

Abstract
In the heart, intracellular Na+ concentration ([Na+]i) is a key modulator of Ca2+ cycling, contractility and cardiac myocyte metabolism. Several Na+ transporters are electrogenic, thus they both contribute to shaping the cardiac action potential and at the same time are affected by it. [Na+]i is controlled by the balance between Na+ influx through various pathways, including the Na+/Ca2+ exchanger and Na+ channels, and Na+ extrusion via the Na+/K+-ATPase. [Na+]i is elevated in HF due to a combination of increased entry through Na+ channels and/or Na+/H+ exchanger and reduced activity of the Na+/K+-ATPase. Here we review the major Na+ transport pathways in cardiac myocytes and how they participate in regulating [Na+]i in normal and failing hearts.
Keywords: intracellular Na+ concentration, myocyte, Na+/K+-ATPase, Na+/Ca2+ exchanger, Na+/H+ exchanger, heart failure
1. Na+ transport and heart function
1.1. [Na+]i and cardiac contractility
Intracellular Na+ concentration ([Na+]i) is an important modulator of cardiac myocyte Ca2+ cycling and contractility [1]. This regulation is mediated mainly by the Na+/Ca2+ exchanger (NCX), which transports three Na+ ions in exchange for one Ca2+ ion, and is the main route for Ca2+ extrusion from cardiac myocytes [2]. During the cardiac cycle, NCX needs to extrude exactly the same amount of Ca2+ that enters through L-type Ca2+ channels (about 8 μmol/L cytosol, [2]) for the myocyte to be in steady-state. Indeed, Bridge et al. [3] have shown that the total charge carried by the nifedipine-sensitive Ca2+ current is twice the total charge carried by the inward NCX current, which means (assuming a 3Na+:1Ca2+ stoichiometry for NCX, [4]) that all Ca2+ entering through Ca2+ channels is extruded by NCX. Thus, small changes in NCX activity may significantly alter Ca2+ homeostasis. NCX is unique among Na+ transporters in that it can work in both Ca2+ extrusion (or forward) and Ca2+ influx (or reverse) mode under physiological conditions, depending on the membrane potential and intracellular and extracellular concentrations of Ca2+ and Na+. High intracellular Ca2+ favors Ca2+ extrusion whereas high [Na+]i and membrane depolarization favor Ca2+ influx. NCX activity is critically regulated by [Na+]i. Even a modest (a few mM) increase in [Na+]i causes the exchanger to extrude less Ca2+, which raises the cellular and sarcoplasmic reticulum (SR) Ca2+ content (Fig. 1). This leads to larger Ca2+ transients and thus larger contractions. At the same time, higher SR Ca2+ load may also lead to spontaneous Ca2+ waves. Part of the Ca2+ released during a wave is extruded through NCX and the depolarizing current that is produced can generate delayed afterdepolarizations (DADs) and triggered arrhythmias. This is the basic mechanism responsible for both the inotropy and arrhythmogenesis produced by cardiac glycosides, specific inhibitors of the Na+/K+-ATPase (NKA) that have been used in the treatment of congestive heart failure for more than two centuries.
Figure 1. [Na+]i regulation and Na+ transport in cardiac myocytes.
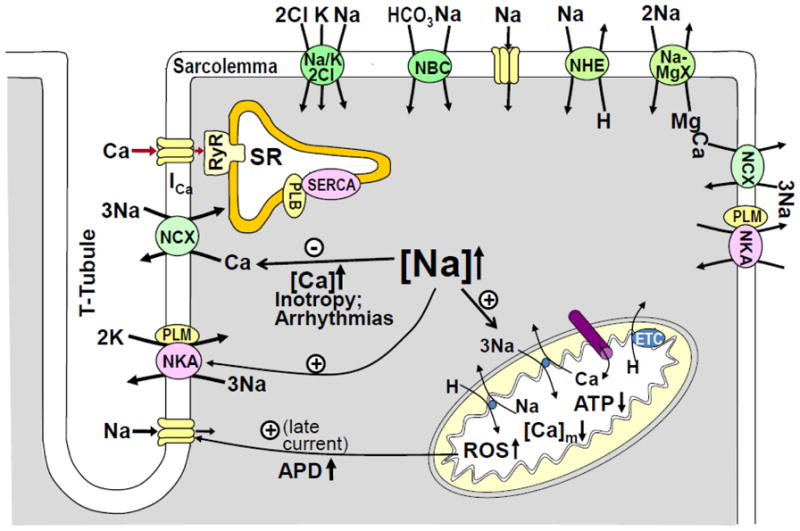
Cartoon showing the main Na+ transporters in cardiac myocytes and the mechanisms by which an increase in [Na+]i affects [Ca2+]i and contractility, AP duration and cardiac metabolism.
DADs occurring in a single myocyte (the source of depolarization) cannot trigger a premature beat in the intact heart because the strong electrotonic coupling between the myocytes acts as a sink for local depolarizing current. Mathematical models have shown that to overcome the source - sink mismatch, DADs have to occur simultaneously in a large number of cells (several hundred thousands) [5]. The requirement for the occurrence of spontaneous Ca release over a broad aggregate of myocardial cells before initiation of an extopic beat was confirmed by optical mapping in ventricular wedge preparations [6]. Such temporal synchronization of spontaneous Ca waves could be attained by localized β-adrenergic stimulation [7]. Reduced gap junction conductance, enhanced NCX, reduced inward rectifier K+ current, fibrosis, and reduced repolarization reserve, factors that are associated with electrical remodeling in heart failure, favor the occurrence of DADs-induced ectopic beats [5,8]. Nevertheless, details about DADs triggering of premature ventricular complexes are incompletely understood.
In ischemia/reperfusion injury, NCX reversal connects an increase in [Na+]i, brought about by activation of the Na+/H+ exchanger (NHE) and Na+/HCO3− cotransporter (NBC), an increase in slowly-inactivating Na+ channels, Na+ entry through connexin hemmichannels and NKA inhibition (reviewed in [9]), to Ca2+ overload. Not surprisingly, cardiac-specific ablation of NCX was found to confer protection against ischemia/reperfusion injury [10].
1.2. [Na+]i and cardiac metabolism
[Na+]i is an important regulator of cardiac metabolism. This is because [Na+]i controls mitochondrial Ca2+ concentration ([Ca2+]m) through the mitochondrial Na+/Ca2+ exchanger (Fig. 1). At high workload, for example with increasing heart rate during adrenergic stimulation, Ca2+ uptake into mitochondria via Ca2+ uniporter is elevated. High [Ca2+]m stimulates several dehydrogenases involved in the tricarboxylic acid cycle [11], which leads to a faster regeneration of NADH and NADPH from NAD+ and NADP+, respectively. This limits and can even prevent decreased [NADH] during increased work driven by larger Ca2+ transients [12]. Faster restoration of the NADH pool allows an increased rate of electron transport and thus augments ATP production. At the same time, reduced NADPH is needed to eliminate the H2O2 produced by dismutation of the superoxide generated at complexes I and III of the electron transport chain [13]. In agreement with this, inhibition of mitochondrial Ca2+ uptake with Ru360 was found to increase the mitochondrial H2O2 production [14]. Thus, elevated [Ca2+]m is also important in controlling the oxidative stress.
In the heart, Ca2+ extrusion from mitochondria occurs mostly through a Na+/Ca2+ exchanger [15]. Although its existence has been accepted for a long time, the molecular identity of the mitochondrial Na+/Ca2+ exchanger has remained elusive until recently, when it was shown that NCLX, the lone member of the Ca2+/cation exchanger (CCX) family found in mammals [16], is in fact the long-sought mitochondrial Na+/Ca2+ exchanger [17,18]. Different from the sarcolemmal NCX, Li+ can substitute for Na+ in NCLX and its activity is inhibited by CGP-37157 [17]. Both features are hallmarks of mitochondrial Na+-dependent Ca2+ efflux. While initially thought to be electroneutral (2Na+:1Ca2+), it seems accepted now that mitochondrial Na+/Ca2+ antiporter exchanges three Na+ ions for one Ca2+ ion [19–21], similar to sarcolemmal NCX. Mitochondrial Na+/Ca2+ exchanger has a half-maximal activity at [Na+]i=5–10 mM, very close to physiological [Na+]i. This makes the exchanger very sensitive to changes in [Na+]i. Elevated [Na+]i accelerates mitochondrial Ca2+ efflux and reduces [Ca2+]m [22–23]. Notably, an electrogenic mitochondrial Na+/Ca2+ exchanger will lower [Ca2+]m following an increase in [Na+]i even in the absence of a Na+ gradient across the inner mitochondrial membrane due to the large, negative mitochondrial membrane potential.
[Na+]i is relatively constant in the normal heart. In isolated cardiac myocytes, [Na+]i increases significantly with increasing the pacing frequency. However, an accelerated heart rate in vivo is generally the result of adrenergic stimulation, which also increases Na+ extrusion through NKA (by relieving its inhibition by the small sarcolemmal protein PLM, [24,25]). The net effect is relatively little change in [Na+]i [26–28]. Combined with larger Ca2+ transients, low [Na+]i facilitates elevated [Ca2+]m and contributes to both increased ATP production to match the higher energy demand and the maintenance of redox homeostasis. [Na+]i is however increased in pathophysiological situations such as ischemia/reperfusion and heart failure (see below). Elevated [Na+]i led to oxidation of NAD(P)H and increased H2O2 formation in myocytes from failing hearts, which could be prevented by pharmacological inhibition of the mitochondrial Na+/Ca2+ exchanger [29]. Higher oxidative stress may further exacerbate the increase in [Na+]i by activating, directly or through Ca2+/calmodulin-dependent kinase II (CaMKII), the slowly inactivating mode of Na+ channels (Fig. 1) [30,31]. Thus, by controlling [Ca2+]m, [Na+]i is an important parameter in matching energy supply and demand and critically regulates the production of mitochondrial reactive oxygen species in cardiac myocytes.
1.3. Na+ transport and action potential waveform
Many Na+ transporters are electrogenic, which means that they both contribute to shaping the action potential (AP) waveform and at the same time are affected by it. Voltage-gated Na+ channels are responsible for the fast upstroke of the AP, however, because they inactivate within a few milliseconds, they have little effect on the AP duration. A few Na+ channels (about 0.5%) either do not inactivate, or close and then reopen, generating a slowly inactivating, or late, Na+ current [32,33]. Some genetic mutations in the SCN5A gene encoding cardiac Na+ channels lead to elevated late Na+ current [34–36], resulting in AP prolongation and long-QT syndrome type 3 (LQT3). Late Na+ current is also increased in pathophysiological conditions such as ischemia/reperfusion [37] and heart failure [38–40], which leads to acquired LQT syndrome. AP prolongation is arrhythmogenic by facilitating the occurrence of early afterdepolarizations, particularly at slow heart rates. At the same time, elevated slowly-inactivating Na+ current may increase [Na+]i and cause DADs and triggered arrhythmias through the mechanism described in section 1.1 [41,42].
By extruding one net positive charge, NKA participates in membrane repolarization and thus may affect both the plateau phase of the AP and the resting membrane potential. At normal heart rates this effect is however rather small. Indeed, acute NKA inhibition depolarizes the myocytes by only 5–6 mV and slightly prolongs the AP (for a review, see [43]). Obviously, long-term NKA blockade will greatly alter Na+ and K+ homeostasis and depolarize the membrane. Surprisingly, a computational model of AP in human ventricular myocytes predicted that an increase of NKA, due to NKA activation by the increasingly higher [Na+]i, current is the predominant factor in AP shortening that occurs with increasing pacing rates [44]. As discussed above, [Na+]i accumulation produced by a physiological increase in the heart rate in vivo is much smaller. However, the adrenergic stimulation that accelerates the heart rate also increases the NKA current by phosphorylating PLM and thus enhancing the NKA affinity for internal [Na+]i [24,25]. Thus, NKA may be a major contributor to AP shortening at high heart rates, particularly in humans and other species (dog, rabbit) with modest delayed rectifier K+ current (IKs).
NCX is also electrogenic, however predicting its effect on the AP is more difficult because the NCX driving force is changing rapidly during an AP. Under physiological conditions, NCX works almost exclusively in the Ca2+ extrusion mode, driven by the negative membrane potential and low [Na+]i at rest (diastole) and the high subsarcolemmal Ca2+ during Ca2+ release [2]. Thus, NCX works to depolarize the sarcolemma and increased NCX activity will prolong the AP. Schouten and Ter Keurs [45] found that reducing the forward mode of NCX by either an increase in external Ca2+ or a decrease in external Na+ shortened the slow phase of repolarization in trabeculae from the rat right ventricle. When [Na+]i is elevated, the balance of ion fluxes through NCX is shifted to reduce Ca2+ extrusion, and thus reduce the depolarizing inward current and shorten the AP [46]. Therefore, through changes in NKA and NCX currents, higher [Na+]i may alleviate the AP prolongation seen in pathophysiological conditions such as heart failure [46].
2. [Na+]i transport in and out of cardiac myocytes
Several transporters facilitate Na+ entry into cardiac myocytes (Fig. 1), with Na+ channels and NCX being the most important quantitatively in the normal heart. Other transporters however may become prominent in producing Na+ overload under pathophysiological conditions. For example, the activity of NHE and NBC is increased greatly during ischemia/reperfusion due to stimulation of proton extrusion by intracellular acidosis. Under mild acidosis (pHi=6.9), NHE and NBC may account for as much as 75% of resting Na+ influx in cardiac myocytes [1]. Other pathways for Na+ entry are the Na+/Mg2+ exchanger, which is involved in Mg2+ extrusion, and Na+/K+/2Cl− cotransporter, which may play a role in volume regulation. [Na+]i is maintained low by active pumping out of the cells by NKA. Here we will briefly review the structure, function and regulation of NCX, Na+ channels and NKA. NHE and NBC are discussed in other articles in this Special Issue.
2.1. Na+/Ca2+ exchanger
NCX1 (970 amino acids, ~110 kDa) is the only NCX isoform present in the heart. NCX1 was initially thought to have 11 transmembrane domains (TM) [47]. More recent topological models suggested nine TMs [48,49]. The newly-uncovered high resolution crystal structure of an NCX from Methanococcus jannaschii (NCX_Mj) identified 10 TMs [50]. Due to the similarity in sequence and hydrophobicity pattern with NCX_Mj, it is likely that NCX1 assumes a similar topology [50]. This NCX1 topology was recently confirmed by a study that re-assessed the topology of the C-terminal TMs of NCX1 [51]. Two highly conserved regions of internal repeats (α1 and α2; ~40 amino acids) are present on the N- and C-termini halves of the molecules. These α repeats are near one another in the folded protein and they contribute to the ion conduction pathways [50,52,53]. The NCX_Mj crystal structure reveals four potential cation binding sites formed exclusively by residues from the α repeats and arranged in a diamond shape [50].
The 10 TMs represent about half of the NCX1 protein. Most of the remaining half (~550 amino acids) is a large, unstructured cytoplasmic loop between TMs 5 and 6. This loop contains two separate Ca2+ binding domains, or β-repeats, CBD1 and CBD2. Elevated [Ca2+]i allows Ca2+ to bind to the CBD domains, which increases the activity of the exchanger (allosteric [Ca2+]i-dependent activation) [54–56]. Allosteric Ca2+ activation occurs at physiological [Ca2+]i (K0.5=22–600 nM) and develops rapidly (within ~100 ms). However, NCX remains active for tens of seconds after [Ca2+]i returns to low levels [57–59]. Thus, NCX is substantially activated in a beating heart, with little beat-to-beat variation. While high [Ca2+]i activates NCX, high [Na+]i inhibits it (Na+-dependent inactivation) in a time and [Na+]i-dependent manner. Na+-dependent inactivation is probably not very important in cardiac myocytes because it occurs at [Na+]i (>30 mM) substantially above the physiological level. [Na+]i-dependent inactivation is eliminated by an increase in membrane PIP2. Moreover, when PIP2 levels are high, elevated [Na+]i induces a mode of exchange activity that does not require allosteric Ca2+ activation [60]. PIP2 may also modulate NCX by affecting its membrane trafficking [61].
In cardiac myocytes, NCX is located mostly at the T-tubules [62–64]. It is however unclear whether/to what extent NCX is present at the junctions between sarcolemma and SR membrane where most of Ca2+-induced Ca2+ release occurs. Some immunofluorescence studies indicate that NCX is not present at the junctions [65,66]. However, a more recent study using high-resolution imaging of transverse myocyte sections found that NCX partially (~27%) co-localizes with ryanodine receptors (RyRs) [67]. This study suggests that NCX is neither excluded from nor concentrated at the junctions. The mean distance between NCX and the nearest RyRs cluster was slightly smaller than would be expected from a uniform NCX distribution. NCX senses an early rise in local [Ca2+]i (vs. bulk [Ca2+]i) during SR Ca2+ release [68,69], suggesting that NCX has some access to Ca2+ in the junctional cleft, in agreement with the results of Jayasinghe et al. [67]. Indeed, Ca2+ entry via reverse-mode NCX can trigger Ca2+ release from the SR at least under certain conditions [70–73], although with low efficiency. Moreover, data from NCX-KO mice [74] and from experiments where Na+ current is manipulated [75] indicate that by priming the junctional cleft with Ca2+, NCX contributes to the normal triggering of Ca2+ release. Of course, this effect is only likely to be relevant during the latency period before an L-type Ca2+ channel opens at a given junction because the local flux is ~1000 times higher for Ca2+ current vs. NCX. Immediately following SR Ca2+ release, Ca2+ extrusion through the fraction of junctionally-located NCX may limit the global cytosolic Ca2+ transient.
2.2. Voltage-gated Na+ channels
Voltage-gated Na+ channels, composed of one pore-forming α subunit and one or two auxiliary β subunits [76], mediate the rapid upstroke of the AP in cardiac myocytes. The α subunit (~240 kDa) consists of four repeat domains (I–IV), each containing six transmembrane segments (S1–S6) and one membrane reentrant domain, connected by internal and external polypeptides loops [77]. Positively charged residues in the S4 segments serve as voltage sensors. Upon depolarization, these charges move outward, triggering activation of the channel [78]. Most Na+ channels switch off Na+ conductance within a few milliseconds upon depolarization (channel inactivation), a process mediated by a hydrophobic isoleucine-phenylalanine-methionine motif located on the short intracellular loop connecting domains III and IV [79]. The tetrodotoxin resistant isoform Nav1.5, encoded by the SCN5A gene, is the predominant α subunit in the heart. Some types of neuronal and skeletal muscle Na+ channels have also been found in the heart (for a review, see [80]). The contribution of such channels to the total myocyte Na+ current is small (<15%) and their physiological role is not fully elucidated. Increasing evidence suggests that these TTX-sensitive Na+ channel isoforms may represent an intrinsic depolarization reserve and play a role in coupling electrical excitation to contraction [75, 81, 82]. However, this is still controversial as, for example, another study found no role for the neuronal Na+ channels in SR Ca2+ release [83]. The auxiliary β subunits (~30–35 kDa, β1–β4 subunits) modulate channel gating, interact with extracellular matrix and play a role as cell adhesion molecules [84].
Nav1.5 Na+ channels are located both in the T-tubules and external sarcolemma [65,85], with a roughly equal distribution [86]. However, some immunofluorescence studies show Nav1.5 mostly at the intercalated disks [85]. Nav1.5 channels interact in complexes with a large number of regulatory proteins (for a review, see [87]). Interaction with ankyrin-G, an anchoring protein that links membrane proteins to the cytoskeleton, is essential for the proper membrane targeting of Na+ channels. Reduced ankyrin-G expression decreases total cellular Nav1.5 expression and its efficient membrane localization, as well as Na+ current [88]. Moreover, a mutation in the ankyrin-binding motif of Nav1.5 that abolishes the binding of ankyrin-G has been shown to induce Brugada syndrome, a condition associated with loss of function of Na+ channels [89]. A recent study suggested that ankyrin-G is an essential functional component of the intercalated discs that connects the voltage-gated Na+ channels, connexin 43, and the cardiac desmosome [90].
While most of cardiac myocyte Na+ channels inactivate quickly, some channels (~0.5%) either do not close, or close and then reopen, generating a slowly-inactivating, late Na+ current [32,33]. Recent evidence suggests that neuronal Na+ channels may also be implicated in the late Na+ current in cardiac myocytes [91]. In normal conditions, this slowly-inactivating Na+ current is too small to significantly affect AP duration and total Na+ influx. However, genetic mutations in the SCN5A gene or pathophysiological conditions such as ischemia [92] and heart failure [33,39] result in elevated late Na+ current, which leads to inherited LQT3 or, respectively, acquired LQT syndrome. In heart failure, late Na+ current may be enhanced by CaMKII-dependent phosphorylation of Nav1.5 channels since CaMKII expression and activity is augmented [93,94] and overexpression of CaMKII increases late Na+ current [95].
2.3. Na+/K+-ATPase
The NKA extrudes three Na+ ions in exchange for two K+ ions using the energy derived from the hydrolysis of one ATP molecule, and thus moves out one net charge per cycle. Internal Na+ and external K+ activate NKA with a half-maximal activation in the range of 10–22 mM and 1–2 mM, respectively (for a review, see [43]). Thus, NKA is ~70% saturated with respect to external K+ at a normal concentration of 4 mM. The ATP concentration for half-maximal NKA activation is 80–150 μM [96,97], therefore under control conditions ATP is not rate limiting for the pump (normal ATP level in myocytes is 5–10 mM).
NKA is composed of three subunits: the catalytic α subunit, the auxiliary β subunit and the tissue-specific regulatory protein FXYD. The NKA-α subunit (~110 kDa) contains the binding sites for Na+, K+, ATP and cardiac glycosides, specific inhibitors of the enzyme. The main function of the smaller β subunit (~50 kDa) is that of a molecular chaperone. Association with β-subunit facilitates the routing to and insertion into the membrane of the α-subunit [98]. The β-subunit also modulates the ATPase activity of NKA. FXYDs are a family of proteins that associate with and modulate NKA in various tissues [99]. The small (72 amino acids) sarcolemmal protein phospholemman (PLM) is the only FXYD protein that is highly expressed in the heart and is an important NKA regulator. Unphosphorylated PLM inhibits NKA, mostly by reducing its affinity for internal Na+ [24,25,100,101] and PLM phosphorylation by PKA or PKC relieves this inhibition [24,101]. This is similar to the way phospholamban modulates SERCA, a P-type ATPase closely related to NKA. PLM phosphorylation and the ensuing NKA disinhibition are an integral part of the sympathetic fight-or-flight response, by enhancing Na+ extrusion to better keep up with the increased Na+ influx at higher heart rates and with larger Ca2+ transients (which drive greater inward NCX current) [26]. This limits the rise in [Na+]i during β-adrenergic stimulation. Thus, NKA regulation by PLM may prevent Ca2+ overload and triggered arrhythmias during sympathetic stimulation of the heart.
The crystal structures of NKA from pig kidney [102] and shark rectal gland [103] have been recently uncovered. The α-subunit has 10 TMs and, like SERCA, three characteristic cytoplasmic domains: the actuator, nucleotide-binding and phosphorylation domains. Two binding sites, common for K+ and Na+ binding, are located ~4Å apart between TMs 4, 5 and 6 [102]. A third Na+ binding site is created by residues from TMs 5, 6 and 9. The TM helix of β-subunit runs nearly parallel to the M7 helix of the α-subunit and comes within interaction distance of NKA-α TM7 and TM10 in multiple places [103]. On the extracellular side, β-subunit makes complex interactions with the loop between TM7 and TM8 of the α-subunit. FXYD proteins (γ-subunit for the pig NKA and FXYD10 for the shark rectal gland NKA) consist of a single TM domain that is in close proximity of NKA-α TM9, on the outside of a groove formed by TM2, TM6, and TM9 [102,103]. The crystal structures identified a number of possible interaction sites between FXYD and NKA-α TM9. In a recent study we demonstrated that interaction between NKA-α-E960 and PLM-F28 sites is critical for the functional effects of PLM on NKA [104]. The conserved FXYD motif is located in the extracellular domain, just prior to the TM domain and seems to interact with both α- and β-subunits. This suggests that the FXYD region may be important for stabilizing α-β-FXYD interaction.
Three α (α1–α3) and three β (β1–β3) NKA subunit isoforms are present in the heart. NKA-α1 is the dominant, ubiquitous isoform, with NKA-α2 and NKA-α3 present in smaller amounts and only in select species. All three isoforms are present in human hearts [105], with NKA-α1 mRNA level slightly higher than that for NKA-α2 and NKA-α3 [106,107]. Fetal and neonatal rodent hearts express NKA-α1 and NKA-α3, and NKA-α3 is replaced by NKA-α2 early in development [108]. Interestingly, the reverse switch occurs in hyperthrophied or failing hearts [109,110]. The enzymatic activity and Na+, K+ and ATP affinities of various NKA-α isoforms are comparable (for a detailed discussion, see [111]). However, different NKA isoforms may function differently, depending on specific membrane localization. It was proposed that NKA-α2 and NKA-α3 are located mainly in the T-tubules, at the junctions with the SR, where they could regulate local NCX and [Ca2+]i, whereas NKA-α1 is central to determining global [Na+]i [112]. This is still incompletely proven in cardiac myocytes as the exact localization of NKA isoforms with respect to the junctions is difficult to assess with the current techniques, and the functional evidence is mostly indirect [113–115]. The functional density of NKA-α2 isoform is ~4-times higher in the T-tubules (vs. external sarcolemma) in myocytes from both rats [113,116] and mice [117], whereas NKA-α1 is more uniformly distributed. Using mice in which NKA-α1 is ouabain-sensitive and NKA-α2 is ouabain-resistant we have recently showed that NKA-α2 inhibition produced a bigger increase in Ca2+ transient amplitude and fractional SR Ca2+ release than blockade of a similar number of NKA-α1 pumps [118]. Thus, NKA-α2 may have a more prominent role (vs. NKA-α1) in modulating Ca2+ release in cardiac myocyte.
The membrane targeting of both NKA and NCX require direct interactions with ankyrin-B, a multivalent “adaptor” protein that targets and tethers select membrane proteins to the cytoskeleton [119]. Loss-of-function mutations in the gene encoding ankyrin-B lead to complex cardiac phenotypes in humans. These phenotypes may include bradycardia, atrial fibrillation, conduction defects, stress- or exercise-induced ventricular arrhythmias and sudden cardiac death (for a review, see [120]). Decreased ankyrin-B function resulted in reduced NKA and NCX targeting to the T-Tubules as well as reduced overall NKA and NCX protein level in cardiac myocytes [121]. Notably, the clinical severity of human ankyrin-B loss-of-function variants has been directly linked with the inability to target NCX and NKA to the cardiac myocyte sarcolemma [122]. Cardiac myocytes from mice heterozygous for a null mutation in ankyrin-B show larger Ca2+ transients and SR Ca2+ content [121]. We have recently found larger Ca2+ spark frequency but unchanged total RyR-mediated SR Ca2+ leak in ventricular myocytes from ankyrin-B heterozygous vs. wild-type mice [123]. Such a bias toward more coordinated RyRs gating is likely due to different RyR regulation at the cytoplasmic side and results in an increased propensity for Ca2+ waves, which may contribute to the increased arrhythmogenicity caused by ankyrin-B loss-of-function.
3. [Na+]i balance in the normal heart
Resting [Na+]i is ~4–8 mM in ventricular myocytes from rabbit, guinea-pig and dog [124–130] and somewhat higher (9–14 mM) for rat and mouse [24,127–129,131,132]. While accurate measurements are lacking, resting [Na+]i in human myocytes is believed to be in the 4–10 mM range. There might be sub-regional differences in [Na+]i in the ventricle [130,133]. Higher [Na+]i was found in epicardial vs. endocardial myocytes in rabbit left ventricle [133], while the opposite was observed in canine ventricle [130]. In rabbit, the NKA expression and current density were similar in the two regions [134], whereas NKA current was significantly larger in epicardial vs. endocardial canine myocytes [130]. Clearly, more data are needed regarding a possible transmural gradient in [Na+]i (and NKA current) and its functional consequences, as well as on [Na+]i regulation in the atria.
The total Na+ influx in resting myocytes is ~1 mM/min [1]. For myocytes to be at steady-state, this Na+ influx has to be matched by Na+ efflux through NKA. Fig. 2A shows the relative contribution of various Na+ entry pathways to this resting Na+ influx, showing rather little variation between different routes. The estimates are based in part on our measurements in rabbit ventricular myocytes [127] and some extrapolations from guinea-pig data [135,136].
Figure 2. The relative contribution of different Na+ entry pathways to Na+ influx.
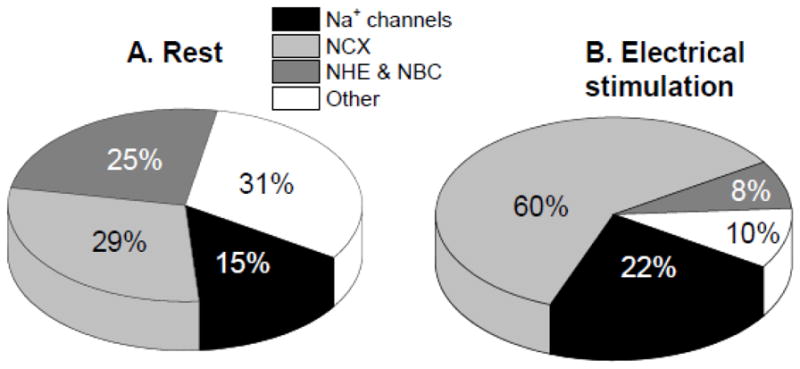
in (A) resting myocytes and (B) myocytes contracting at 1 Hz. The estimates are based in part on our measurements in rabbit ventricular myocytes [124] and some extrapolations from guinea-pig data [132,133] and correspond to mammalian species with lower resting [Na+]i (i.e. other than rat and mouse).
In isolated myocytes, [Na+]i increases during stimulation in a frequency dependent manner. This is mainly because Na+ influx per unit time is higher due to more frequent activation of NCX and Na+ channels. During electrical stimulation, NCX is likely the dominant route for Na+ entry, bringing in almost three times more Na+ than Na+ channels or other pathways combined (Fig. 2B). While at rest ion fluxes through NCX are relatively small (mainly due to low diastolic [Ca2+]i), during electrical stimulation NCX has to remove ~8 μM Ca2+ that enters via Ca2+ current during each AP. For a myocyte contracting at 1 Hz, this means that NCX brings in ~1.5 mM/min Na+, about 5–6 fold more than in a resting cell. Na+ channels also bring in considerably more Na+ during electrical pacing than at rest. The activation and availability curves of Na+ channels overlap such that there is almost no steady-state availability at potentials where activation is appreciable. We have measured a tetrodotoxin-sensitive resting Na+ influx of 0.18 mM/min in rabbit ventricular myocytes [137], which would require a window Na+ current ~0.01% of the peak current [1]. During the AP upstroke, Na+ current is very large, but very brief, resulting in an additional Na+ influx of ~8 μM associated with each AP [1]. For a cell contracting at 1 Hz, this means that Na+ channel activation contributes about 0.5 mM/min to the rate of Na+ influx. Thus, Na+ influx in contracting myocytes is about 3 mM/min, surprisingly only three times larger than at rest. A new steady-state [Na+]i is reached when this higher Na+ influx is matched by an elevated NKA-mediated Na+ extrusion. During pacing, NKA is activated mainly by the increased [Na+]i but could also be stimulated via a nitric oxide and phospholemman-dependent mechanism [138].
4. [Na+]i balance in the failing heart
[Na+]i is elevated in heart failure (HF), both in humans and in animal models [137,139–142]. By favoring more Ca2+ influx via NCX, elevated [Na+]i may limit the contractile dysfunction in HF. Longer AP and smaller Ca2+ transients also favor Ca2+ influx via NCX in HF, but the effect seems to be less important quantitatively than that produced by higher [Na+]i [137]. However, high [Na+]i may also negatively affect the cardiac metabolism and oxidative state.
Both larger Na+ influx and reduced Na+ extrusion may contribute to elevate [Na+]i in HF. Protein expression of α1-, α3- and β1-subunits of NKA are reduced in failing human myocardium [143], with no changes in the mRNA levels [144]. Protein and mRNA levels of the dominant NKA-α1 isoform are unchanged in most rat HF models, whereas NKA-α2 is generally reduced and NKA-α3 is increased [145]. All three NKA α-subunit isoforms showed lower protein expression in myocytes from rabbits with pressure and volume overload-induced HF [146]. It is difficult however to directly correlate such biochemical findings to NKA function in myocytes, as protein expression measurements cannot differentiate between internalized vs. sarcolemmal NKA nor between functional and inactive pumps. Moreover, NKA may be regulated differently in HF. Indeed, we found reduced NKA expression (by ~36%, [146]) but unchanged function (both maximal Na+ transport rate and apparent affinity for internal Na+) [137] in myocytes from failing rabbit hearts. This apparent paradox is likely caused by reduced PLM protein expression and increased phosphorylation of PLM, which disinhibits NKA [146]. In this context, it is surprising that very few studies address NKA function in HF. Myocytes from rats with HF following myocardial infarction showed a reduction in the maximal Na+ extrusion rate (mainly through NKA-α2 isoform) but unchanged [Na+]i-affinity [110,147]. In contrast, myocytes from dogs with chronic atrioventricular block and hypertrophy have unaltered maximal NKA pump current and reduced [Na+]i-affinity [148]. Smaller maximal NKA current, caused mainly by a reduction in the current carried by the α2 isoform seems to underlie the [Na+]i increase in mice with end-stage HF following SERCA2 knockout [141].
Larger Na+ influx could also cause elevated [Na+]i in HF. We [137] and others [140] found this to be the case for myocytes from rabbits with pressure and volume-overload induced HF. We found that the initial slope of [Na+]i rise upon NKA inhibition was twice as large as in myocytes from normal hearts [137]. Most of this elevated Na+ entry was TTX-sensitive, suggesting a role for Na+ channels. As discussed in section 2.2, late Na+ current is increased in HF and this may contribute to the increased Na+ influx. Indeed, ranolazine, a late Na+ current inhibitor, was found to reduce [Na+]i and diastolic Ca2+ overload in failing human hearts [40]. NHE upregulation may also participate in elevated Na+ influx in HF [140]. Moreover, chronic treatment with cariporide, an NHE inhibitor, prevented the onset of HF in rabbits with pressure and volume overload [149]. One of the difficulties in assessing the contribution of different pathways to Na influx in HF lies in the uncomplete specificity of the pharmacological inhibitors. For example, cariporide also partially inhibits late Na+ channels [150] at concentrations used to block NHE.
5. Conclusions
In summary, [Na+]i and Na+ transport are key factors for regulation of Ca2+ cycling, contractility, action potential waveform and metabolism in cardiac myocytes. [Na+]i is the result of a delicate balance between Na+ influx and efflux. Perturbation of this balance in HF results in elevated [Na+]i, with important consequences on cardiac myocyte function. While we have learned a great deal about Na+ regulation in the heart, more data are needed regarding Na+ regulation in the human heart, regional differences in [Na+]i and Na+ transport (ventricle vs. atria, endocardial vs. epicardial) and the mechanisms responsible for [Na+]i dysregulation in HF.
Highlights.
We review the major Na transport pathways in cardiac myocytes
[Na]i modulates myocyte Ca cycling, contractility, action potential and metabolism
We discuss the balance of Na fluxes in normal hearts and in heart failure
[Na]i is elevated in heart failure
Acknowledgments
6. Funding Sources
This work was supported in part by NIH (grants HL-109501 to SD; HL-81526 to DMB).
Abbreviations list
- AP
action potential
- [Ca2+]i
concentration of free Ca2+ in the cytosol
- [Ca2+]m
concentration of free Ca2+ in the mitochondrial matrix
- CaMKII
Ca2+/calmodulin dependent kinase II
- DAD
delayed afterdepolarization
- FXYD
family of proteins that are specific Na/K-ATPase regulators
- [Na+]i
intracellular Na+ concentration
- NBC
Na+/HCO3− cotransporter
- NCX
Na+/Ca2+ exchanger
- NHE
Na+/H+ exchanger
- NKA
Na+/K+-ATPase
- PLM
phospholemman
- RyR
ryanodine receptor
- SR
sarcoplasmic reticulum
- TM
transmembrane domain
Footnotes
DISCLOSURES
Sanda Despa, None; Donald M. Bers, None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bers DM, Barry WH, Despa S. Intracellular Na+ regulation in cardiac myocytes. Cardiovasc Res. 2003;57:897–912. doi: 10.1016/s0008-6363(02)00656-9. [DOI] [PubMed] [Google Scholar]
- 2.Bers DM. Excitation-contraction coupling and cardiac contractile force. 2. Kluwer Academic Publishers; 2001. p. 427. [Google Scholar]
- 3.Bridge JH, Smolley JR, Spitzer KW. The relationship between charge movements associated with ICa and INa-Ca in cardiac myocytes. Science. 1990;248:376–378. doi: 10.1126/science.2158147. [DOI] [PubMed] [Google Scholar]
- 4.Reeves JP, Hale CC. The stoichiometry of the cardiac sodium-calcium exchange system. J Biol Chem. 1984;259:7733–7739. [PubMed] [Google Scholar]
- 5.Xie Y, Sato D, Garfinkel A, Qu Z, Weiss JN. So little source, so much sink: requirements for afterdepolarizations to propagate in tissue. Biophys J. 2010;99:1408–1415. doi: 10.1016/j.bpj.2010.06.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hoeker GS, Katra RP, Wilson LD, Plummer BN, Laurita KR. Spontaneous calcium release in tissue from the failing canine heart. Am J Physiol Heart Circ Physiol. 2009;297:H1235–H1242. doi: 10.1152/ajpheart.01320.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Myles RC, Wang L, Kang C, Bers DM, Ripplinger CM. Local β-adrenergic stimulation overcomes source-sink mismatch to generate focal arrhythmia. Circ Res. 2012;110:1454–1464. doi: 10.1161/CIRCRESAHA.111.262345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pogwizd SM, Schlotthauer K, Li L, Yuan W, Bers DM. Arrhythmogenesis and contractile dysfunction in heart failure: Roles of sodium-calcium exchange, inward rectifier potassium current, and residual beta-adrenergic responsiveness. Circ Res. 2001;88:1159–1167. doi: 10.1161/hh1101.091193. [DOI] [PubMed] [Google Scholar]
- 9.Murphy E, Eisner DA. Regulation of intracellular and mitochondrial sodium in health and disease. Circ Res. 2009;104:292–303. doi: 10.1161/CIRCRESAHA.108.189050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Imahashi K, Pott C, Goldhaber JI, Steenbergen C, Philipson KD, Murphy E. Cardiac-specific ablation of the Na+-Ca2+ exchanger confers protection against ischemia/reperfusion injury. Circ Res. 2005;97:916–921. doi: 10.1161/01.RES.0000187456.06162.cb. [DOI] [PubMed] [Google Scholar]
- 11.McCormack JG, Halestrap AP, Denton RM. Role of calcium ions in regulation of mammalian intramitochondrial metabolism. Physiol Rev. 1990;70:391–425. doi: 10.1152/physrev.1990.70.2.391. [DOI] [PubMed] [Google Scholar]
- 12.Brandes R, Bers DM. Intracellular Ca2+ increases the mitochondrial NADH concentration during elevated work in intact cardiac muscle. Circ Res. 1997;80:82–87. doi: 10.1161/01.res.80.1.82. [DOI] [PubMed] [Google Scholar]
- 13.Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell. 2005;120:483–495. doi: 10.1016/j.cell.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 14.Kohlhaas M, Liu T, Knopp A, Zeller T, Ong MF, Böhm M, O’Rourke B, Maack C. Elevated cytosolic Na+ increases mitochondrial formation of reactive oxygen species in failing cardiac myocytes. Circulation. 2010;121:1606–1613. doi: 10.1161/CIRCULATIONAHA.109.914911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Griffiths EJ. Mitochondrial calcium transport in the heart: physiological and pathological roles. J Mol Cell Cardiol. 2009;46:789–803. doi: 10.1016/j.yjmcc.2009.03.001. [DOI] [PubMed] [Google Scholar]
- 16.Lytton J. Na+/Ca2+ exchangers: three mammalian gene families control Ca2+ transport. Biochem J. 2007;406:365–382. doi: 10.1042/BJ20070619. [DOI] [PubMed] [Google Scholar]
- 17.Palty R, Silverman WF, Hershfinkel M, Caporale T, Sensi SL, Parnis J, Nolte C, Fishman D, Shoshan-Barmatz V, Herrmann S, Khananshvili D, Sekler I. NCLX is an essential component of mitochondrial Na+/Ca2+ exchange. Proc Natl Acad Sci USA. 2010;107:436–441. doi: 10.1073/pnas.0908099107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Palty R, Sekler I. The mitochondrial Na+/Ca2+ exchanger. Cell Calcium. 2012;52:9–15. doi: 10.1016/j.ceca.2012.02.010. [DOI] [PubMed] [Google Scholar]
- 19.Jung DW, Baysal K, Brierley GP. The sodium-calcium antiport of heart mitochondria is not electroneutral. J Biol Chem. 1995;270:672–678. doi: 10.1074/jbc.270.2.672. [DOI] [PubMed] [Google Scholar]
- 20.Baysal K, Jung DW, Gunter KK, Gunter TE, Brierley GP. Na+-dependent Ca2+ efflux mechanism of heart mitochondria is not a passive Ca2+/2Na+ exchanger. Am J Physiol. 1994;266:C800–C808. doi: 10.1152/ajpcell.1994.266.3.C800. [DOI] [PubMed] [Google Scholar]
- 21.Kim B, Matsuoka S. Cytoplasmic Na+-dependent modulation of mitochondrial Ca2+ via electrogenic mitochondrial Na+-Ca2+ exchange. J Physiol. 2008;586:1683–1697. doi: 10.1113/jphysiol.2007.148726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cox DA, Matlib MA. A role for the mitochondrial Na+-Ca2+ exchanger in the regulation of oxidative phosphorylation in isolated heart mitochondria. J Biol Chem. 1993;268:938–947. [PubMed] [Google Scholar]
- 23.Maack C, Cortassa S, Aon MA, Ganesan AN, Liu T, O’Rourke B. Elevated cytosolic Na+ decreases mitochondrial Ca2+ uptake during excitation-contraction coupling and impairs energetic adaptation in cardiac myocytes. Circ Res. 2006;99:172–182. doi: 10.1161/01.RES.0000232546.92777.05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Despa S, Bossuyt J, Han F, Ginsburg KS, Jia LG, Kutchai H, Tucker AL, Bers DM. Phospholemman-phosphorylation mediates the β-adrenergic effects on Na/K pump function in cardiac myocytes. Circ Res. 2005;97:252–259. doi: 10.1161/01.RES.0000176532.97731.e5. [DOI] [PubMed] [Google Scholar]
- 25.Silverman BZ, Fuller W, Eaton P, Deng J, Moorman JR, Cheung JY, James AF, Shattock MJ. Serine 68 phosphorylation of phospholemman: acute isoform-specific activation of cardiac Na/K ATPase. Cardiovasc Res. 2005;65:93–103. doi: 10.1016/j.cardiores.2004.09.005. [DOI] [PubMed] [Google Scholar]
- 26.Despa S, Tucker AL, Bers DM. Phospholemman-mediated activation of Na/K-ATPase limits [Na]i and inotropic state during β-adrenergic stimulation in mouse ventricular myocytes. Circulation. 2008;117:1849–1855. doi: 10.1161/CIRCULATIONAHA.107.754051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bers DM, Despa S. Na/K-ATPase – an integral player in the adrenergic fight-or-flight response. Trends Cardiovasc Med. 2009;19:111–118. doi: 10.1016/j.tcm.2009.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang J, Gao E, Song J, Zhang XQ, Li J, Koch WJ, Tucker AL, Philipson KD, Chan TO, Feldman AM, Cheung JY. Phospholemman and β-adrenergic stimulation in the heart. Am J Physiol Heart Circ Physiol. 2010;298:H807–H815. doi: 10.1152/ajpheart.00877.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu T, O’Rourke B. Enhancing mitochondrial Ca2+ uptake in myocytes from failing hearts restores energy supply and demand matching. Circ Res. 2008;103:279–288. doi: 10.1161/CIRCRESAHA.108.175919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wagner S, Ruff HM, Weber SL, Bellmann S, Sowa T, Schulte T, Anderson ME, Grandi E, Bers DM, Backs J, Belardinelli L, Maier LS. Reactive oxygen species-activated Ca/calmodulin kinase IIδ is required for late INa augmentation leading to cellular Na and Ca overload. Circ Res. 2011;108:555–565. doi: 10.1161/CIRCRESAHA.110.221911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang XQ, Yamada S, Barry WH. Ranolazine inhibits an oxidative stress-induced increase in myocyte sodium and calcium loading during simulated-demand ischemia. J Cardiovasc Pharmacol. 2008;51:443–449. doi: 10.1097/FJC.0b013e318168e711. [DOI] [PubMed] [Google Scholar]
- 32.Saint DA, Ju YK, Gage PW. A persistent sodium current in rat ventricular myocytes. J Physiol. 1992;453:219–231. doi: 10.1113/jphysiol.1992.sp019225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Maltsev VA, Sabbah HN, Higgins RS, Silverman N, Lesch M, Undrovinas AI. Novel, ultraslow inactivating sodium current in human ventricular cardiomyocytes. Circulation. 1998;98:2545–2552. doi: 10.1161/01.cir.98.23.2545. [DOI] [PubMed] [Google Scholar]
- 34.Huang H, Priori SG, Napolitano C, O’Leary ME, Chahine M. Y1767C, a novel SCN5A mutation, induces a persistent Na+ current and potentiates ranolazine inhibition of Nav1.5 channels. Am J Physiol Heart Circ Physiol. 2011;300:H288–H299. doi: 10.1152/ajpheart.00539.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ruan Y, Liu N, Bloise R, Napolitano C, Priori SG. Gating properties of SCN5A mutations and the response to mexiletine in long-QT syndrome type 3 patients. Circulation. 2007;116:1137–1144. doi: 10.1161/CIRCULATIONAHA.107.707877. [DOI] [PubMed] [Google Scholar]
- 36.Nuyens D, Stengl M, Dugarmaa S, Rossenbacker T, Compernolle V, Rudy Y, Smits JF, Flameng W, Clancy CE, Moons L, Vos MA, Dewerchin M, Benndorf K, Collen D, Carmeliet E, Carmeliet P. Abrupt rate accelerations or premature beats cause life-threatening arrhythmias in mice with long-QT3 syndrome. Nat Med. 2001;7:1021–1027. doi: 10.1038/nm0901-1021. [DOI] [PubMed] [Google Scholar]
- 37.Saint DA. The role of the persistent Na+ current during cardiac ischemia and hypoxia. J Cardiovasc Electrophysiol. 2006;17 (Suppl 1):S96–S103. doi: 10.1111/j.1540-8167.2006.00390.x. [DOI] [PubMed] [Google Scholar]
- 38.Undrovinas AI, Maltsev VA, Sabbah HN. Repolarization abnormalities in cardiomyocytes of dogs with chronic heart failure: role of sustained inward current. Cell Mol Life Sci. 1999;55:494–505. doi: 10.1007/s000180050306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Valdivia CR, Chu WW, Pu J, Foell JD, Haworth RA, Wolff MR, Kamp TJ, Makielski JC. Increased late sodium current in myocytes from a canine heart failure model and from failing human heart. J Mol Cell Cardiol. 2005;38:475–483. doi: 10.1016/j.yjmcc.2004.12.012. [DOI] [PubMed] [Google Scholar]
- 40.Sossalla S, Wagner S, Rasenack ECL, Ruff H, Weber SL, Schondube FA, Tirilomis T, Tenderich G, Hasenfuss G, Belardinelli L, Maier LS. Ranolazine improves diastolic dysfunction in isolated myocardium from failing human hearts - Role of late sodium current and intracellular ion accumulation. J Mol Cell Cardiol. 2008;45:32–43. doi: 10.1016/j.yjmcc.2008.03.006. [DOI] [PubMed] [Google Scholar]
- 41.Fredj S, Lindegger N, Sampson KJ, Carmeliet P, Kass RS. Altered Na+ channels promote pause-induced spontaneous diastolic activity in long QT syndrome type 3 myocytes. Circ Res. 2006;99:1225–1232. doi: 10.1161/01.RES.0000251305.25604.b0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Song Y, Shryock JC, Belardinelli L. An increase of late sodium current induces delayed after depolarizations and sustained triggered activity in atrial myocytes. Am J Physiol Heart Circ Physiol. 2008;294:H2031–H2039. doi: 10.1152/ajpheart.01357.2007. [DOI] [PubMed] [Google Scholar]
- 43.Glitsch HG. Electrophysiology of the sodium-potassium-ATPase in cardiac cells. Physiol Rev. 2001;81:1791–826. doi: 10.1152/physrev.2001.81.4.1791. [DOI] [PubMed] [Google Scholar]
- 44.Grandi E, Pasqualini FS, Bers DM. A novel computational model of the human ventricular action potential and Ca transient. J Mol Cell Cardiol. 2010;48:112–1121. doi: 10.1016/j.yjmcc.2009.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schouten VJ, ter Keurs HE. The slow repolarization phase of the action potential in rat heart. J Physiol. 1985;360:13–25. doi: 10.1113/jphysiol.1985.sp015601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Armoundas AA, Hobai IA, Tomaselli GF, Winslow RL, O’Rourke B. Role of sodium-calcium exchanger in modulating the action potential of ventricular myocytes from normal and failing hearts. Circ Res. 93:46–53. doi: 10.1161/01.RES.0000080932.98903.D8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Philipson KD, Nicoll DA. Molecular and kinetic aspects of sodium-calcium exchange. Int Rev Cytol. 1993;137C:199–227. [PubMed] [Google Scholar]
- 48.Nicoll DA, Ottolia M, Lu L, Lu Y, Philipson KD. A new topological model of the cardiac sarcolemmal Na+-Ca2+ exchanger. J Biol Chem. 1999;274:910–917. doi: 10.1074/jbc.274.2.910. [DOI] [PubMed] [Google Scholar]
- 49.Nicoll DA, Ren X, Ottolia M, Phillips M, Paredes AR, Abramson J, Philipson KD. What we know about the structure of NCX1 and how it relates to its function. Ann NY Acad Sci. 2007;1099:1–6. doi: 10.1196/annals.1387.014. [DOI] [PubMed] [Google Scholar]
- 50.Liao J, Li H, Zeng W, Sauer DB, Belmares R, Jiang Y. Structural insight into the ion-exchange mechanism of the sodium/calcium exchanger. Science. 2012;335:686–690. doi: 10.1126/science.1215759. [DOI] [PubMed] [Google Scholar]
- 51.Ren X, Philipson KD. The topology of the cardiac Na+/Ca2+ exchanger, NCX1. J Mol Cell Cardiol. 2013;57:68–71. doi: 10.1016/j.yjmcc.2013.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Qiu Z, Nicoll DA, Philipson KD. Helix packing of functionally important regions of the cardiac Na+-Ca2+ exchanger. J Biol Chem. 2001;276:194–199. doi: 10.1074/jbc.M005571200. [DOI] [PubMed] [Google Scholar]
- 53.Ren X, Nicoll DA, Philipson KD. Helix packing of the cardiac Na+-Ca2+ exchanger: proximity of transmembrane segments 1, 2, and 6. J Biol Chem. 2006;281:22808–22814. doi: 10.1074/jbc.M604753200. [DOI] [PubMed] [Google Scholar]
- 54.Levitsky DO, Nicoll DA, Philipson KD. Identification of the high affinity Ca2+-binding domain of the cardiac Na+-Ca2+ exchanger. J Biol Chem. 1994;269:22847–22852. [PubMed] [Google Scholar]
- 55.Matsuoka S, Nicoll DA, Hrysko LV, Levitsky DO, Weiss JN, Philipson KD. Regulation of the cardiac Na+-Ca2+ exchanger by Ca2+. Mutational analysis of the Ca2+-binding domain. J Gen Physiol. 1995;105:403–420. doi: 10.1085/jgp.105.3.403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mercado Besserer G, Ottolia M, Nicoll DA, Chaptal V, Cascio D, Philipson KD, Abramson J. The second Ca2+ - binding domain of the Na+-Ca2+ exchanger is essential for regulation: Crystal structures and mutational analysis. Proc Natl Acad Sci. 2007;104:18467–18472. doi: 10.1073/pnas.0707417104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Reeves JP, Condrescu M. Allosteric activation of sodium-calcium exchnage activity by calcium: persistence at low calcium concentrations. J Gen Physiol. 2003;122:621–639. doi: 10.1085/jgp.200308915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bers DM, Ginsburg KS. Na:Ca stoichiometry and cytosolic Ca-dependent activation of NCX in intact cardiomyocytes. Ann NY Acad Sci. 2007;1099:326–338. doi: 10.1196/annals.1387.060. [DOI] [PubMed] [Google Scholar]
- 59.Ginsburg KS, Weber CR, Bers DM. Cardiac Na:Ca exchanger: Dynamics of Ca-dependent activation and deactivation in intact myocytes. J Physiol. 2013 Feb 11; doi: 10.1113/jphysiol.2013.252080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Urbanczyk J, Chernysh O, Condrescu M, Reeves JP. Sodium-calcium exchange does not require allosteric calcium activation at high cytosolic sodium concentrations. J Physiol. 2006;575:693–705. doi: 10.1113/jphysiol.2006.113910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yaradanakul A, Feng S, Shen C, Lariccia V, Lin MJ, Yang J, Kang TM, Dong P, Yin HL, Albanesi JP, Hilgemann DW. Dual control of cardiac Na+/Ca2+ exchange by PIP2: electrophysiological analysis of direct and indirect mechanisms. J Physiol. 2007;582:991–1010. doi: 10.1113/jphysiol.2007.132712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Frank HS, Mottino J, Reid D, Molday RS, Philipson KD. Distribution of the Na+-Ca2+ exchanger protein in mammalian cardiac myocytes; An immunofluorescence and immunoco-lloidal glod-labeling study. J Biol Chem. 1992;117:337–345. doi: 10.1083/jcb.117.2.337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yang Z, Pascarel C, Steele DS, Komukai K, Brette F, Orchard CH. Na+-Ca2+ exchange activity is localized in the T-tubules of rat ventricular myocytes. Circ Res. 2002;91:315–322. doi: 10.1161/01.res.0000030180.06028.23. [DOI] [PubMed] [Google Scholar]
- 64.Despa S, Brette F, Orchard CH, Bers DM. Na/Ca exchange and Na/K-ATPase function are equally concentrated in transverse tubules of rat ventricular myocytes. Biophys J. 2003;85:3388–3396. doi: 10.1016/S0006-3495(03)74758-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Scriven DRL, Dan P, Moore EDW. Distribution of proteins implicated in excitation-contraction coupling in rat ventricular myocytes. Biophys J. 2000;79:2682–2691. doi: 10.1016/S0006-3495(00)76506-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mohler PJ, Davis JQ, Bennet V. Ankyrin-B coordinates the Na/K ATPase, Na/Ca exchanger, and InsP3 receptor in a cardiac T-tubule/SR microdomain. PLoS Biol. 2005;3(12):e423. doi: 10.1371/journal.pbio.0030423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jayasinghe ID, Cannell MB, Soeller C. Organization of ryanodine receptors, transverse tubules, and sodium-calcium exchanger in rat myocytes. Biophys J. 2009;97:2664–2673. doi: 10.1016/j.bpj.2009.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Trafford AW, Diaz ME, O’Neill SC, Eisner DA. Comparison of subsarcolemmal and bulk calcium concentration during spontaneous calcium release in rat ventricular myocytes. J Physiol. 1995;488:577–586. doi: 10.1113/jphysiol.1995.sp020991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Weber CR, Piacentino V, III, Ginsburg KS, Houser SR, Bers DM. Na/Ca exchange current and submembrane [Ca] during the cardiac action potential. Circ Res. 2002;90:182–189. doi: 10.1161/hh0202.103940. [DOI] [PubMed] [Google Scholar]
- 70.Acsai K, Antoons G, Livshitz L, Rudy Y, Sipido KR. Microdomain [Ca2+] near ryanodine receptors as reported by L-type Ca2+ and Na+/Ca2+ exchange currents. J Physiol. 2011;589:2569–2583. doi: 10.1113/jphysiol.2010.202663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wasserstrom JA, Vites AM. The role of Na+-Ca2+ exchange in activation of excitation-contraction coupling in rat ventricular myocytes. J Physiol. 1996;493:529–542. doi: 10.1113/jphysiol.1996.sp021401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sipido KR, Maes M, Van de Werf F. Low efficiency of Ca2+ entry through the Na+-Ca2+ exchanger as trigger for Ca2+ release from the sarcoplasmic reticulum. A comparison between L-type Ca2+ current and reverse-mode Na+-Ca2+ exchange. Circ Res. 1997;81:1034–1044. doi: 10.1161/01.res.81.6.1034. [DOI] [PubMed] [Google Scholar]
- 73.Litwin SE, Li J, Bridge JH. Na-Ca exchange and the trigger for sarcoplasmic reticulum Ca release: studies in adult rabbit ventricular myocytes. Biophys J. 1998;75:359–371. doi: 10.1016/S0006-3495(98)77520-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Neco P, Rose B, Huynh N, Zhang R, Bridge JH, Philipson KD, Goldhaber JI. Sodium-calcium exchange is essential for effective triggering of calcium release in mouse heart. Biophys J. 2010;99:755–764. doi: 10.1016/j.bpj.2010.04.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Torres NS, Larbig R, Rock A, Goldhaber JI, Bridge JH. Na+ currents are required for efficient excitation-contraction coupling in rabbit ventricular myocytes: a possible contribution of neuronal Na+ channels. J Physiol. 2010;588:4249–4260. doi: 10.1113/jphysiol.2010.194688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Abriel H, Kass RS. Regulation of the voltage-gated cardiac sodium channel Nav1.5 by interacting proteins. TCM. 2005;15:35–40. doi: 10.1016/j.tcm.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 77.Catterall WA. Cellular and molecular biology of voltage-gated Na channels. Physiol Rev. 1992;72:S15–S48. doi: 10.1152/physrev.1992.72.suppl_4.S15. [DOI] [PubMed] [Google Scholar]
- 78.Stühmer W, Conti F, Suzuki H, Wang XD, Nada M, Yahagi N, Kubo H, Numa S. Structural parts involved in activation and inactivation of the sodium channel. Nature. 1989;339:597–603. doi: 10.1038/339597a0. [DOI] [PubMed] [Google Scholar]
- 79.Balser JR. The cardiac sodium channel: gating function and molecular pharmacology. J Mol Cell Cardiol. 2001;33:599–613. doi: 10.1006/jmcc.2000.1346. [DOI] [PubMed] [Google Scholar]
- 80.Haufe V, Chamberland C, Dumaine R. The promiscuous nature of the cardiac sodium current. J Mol Cell Cardiol. 2007;42:469–477. doi: 10.1016/j.yjmcc.2006.12.005. [DOI] [PubMed] [Google Scholar]
- 81.Maier SK, Westenbroek RE, Schenkman KA, Feigl EO, Scheuer T, Catterall WA. An unexpected role for brain-type sodium channels in coupling of cell surface depolarization to contraction in the heart. Proc Natl Acad Sci U S A. 2002;99:4073–4078. doi: 10.1073/pnas.261705699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Noujaim SF, Kaur K, Milstein M, Jones JM, Furspan P, Jiang D, Auerbach DS, Herron T, Meisler MH, Jalife J. A null mutation of the neuronal sodium channel NaV1.6 disrupts action potential propagation and excitation-contraction coupling in the mouse heart. FASEB J. 2012;26:63–72. doi: 10.1096/fj.10-179770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Brette F, Orchard CH. No apparent requirement for neuronal sodium channels in excitation-contraction coupling in rat ventricular myocytes. Circ Res. 2006;98:667–674. doi: 10.1161/01.RES.0000209963.02720.70. [DOI] [PubMed] [Google Scholar]
- 84.Isom LL, De Jongh KS, Catterall WA. Auxiliary subunits of voltage-gated ion channels. Neuron. 1994;12:1183–1194. doi: 10.1016/0896-6273(94)90436-7. [DOI] [PubMed] [Google Scholar]
- 85.Kucera JP, Rohr S, Rudy Y. Localization of sodium channels in intercalated disks modulates cardiac conduction. Circ Res. 2002;91:1176–1182. doi: 10.1161/01.res.0000046237.54156.0a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Brette F, Orchard CH. Density and sub-cellular distribution of cardiac and neuronal sodium channel isoforms in rat ventricular myocytes. Biochem Biophys Res Commun. 2006;348:1163–1166. doi: 10.1016/j.bbrc.2006.07.189. [DOI] [PubMed] [Google Scholar]
- 87.Abriel H. Cardiac sodium channel Nav1.5 and interacting proteins: Physiology and pathophysiology. J Mol Cell Cardiol. 2010;48:2–11. doi: 10.1016/j.yjmcc.2009.08.025. [DOI] [PubMed] [Google Scholar]
- 88.Lowe JS, Palygin O, Bhasin N, Hund TJ, Boyden PA, Shibata E, Anderson ME, Mohler PJ. Voltage-gated Nav channel targeting in the heart requires an ankyrin-G dependent cellular pathway. J Cell Biol. 2008;180:173–186. doi: 10.1083/jcb.200710107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Priori SG, Napolitano C, Gasparini M, Pappone C, Della Bella P, Brignole M, Giordano U, Giovannini T, Menozzi C, Bloise R, Crotti L, Terreni L, Schwartz PJ. Clinical and genetic heterogeneity of right bundle branch block and ST-segment elevation syndrome: A prospective evaluation of 52 families. Circulation. 2000;102:2509–2515. doi: 10.1161/01.cir.102.20.2509. [DOI] [PubMed] [Google Scholar]
- 90.Sato PY, Coombs W, Lin X, Nekrasova O, Green KJ, Isom LL, Taffet SM, Delmar M. Interactions between ankyrin-G, Plakophilin-2, and Connexin43 at the cardiac intercalated disc. Circ Res. 2011;109:193–201. doi: 10.1161/CIRCRESAHA.111.247023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Biet M, Barajas-Martínez H, Ton AT, Delabre JF, Morin N, Dumaine R. About half of the late sodium current in cardiac myocytes from dog ventricle is due to non-cardiac-type Na+ channels. J Mol Cell Cardiol. 2012;53:593–598. doi: 10.1016/j.yjmcc.2012.06.012. [DOI] [PubMed] [Google Scholar]
- 92.Ju YK, Saint DA, Gage PW. Hypoxia increases persistent sodium current in rat ventricular myocytes. J Physiol. 1996;497:337–347. doi: 10.1113/jphysiol.1996.sp021772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kirchhefer U, Schmitz W, Scholz H, Neumann J. Activity of cAMP-dependent protein kinase and Ca2+/calmodulin-dependent protein kinase in failing and nonfailing human hearts. Cardiovasc Res. 1999;42:254–261. doi: 10.1016/s0008-6363(98)00296-x. [DOI] [PubMed] [Google Scholar]
- 94.Ai X, Curran JW, Shannon TR, Bers DM, Pogwizd SM. Ca2+/Calmodulin-Dependent Protein Kinase Modulates Cardiac Ryanodine Receptor Phosphorylation and Sarcoplasmic Reticulum Ca2+ Leak in Heart Failure. Circ Res. 2005;97:1314–1322. doi: 10.1161/01.RES.0000194329.41863.89. [DOI] [PubMed] [Google Scholar]
- 95.Wagner S, Dybkova N, Rasenack ECL, Jacobshagen C, Fabritz L, Kirchhof P, Maier SKG, Zhang T, Hasenfuss G, Brown JH, Bers DM, Maier LS. Ca2+/calmodulin-dependent protein kinase II regulates cardiac Na+ channels. J Clin Invest. 2006;116:3127–3138. doi: 10.1172/JCI26620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Hilgemann DW, Nagel GA, Gadsby DC. Na/K pump current in giant membrane patches excised from ventricular myocytes. In: Kaplan JH, De Weer P, editors. The sodium pump: recent developments. New York: Rockefeller Univ Press; 1991. pp. 543–547. [Google Scholar]
- 97.Friedrich T, Bamberg E, Nagel G. Na+,K+-ATPase pump currents in giant excised patches activated by an ATP concentration jump. Biophys J. 1996;71:2486–2500. doi: 10.1016/S0006-3495(96)79442-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Geering K. Functional roles of Na,K-ATPase subunits. Curr Opin Nephrol Hypertens. 2008;17:526–532. doi: 10.1097/MNH.0b013e3283036cbf. [DOI] [PubMed] [Google Scholar]
- 99.Sweadner KJ, Rael E. The FXYD gene family of small ion transport regulators or channels: cDNA sequence, protein signature sequence, and expression. Genomics. 2000;68:41–56. doi: 10.1006/geno.2000.6274. [DOI] [PubMed] [Google Scholar]
- 100.Crambert G, Fuzesi M, Garty H, Karlish S, Geering K. Phospholemman (FXYD1) associates with Na+,K+-ATPase and regulates its transport properties. Proc Natl Acad Sci USA. 2002;99:11476–11481. doi: 10.1073/pnas.182267299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Bibert S, Roy S, Schaer D, Horisberger J-D, Geering K. Phosphorylation of phospholemman (FXYD1) by protein kinases A and C modulates distinct Na,K-ATPase isozymes. J Biol Chem. 2008;283:476–486. doi: 10.1074/jbc.M705830200. [DOI] [PubMed] [Google Scholar]
- 102.Morth JP, Pedersen BP, Toustrup-Jensen MS, Sorensen TLM, Petersen J, Andersen JP, Vilsen B, Nissen P. Crystal structure of the sodium-potassium pump. Nature. 2007;450:1043–1049. doi: 10.1038/nature06419. [DOI] [PubMed] [Google Scholar]
- 103.Shinoda T, Ogawa H, Cornelius F, Toyoshima C. Crystal structure of the sodium-potassium pump at 2.4 A resolution. Nature. 2009;459:446–450. doi: 10.1038/nature07939. [DOI] [PubMed] [Google Scholar]
- 104.Khafaga M, Bossuyt J, Mamikonian L, Li JC, Lee LL, Yarov-Yarovoy V, Despa S, Bers DM. Na+/K+-ATPase E960 and Phospholemman F28 are critical for their functional interaction. Proc Natl Acad Sci USA. 2012;109:20756–20761. doi: 10.1073/pnas.1207866109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Sweadner KJ, Herrera VLM, Amato S, Moellmann A, Gibbons DK, Repke KRH. Immunologic identification of Na,K-ATPase isoforms in myocardium. Circ Res. 1994;74:669–678. doi: 10.1161/01.res.74.4.669. [DOI] [PubMed] [Google Scholar]
- 106.Shamraj OI, Melvin D, Lingrel JB. Expression of Na,K-ATPase isoforms in human heart. Biochem Biophys Res Commun. 1991;179:1434–1440. doi: 10.1016/0006-291x(91)91733-s. [DOI] [PubMed] [Google Scholar]
- 107.Zahler R, Gilmore-Herbert M, Baldwin JC, Franco K, Benz EJ., Jr Expression of alpha isoforms of the Na,K-ATPase in human heart. Biochim Biophys Acta. 1993;1149:189–194. doi: 10.1016/0005-2736(93)90200-j. [DOI] [PubMed] [Google Scholar]
- 108.Lucchesi PA, Sweadner KJ. Postnatal changes in Na,K-ATPase isoform expression in rat cardiac ventricle. J Biol Chem. 1991;266:9327–9331. [PubMed] [Google Scholar]
- 109.Charlemagne D, Orlowski J, Oliviero P, Rannou F, Sainte Beuve C, Swynghedauw B, Lane LK. Alteration of Na,K-ATPase subunit mRNA and protein levels in hypertrophied rat heart. J Biol Chem. 1994;269:1541–1547. [PubMed] [Google Scholar]
- 110.Semb SO, Lunde PK, Holt E, Tonnessen T, Christensen G, Sejersted OM. Reduced myocardial Na-K pump capacity in congestive heart failure following myocardial infarction in rats. J Mol Cell Cardiol. 1998;30:1311–1328. doi: 10.1006/jmcc.1998.0696. [DOI] [PubMed] [Google Scholar]
- 111.Blanco G, Mercer RW. Isozymes of the Na-K-ATPase: heterogeneity in structure, diversity in function. Am J Physiol. 1998;275:F633–F650. doi: 10.1152/ajprenal.1998.275.5.F633. [DOI] [PubMed] [Google Scholar]
- 112.Juhaszova M, Blaustein MP. Na+ pump low and high ouabain affinity alpha subunit isoforms are differently distributed in cells. Proc Natl Acad Sci USA. 1997;94:1800–1805. doi: 10.1073/pnas.94.5.1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Swift F, Tovsrud N, Enger UH, Sjaastad I, Sejersted OM. The Na+,K+-ATPase α2-isoform regulates cardiac contractility in rat cardiomyocytes. Cardiovasc Res. 2007;75:109–117. doi: 10.1016/j.cardiores.2007.03.017. [DOI] [PubMed] [Google Scholar]
- 114.Yamamoto T, Su Z, Moseley AE, Kadono T, Zhang J, Cougnon M, Li F, Lingrel JB, Barry WH. Relative abundance of α2 Na+ pump isoform influences Na+-Ca2+ exchanger currents and Ca2+ transients in mouse ventricular myocytes. J Mol Cell Cardiol. 2005;39:113–120. doi: 10.1016/j.yjmcc.2005.03.023. [DOI] [PubMed] [Google Scholar]
- 115.Swift F, Tovsrud N, Sjaastad I, Sejersted OM, Niggli E, Egger M. Functional coupling of α2-isoform Na+/K+-ATPase and Ca2+ extrusion through the Na+/Ca2+-exchanger in cardiomyocytes. Cell Calcium. 2010;48:54–60. doi: 10.1016/j.ceca.2010.06.006. [DOI] [PubMed] [Google Scholar]
- 116.Despa S, Bers DM. Functional analysis of Na+/K+-ATPase isoform distribution in rat ventricular myocytes. Am J Physiol Cell Physiol. 2007;293:C321–C327. doi: 10.1152/ajpcell.00597.2006. [DOI] [PubMed] [Google Scholar]
- 117.Berry RG, Despa S, Fuller W, Bers DM, Shattock MJ. Differential distribution and regulation of mouse Na+/K+-ATPase α1 and α2-subunits in T-tubule and surface sarcolemmal membranes. Cardiovasc Res. 2007;73:92–100. doi: 10.1016/j.cardiores.2006.11.006. [DOI] [PubMed] [Google Scholar]
- 118.Despa S, Lingrel JB, Bers DM. Na/K-ATPase α2-subunit preferentially affects sarcoplasmic reticulum Ca release in mouse cardiac myocytes. Cardiovasc Res. 2012;95:480–486. doi: 10.1093/cvr/cvs213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Hashemi SM, Hund TJ, Mohler PJ. Cardiac ankyrins in health and disease. J Mol Cell Cardiol. 2009;47:203–209. doi: 10.1016/j.yjmcc.2009.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Cunha SR, Bhasin N, Mohler PJ. Targeting and stability of Na/Ca exchanger 1 in cardiomyocytes requires direct interaction with the membrane adaptor protein ankyrin-B. J Biol Chem. 2007;282:4875–4883. doi: 10.1074/jbc.M607096200. [DOI] [PubMed] [Google Scholar]
- 121.Mohler PJ, Schott JJ, Gramolini AO, Dilly KW, Guatimosin S, duBell WH, Song LS, Haurogné K, Kyndt F, Ali ME, Rogers TB, Lederer WJ, Escande D, Le Marec H, Bennet V. Ankyrin-B mutation causes type 4 long-QT cardiac arrhythmia and sudden cardiac death. Nature. 2003;421:634–639. doi: 10.1038/nature01335. [DOI] [PubMed] [Google Scholar]
- 122.Mohler PJ, Le Scouarnec S, Denjoy I, Lowe JS, Guicheney P, Caron L, Driskell IM, Schott JJ, Norris K, Leenhardt A, Kim RB, Escande D, Roden DM. Defining the cellular phenotype of “ankyrin-B syndrome” variants: human ANK2 variants associated with clinical phenotypes display a spectrum of activities in cardiomyocytes. Circulation. 2007;115(4):432–441. doi: 10.1161/CIRCULATIONAHA.106.656512. [DOI] [PubMed] [Google Scholar]
- 123.Camors E, Mohler PJ, Bers DM, Despa S. Ankyrin-B reduction alters Na and Ca transport promoting cardiac myocyte arrhythmic activity. J Mol Cell Cardiol. 2012;52:1240–1248. doi: 10.1016/j.yjmcc.2012.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Gray RP, McIntyre H, Sheridan DS, Fry CH. Intracellular sodium and contractile function in hypertrophied human and guinea-pig myocardium. Pflugers Arch. 2001;442:117–123. doi: 10.1007/s004240000512. [DOI] [PubMed] [Google Scholar]
- 125.Yao A, Su Z, Nonaka A, Zubair I, Spitzer KW, Bridge JH, Muelheims G, Ross J, Jr, Barry WH. Abnormal myocyte Ca2+ homeostasis in rabbits with pacing-induced heart failure. Am J Physiol. 1998;275(4 Pt 2):H1441–H1448. doi: 10.1152/ajpheart.1998.275.4.H1441. [DOI] [PubMed] [Google Scholar]
- 126.Harrison SM, McCall E, Boyet MR. The relationship between contraction and intracellular sodium in rat and guinea-pig ventricular myocytes. J Physiol. 1992;449:517–550. doi: 10.1113/jphysiol.1992.sp019100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Despa S, Islam MA, Pogwizd SM, Bers DM. Intracellular [Na+]i and Na+-pump rate in rat and rabbit ventricular myocytes. J Physiol. 2002;539(Pt 1):133–143. doi: 10.1113/jphysiol.2001.012940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Levi AJ, Lee CO, Brooksby P. Properties of the fluorescent sodium indicator SBFI in rat and rabbit cardiac myocytes. J Cardiovasc Electrophysiol. 1994;5:241–257. doi: 10.1111/j.1540-8167.1994.tb01161.x. [DOI] [PubMed] [Google Scholar]
- 129.Shattock MJ, Bers DM. Rat vs. rabbit ventricle: Ca flux and intracellular Na assessed by ion-selective microelectrodes. Am J Physiol. 1989;256:C813–C822. doi: 10.1152/ajpcell.1989.256.4.C813. [DOI] [PubMed] [Google Scholar]
- 130.Gao J, Wang W, Cohen IS, Mathias RT. Transmural gradients in Na/K pump activity and [Na+]i in canine ventricle. Biophys J. 2005;89:1700–1709. doi: 10.1529/biophysj.105.062406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Donoso P, Mill JG, O’Neil SC, Eisner DA. Fluorescence measurements of cytoplasmic and mitochondrial sodium concentration in rat ventricular myocytes. J Physiol. 1992;448:493–509. doi: 10.1113/jphysiol.1992.sp019053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Yao AS, Su Z, Nonaka A, Zubair I, Lu L, Philipson KD, Bridge JH, Barry WH. Effects of overexpression of the Na+-Ca2+ exchanger on [Ca2+] transients in murine ventricular myocytes. Circ Res. 1998;82(6):657–665. doi: 10.1161/01.res.82.6.657. [DOI] [PubMed] [Google Scholar]
- 133.Cook SJ, Chamunorwa JP, Lancaster MK, O’Neill SC. Regional differences in the regulation of intracellular sodium and in action potential configuration in rabbit left ventricle. Pflugers Arch. 1997;433:515–522. doi: 10.1007/s004240050307. [DOI] [PubMed] [Google Scholar]
- 134.Lancaster MK, Bennet DL, Cook SJ, O’Neill SC. Na/K pump α subunit expression in rabbit ventricle and regional variations of intracellular Na regulation. Pflugers Arch. 2000;440:735–739. doi: 10.1007/s004240000328. [DOI] [PubMed] [Google Scholar]
- 135.Leem CH, Lagadic-Gossmann D, Vaughan-Jones RD. Characterization of intracellular pH regulation in the guinea-pig ventricular myocytes. J Physiol. 1999;509:487–496. doi: 10.1111/j.1469-7793.1999.0159z.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Fry CH. Measurement and control of intracellular magnesium ion concentration in guinea pig and ferret ventricular myocardium. Magnesium. 1986;5:306–316. [PubMed] [Google Scholar]
- 137.Despa S, Islam MA, Weber CR, Pogwizd SM, Bers DM. Intracellular Na+ concentration is elevated in heart failure, but Na/K-pump function is unchanged. Circulation. 2002;105:2543–2548. doi: 10.1161/01.cir.0000016701.85760.97. [DOI] [PubMed] [Google Scholar]
- 138.Pavlovic D, Hall AR, Fuller W, Shattock MJ. Rapid Pacing Stimulates Na/K ATPase in Rat Ventricular Myocytes via a Nitric Oxide and Phospholemman-dependent Mechanism. Circulation. 2010;122:A16046. [Google Scholar]
- 139.Pieske B, Maier LS, Piacentino V, 3rd, Weisser J, Hasenfuss G, Houser S. Rate dependence of [Na+]i and contractility in nonfailing and failing human myocardium. Circulation. 2002;106:447–453. doi: 10.1161/01.cir.0000023042.50192.f4. [DOI] [PubMed] [Google Scholar]
- 140.Baartscheer A, Schumacher CA, van Borren MM, Belterman CN, Coronel R, Fiolet JW. Increased Na+/H+-exchange activity is the cause of increased [Na+]i and underlies disturbed calcium handling in the rabbit pressure and volume overload heart failure model. Cardiovasc Res. 2003;57:1015–1024. doi: 10.1016/s0008-6363(02)00809-x. [DOI] [PubMed] [Google Scholar]
- 141.Louch WE, Hougen K, Mørk HK, Swift F, Aronsen JM, Sjaastad I, Reims HM, Roald B, Andersson KB, Christensen G, Sejersted OM. Sodium accumulation promotes diastolic dysfunction in end-stage heart failure following Serca2 knockout. J Physiol. 2010;588:465–478. doi: 10.1113/jphysiol.2009.183517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Schillinger W, Teucher N, Christians C, Kohlhaas M, Sossalla S, Van Nguyen P, Schmidt AG, Schunck O, Nebendahl K, Maier LS, Zeitz O, Hasenfuss G. High intracellular Na+ preserves myocardial function at low heart rates in isolated myocardium from failing hearts. Eur J Heart Fail. 2006;8:673–680. doi: 10.1016/j.ejheart.2006.01.013. [DOI] [PubMed] [Google Scholar]
- 143.Schwinger RH, Wang J, Frank K, Müller-Ehmsen J, Brixus K, McDonough AA, Erdmann E. Reduced sodium pump α1, α3 and β1-isoform protein levels and Na+,K+-ATPase activity but unchanged Na+-Ca2+ exchanger protein levels in human heart failure. Circulation. 1999;99:2105–2112. doi: 10.1161/01.cir.99.16.2105. [DOI] [PubMed] [Google Scholar]
- 144.Allen D, Schimdt TA, Marsh JD, Kieldsen K. Na,K-ATPase expression in normal and failing human left ventricle. Basic Res Cardiol. 1992;87 (Suppl 1):87–94. doi: 10.1007/978-3-642-72474-9_7. [DOI] [PubMed] [Google Scholar]
- 145.Verdonck F, Volders PGA, Vos MA, Sipido KR. Intracellular Na+ and altered Na+ transport mechanisms in cardiac hyperhtrophy and failure. J Mol Cell Cardiol. 2003;35(1):5–25. doi: 10.1016/s0022-2828(02)00280-8. [DOI] [PubMed] [Google Scholar]
- 146.Bossuyt J, Ai X, Moorman RJ, Pogwizd SM, Bers DM. Expression and phosphorylation of the Na+-pump regulatory subunit phospholemman in heart failure. Circ Res. 2005;97(6):558–565. doi: 10.1161/01.RES.0000181172.27931.c3. [DOI] [PubMed] [Google Scholar]
- 147.Swift F, Birkeland JA, Tovsrud N, Enger UH, Aronsen JM, Louch WE, Sjaastad I, Sejersted OM. Altered Na+/Ca2+-exchanger activity due to downregulation of Na+/K+-ATPase α2-isoform in heart failure. Cardiovasc Res. 2008;78:71–78. doi: 10.1093/cvr/cvn013. [DOI] [PubMed] [Google Scholar]
- 148.Verdonck F, Volders PG, Vos MA, Sipido KR. Increased Na+ concentration and altered Na+/K+ pump activity in hypertrophied canine ventricular cells. Cardiovasc Res. 2003;57(4):1035–1043. doi: 10.1016/s0008-6363(02)00734-4. [DOI] [PubMed] [Google Scholar]
- 149.Baartscheer A, Schumacher CA, van Borren MM, Belterman CN, Coronel R, Opthof T, Fiolet JW. Chronic inhibition of Na+/H+-exchanger attenuates cardiac hypertrophy and prevents cellular remodeling in heart failure. Cardiovasc Res. 2005;65:83–92. doi: 10.1016/j.cardiores.2004.09.024. [DOI] [PubMed] [Google Scholar]
- 150.Chattou S, Coulombe A, Diacono J, Le Grand B, John G, Feuvray D. Slowly inactivating component of sodium current in ventricular myocytes is decreased by diabetes and partially inhibited by known Na+-H+ Exchange blockers. J Mol Cell Cardiol. 2000;32:1181–1192. doi: 10.1006/jmcc.2000.1151. [DOI] [PubMed] [Google Scholar]