied extensively. Dalcetrapib administration with statins, simvastatin, rosuvastatin, and pravastatin was well-tolerated[47]. Dalcetrapib exposure was significantly lower when co-administered with simvastatin and rosuvastatin but it was not different when co-administered with pravastatin[47]. Statin exposure was not influenced by dalcetrapib co-administration except for lower exposure to rosuvastatin[47]. Co-administration of dalcetrapib and atorvastatin did not result in clinically meaningful changes in the pharmacokinetics of either drug [48]. Moreover, administration of ezetimibe with dalcetrapib did not generate significant drug-drug interaction[49]. Dalcetrapib did not interact with monophasic oral contraceptives Microgynon® 30 (ethinylestradiol 0.03 mg/levonorgestrel 0.15 mg)[50]. Co-administration of a uridine 5′-diphospho-glucuronosyltransferase (UDP)-glucuronosyltransferase (UGT) inhibitor probenecid, with dalcetrapib increased the AUC from time zero to infinity (AUC∞) and Cmax of dalcetrapib-thiol by 14% and 21%, respectively[51].
2.2.2 Clinical trials
The dal-HEART program included a series of Phase 2 and 3 clinical studies aiming to evaluate the efficacy and safety of dalcetrapib in humans. The dal-VESSEL[52] and dal-PLAQUE[53] were the most notable trials in this program.
The dal-PLAQUE trial evaluated the effect of dalcetrapib on atherosclerotic plaques in 130 patients with CAD or high risk for a cardiovascular disease. Patients randomized to take dalcetrapib 600 mg daily or placebo in addition to standard medical treatment for 24 months. Total vessel area, wall area, normalized carotid artery wall index, and arterial inflammation within an index vessel was not different between groups. However, dalcetrapib significantly reduced the Magnetic Resonance Imaging-derived change in total vessel area compared with placebo. Arterial inflammation was evaluated by the use of 18F-fluorodeoxyglucose (18F-FDG), a radiopharmaceutical commonly used in imaging with positron emission tomography. While dalcetrapib did not have any significant effect on arterial inflammation, a post hoc analysis, limited to carotid artery, showed dalcetrapib significantly reduced arterial inflammation in the most diseased segments[53].
The dal-VESSEL trial evaluated dalcetrapib safety profile and the effect of dalcetrapib on endothelial function in 476 patients with CAD or cardiovascular risk equivalent for 36 weeks. Patients were randomized to receive dalcetrapib or placebo in addition to standard medical treatment. Changes in flow-mediated dilation (FMD percent) of right brachial artery and 24 hour ambulatory blood pressure monitoring, markers of inflammation, oxidative stress, and coagulation were evaluated at different time during the 36 weeks. At the study completion, HDL-C levels increased by 31% but LDL-C levels did not change significantly. When compared with the placebo, dalcetrapib had no effect on FMD after either 12 or 36 weeks of treatment. Dalcetrapib had no effect on ambulatory blood pressure up to 36 weeks of treatment. Biomarkers of inflammation, oxidative stress and coagulation were unaffected by dalcetrapib, although the concentration of lipoprotein-associated phospholipase A2 (Lp-PLA2) were increased by 17% in those taking dalcetrapib[52].
The dal-OUTCOMES were a series of Phase 3 clinical studies involving 15,600 patients aiming to evaluate the efficacy and safety of dalcetrapib in patients with acute coronary syndrome[15]. Patients were randomized to take dalcetrapib or placebo. This trial was designed to continue until 1,600 primary outcomes have occurred with an anticipated conclusion in 2013. However, it was reported in May of 2012 that the dal-OUTCOMES trial had been prematurely terminated by an independent DSMB because of an apparent lack of efficacy[21]. Subsequently, Hoffmann-La Roche has discontinued the entire dalcetrapib development program (dal-HEART).
The recently published dal-OUTCOMES trial showed that dalcetrapib significantly increased HDL-C and apoA1 levels but had no effect on LDL-C concentration[54]. Dalcetrapib did not alter the risk of major cardiovascular events since cumulative event rates were 8.0% and 8.3%, for dalcetrapib and placebo, respectively (p=0.52). Although, dalcetrapib had an acceptable adverse-effect profile, the drug significantly increased systolic blood pressure and the concentration of C-reactive protein[54].
Three probable reasons for termination of the dal-OUTCOMES trial has been proposed: (i) the increase in HDL-C level is not accompanied by an improvement of the HDL-C atheroprotective effects (ii) the HDL capacity to bind It is possible that the favourable effect of dalcetrapib on HDL-C levels were counterbalanced by adverse events on blood pressure and its pro-inflammatory effect (iii) the atheroprotective effect of HDL-C observed in clinical studies is an epiphenomenon rather than its protective effect against coronary vascular disease
2.3 Anacetrapib
Similar to torcetrapib, anacetrapib inhibits both heterotypic and homotypic CE transfer. Inhibition of homotypic CE transfer by anacetrapib may restrict the elevation of HDL-C mediated reverse cholesterol transport. However, the clinical importance of this mechanism is unclear[22]. Anacetrapib is currently undergoing clinical development by Merck & Co.
2.3.1 Pharmacokinetics
Oral absorption of anacetrapib was rapid with a tmax of ~4 hour[55] similarly rapid inhibition of serum CETP activity was observed reaching maximum inhibition at ~4 h post dose. The activity of CETP was inhibited by approximately 80% by 24 hours post dose[56]. Feeding status significantly influenced anacetrapib exposure resulting in 2–3 fold increase in the exposure after low fat meal and 6–8 fold increase after high fat meal[55]. The values of elimination half-life were 9 to 62 hour in the fasted volunteers and 42–83 hour in fed volunteers[55]. There was an apparent plateau in the oral absorption of higher doses[55]. Pharmacokinetic and pharmacodynamic properties of anacetrapib were comparable with respect to age, gender, and obesity[55].
Pre-clinical pharmacokinetic studies of anacetrapib in rats and rhesus monkeys showed an oral bioavailability of 38% and 13%, respectively. The AUC was not dose proportional between 1 to 500 mg/kg possibly related to limited water solubility at higher doses[57]. After oral administration of [14C] anacetrapib, ~90% of the dose was recovered within 48 hours.
The recovered anacetrapib was mainly unchanged in feces and via biliary excretion (~15%) and urine (<2%). Metabolism included the formation of oxidative and glucuronidated metabolites. The main metabolite included of O-demethylated M1, hydroxylated on the biphenyl moiety M2 and hydroxylated on the isopropyl side chain M3 followed by glucuronidation of oxidative metabolites[57].
A mass balance study in six healthy male volunteers using 150 mg of [14C]anacetrapib was reported by Kumar et al.[58]. Similar to pre-clinical studies, fecal excretion was the main route of elimination. In general, anacetrapib seem to have low to moderate oral absorption and the fraction of the drug reaching the systemic circulation is mainly eliminated as oxidative metabolites via biliary/fecal route[58].
Anacetrapib drug interactions with simvastatin[59], digoxin[60], warfarin[61], the CYP3A4 substrate midazolam[62] and the CYP3A4 inhibitor ketoconazole[62] were studied. Anacetrapib (150 mg) did not influence midazolam clearance[62]. However, anacetrapib exposure was increased by 4.5-fold when it was given with 400 mg ketoconazole thus indicating that anacetrapib is a substrate but not an inhibitor of CYP3A4[62]. Co-administration of anacetrapib with simvastatin (40 mg), that is also a CYP3A4 substrate, resulted in ~30% increase in exposure to simvastatin lactone (the administered form) and simvastatin acid (active drug)[59]. Anacetrapib did not meaningfully affect the exposure to digoxin, a cardiovascular agent, and a known P-glycoprotein substrate[60]. Moreover, anacetrapib did not influence the pharmacokinetics or pharmacodynamics of the anticoagulant agent warfarin that is a CYP2C9 substrate with narrow therapeutic index[61].
Population pharmacokinetic and pharmacodynamic modelling revealed that a maximum effect model (Emax) describes the relationship between anacetrapib concentration and changes in HDL-C and LDL-C. The estimated Emax for increase in HDL-C was 160% and EC50 (effective concentration for half-maximum response) was 0.22 μmol/L[56].
2.3.2 Clinical trials
In patients with dyslipidemia, anacetrapib monotherapy increased HDL-C by 129% and reduced LDL-C by 38% dose-dependently without affecting ambulatory blood pressure[16]. Yvan-Charvet et al. evaluated the effects of niacin and anacetrapib on lipid profile and the ability of HDL to promote net cholesterol efflux[63]. Anacetrapib effectively increased HDL-C, and decreased LDL-C levels and improved HDL cholesterol efflux capacity from macrophages[63].
The efficacy and safety of anacetrapib was evaluated in a clinical trial named DEFINE[64]. A total of 1,623 patients with CAD or high risk for CAD on statin therapy were randomized to receive anacetrapib or placebo for 18 months. After 24 weeks of treatment, anacetrapib decreased LDL-C by 40% and increased HDL-C by 138% as compared with placebo. Despite pronounced elevation of HDL-C, C-reactive protein levels did not change significantly. Furthermore, anacetrapib had no “off-target” adverse effects like torcetrapib through 76 weeks of treatment[64]. Moreover, administration of anacetrapib at 150 mg daily to 30 healthy individuals resulted in a significantly lower concentration of medium and small VLDL, and medium and small LDL (LDL2a, 2b, and 3a)[65]. The presence of small, dense LDL particles is associated with the risk of development of ischemic heart disease[66].
The REVEAL study (ClinicalTrials.gov Identifier: NCT01252953) is a Phase 3 clinical trial aiming to recruit approximately 30,000 patients with CAD. Its objective is to evaluate the effectiveness of anacetrapib on cardiovascular events through four years of follow up. This study was started in 2011 with an excepted completion date of 2017.
2.4 Evacetrapib
Evacetrapib is a CETP inhibitor currently undergoing development by Eli Lilly and Company. It has a similar structure and mechanism of action to torcetrapib but based on IC50 values appear to be a more potent CETP inhibitor than torcetrapib or anacetrapib[17, 67]. However, the underlying mechanism of evacetrapib effect on HDL-C mediated reverse cholesterol transport and heterotypic and homotypic CE transfer have not been elucidated[22].
2.4.1 Pharmacokinetics
Currently no information is available in the published literature on evacetrapib pharmacokinetics.
2.4.2 Clinical trials
The efficacy and safety of evacetrapib was evaluated in a Phase 2 clinical study involving 398 dyslipidemic patients with elevated LDL-C or low HDL-C levels (ClinicalTrials.gov Identifier: NCT01105975). Evacetrapib was taken either alone or in combination therapy with statins. The patients were randomized to receive evacetrapib monotherapy at doses of 30, 100 and 500 mg daily or placebo for 3 months. Moreover, the effect of evacetrapib 100 mg daily was evaluated in 239 patients who were taking statins. In comparison with the placebo, evacetrapib monotherapy reduced LDL-C levels by 14–36% and increased HDL-C levels by 54–129% in a dose dependent manner. In addition, evacetrapib in combination therapy with statins elevated HDL-C plasma concentrations similar to evacetrapib monotherapy but had an additional reduction of LDL-C levels by 11–14% compared to statin monotherapy. Evacetrapib did not show off-target adverse effects similar to torcetrapib[68]. Recently, Eli Lilly and Company has started a Phase 3 clinical trial named “A Study of Evacetrapib in High-Risk Vascular Disease, ACCELERATE” with an estimated enrolment of 11,000 patients (ClinicalTrials.gov Identifier: NCT01687998).
3. Is there a future for CETP inhibitors?
The failure of torcetrapib challenged the notion of favourable effect of CETP inhibitors on cardiovascular events mediated by increasing the concentration of HDL-C. However, “off-target” effects of torcetrapib that are unrelated to CETP inhibition could potentiate these side effects. A decisive argument supporting this hypothesis is that other CETP inhibitors do not exhibit torcetrapib-like adverse effects[39].
In addition, based on prior knowledge, it could not be predicted that a seemingly safe molecule like dalcetrapib could lack adequate efficacy. Dalcetrapib induced a modest increase in HDL-C level but did not decrease the LDL-C level. Individuals with the CETP genetic variation and lower risk of CAD, often have higher concentration of HDL-C and lower LDL-C [69]. Therefore, it is possible that for achieving a desired clinical outcome, it is necessary to select a CETP inhibitor that affect both HDL-C and LDL-C concurrently.
Dalcetrapib has a distinct structure and unique mechanism of action. It only raises HDL-C but does not affect the concentration of LDL-C. Some aspects of dalcetrapib pharmacokinetics including the need for the bioactivation of the molecule to the thiol and relatively short elimination half life of dalcetrapib-thiol may render the drug ineffective in a portion of the population thus may explain the failure of this drug in Phase 3 clinical trials.
Anacetrapib and evacetrapib are parent drug without the need for the bioactivation. Both agents not only increase HDL-C effectively but also decrease LDL-C concentration by more than 30%. This may provide a strong motivation for carrying out of clinical trials with these CETP inhibitors. Finally, we should consider that even if positive results are demonstrated in future studies it may be difficult to determine that the beneficial clinical outcome is due to HDL-C increase or LDL-C lowering or other effects.
4. Is HDL-C level still a real risk factor and practical therapeutic target?
Patients with atherosclerosis typically present with low concentration of HDL-C[1, 70]. However, it is not clear whether a fundamental association exists between HDL-C and the risk of CAD.
Some studies reported genetic variants related to HDL-C levels correlated with cardiovascular disease[71–73]. Recently, a Mendelian randomization study was carried out to evaluate the association between plasma HDL-C and the risk of cardiovascular events[69]. This study reported that some genetic mechanisms including polymorphism in endothelial lipase gene and 14 other SNPs commonly associated with high HDL-C level did not reduce the risk of cardiovascular events[69]. These data challenge the concept that low HDL-C level is a real risk factor for CAD.
The most crucial role for HDL-C is cholesterol efflux capacity that consists of several pathways such as ABCA1, ABCG1, scavenger receptor class B member 1 (SR-B1), and aqueous diffusion. Through these pathways, HDL-C could reverse transport cholesterol from foam cells in atherosclerotic plaques. HDL functionality is defined as the capacity of HDL to promote cholesterol reverse transport by accepting cholesterol from foam cells.
Functionality of HDL, independent of its concentration could determine atherosclerotic burden[6, 74]. Recently, Khera et al. [6] carried out a clinical study for evaluation of association between HDL-C functionality and development of atherosclerotic plaques. Atherosclerosis development was evaluated by carotid intima-media thickness and coronary artery angiography. They reported a strong inverse association between atherosclerotic development and HDL-C functionality that determined by cholesterol efflux capacity from macrophages and this association was independent of the HDL-C level. Therefore, application of this functional biomarker may be a more informative than the concentration of plasma HDL-C [75, 76].
In view of the heterogeneity in HDL-C particle size, charge, and composition, the mere concentration of HDL-C is not a good surrogate marker for HDL functionality[6]. Moreover, cholesterol efflux is also carried out by several other pathways including aqueous diffusion, ABCG1, SR-B1 and ABCA1[77] and the cholesterol efflux capacity assay used in Khera et al. measures the total cholesterol efflux from macrophages[6].
Several studies showed significant and reproducible association between cardiovascular disease risk factors and HDL-C related esterase/lactonase paraoxonase 1 (PON1)[78, 79]. This pathway is associated to the anti-inflammatory and antioxidant properties of HDL-C. Genetic studies that showed an association between PON1 and CAD risk and oxidative stress support its selection as an HDL-C functionality assay[6, 79]. Because HDL-C has a variety of functions such as antioxidant, anti-inflammatory, anti-thrombogenic and atheroprotective activities although not all of them are related to atheroprotection[80]. Therefore, mechanistic approach for determination of HDL-C functionality may be useful for future clinical study and better evaluation of HDL-C role in CAD. In the future clinical studies, it is advisable to use a measure of HDL cholesterol efflux capacity, in addition to HDL-C level, as a biomarker of effect for CETP inhibitors. The pragmatic issues associated with conducting a functional assay in large scale will remain an obvious challenge of the use of this biomarker.
5. Concluding remarks
No other class of lipid modulators has been introduced in approximately 30 years since the introduction of the first statin, lovastatin[81]. Inhibition of CETP pathway remains an attractive mechanism of action for a novel class of cardiovascular agents. However, failure of the first two drugs in this category questions the future of this class of drugs. It transpires that torcetrapib failure was because of off-target effects, not because of CETP inhibition. Dalcetrapib failure could be attributed to several factors including transient CETP modulation by dalcetrapib-thiol and the pharmacokinetic variability associated with dalcetrapib activation. Two other agents in this class are undergoing clinical development and they have shown promising results in Phase 1 and 2 clinical trials. The success of anacetrapib and evacetrapib will remain to be confirmed after the completion of REVEAL and ACCELERATE studies, in 2017 and 2015, respectively.
Acknowledgments
Support of grant # R15 GM101599 from the National Institutes of Health is gratefully acknowledged.
Footnotes
The authors (Mohammadpour AH and Akhlaghi F) declare no conflicts of interest.
Reference List
- 1.Gordon T, Castelli WP, Hjortland MC, et al. High density lipoprotein as a protective factor against coronary heart disease. the Framingham study. Am J Med. 1977 May;62(5):707–14. doi: 10.1016/0002-9343(77)90874-9. [DOI] [PubMed] [Google Scholar]
- 2.Castelli WP, Garrison RJ, Wilson PW, et al. Incidence of coronary heart disease and lipoprotein cholesterol levels. The Framingham Study. JAMA. 1986 Nov 28;256(20):2835–8. [PubMed] [Google Scholar]
- 3.Corti MC, Guralnik JM, Salive ME, et al. HDL cholesterol predicts coronary heart disease mortality in older persons. JAMA. 1995 Aug 16;274(7):539–44. [PubMed] [Google Scholar]
- 4.Gordon DJ, Probstfield JL, Garrison RJ, et al. High-density lipoprotein cholesterol and cardiovascular disease. Four prospective American studies. Circulation. 1989 Jan;79(1):8–15. doi: 10.1161/01.cir.79.1.8. [DOI] [PubMed] [Google Scholar]
- 5.Kuvin JT, Alsheikh-Ali AA, Karas RH. High-density lipoprotein cholesterol-raising strategies. J Cardiovasc Pharmacol. 2006 Feb;47(2):196–204. doi: 10.1097/01.fjc.0000199684.20578.7c. [DOI] [PubMed] [Google Scholar]
- 6.Khera AV, Cuchel M, de la Llera-Moya M, et al. Cholesterol efflux capacity, high-density lipoprotein function, and atherosclerosis. N Engl J Med. 2011 Jan 13;364(2):127–35. doi: 10.1056/NEJMoa1001689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Staels B, Dallongeville J, Auwerx J, et al. Mechanism of action of fibrates on lipid and lipoprotein metabolism. Circulation. 1998 Nov 10;98(19):2088–93. doi: 10.1161/01.cir.98.19.2088. [DOI] [PubMed] [Google Scholar]
- 8.Rubins HB, Robins SJ, Collins D, et al. Gemfibrozil for the secondary prevention of coronary heart disease in men with low levels of high-density lipoprotein cholesterol. Veterans Affairs High-Density Lipoprotein Cholesterol Intervention Trial Study Group. N Engl J Med. 1999 Aug 5;341(6):410–8. doi: 10.1056/NEJM199908053410604. [DOI] [PubMed] [Google Scholar]
- 9.Shepherd J. Fibrates and statins in the treatment of hyperlipidaemia: an appraisal of their efficacy and safety. Eur Heart J. 1995 Jan;16(1):5–13. doi: 10.1093/eurheartj/16.1.5. [DOI] [PubMed] [Google Scholar]
- 10.Bruckert E, Labreuche J, Amarenco P. Meta-analysis of the effect of nicotinic acid alone or in combination on cardiovascular events and atherosclerosis. Atherosclerosis. 2010 Jun;210(2):353–61. doi: 10.1016/j.atherosclerosis.2009.12.023. [DOI] [PubMed] [Google Scholar]
- 11.Singh IM, Shishehbor MH, Ansell BJ. High-density lipoprotein as a therapeutic target: a systematic review. JAMA. 2007 Aug 15;298(7):786–98. doi: 10.1001/jama.298.7.786. [DOI] [PubMed] [Google Scholar]
- 12.Lagrost L, Gambert P, Dangremont V, et al. Role of cholesteryl ester transfer protein (CETP) in the HDL conversion process as evidenced by using anti-CETP monoclonal antibodies. J Lipid Res. 1990 Sep;31(9):1569–75. [PubMed] [Google Scholar]
- 13.Okamoto H, Yonemori F, Wakitani K, et al. A cholesteryl ester transfer protein inhibitor attenuates atherosclerosis in rabbits. Nature. 2000 Jul 13;406(6792):203–7. doi: 10.1038/35018119. [DOI] [PubMed] [Google Scholar]
- 14.Clark RW, Ruggeri RB, Cunningham D, et al. Description of the torcetrapib series of cholesteryl ester transfer protein inhibitors, including mechanism of action. J Lipid Res. 2006 Mar;47(3):537–52. doi: 10.1194/jlr.M500349-JLR200. [DOI] [PubMed] [Google Scholar]
- 15.Schwartz GG, Olsson AG, Ballantyne CM, et al. Rationale and design of the dal-OUTCOMES trial: efficacy and safety of dalcetrapib in patients with recent acute coronary syndrome. Am Heart J. 2009 Dec;158(6):896–901. e3. doi: 10.1016/j.ahj.2009.09.017. [DOI] [PubMed] [Google Scholar]
- 16.Krishna R, Anderson MS, Bergman AJ, et al. Effect of the cholesteryl ester transfer protein inhibitor, anacetrapib, on lipoproteins in patients with dyslipidaemia and on 24-h ambulatory blood pressure in healthy individuals: two double-blind, randomised placebo-controlled phase I studies. Lancet. 2007 Dec 8;370(9603):1907–14. doi: 10.1016/S0140-6736(07)61813-3. [DOI] [PubMed] [Google Scholar]
- 17.Cao G, Beyer TP, Zhang Y, et al. Evacetrapib is a novel, potent, and selective inhibitor of cholesteryl ester transfer protein that elevates HDL cholesterol without inducing aldosterone or increasing blood pressure. J Lipid Res. 2011 Dec;52(12):2169–76. doi: 10.1194/jlr.M018069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Weber O, Willmann S, Bischoff H, et al. Prediction of a potentially effective dose in humans for BAY 60-5521, a potent inhibitor of cholesteryl ester transfer protein (CETP) by allometric species scaling and combined pharmacodynamic and physiologically-based pharmacokinetic modelling. Br J Clin Pharmacol. 2012 Feb;73(2):219–31. doi: 10.1111/j.1365-2125.2011.04064.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sarich TC, Connelly MA, Schranz DB, et al. A Phase 0 study of the inhibition of cholesteryl ester transfer protein activity by JNJ-28545595 in plasma from normolipidemic and dyslipidemic humans. Int J Clin Pharmacol Ther. 2012 May 14; doi: 10.5414/CP201627. [DOI] [PubMed] [Google Scholar]
- 20.Tall AR, Yvan-Charvet L, Wang N. The failure of torcetrapib: was it the molecule or the mechanism? Arterioscler Thromb Vasc Biol. 2007 Feb;27(2):257–60. doi: 10.1161/01.ATV.0000256728.60226.77. [DOI] [PubMed] [Google Scholar]
- 21.Sweetlove M. Phase III trial of dalcetrapib: discontinued due to lack of efficacy. Pharmaceutical Medicine. 2012;26(4):253–6. doi: 10.2165/11631240-000000000-00000. [DOI] [Google Scholar]
- 22.Shinkai H. Cholesteryl ester transfer-protein modulator and inhibitors and their potential for the treatment of cardiovascular diseases. Vasc Health Risk Manag. 2012;8:323–31. doi: 10.2147/VHRM.S25238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Masson D, Jiang XC, Lagrost L, et al. The role of plasma lipid transfer proteins in lipoprotein metabolism and atherogenesis. J Lipid Res. 2009 Apr;50( Suppl):S201–6. doi: 10.1194/jlr.R800061-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Barter PJ, Rye KA. Cholesteryl ester transfer protein (CETP) inhibition as a strategy to reduce cardiovascular risk. J Lipid Res. 2012 May 1; doi: 10.1194/jlr.R024075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Boekholdt SM, Kuivenhoven JA, Hovingh GK, et al. CETP gene variation: relation to lipid parameters and cardiovascular risk. Curr Opin Lipidol. 2004 Aug;15(4):393–8. doi: 10.1097/01.mol.0000137226.54278.60. [DOI] [PubMed] [Google Scholar]
- 26.Thompson A, Di Angelantonio E, Sarwar N, et al. Association of cholesteryl ester transfer protein genotypes with CETP mass and activity, lipid levels, and coronary risk. JAMA. 2008 Jun 18;299(23):2777–88. doi: 10.1001/jama.299.23.2777. [DOI] [PubMed] [Google Scholar]
- 27.Brown ML, Inazu A, Hesler CB, et al. Molecular basis of lipid transfer protein deficiency in a family with increased high-density lipoproteins. Nature. 1989 Nov 23;342(6248):448–51. doi: 10.1038/342448a0. [DOI] [PubMed] [Google Scholar]
- 28.Inazu A, Brown ML, Hesler CB, et al. Increased high-density lipoprotein levels caused by a common cholesteryl-ester transfer protein gene mutation. N Engl J Med. 1990 Nov 1;323(18):1234–8. doi: 10.1056/NEJM199011013231803. [DOI] [PubMed] [Google Scholar]
- 29.Boekholdt SM, Thompson JF. Natural genetic variation as a tool in understanding the role of CETP in lipid levels and disease. J Lipid Res. 2003 Jun;44(6):1080–93. doi: 10.1194/jlr.R200018-JLR200. [DOI] [PubMed] [Google Scholar]
- 30.Ridker PM, Pare G, Parker AN, et al. Polymorphism in the CETP gene region, HDL cholesterol, and risk of future myocardial infarction: Genomewide analysis among 18 245 initially healthy women from the Women’s Genome Health Study. Circ Cardiovasc Genet. 2009 Feb;2(1):26–33. doi: 10.1161/CIRCGENETICS.108.817304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wolk R, Chen D, Clark RW, et al. Pharmacokinetic, pharmacodynamic, and safety profile of a new cholesteryl ester transfer protein inhibitor in healthy human subjects. Clin Pharmacol Ther. 2009 Oct;86(4):430–7. doi: 10.1038/clpt.2009.120. [DOI] [PubMed] [Google Scholar]
- 32.Dalvie D, Chen W, Zhang C, et al. Pharmacokinetics, metabolism, and excretion of torcetrapib, a cholesteryl ester transfer protein inhibitor, in humans. Drug Metab Dispos. 2008 Nov;36(11):2185–98. doi: 10.1124/dmd.108.023176. [DOI] [PubMed] [Google Scholar]
- 33.Prakash C, Chen W, Rossulek M, et al. Metabolism, pharmacokinetics, and excretion of a cholesteryl ester transfer protein inhibitor, torcetrapib, in rats, monkeys, and mice: characterization of unusual and novel metabolites by high-resolution liquid chromatography-tandem mass spectrometry and 1H nuclear magnetic resonance. Drug Metab Dispos. 2008 Oct;36(10):2064–79. doi: 10.1124/dmd.108.022277. [DOI] [PubMed] [Google Scholar]
- 34.Nissen SE, Tardif JC, Nicholls SJ, et al. Effect of torcetrapib on the progression of coronary atherosclerosis. N Engl J Med. 2007 Mar 29;356(13):1304–16. doi: 10.1056/NEJMoa070635. [DOI] [PubMed] [Google Scholar]
- 35.Kastelein JJ, van Leuven SI, Burgess L, et al. Effect of torcetrapib on carotid atherosclerosis in familial hypercholesterolemia. N Engl J Med. 2007 Apr 19;356(16):1620–30. doi: 10.1056/NEJMoa071359. [DOI] [PubMed] [Google Scholar]
- 36.Bots ML, Visseren FL, Evans GW, et al. Torcetrapib and carotid intima-media thickness in mixed dyslipidaemia (RADIANCE 2 study): a randomised, double-blind trial. Lancet. 2007 Jul 14;370(9582):153–60. doi: 10.1016/S0140-6736(07)61088-5. [DOI] [PubMed] [Google Scholar]
- 37.Barter PJ, Caulfield M, Eriksson M, et al. Effects of torcetrapib in patients at high risk for coronary events. N Engl J Med. 2007 Nov 22;357(21):2109–22. doi: 10.1056/NEJMoa0706628. [DOI] [PubMed] [Google Scholar]
- 38.Rader DJ. Illuminating HDL--is it still a viable therapeutic target? N Engl J Med. 2007 Nov 22;357(21):2180–3. doi: 10.1056/NEJMe0707210. [DOI] [PubMed] [Google Scholar]
- 39.Hewing B, Fisher EA. Rationale for cholesteryl ester transfer protein inhibition. Curr Opin Lipidol. 2012 Apr 18; doi: 10.1097/MOL.0b013e328353ef1d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bentley D, Young AM, Rowell L, et al. Evidence of a drug-drug interaction linked to inhibition of ester hydrolysis by orlistat. J Cardiovasc Pharmacol. 2012 Jul 19; doi: 10.1097/FJC.0b013e31826731ff. [DOI] [PubMed] [Google Scholar]
- 41.Vergeer M, Stroes ES. The pharmacology and off-target effects of some cholesterol ester transfer protein inhibitors. Am J Cardiol. 2009 Nov 16;104(10 Suppl):32E–8E. doi: 10.1016/j.amjcard.2009.09.017. [DOI] [PubMed] [Google Scholar]
- 42.Derks M, Anzures-Cabrera J, Turnbull L, et al. Safety, tolerability and pharmacokinetics of dalcetrapib following single and multiple ascending doses in healthy subjects: a randomized, double-blind, placebo-controlled, phase I study. Clin Drug Investig. 2011;31(5):325–35. doi: 10.1007/BF03256931. [DOI] [PubMed] [Google Scholar]
- 43.Niesor EJ, Magg C, Ogawa N, et al. Modulating cholesteryl ester transfer protein activity maintains efficient pre-beta-HDL formation and increases reverse cholesterol transport. J Lipid Res. 2010 Dec;51(12):3443–54. doi: 10.1194/jlr.M008706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kuhlmann O, Heinig K. Dalcetrapib pharmacokinetics and metabolism in the cynomolgus monkey. Xenobiotica. 2011 May;41(5):430–6. doi: 10.3109/00498254.2010.551223. [DOI] [PubMed] [Google Scholar]
- 45.Derks M, Kawamura H, Abt M, et al. Effects of food intake on the pharmacokinetic properties of dalcetrapib: findings from three phase I, single-dose crossover studies in healthy volunteers. Clin Ther. 2011 Jun;33(6):754–65. doi: 10.1016/j.clinthera.2011.05.046. [DOI] [PubMed] [Google Scholar]
- 46.Xiao D, Shi D, Yang D, et al. Carboxylesterase-2 is a highly sensitive target of the antiobesity agent orlistat with profound implications in the activation of anticancer prodrugs. Biochem Pharmacol. 2013 Feb 1;85(3):439–47. doi: 10.1016/j.bcp.2012.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Derks M, Abt M, Phelan M, et al. Coadministration of dalcetrapib with pravastatin, rosuvastatin, or simvastatin: no clinically relevant drug-drug interactions. J Clin Pharmacol. 2010 Oct;50(10):1188–201. doi: 10.1177/0091270009358709. [DOI] [PubMed] [Google Scholar]
- 48.Derks M, Abt M, Parr G, et al. No clinically relevant drug-drug interactions when dalcetrapib is co-administered with atorvastatin. Expert Opin Investig Drugs. 2010 Oct;19(10):1135–45. doi: 10.1517/13543784.2010.509342. [DOI] [PubMed] [Google Scholar]
- 49.Derks M, Abt M, Phelan M. Lack of clinically relevant drug-drug interactions when dalcetrapib is co-administered with ezetimibe. Br J Clin Pharmacol. 2010 Dec;70(6):825–33. doi: 10.1111/j.1365-2125.2010.03763.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Young A, Anzures-Cabrera J, Derks M. No clinically relevant drug-drug interactions when dalcetrapib is co-administered with a monophasic oral contraceptive (Microgynon(R) 30) Int J Clin Pharmacol Ther. 2012 Apr;50(4):248–56. doi: 10.5414/cp201647. [DOI] [PubMed] [Google Scholar]
- 51.Baldo PA, Anzures-Cabrera J, Bentley D. In vivo evaluation of drug-drug interactions linked to UGT inhibition: the effect of probenecid on dalcetrapib pharmacokinetics. Int J Clin Pharmacol Ther. 2013 Jan 29; doi: 10.5414/CP201766. [DOI] [PubMed] [Google Scholar]
- 52.Luscher TF, Taddei S, Kaski JC, et al. Vascular effects and safety of dalcetrapib in patients with or at risk of coronary heart disease: the dal-VESSEL randomized clinical trial. Eur Heart J. 2012 Apr;33(7):857–65. doi: 10.1093/eurheartj/ehs019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fayad ZA, Mani V, Woodward M, et al. Safety and efficacy of dalcetrapib on atherosclerotic disease using novel non-invasive multimodality imaging (dal-PLAQUE): a randomised clinical trial. Lancet. 2011 Oct 29;378(9802):1547–59. doi: 10.1016/S0140-6736(11)61383-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schwartz GG, Olsson AG, Abt M, et al. Effects of Dalcetrapib in Patients with a Recent Acute Coronary Syndrome. N Engl J Med. 2012 Nov 5; doi: 10.1056/NEJMoa1206797. [DOI] [PubMed] [Google Scholar]
- 55.Krishna R, Garg A, Panebianco D, et al. Single-dose pharmacokinetics and pharmacodynamics of anacetrapib, a potent cholesteryl ester transfer protein (CETP) inhibitor, in healthy subjects. Br J Clin Pharmacol. 2009 Oct;68(4):535–45. doi: 10.1111/j.1365-2125.2009.03465.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Krishna R, Bergman AJ, Jin B, et al. Multiple-dose pharmacodynamics and pharmacokinetics of anacetrapib, a potent cholesteryl ester transfer protein (CETP) inhibitor, in healthy subjects. Clin Pharmacol Ther. 2008 Dec;84(6):679–83. doi: 10.1038/clpt.2008.109. [DOI] [PubMed] [Google Scholar]
- 57.Tan EY, Hartmann G, Chen Q, et al. Pharmacokinetics, metabolism, and excretion of anacetrapib, a novel inhibitor of the cholesteryl ester transfer protein, in rats and rhesus monkeys. Drug Metab Dispos. 2010 Mar;38(3):459–73. doi: 10.1124/dmd.109.028696. [DOI] [PubMed] [Google Scholar]
- 58.Kumar S, Tan EY, Hartmann G, et al. Metabolism and excretion of anacetrapib, a novel inhibitor of the cholesteryl ester transfer protein, in humans. Drug Metab Dispos. 2010 Mar;38(3):474–83. doi: 10.1124/dmd.109.028704. [DOI] [PubMed] [Google Scholar]
- 59.Krishna R, Garg A, Jin B, et al. Assessment of a pharmacokinetic and pharmacodynamic interaction between simvastatin and anacetrapib, a potent cholesteryl ester transfer protein (CETP) inhibitor, in healthy subjects. Br J Clin Pharmacol. 2009 May;67(5):520–6. doi: 10.1111/j.1365-2125.2009.03385.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Krishna R, Stypinski D, Ali M, et al. Lack of an effect of anacetrapib on the pharmacokinetics of digoxin in healthy subjects. Biopharm Drug Dispos. 2011 Dec;32(9):525–9. doi: 10.1002/bdd.776. [DOI] [PubMed] [Google Scholar]
- 61.Krishna R, Stypinski D, Ali M, et al. Lack of a meaningful effect of anacetrapib on the pharmacokinetics and pharmacodynamics of warfarin in healthy subjects. Br J Clin Pharmacol. 2012 Jul;74(1):116–24. doi: 10.1111/j.1365-2125.2012.04171.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Krishna R, Bergman AJ, Jin B, et al. Assessment of the CYP3A-mediated drug interaction potential of anacetrapib, a potent cholesteryl ester transfer protein (CETP) inhibitor, in healthy volunteers. J Clin Pharmacol. 2009 Jan;49(1):80–7. doi: 10.1177/0091270008326718. [DOI] [PubMed] [Google Scholar]
- 63.Yvan-Charvet L, Kling J, Pagler T, et al. Cholesterol efflux potential and antiinflammatory properties of high-density lipoprotein after treatment with niacin or anacetrapib. Arterioscler Thromb Vasc Biol. 2010 Jul;30(7):1430–8. doi: 10.1161/ATVBAHA.110.207142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cannon CP, Shah S, Dansky HM, et al. Safety of anacetrapib in patients with or at high risk for coronary heart disease. N Engl J Med. 2010 Dec 16;363(25):2406–15. doi: 10.1056/NEJMoa1009744. [DOI] [PubMed] [Google Scholar]
- 65.Krauss RM, Wojnooski K, Orr J, et al. Changes in lipoprotein subfraction concentration and composition in healthy individuals treated with the CETP inhibitor anacetrapib. J Lipid Res. 2012 Mar;53(3):540–7. doi: 10.1194/jlr.M018010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lamarche B, Tchernof A, Moorjani S, et al. Small, dense low-density lipoprotein particles as a predictor of the risk of ischemic heart disease in men. Prospective results from the Quebec Cardiovascular Study. Circulation. 1997 Jan 7;95(1):69–75. doi: 10.1161/01.cir.95.1.69. [DOI] [PubMed] [Google Scholar]
- 67.Fernandez MC, Escribano A, Mateo AI, et al. Design, synthesis and structure-activity-relationship of 1,5-tetrahydronaphthyridines as CETP inhibitors. Bioorg Med Chem Lett. 2012 May 1;22(9):3056–62. doi: 10.1016/j.bmcl.2012.03.075. [DOI] [PubMed] [Google Scholar]
- 68.Nicholls SJ, Brewer HB, Kastelein JJ, et al. Effects of the CETP inhibitor evacetrapib administered as monotherapy or in combination with statins on HDL and LDL cholesterol: a randomized controlled trial. JAMA. 2011 Nov 16;306(19):2099–109. doi: 10.1001/jama.2011.1649. [DOI] [PubMed] [Google Scholar]
- 69.Voight BF, Peloso GM, Orho-Melander M, et al. Plasma HDL cholesterol and risk of myocardial infarction: a mendelian randomisation study. Lancet. 2012 May 17; doi: 10.1016/S0140-6736(12)60312-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wilson PW, Abbott RD, Castelli WP. High density lipoprotein cholesterol and mortality. The Framingham Heart Study. Arteriosclerosis. 1988 Nov-Dec;8(6):737–41. doi: 10.1161/01.atv.8.6.737. [DOI] [PubMed] [Google Scholar]
- 71.Franceschini G, Sirtori CR, Capurso A, 2nd, et al. A-IMilano apoprotein. Decreased high density lipoprotein cholesterol levels with significant lipoprotein modifications and without clinical atherosclerosis in an Italian family. J Clin Invest. 1980 Nov;66(5):892–900. doi: 10.1172/JCI109956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Calabresi L, Baldassarre D, Castelnuovo S, et al. Functional lecithin: cholesterol acyltransferase is not required for efficient atheroprotection in humans. Circulation. 2009 Aug 18;120(7):628–35. doi: 10.1161/CIRCULATIONAHA.108.818143. [DOI] [PubMed] [Google Scholar]
- 73.Frikke-Schmidt R, Nordestgaard BG, Stene MC, et al. Association of loss-of-function mutations in the ABCA1 gene with high-density lipoprotein cholesterol levels and risk of ischemic heart disease. JAMA. 2008 Jun 4;299(21):2524–32. doi: 10.1001/jama.299.21.2524. [DOI] [PubMed] [Google Scholar]
- 74.Tall AR. Cholesterol efflux pathways and other potential mechanisms involved in the athero-protective effect of high density lipoproteins. J Intern Med. 2008 Mar;263(3):256–73. doi: 10.1111/j.1365-2796.2007.01898.x. [DOI] [PubMed] [Google Scholar]
- 75.Linsel-Nitschke P, Jansen H, Aherrarhou Z, et al. Macrophage cholesterol efflux correlates with lipoprotein subclass distribution and risk of obstructive coronary artery disease in patients undergoing coronary angiography. Lipids Health Dis. 2009;8:14. doi: 10.1186/1476-511X-8-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mikkola TS, Anthony MS, Clarkson TB, et al. Serum cholesterol efflux potential is an independent predictor of coronary artery atherosclerosis. Atherosclerosis. 2003 Sep;170(1):31–8. doi: 10.1016/s0021-9150(03)00247-8. [DOI] [PubMed] [Google Scholar]
- 77.Low H, Hoang A, Sviridov D. Cholesterol efflux assay. J Vis Exp. 2012;(61):e3810. doi: 10.3791/3810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Daniil G, Phedonos AA, Holleboom AG, et al. Characterization of antioxidant/anti-inflammatory properties and apoA-I-containing subpopulations of HDL from family subjects with monogenic low HDL disorders. Clin Chim Acta. 2011 Jun 11;412(13–14):1213–20. doi: 10.1016/j.cca.2011.03.011. [DOI] [PubMed] [Google Scholar]
- 79.Bhattacharyya T, Nicholls SJ, Topol EJ, et al. Relationship of paraoxonase 1 (PON1) gene polymorphisms and functional activity with systemic oxidative stress and cardiovascular risk. JAMA. 2008 Mar 19;299(11):1265–76. doi: 10.1001/jama.299.11.1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Barter PJ, Nicholls S, Rye KA, et al. Antiinflammatory properties of HDL. Circ Res. 2004 Oct 15;95(8):764–72. doi: 10.1161/01.RES.0000146094.59640.13. [DOI] [PubMed] [Google Scholar]
- 81.Krukemyer JJ, Talbert RL. Lovastatin: a new cholesterol-lowering agent. Pharmacotherapy. 1987;7(6):198–210. doi: 10.1002/j.1875-9114.1987.tb03524.x. [DOI] [PubMed] [Google Scholar]
- 82.Zhao XQ, Morse JS, Dowdy AA, et al. Safety and Tolerability of Simvastatin Plus Niacin in Patients With Coronary Artery Disease and Low High-Density Lipoprotein Cholesterol (The HDL Atherosclerosis Treatment Study) Am J Cardiol. 2004;93:307–312. doi: 10.1016/j.amjcard.2003.10.009. [DOI] [PubMed] [Google Scholar]