

Abstract
BACKGROUND
The androgen receptor (AR) signaling continues to be essential in castrate-resistant prostate cancer (CRPC). Taxel-based chemotherapy is the current standard treatment for CRPC patients. Unfortunately, almost all patients eventually develop resistance toward this chemotherapy. Significantly, it was recently found that the anti-tumor effect of paclitaxel in CRPC is due to its inhibition of AR activity via its inhibition of microtubule dynamics. Polo-like kinase 1 (Plk1), a critical regulator in many cell cycle events, is elevated in prostate cancer (PCa) and linked to tumor grades. Of note, we have previously shown that Plk1 phosphorylates CLIP-170 and p150Glued, two important regulators of microtubule dynamics.
METHODS
We compared paclitaxel-mediated phenotypes (inhibition of the AR signaling, decrease of microtubule dynamics and cell death) of PCa cells expressing different forms of CLIP-170 and p150Glued with different Plk1 phosphorylation states.
RESULTS
we show that Plk1 phosphorylation of CLIP-170 and p150Glued affects cellular responses to paclitaxel. Expression of Plk1-unphosphorylatable mutants of CLIP-170 and p150Glued results in increased paclitaxel-induced apoptosis, increased protein degradation of the AR, and decreased nuclear accumulation of the AR in response to androgen in prostate cancer cells. Finally, we show that cells expressing unphosphorylatable mutants of CLIP-170 have defective microtubule dynamics, thus providing a new mechanism to understand how Plk1-associated kinase activity promotes constitutive activation of AR signaling in CRPC.
CONCLUSIONS
Our data suggest that a combination of inhibition of Plk1 and paclitaxel might be a novel avenue for treatment of CRPC.
Keywords: Prostate cancer, Plk1, paclitaxel, androgen receptor, microtubule dynamics
INTRODUCTION
It has been well established that the androgen receptor (AR) plays a critical role in initiation and progression of prostate cancer (PCa). In response to androgen, the activated AR is translocated from the cytoplasm into the nucleus, acting as a transcription factor that activates many downstream proteins, such as prostate-specific antigen (PSA). Enough evidence suggests that continued AR signaling is essential for PCa development even after castration, one major approach for treatment of late-stage PCa. Therefore, targeted inhibition of AR signaling remains a promising approach for patients with castrate-resistant prostate cancer (CRPC) [1]. Significantly, paclitaxel, the major chemotherapeutic agent currently used for CRPC, was recently found to inhibit the AR [2, 3]. At the cellular level, paclitaxel inhibits microtubule depolymerization, resulting in mitotic arrest, followed by apoptosis [4]. However, PCa cells in patients actually divide much more slowly than PCa cells in culture, indicating that the clinical effect of paclitaxel likely results from both mitotic catastrophe and interphase effects.
Microtubule dynamics consists of alternating phases of growth and shortening, a pattern of behavior known as dynamic instability [5]. Of significance, microtubule dynamics is essential for AR trafficking into the nucleus during interphase. Paclitaxel prevents nuclear localization of the AR via inhibition of microtubule dynamics [2]. Therefore, targeting inhibition of microtubule dynamics is expected to prevent paclitaxel resistance in CRPC. Microtubule dynamics is regulated by proteins that bind specifically to the plus ends of the growing microtubules (+TIPs). CLIP-170, the founding member of the microtubule plus-end family [6], is composed of three separate regions: N terminus, central coiled-coil region, and C terminus (Fig 1A). In addition to two conserved cytoskeleton-associated protein glycine-rich (CAP-Gly) domains, the N terminus has three serine-rich regions. The N-terminal domain plays an essential role in microtubule targeting, the long central coiled-coil domain is responsible for dimerization of the protein, and the C terminus, which contains two zinc-finger domains, interferes with microtubule binding by interacting with the N terminus. Enough evidence has accumulated to support the notion that CLIP-170 plays an important role in microtubule dynamics. p150Glued, a component of the dynein/dynactin complex, is a major CLIP-170-interacting partner (Fig 1B). Localization of p150Glued at microtubule plus ends is CLIP-170 dependent [7]. Mapping experiments indicat that the C terminus of CLIP-170 directly interacts with the N terminus of p150Glued (Fig 1C) [7].
Fig. 1.

We hypothesize that Plk1 phosphorylation of CLIP-170 and p150Glued increases microtubule dynamics, thus enhancing AR signaling even in the presence of paclitaxel. A: Schematic representation of CLIP-170 domains. S195, located in the N-terminal domain, is targeted by Plk1, whereas S1318, located in the C-terminal domain, is phosphorylated by CK2 [13]. Cdc2 phosphorylates T287 [18]. B: Schematic representation of p150Glued domains. S179 is phosphorylated by Plk1 [14]. C: Possible interactions between CLIP-170, dynactin, dynein, and LIS1 at the plus end of a growing microtubule. It has been proposed that the C-terminal zinc-finger domain of CLIP-170 interacts with the N-terminal CAP-Gly domain of p150Glued, and the N-terminal CAP-Gly domain of CLIP-170 binds to microtubule plus ends. LIS1 interacts with both dynein and CLIP-170. D: Plk1 is elevated in prostate cancer cells. Different prostate cells were either unsynchronized or treated with thymidine to arrest at the G1/S boundary, followed by immunoblotting (IB) with antibodies against Plk1. RWPE1 cells, established by transfection with a single copy of the human papilloma virus 18 into epithelial cells derived from the peripheral zone of a histologically normal adult human prostate, were used as non-transformed control cells. LNCaP, C4-2 and 22RV1 are AR positive PCa cells, whereas ARCaP, DU145 and PC-3 are AR negative PCa cells. E: A working model supported by the data presented here. Paclitaxel-induced blockage of AR signaling is due to its inhibition of microtubule dynamics, which is essential for AR trafficking into the nucleus [2].
Polo-like kinase 1 (Plk1) is a regulator of many cell cycle events, such as mitotic entry, bipolar spindle formation, and cytokinesis [8]. Plk1 expression correlates with cellular proliferation and prognosis of patients with various cancers [9], including PCa [10]. Targeted inhibition of Plk1 causes mitotic catastrophe and induction of apoptosis in PCa cells, suggesting that Plk1 might be a potential therapeutic target for PCa [11] [12]. However, whether inhibition of Plk1 can be a novel approach to overcome paclitaxel resistance in CRPC is still unknown. Of note, we recently showed that Plk1 directly phosphorylates CLIP-170 [13] and p150Glued [14]. Significantly, Plk1 can be easily detected in various PCa cells when they are either randomly growing or treated with thymidine to arrest at late G1 (Fig 1D), further supporting the notion that Plk1 might have cancer cell-specific functions in interphase.
In this communication, we show that Plk1-associated kinase activity toward CLIP-170 and p150Glued enhances microtubule dynamics, thus promoting AR signaling even in the presence of paclitaxel, eventually contributing to development of paclitaxel resistance in CRPC (Fig 1E).
MATERIALS AND METHODS
Cell culture
PC3 and U2OS cells were cultured in Dulbecco modified Eagle medium DMEM) supplemented with 10% (vol/vol) fetal bovine serum (FBS), 100 units/ml penicillin, and 100 units/ml streptomycin at 37°C in 8% CO2. LNCaP, C4-2 and 22Rv1 cells were cultured in RPMI-1640 (ATCC) supplemented with 10% FBS (vol/vol), 100 units/ml penicillin, and 100 units/ml streptomycin at 37°C in 8% CO2. Paclitaxel-resistant PC3 cells were kindly provided by Drs. Mikio Namiki and Evan Keller [15].
Transfection and stable cell line generation
PC3 and LNCaP cells were transfected with GFP-CLIP-170 constructs (WT, S195A, S195A/S1318A, T286A, S195E, S195E/1318D, T286D) and GFP-p150glued constructs (WT, S179A, S179D) with MegaTran (ORIGENE). After 24 hrs post-transfection, cells were selected with 400 μg/ml of G418 for 2 weeks and amplified.
Western blotting
Cell lysates prepared in TBSN buffer (20 mM Tris, pH 8.0, 150 mM NaCl, 1.5 mM EDTA, 5 mM EGTA, 0.5% Nonidet P-40, 0.5 mM Na3VO4) supplemented with proteinase inhibitors were resolved by SDS-PAGE, transferred to Whatman Westran PVDF membrane (Sigma, Z671088), and subjected to immunoblotting (IB) with antibodies against Plk1 (Santa Cruz, sc-17783), AR (Santa Cruz, sc-7305), γ-tubulin (Sigma, T3559) and β-actin (Sigma, A5441), followed by incubation with horseradish peroxidase-linked secondary antibodies (GE healthcare).
Isolation of cytoplasmic and nuclear fractions
Cytoplasmic and nuclear fractions were prepared with a kit from ActiveMotif (40410) according to the manufacturer's instructions.
Immunofluorescence (IF) staining
Cells were fixed with 4% paraformaldehyde for 10 minutes, washed with 0.1% Triton X-100 PBS, and permeabilized with methanol for 2 minutes. After being washed with 0.1% Triton X-100 PBS, cells were blocked with 3% bovine serum albumin (BSA) in PBS for 10 minutes and incubated with anti-AR antibody for 2 h at room temperature, followed by incubation with Alexa Fluor® 555 Goat Anti-Mouse IgG (H+L) secondary antibody (Invitrogen, Cat.: A21424) and 4,6-diamidino-2-phenylindole (DAPI) for 1 hour.
Recording and measurement of GFP-tagged protein signal
U2OS cells stably expressing GFP-CLIP-170 constructs (WT, S195A, S195A/S1318A, T286A) or GFP-p150Glued constructs (WT, S179A) were grown on coverslips. Variable-angle epifluorescence microscopy (VAEM) was used to record GFP-tagged protein activities. Exposure time was 200 ms and time-lapse was 10 seconds. Ten random cells from each stable cell line were recorded and 5 randomly assembled CLIP-170 or p150Glued proteins from each cell were measured. The maximum length of GFP-tagged protein, the growth rate of GFP-tagged protein and the lifespan of GFP-tagged protein from appearance to disappearance were measured as described previously [16].
Reverse transcription-PCR and quantitative real-time PCR
After LNCaP cells stably expressing p150Glued (WT, S179A, S179D) and CLIP-170 (WT, S195A, S195A/S1318A) constructs were treated with 100 nM of DHT, 100 nM of paclitaxel or both drugs for 2 hrs, total mRNA was extracted using TriZOL method and subjected to reverse transcription according to the protocol by manufacturer (Applied Biosystems). Quantitative real time PCR (Qiagen, RT2 SYBR® Green qPCR Mastermixes) was performed for β-actin, PSA, maspin using cDNA products from 250 ng of mRNA. The mRNA levels of PSA and maspin were calculated using 2ΔΔCT method and normalized to β-actin. PCR was performed using primers specific for PSA (forward 5'-TGTGCTTCAAGGTATCACGT-3' and reverse 5'-GCAACCCTGGACCTCACA-3'), maspin (forward 5'-ATGGTGGGGATTCCATAGAG- 3' and reverse 5'-CCTGATCCAGCAACATTAGC-3'), and β-actin (forward 5'-AGATCACTGCCCTGGCA-3' and reverse 5'-GTCAAGAAAGGGTGTAACGC-3').
RESULTS
Inhibition of Plk1 increases the cellular response to paclitaxel
With the goal to test whether Plk1-associated kinase activity contributes to development of paclitaxel resistance in CRPC, we asked whether Plk1 affects the cellular response of PCa cells to paclitaxel. We first used the previously established paclitaxel-resistant PC-3 cells (PC-3-TxR) [15]. As indicated, in comparison to the parental PC-3 cells, the level of Plk1 protein is higher in PC-3-TxR cells (Fig 2A). To determine whether PC-3-TxR cells are more sensitive to inhibition of Plk1, we treated these cells with BI2536, an inhibitor of Plk1 [17]. Interestingly, the cellular response to BI2536 corresponded to the level of Plk1, as PC-3-TxR cells showed a much higher percentage of phospho-H3-positive cells upon BI2536 treatment (Fig 2B). In agreement, the IC50 value for BI2536 in PC-3-TxR cells (100 nM) is lower than that in PC-3 cells (320 nM). Furthermore, a combination of BI2536 and paclitaxel led to rapid cell death in PC3-TxR cells, as indicated by a 6.2-fold increase of cleaved PARP signal (Fig 2C). Finally, a combination of BI2536 and paclitaxel also resulted in a significantly more severe cell death than BI2536 or paclitaxel alone in PC3 cells. Although the intensity of uncleaved PARP did not change dramatically, increase of intensity of cleaved PARP upon dual treatment of paclitaxel and BI2536 was obvious (Fig 2D).
Fig. 2.
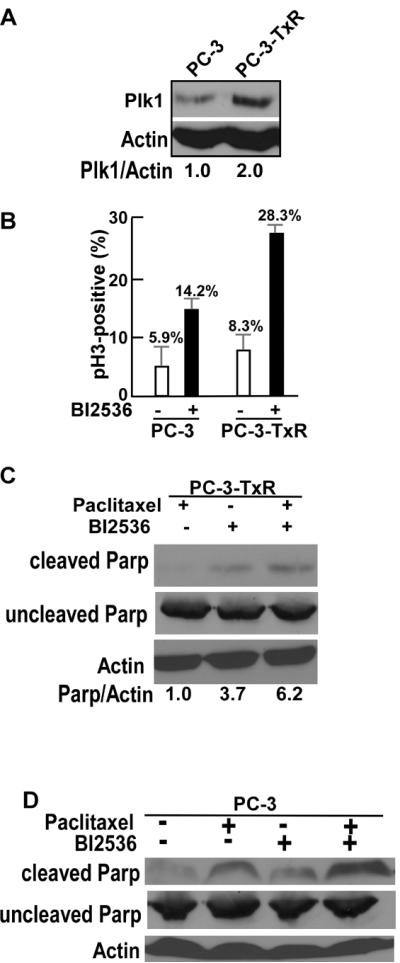
Plk1-associated kinase activity contributes to paclitaxel resistance in PC-3 cells. A: Plk1 is overexpressed in paclitaxel-resistant PC-3 (PC-3-TxR) cells in comparison to non-resistant PC-3 cells. Total lysates from PC-3 and PC-3-TxR cells were subjected to IB with antibodies against Plk1 and β-actin, followed by quantification with ImageJ software. The ratio of Plk1/β-actin of control PC-3 cells was set to 1.0. B: PC-3-TxR cells are more sensitive to inhibition of Plk1. PC-3 or PC-3-TxR cells were treated with or without BI2536 (100 nM) for 10 hrs and subjected to immunofluorescence (IF) staining with antibodies against phospho-histone 3 (pH3). At least 400 cells were counted for quantification. C and D: Inhibition of Plk1 results in apoptosis of PC-3-TxR (C) and PC-3 (D) cells. Cells were treated with paclitaxel (20 nM), BI2536 (200 nM) or both drugs for 10 hrs, and harvested for IB with indicated antibodies. Parp: poly(ADP-ribosyl) polymerase.
Plk1 phosphorylation of p150Glued and CLIP-170 prevents paclitaxel-induced apoptosis
We previously showed that Plk1 phosphorylates p150Glued-S179 and CLIP-170-S195. [13, 14]. So, we asked whether expression of p150Glued and CLIP-170 constructs carrying various mutations at the Plk1 sites affects cellular responses to the microtubule poisons, nocodazole and paclitaxel. Expression of p150Glued-S179A in U2OS cells potentiated paclitaxel-induced cell death, indicated by a 2.3-fold increase of the cleaved PARP signal in comparison to control cells without drug treatment (Fig 3A). Similarly, in PC-3 cells expressing p150Glued-S179A, paclitaxel treatment increased cleaved PARP intensity 1.4-fold. In contrast, expression of p150Glued-S179D, the phosphomimetic mutant, inhibited paclitaxel-induced cell death in PC-3 cells (Fig 3B). Therefore, Plk1 phosphorylation of p150Glued affects the cellular response to paclitaxel.
Fig. 3.

Plk1 phosphorylation of p150Glued and CLIP-170 affects the cellular response to paclitaxel. A: U2OS cells expressing p150Glued-S179A are more sensitive to paclitaxel. U2OS cells stably expressing p150Glued constructs (WT or S179A) were treated with 200 ng/ml of nocodazole (Noc.) or 50 nM of paclitaxel (PTX) for 10 hrs and analyzed by anti-cleaved Parp IB. The ratio of cleaved Parp/β-actin is indicated at the bottom. B: PC-3 cells expressing p150Glued-S179A or -S179D are more sensitive or resistant to paclitaxel, respectively. PC-3 cells stably expressing p150Glued constructs (WT, S179A or S179D) were treated with 200 ng/ml of nocodazole or 100 nM of paclitaxel for 10 hrs, harvested for anti-cleaved Parp IB, and quantified with ImageJ. C: U2OS cells expressing CLIP-170-S195A/S1318A are more sensitive to paclitaxel. U2OS cells stably expressing CLIP-170 constructs (WT, S195A, S195A/1318A or T287A) were treated with nocodazole or paclitaxel as in A, and subjected to IB. D: PC-3 cells expressing CLIP-170-S195A/S1318A are more sensitive to paclitaxel. PC-3 cells stably expressing CLIP-170 constructs (WT, S195A, S195A/1318A or T287A) were treated with nocodazole or paclitaxel as in B, subjected to IB with the indicated antibodies, and quantified. For comparison, the ratio of cleaved Parp/β-actin of the cells without drug treatment was set to 1.0 within each group that expresses the same construct. WT, wild type.
In addition to Plk1, CLIP-170 is targeted by both casein kinase 2 (CK2) and Cdc2/cyclin (Fig 1A). We previously showed that Plk1 phosphorylation of CLIP-170 at S195 creates a docking site to recruit CK2, which subsequently phosphorylates CLIP-170 at S1318, and that phosphorylation of CLIP-170 by both Plk1 and CK2 promotes timely formation of kinetochore-microtubule attachments in metaphase [13]. We also showed that Cdc2/cyclin B phosphorylation of CLIP-170 at T287 regulates its binding pattern with tubulin [18]. Here, we asked whether expression of CLIP-170 constructs with various mutations at these phosphorylation sites affects the cellular response to microtubule poisons. As indicated, expression of both CLIP-170-S195A and CLIP-170-S195A/S1318A double mutant, but not CLIP-170-T287A, rendered the cells sensitive to microtubule poisons. In particular, paclitaxel treatment induced a 1.7- and 1.8-fold increase of cleaved PARP signals in U2OS cells expressing CLIP-170-S195A and CLIP-170-S195A/S1318A, respectively (Fig 3C). To test whether this observation is a general phenomenon or cell-type specific, we performed similar experiments in PC-3 cells. Again, in PC-3 cells expressing CLIP-170-S195A and CLIP-170-S195A/S1318A, paclitaxel treatment induced a 1.4- and 2.4-fold increase of cleaved PARP signal, respectively (Fig 3D). Therefore, we conclude that Plk1 phosphorylation of CLIP-170 inhibits paclitaxel-induced cell death as well.
Plk1 phosphorylation of CLIP-170 and p150Glued enhances AR signaling
Because the anti-tumor effect of paclitaxel in CRPC is at least partially due to its inhibition of AR signaling [3] and because AR trafficking to the nucleus is mediated by the microtubule-motor protein dynein [2], we asked whether Plk1-associated kinase activity toward CLIP-170 and p150Glued affects AR signaling. Toward that end, we used LNCaP cells stably expressing CLIP-170 and p150Glued constructs with various mutations at critical phosphorylation sites. As indicated, cells expressing CLIP-170-S195A and CLIP-170-S195A/S1318A had reduced levels of AR in comparison to cells expressing WT CLIP-170. In contrast, expression of CLIP-170-S195E, -S195E/SD1318D and -T287D mutants increased the protein levels of the AR by 5.7-, 4.4- and 3.1-fold, respectively, indicating that phosphorylation of CLIP-170 by Plk1/CK2/cdc2 stabilizes the AR (Fig 4A). To confirm this observation, we treated cells with cycloheximide, an inhibitor of protein translation, for different times and analyzed the protein levels of AR. As we expected, degradation of the AR was significantly faster in cells expressing CLIP-170-S195A/S1318A than in cells expressing WT CLIP-170 (Fig 4B). Because activation of the AR is androgen-dependent and because paclitaxel antagonizes androgen-induced AR activation, we also asked whether expression of unphosphorylatable CLIP-170 mutants affects androgenand paclitaxel-mediated AR modulation. In cells expressing CLIP-170-WT, -S195A and -T287A, treatment with dihydrotestosterone (DHT) and paclitaxel increased and decreased the protein levels of the AR, respectively. However, in cells expressing CLIP-170-S195A/S1318A, DHT treatment failed to stabilize the AR to a level detectable by Western blotting (Fig 4C, compare quantification results of lane 2 versus lane 10). Similar experiments were performed for cells expressing p150Glued constructs (WT, S179A, S179D) to determine whether Plk1 phosphorylation of p150Glued also regulates AR signaling. The protein level of the AR in cells expressing p150Glued-S179A was about 10% of that in cells expressing WT p150Glued (Fig 4D). Furthermore, DHT-induced AR elevation was also partially inhibited in cells expressing p150Glued-S179A in comparison to cells expressing WT p150Glued (Fig 4E, compare quantification results of lane 2 versus 6, lane 4 versus 8). Therefore, Plk1 phosphorylation of both CLIP-170 and p150Glued promotes the stabilization of the AR protein.
Fig. 4.

Plk1 phosphorylation of CLIP-170 and p150Glued increases the stability of the AR. A: Expression of CLIP-170-S195E or -S195E/S1318D increases the level of the AR. Total lysates from LNCaP cells stably expressing CLIP-170 constructs (WT, S195A, S195A/S1318A, T286A, S195E, S195E/1318D or T287D) were subjected to IB with antibodies against the AR or β-actin, and quantified. For comparison, the ratio of AR/β-actin of cells expressing WT CLIP-170 was set to 1.0. B: Expression of CLIP-170-S195A/S1318A enhances degradation of the AR. LNCaP cells stably expressing CLIP-170 constructs (WT or S195A/S1318A) were treated with 75 μg/ml of cycloheximide (CHX) for different times (0, 10, 20, 30 minutes), and harvested for IB. C: Expression of CLIP-170-S195A/S1318A prevents DHT-induced AR activation. LNCaP cells stably expressing CLIP-170 constructs were treated with DHT (100 nM), paclitaxel (100 nM) or both drugs (D+T) for 2 hrs and harvested for IB. DHT, dihydrotestosterone. D: Expression of p150Glued-S179A reduces the protein level of the AR. Total lysates from LNCaP cells stably expressing p150Glued constructs (WT, S179A or S179D) were analyzed by IB. E, expression of p150Glued-S179A inhibits DHT-induced AR activation. LNCaP cells stably expressing p150Glued constructs (WT or S179A) were treated with DHT or PTX as in C and analyzed by IB.
Androgen induces translocation of the AR from the cytoplasm to the nucleus, where it acts as a transcription factor to regulate transcription of many downstream targets. Paclitaxel treatment reduces androgen-induced nuclear accumulation of the AR. Thus, we asked whether Plk1 phosphorylation of p150Glued and CLIP-170 affects the nuclear accumulation of the AR upon androgen and paclitaxel treatment. As indicated, DHT treatment caused apparent nuclear accumulation of the AR in cells expressing p150Glued-WT and -S179D even in the presence of paclitaxel. In contrast, no apparent nuclear accumulation of the AR was observed by Western blotting in cells expressing p150Glued-S179A in the presence of DHT and paclitaxel. Quantification indicates that AR levels are undetectable under this condition (Fig 5A). Similar experiments were performed for cells expressing CLIP-170 with various mutations. Quantification of Western blotting results indicates that cells expressing CLIP-170-S195A and -S195A/S1318A showed reduced nuclear accumulation of the AR in the presence of DHT and paclitaxel in comparison to cells expressing CLIP-170-WT and -T287A (Fig 5B). Therefore, we conclude that Plk1 phosphorylation of p150Glued and CLIP-170 contributes to androgen-induced AR translocation from the cytoplasm into the nucleus.
Fig. 5.
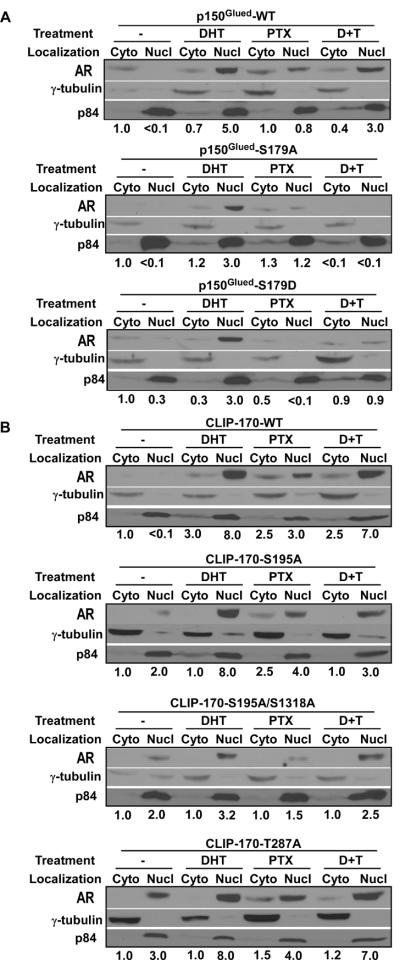
Expression of unphosphorylatable mutants of p150Glued and CLIP-170 affects cytoplasmic/nuclear shuttling of the AR. A: Expression of p150Glued-S179A inhibits DHT-induced nuclear accumulation of the AR and promotes paclitaxel-mediated nuclear exclusion of the AR. LNCaP cells stably expressing p150Glued constructs (WT, S179A or S179D) were treated with 100 nM of DHT, 100 nM of paclitaxel or both drugs (D+T) for 2 hrs and harvested for subcellular fractionation to separate cytoplasmic (Cyto) and nuclear fractions (Nucl), followed by IB with indicated antibodies. The antibodies against γ-tubulin and p84 were used as cytoplasmic and nuclear markers, respectively. Only half of cytoplasmic fraction was loaded in comparison to nuclear fraction. B: Expression of CLIP-170-S195A/S1318A reduces DHT-induced nuclear accumulation of the AR and enhances paclitaxel-mediated nuclear exclusion of the AR. LNCaP cells stably expressing CLIP-170 constructs (WT, S195A, S195A/S1318A or T287A) were treated with DHT or paclitaxel and processed as in A. To quantify, the cytosolic AR levels without drug treatment in each experiment were set to be 1.0.
To confirm these findings, which had been obtained by Western blotting, we also performed immunofluorescence (IF) staining with antibodies against the AR to directly visualize its subcellular localization under different conditions. To quantify the results, we divided the cells into three categories; high, medium and low nuclear AR signals. The overall conclusion is consistent with what we observed with Western blotting in Fig 5. The percentages of cells expressing p150Glued-WT, -S179A and -S179D with high AR signals were 5%, 3% and 8%, respectively. Upon DHT treatment, the percentages of cells expressing p150Glued-WT, -S179A and -S179D with high AR signals were 7%, 1% and 11%, respectively. Of note, 67% of DHT- treated, p150Glued-S179A-expressing cells showed very low nuclear AR intensity. In contrast, only 42% and 36% of p150Glued-WT- and p150Glued-S179D-expressing cells showed low nuclear AR intensities. In the case of CLIP-170, introduction of the S195A/S1318A mutation reduced high nuclear AR signal-expressing cells from 6% to 2%. Such a difference was much more evident upon DHT treatment. 16% of cells expressing CLIP-170-WT showed high nuclear AR signals, whereas only 4% of cells expressing CLIP-170-S195A/S1318A had high nuclear AR signals, indicating that expression of CLIP-170-S195A/S1318A likely inhibits the cytoplasmic/nuclear shuttling of the AR. Paclitaxel treatment reduced nuclear AR signals in all cells, regardless of which construct was expressed. Finally, in cells treated with DHT and paclitaxel, 3% of cells expressing CLIP-170-S195A/S1318A showed high nuclear AR signals in comparison to 6% of cells expressing CLIP-170-WT. Furthermore, 43% of cells expressing CLIP-170-S195A/S1318A showed low nuclear AR signals in comparison to 37% of cells expressing CLIP-170-WT, consistent with the Western blotting data presented in Fig 5.
Finally, we asked whether Plk1 phosphorylation of CLIP-170 and p150Glued affects the expression of AR downstream target genes. Introduction of Plk1-unphosphorylatable mutation (S195A) in CLIP-170 prevented androgen-induced elevation of both mRNA and protein levels of PSA, whose expression is positively regulated by the AR (Fig 6A, 6C). In contrast, cells expressing CLIP-170-S195A showed a higher level of maspin, whose expression is negatively regulated by the AR (Fig 6B). Compared to cells expressing p150Glued-WT and -S179D, cells expressing p150Glued-S179A also showed reduced levels of PSA and Nkx3.1, whose expression is also positively regulated by AR (Fig 6D, 6F), but elevated levels of maspin (Fig 6E, 6F). In summary, we conclude that Plk1 phosphorylation of CLIP-170 and p150Glued promotes the cytoplasmic/nuclear shuttling of the AR, eventually resulting in regulation of AR target genes, such as PSA, Nkx3.1 and maspin.
Fig. 6.

Plk1 phosphorylation of CLIP-170 and p150Glued affects the AR signaling events. A–C: Plk1/CK2 phosphorylation of CLIP-170 enhances expression of AR target genes. LNCaP cells stably expressing CLIP-170 constructs (WT, S195A, S195A/S1318A) were treated with DHT, paclitaxel or both drugs, and harvested for mRNA and protein analysis. The expression levels of PSA and maspin mRNA were examined by semiquantitative RT-PCR analysis (A, B) and the expression level of PSA protein was also analyzed by Western blot (C). (**, P < 0.01). D–F: Plk1 phosphorylation of p150Glued increases expression of AR target genes. LNCaP cells stably expressing p150Glued constructs (WT, S179A, S179D) were treated with DHT, paclitaxel or both drugs, and harvested for mRNA and protein analysis. The expression levels of PSA and maspin mRNA were examined by semiquantitative RT-PCR analysis (D, E) and the expression levels of Nkx3.1 and maspin protein were analyzed by Western blot (F).
Plk1 phosphorylation of CLIP-170 promotes microtubule dynamics
We previously showed that Plk1 phosphorylation of p150Glued affects its association with microtubules during the G2/M transition, consequently regulating nuclear envelop breakdown [14]. We further investigated how Plk1 phosphorylation of CLIP-170 affects cytoplasmic/nuclear shuttling of the AR. One likely explanation is that Plk1-associated kinase activity towards CLIP-170 promotes microtubule dynamics, a process that controls AR trafficking. We thus monitored microtubule dynamics in live cells stably expressing GFP-CLIP-170 constructs (WT, S195A, S195A/S1318A, T287A) by variable-angle epifluorescence microscopy (VAEM). Images were taken every 10 seconds and 100 images were combined to generate one movie. These movies were further used to measure various parameters of microtubule dynamics. Compared to cells expressing CLIP-170-WT, cells expressing CLIP-170-S195A, -S195A/S1318A and -T287A showed decreased time of duration (Fig 7A), reduced maximal microtubule length (Fig 7B), and slower rate of microtubule growth (Fig 7C). Therefore, phosphorylation of CLIP-170 by Plk1, CK2 and cdc2/cyclin B promotes microtubule dynamics.
Fig. 7.
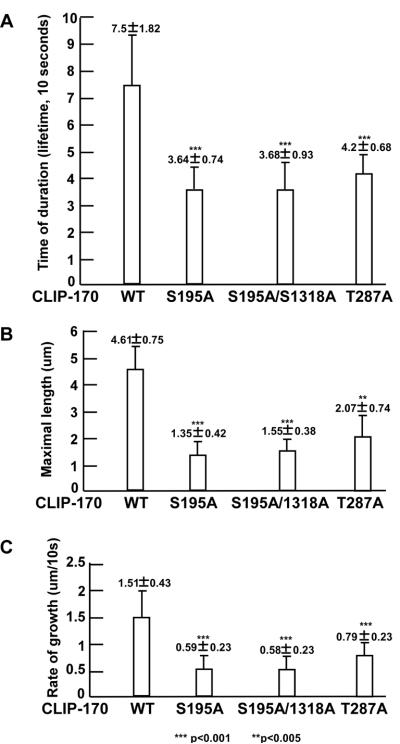
Expression of unphosphorylatable mutants of CLIP-170 inhibits microtubule dynamics. U2OS cells stably expressing GFP-CLIP-170 constructs (WT, S195A, S195A/S1318A or T287A) were grown on coverslips and analyzed for GFP signal by variable-angle epifluorescence microscopy (VAEM). Images were taken every 10 seconds. Ten cells from each group were recorded and 5 assembled microtubules from each cell were measured for time of duration (lifetime) of assembled CLIP-170 protein (A), the maximum length of microtubules (B), and the rate of CLIP-170 protein assembly (growth) (C). In comparison with cells expressing GFP-CLIP170-WT, statistical significance of cells expressing CLIP-170-S195A, -S195/S1318A and -T287A was calculated using SPSS Statistics 17.0 (t test). P < 0.01 was considered statistically significant. For all panels, statistical values are **, P < 0.005; ***, P < 0.001.
DISCUSSION
Different approaches have been developed to understand and overcome gradual development of resistance of cancer cells after initial response to chemotherapy [19]. Since one obstacle of chemotherapy is the expression of multidrug-efflux pumps (e.g., P-glycoprotein) that lower intracellular drug levels, inhibition of P-glycoprotein is one approach to increase drug efficacy [20]. In support, knockdown of P-glycoprotein, which was up-regulated in paclitaxel-resistant DU145 (DU145-TxR) PCa cells but not in PC-3-TxR cells, by MDR-1 (multiple drug resistance) siRNA restored paclitaxel sensitivity in DU145-TxR but not in PC-3-TxR, indicating that upregulation of P-glycoprotein is not always the main cause of paclitaxel resistance [15]. Furthermore, development of P-glycoprotein inhibitors has been a challenge for medicinal chemists due to undesired drug interactions and limited in vivo activities [21]. Using an Eμ-myc-driven lymphoma mouse model, Lowe and coworkers demonstrated that anti-apoptotic Bcl-2 functions as a potent multidrug resistance protein [22]. However, whether the Eμ-myc lymphoma model, which is highly sensitive to apoptosis [23], is representative for human tumors at large, which are >90% of epithelial origin, has been questioned [24, 25].
The research described in this communication is novel, in our opinion, for three reasons: First, it focuses on a druggable target, Plk1. Indeed, several specific Plk1 inhibitors have been developed and are already in clinical trials [8]. Second, it utilizes well-established PCa cells of epithelial origin. Third, it reveals a novel Plk1 function in interphase: how it regulates microtubule dynamics [26]. Our data show that inhibition of Plk1 significantly potentiates paclitaxel-mediated cell death, suggesting that this novel approach will be effective in modulating paclitaxel-mediated cytotoxicity in PCa (Fig 2). It was previously reported that treatment of breast cancer cells with siRNAs targeting Plk1 improved sensitivity toward paclitaxel in a synergistic manner [27]. Moreover, in a human xenograft experiment using MDA-MB-435 cells, the combination of Plk1 antisense oligonucleotides with paclitaxel led to synergistic reduction of tumor growth after three weeks of treatment compared with either agent alone [28]. Therefore, a combination of inhibition of Plk1 and paclitaxel to induce cell death acts in a synergistic manner in multiple cancer types but the mechanisms are likely different. In PCa, we propose that Plk1-mediated activation of AR signaling contributes to development of paclitaxel resistance (Fig 1E). Such a hypothesis is based upon 1) the observation that inhibition of AR signaling is a mechanism of paclitaxel chemotherapy in PCa [3], 2) the finding that the cytoplasmic/nuclear shuttling of the AR is microtubule dynamics dependent [2], 3) the fact that paclitaxel prevents microtubule depolymerization, and 4) our findings that Plk1 phosphorylation of CLIP-170 and p150Glued directly affects microtubule dynamics, consequently AR signaling. Our data challenges the traditional view that Plk1 functions only on typical mitotic events. This new and substantively different departure from the status quo is expected to overcome problems that have been associated with other potential means of preventing cellular resistance to paclitaxel, finally resulting in an efficacious approach to CRPC therapy.
ACKNOWLEDGEMENTS
We appreciate Eleanor Erikson for critical reading of the manuscript. We thank Drs. Miko Namiki and Evan Keller for paclitaxel-resistant PC-3 and DU145 cell lines. This work was supported by the China Scholarship Council (X.H.), NIH grant R01CA157429 (X.L.), and NSF grant MCB-1049693 (X.L.).
REFERENCES
- 1.Schrijvers D, Van Erps P, Cortvriend J. Castration-refractory prostate cancer: New drugs in the pipeline. Advances in therapy. 2010;27(5):285–96. doi: 10.1007/s12325-010-0038-1. [DOI] [PubMed] [Google Scholar]
- 2.Darshan MS, et al. Taxane-induced blockade to nuclear accumulation of the androgen receptor predicts clinical responses in metastatic prostate cancer. Cancer research. 2011;71(18):6019–29. doi: 10.1158/0008-5472.CAN-11-1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gan L, et al. Inhibition of the androgen receptor as a novel mechanism of taxol chemotherapy in prostate cancer. Cancer research. 2009;69(21):8386–94. doi: 10.1158/0008-5472.CAN-09-1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jordan MA, Wilson L. Microtubules as a target for anticancer drugs. Nature reviews. Cancer. 2004;4(4):253–65. doi: 10.1038/nrc1317. [DOI] [PubMed] [Google Scholar]
- 5.Mitchison T, Kirschner M. Dynamic instability of microtubule growth. Nature. 1984;312(5991):237–42. doi: 10.1038/312237a0. [DOI] [PubMed] [Google Scholar]
- 6.Perez F, et al. CLIP-170 highlights growing microtubule ends in vivo. Cell. 1999;96(4):517–27. doi: 10.1016/s0092-8674(00)80656-x. [DOI] [PubMed] [Google Scholar]
- 7.Lansbergen G, et al. Conformational changes in CLIP-170 regulate its binding to microtubules and dynactin localization. J Cell Biol. 2004;166(7):1003–14. doi: 10.1083/jcb.200402082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Strebhardt K. Multifaceted polo-like kinases: drug targets and antitargets for cancer therapy. Nature reviews. Drug discovery. 2010;9(8):643–60. doi: 10.1038/nrd3184. [DOI] [PubMed] [Google Scholar]
- 9.Eckerdt F, Yuan J, Strebhardt K. Polo-like kinases and oncogenesis. Oncogene. 2005;24(2):267–76. doi: 10.1038/sj.onc.1208273. [DOI] [PubMed] [Google Scholar]
- 10.Weichert W, et al. Polo-like kinase 1 is overexpressed in prostate cancer and linked to higher tumor grades. The Prostate. 2004;60(3):240–5. doi: 10.1002/pros.20050. [DOI] [PubMed] [Google Scholar]
- 11.Reagan-Shaw S, Ahmad N. Silencing of polo-like kinase (Plk) 1 via siRNA causes induction of apoptosis and impairment of mitosis machinery in human prostate cancer cells: implications for the treatment of prostate cancer. FASEB J. 2005;19(6):611–3. doi: 10.1096/fj.04-2910fje. [DOI] [PubMed] [Google Scholar]
- 12.Liu XS, et al. Polo-like kinase 1 facilitates loss of Pten tumor suppressor-induced prostate cancer formation. J Biol Chem. 2011;286(41):35795–800. doi: 10.1074/jbc.C111.269050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li H, et al. Phosphorylation of CLIP-170 by Plk1 and CK2 promotes timely formation of kinetochore-microtubule attachments. The EMBO journal. 2010;29(17):2953–65. doi: 10.1038/emboj.2010.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li H, et al. Polo-like kinase 1 phosphorylation of p150Glued facilitates nuclear envelope breakdown during prophase. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(33):14633–8. doi: 10.1073/pnas.1006615107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Takeda M, et al. The establishment of two paclitaxel-resistant prostate cancer cell lines and the mechanisms of paclitaxel resistance with two cell lines. The Prostate. 2007;67(9):955–67. doi: 10.1002/pros.20581. [DOI] [PubMed] [Google Scholar]
- 16.Staiger CJ, et al. Actin filament dynamics are dominated by rapid growth and severing activity in the Arabidopsis cortical array. The Journal of cell biology. 2009;184(2):269–80. doi: 10.1083/jcb.200806185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Steegmaier M, et al. BI 2536, a potent and selective inhibitor of polo-like kinase 1, inhibits tumor growth in vivo. Current biology : CB. 2007;17(4):316–22. doi: 10.1016/j.cub.2006.12.037. [DOI] [PubMed] [Google Scholar]
- 18.Yang X, et al. Cdc2-mediated phosphorylation of CLIP-170 is essential for its inhibition of centrosome reduplication. The Journal of biological chemistry. 2009;284(42):28775–82. doi: 10.1074/jbc.M109.017681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moreno-Aspitia A, Perez EA. Anthracycline- and/or taxane-resistant breast cancer: results of a literature review to determine the clinical challenges and current treatment trends. Clin Ther. 2009;31(8):1619–40. doi: 10.1016/j.clinthera.2009.08.005. [DOI] [PubMed] [Google Scholar]
- 20.Gottesman MM, Fojo T, Bates SE. Multidrug resistance in cancer: role of ATP-dependent transporters. Nat Rev Cancer. 2002;2(1):48–58. doi: 10.1038/nrc706. [DOI] [PubMed] [Google Scholar]
- 21.Baumert C, Hilgeroth A. Recent advances in the development of P-gp inhibitors. Anticancer Agents Med Chem. 2009;9(4):415–36. doi: 10.2174/1871520610909040415. [DOI] [PubMed] [Google Scholar]
- 22.Schmitt CA, Rosenthal CT, Lowe SW. Genetic analysis of chemoresistance in primary murine lymphomas. Nat Med. 2000;6(9):1029–35. doi: 10.1038/79542. [DOI] [PubMed] [Google Scholar]
- 23.Cory S, et al. Insights from Bcl-2 and Myc: malignancy involves abrogation of apoptosis as well as sustained proliferation. Cancer Res. 1999;59(7 Suppl):1685s–1692s. [PubMed] [Google Scholar]
- 24.Brown JM, Attardi LD. The role of apoptosis in cancer development and treatment response. Nat Rev Cancer. 2005;5(3):231–7. doi: 10.1038/nrc1560. [DOI] [PubMed] [Google Scholar]
- 25.Brown JM, Wilson G. Apoptosis genes and resistance to cancer therapy: what does the experimental and clinical data tell us? Cancer Biol Ther. 2003;2(5):477–90. doi: 10.4161/cbt.2.5.450. [DOI] [PubMed] [Google Scholar]
- 26.Liu XS, Song B, Liu X. The substrates of Plk1, beyond the functions in mitosis. Protein & cell. 2010;1(11):999–1010. doi: 10.1007/s13238-010-0131-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Spankuch B, et al. Rational combinations of siRNAs targeting Plk1 with breast cancer drugs. Oncogene. 2007;26(39):5793–807. doi: 10.1038/sj.onc.1210355. [DOI] [PubMed] [Google Scholar]
- 28.Spankuch B, et al. Down-regulation of Polo-like kinase 1 elevates drug sensitivity of breast cancer cells in vitro and in vivo. Cancer research. 2006;66(11):5836–46. doi: 10.1158/0008-5472.CAN-06-0343. [DOI] [PubMed] [Google Scholar]