

SUMMARY
We have successfully expressed and purified active human glycogen synthase-1 (hGYS1). Successful production of the recombinant hGYS1 protein was achieved by co-expression of hGYS1 and rabbit glycogenin (rGYG1) using the MultiBac baculovirus expression system (BEVS). Functional measurements of activity ratios of hGYS1 in the absence and presence of glucose-6-phosphate and treatment with phosphatase indicate that the expressed protein is heavily phosphorylated. We used mass spectrometry to further characterize the sites of phosphorylation, which include most of the known regulatory phosphorylation sites, as well as several sites unique to the insect cell over-expression. Obtaining large quantities of functional hGYS1 will be invaluable for future structural studies as well as detailed studies on the effects on specific sites of phosphorylation.
Keywords: human glycogen synthase-1
Introduction
Glycogen is a branched polymer that serves as the major storage form of glucose in fungi and mammals[1]. The branched structure of glycogen permits rapid mobilization of glucose when needed, making it an ideal source of energy for short-term metabolic demands[2]. The pathway for bulk glycogen synthesis is conserved from bacterial to man and includes glycogen synthase and the branching enzyme, which are responsible for forming the α-1,4 and α-1,6 glycosidic linkages, respectively. In mammals, there are two forms of glycogen synthase; GYS2, which is expressed exclusively in liver and GYS1, which is expressed in skeletal muscle and most other tissues. As a major energy reserve, the regulation of glycogen synthesis and degradation is crucial and is mediated both by changes in covalent phosphorylation and by allosteric regulators, chief amongst these is the activator glucose-6-phosphate (G6P)[3]. Glycogen synthase is inhibited by phosphorylation[3]. There are nine known sites of phosphorylation in hGYS1 and they are predominantly clustered on the C-terminal end of the protein (Fig. 1). Several sites are known to be hierarchal in nature, meaning that some sites of phosphorylation are dependent on prior phosphorylations[4,5]. This complex pattern of phosphorylation creates multiple levels of control on the activity of glycogen synthase.
Fig. 1.

Cartoon depicting the known sites of phosphorylation of hGYS1
In conjunction with glycogen synthase and the branching enzyme, glycogenin is another key enzyme involved in glycogen synthesis. Glycogenin mediates the initiation of the glycogen particle by forming the primer upon which glycogen synthase can act[6,7]. Glycogenin uses UDP-glucose as the donor nucleotide sugar to create a linear glucose chain attached to a conserved tyrosine residue via α-1,4-glycosidic linkages[3]. Several studies have implicated the C-terminus of glycogenin in mediating direct interactions with glycogen synthase[6,7]. In fact, the C-terminal 33-amino acids of glycogenin can be used to extract glycogen synthase from tissue lysates[8]. In addition, glycogenin was found to interact with glycogen synthase in the cell and, when over-expressed with glycogen synthase, can redirect glycogen synthase from the insoluble to soluble fraction[9].
Our goal has been to produce sufficient quantities of human skeletal muscle glycogen synthase (hGYS1) for crystallographic and structure-function studies. Although, glycogen synthase from yeast is easily over-expressed in E. coli systems[10,11], efforts to produce metazoan forms of glycogen synthase from C. elegans to H. sapiens have failed to achieve high-level expression of an active enzyme, even when co-expressed with mammalian glycogenin (S. Perez-Miller, unpublished). Similarly, expression of human GYS1 alone in baculoviral systems produced inconsistent results (S. Perez-Miller, unpublished). We were only able to reproducibly produce milligram quantities of functional glycogen synthase through co-expression with glycogenin in Hi5 insect cells. The scheme of this preparation and implications for this co-expression, as well as levels of post-translational phosphorylation, are discussed.
Materials and methods
Expression and Purification of hGYS1
Human glycogen synthase (hGYS1) clone was obtained from Open Biosystems, amplified and sub-cloned into an IMPACT E.coli expression vector pTXB1 (NEB Inc, MA) using NdeI and SpeI with an intein tag and a chitin-binding domain (CBD) at the C-terminus of hGYS1. The hGYS1-intein-CBD was later sub-cloned in a pFL MultiBac transfer vector[12] using the XhoI and SphI sites; the gene encoding rabbit Glycogenin (rGYG1) was amplified by PCR and sub-cloned into the BamHI and HindIII sites of pFL-CBD-intein-hGYS1, yielding the final transfer vector, pFL-CBD-intein-hGYS1-rGYG1 (Fig. 2). Expression of hGYS1 is driven by transcription of the p10 promoter, while glycogenin is driven by the polh promoter thus allowing for simultaneous expression of both proteins (Fig. 2). High titer expression baculovirus harboring both the intein-CBD tagged hGYS1 and rGYG1 was generated using the transfer vector, pFL-intein-hGYS1-rGYG1 in Sf9 cells as previously described[12]. The expression protocol for protein production in Hi5 cells has been described elsewhere[13]. 96 hours after infection, the cells were separated from the growth media by centrifugation at 30,000g and the pellet was resuspended in 50 mL of lysis buffer (20 mM Tris-HCl pH 8.5, 500 mM NaCl, 1 mM EDTA, and 0.1% Triton x-100). Cells were then lysed at 4°C by gentle stirring for one hour. The clarified lysate, obtained by centrifugation for 30 min at 30,000 rpm, was mixed with 10 mL of chitin beads. After incubation for one hour at 4°C, the beads were washed with lysis buffer (excluding Triton), and elution of the protein from the chitin beads was performed by incubating with cleavage buffer (20 mM Tris-HCl pH 8.5, 500 mM NaCl, 50 mM dithiothreitol (DTT), and 1 mM EDTA) overnight at 4°C and beads were separated by loading on a column and collecting the flow-through. Following elution, the protein was extensively dialyzed against 20 mM Tris pH 8.5, 50 mM NaCl, and 1 mM DTT and concentrated to 4 mg/mL using Amicon ultra-15 centrifugal filters (Millipore, MA) prior to flash freezing in liquid nitrogen and stored at -80°C.
Fig. 2.
Cloning of hGYS1. Scheme showing pFL vector with sites of insertion and promoters used for expression of hGYS1 and rGYG1. Coomassie stained polyacrylamide gel shows purity of hGYS1 following purification from Hi5 cells.
Analysis of glycogen synthase phosphorylation by Western Blot
Prior studies have demonstrated that the phosphorylated forms of glycogen synthase can be separated from the dephosphorylated forms by SDS-PAGE[14]. Serial dilutions of purified recombinant hGYS1 were loaded onto a 4-20% gradient polyacrylamide gel (Life Technologies, CA) with and without treatment using lambda phosphatase (New England Biolabs, MA). Following electrophoresis, the proteins were transferred to a Hybond-ECL nitrocellulose membrane (GE Healthcare). The membrane was blocked with 1% fish gelatin overnight. The protein bound to the nitrocellulose paper is then incubated with primary monoclonal mouse antibody against glycogen synthase-1 (GS-7H5) from Santa Cruz (sc-81173) at a dilution of 1:1000 overnight at 4°C. The secondary antibody is a fluorescent anti-mouse IgG antibody IRDye800 (Rockland, PA) and was used at a 1:10,000 dilution. The blot was washed with TBS-T and incubated with secondary antibody for an additional 45 minutes. The amount of fluorescence was then monitored using the Odyssey by LICOR.
Glycogen synthase activity assays
Glycogen synthase activity was determined by the method of Thomas et al.[15]. Reactions were carried out at 30°C for 15 min 4.4 mM UDP-glucose in the presence or absence of 7.2 mM G6P for samples directly purified from Hi5 cells as well as samples treated with lambda phosphatase which releases phosphates from phosphorylated serine, threonine and tyrosine residues. The concentration for G6P activation (Ka) was determined by varying the concentration of G6P between 0.01 mM and 10 mM in the presence of 4.4 mM UDP-glucose[9]. As lambda phosphatase will also degrade G6P and thereby interfere with this assay, we ensured complete removal of the phosphatase by performing the dephosphorylation reaction prior to elution of glycogen synthase from the chitin beads. Following the addition of lambda phosphatase, the beads were incubated at 37 °C for 1 hour, then the beads were extensively washed and the dephosphorylated hGYS1 was eluted with cleavage buffer as described above.
Analysis of Phosphorylation by Mass spectrometry
Approximately 5 μg of purified recombinant hGYS1 was precipitated by addition of 20% trichloroacetic acid (TCA) and then the protein was reductively alkylated using tris (2-carboxyethyl) phosphine (TCEP) followed by chloroacetamide. Human GYS1 prepared in this manner was then subjected to overnight digestion with endoprotease Lys-C (Roche Diagnostics) in 8M Urea, 100mM Tris-HCl, pH 8.5. Following dilution of the sample to 2M Urea, the protein was further digested overnight using trypsin (Promega) at 37oC. Multidimensional Protein Identification Mass Spectrometry (MudPIT) was used to analyze the resulting peptides as previously described[16-18]. The acquired data was further processed and analyzed using the software packages SEQUEST using Proteome Discoverer 1.3 (Thermo), X! Tandem[19] using Proteome Discoverer 1.3 (Thermo), as well as Scaffold (Proteome Software). PhophoRS, a probability-based program within Proteome Discoverer 1.3, was used to calculate and score sites for their probability of phosphorylation[20].
Results and Discussion
Protein Expression and Purification
Heterologous expression of numerous metazoan glycogen synthases had been attempted in E. coli with and without co-expression of multiple proteins partners, kinases and chaperones with limited success (unpublished data). Few of these approaches led to soluble or active protein. Reasoning that eukaryotic factors involved in post-translational events may be necessary for robust expression, we attempted expression of hGYS1 in insect cells using BEVS wherein these events might be achieved. Insect cells represent an ideal system since they can be easily cultured and transfected, have eukaryotic protein folding system, are amenable to multi-protein complexes, have many of the same post-translational processes as higher eukaryotes, and can be grown in large volumes for large protein expression[21,22]. However, attempts to express human GYS1 alone in insect cells frequently resulted in insoluble expression of an inactive enzyme; suggesting that a binding partner might be necessary for robust expression. Our recent structural work indicates that the surface of glycogen synthase contains as many as four distinct glycogen binding sites on the surface of each subunit[11]. In addition, mammalian glycogen synthase is stoichiometrically associated with glycogen particles in cells, such that precipitation of glycogen is generally the first step for high yield purification of glycogen synthase from mammalian tissues[14]. Consequently, we reasoned that there may insufficient numbers of glycogen particles in the cultured insect cells to completely solubilize the over-expressed enzyme. Since glycogenin is both a binding partner of glycogen synthase and the initiator for the production of glycogen particles, we reasoned that co-expression of both glycogen synthase and glycogenin might improve soluble expression of glycogen synthase in insects in a manner similar to that demonstrated for mammalian cell culture[9].
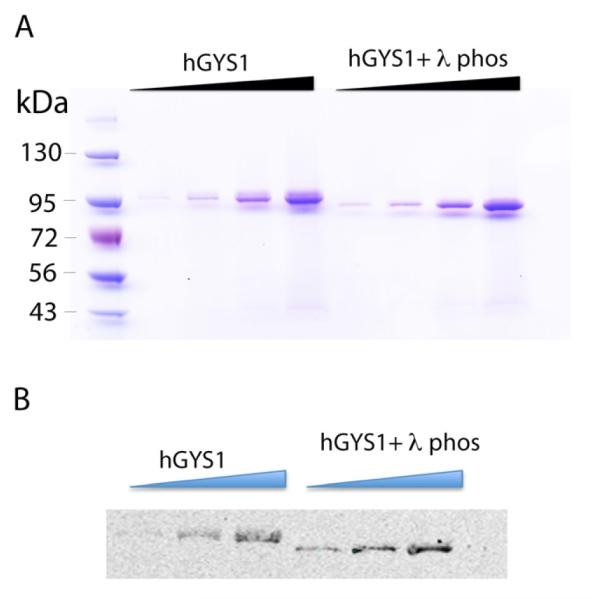
To test our hypothesis, we decided to express the hGYS1 protein and glycogenin together in the MultiBac expression system, which is specficically tailored for expression of mulitprotein complexes[21]. The coding regions for both hGYS1 and rGYG1 were cloned into the multicloning sites 1 and 2 of the pFL baculovirus transfer vector (Fig. 2) [22]. The multigene construct containing hGYS1 and rGYG1 gene was integrated into the engineered MultiBac genome by Tn7 transposase using transposition sites Tn7L and Tn7R (Fig. 2), followed by production of baculovirus in Sf9 cells. Proteins were co-expressed using Hi5 cells, the latter insect cells are used for protein expression. The CBD binds with high affinity to chitin binding beads and following extensive washing steps; the hGYS1 protein is eluted by addition of DTT. Since the regulation of glycogen synthase includes interactions of its N- and C-terminal regulatory extensions[3], an additional feature of this purification strategy is that the intein cleavage site between the CBD and hGYS1 generates a construct lacking any purification tags that may affect its regulation by G6P or phosphorylation. Using this approach, we obtain high yields of hGYS1 (10 mg/L of culture) protein that is greater than 95% pure (Fig. 2B). Western blot analysis of the protein at 95 kDa using a glycogen synthase-specific antibody confirmed that the purified protein is indeed hGYS1 (Fig. 2B).
As Hi5 cells are capable of post-translational modifications to the hGYS1 enzyme, we next examined whether hGYS1 produced in this manner was phosphorylated. Prior work had demonstrated that phosphorylated and dephosphorylated glycogen synthase can be separated by SDS-PAGE[23]. Consequently, purified hGYS1 was treated with lambda phosphatase, a non-specific phosphatase capable of removing most protein phosphorylations, and resulting protein resolved on a polyacrylamide gel and compared to untreated hGYS1 (Fig. 2B). Consistent with prior work[14], phosphatase treatment resulted in an increased mobility of hGYS1 (Fig. 3A,B), implying that the protein produced in Hi5 cells is phosphorylated. Western blot analysis of the protein using a glycogen synthase-specific antibody confirms that the purified protein at 95 kDa is hGYS1 and confirms electrophoretic mobility upon incubation with the phosphatase (Fig. 3B).
Fig. 3.
hYGS1 purification. A) Panel shows scan of coomassie stained polyacrylamide gel of increasing concentrations of hGYS1 (ranging from 0.2 μg to 2 μg in four lanes) in the absence and presence of lambda phosphatase. B) Panel shows Western blot analysis of hyGS1 (0.4 μg to 2 μg in three lanes) using a glycogen synthase-specific antibody. All gels were run on a 4-20% polyacrylamide gel (Life technologies). The Marker lane contains 10 uL of pre-stained protein marker EZ-Run pre-stained Rec protein ladder (ThermoFisher). All samples treated with lambda phosphatase are referred to as hGYS1 + λ phos.
Functional Assay
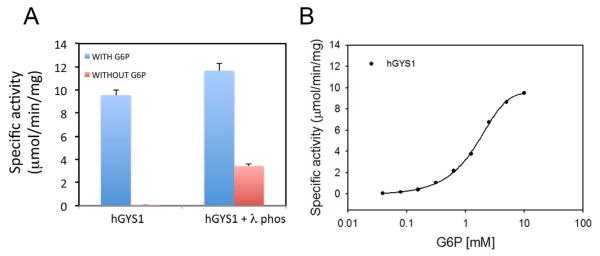
As the physical properties of the recombinant hGYS1 suggested that the protein was phosphorylated, we characterized the enzyme activity in the presence and absence of its allosteric activator, G6P. G6P can overcome the inhibitory effects of phosphorylation and restore full catalytic activity to glycogen synthase[3]. Because of this property, it is possible to assess the activity state of glycogen synthase by measuring activity in the absence and presence of G6P[3]. The ratio of the activity in the absence of G6P to that in the presence of G6P is a measure of its phosphoryation state and this activity ratio can vary from less than 0.01 (the phosphoryated enzyme having less than 1% activity compared to the fully active form) to approximately 0.6[24]. We measured the specific activity of Hi5-purified hGYS1, as well as hGYS1 treated with the lambda phosphatase enzyme in the presence and absence of G6P (Fig. 4A).
Fig. 4.

Glycogen Synthase activity. A) Glycogen synthase activity of hGSY1 (+/− lambda phosphatase) in the presence and absence of glucose 6-phosphate (G6P). B) Half-maximal activation is determined using measurements of specific activity for hGYS1 in the presence of varying concentration of G6P (0.01 to 10 mM). All activity shown are measured in μmol/min/mg.
In the presence of G6P, the activity was measured to be 9.5 μmol/min/mg for hGYS1 (n=3) and 11.6 μmol/min/mg for the lambda phosphatase treated hGYS1 (n=3). In the absence of G6P, there was little detectable activity for hGYS1 (0.029 μmol/min/mg), but the specific activity for the form incubated with lambda phosphatase was 3.42 μmol/min/mg (Fig. 4A). The activity ratio (−G6P/+G6P) was calculated to be 0.003 for purified recombinant hGYS1 and 0.29 for the lambda phosphatase treated form of hGYS1. This low activity ratio (0.003) is indicative of a highly phosphorylated form of the enzyme, consistent with the change in mobility on SDS-PAGE upon phosphatase treatment. We compared these activity ratios to those previously measured for glycogen synthase expressed different cell types such as COS and Rat-1 cells[25]. Skurat et. al. measured activity ratios for wild type (wt) protein as well as enzyme forms where every known phosphorylation site was altered by site-directed mutagenesis. The activity ratio for synthase in COS cells was measured at 0.009 whereas a form of glycogen synthase with mutations at both sites 2 and 3a exhibited the highest activity ratio of 0.590[4]. Similar studies in Rat-1 cells demonstrated activity ratios between 0.014 for the native enzyme and 0.42 for a form of glycogen synthase where all potential phosphorylation sites were changed to alanine except site 2a[4]. The activity ratio we obtained for hGYS1 purified in insect cells is in the same range as that found for synthase in COS and Rat-1 cells. However, our lower activity ratios for the phosphatase treated enzyme may reflect its different sites of phosphorylation and/or incomplete removal of regulatory phosphates by lambda phosphatase.
A dose-dependent titration of G6P to measure the concentration of G6P activation was also performed for purified recombinant hGYS1 (Fig. 3B). We obtained a Ka value for G6P of 1.8 mM (Fig. 3B) is nearly identical to the value obtained for glycogen synthase purified from rabbit muscle of 1.6 mM[26].
It is clear from the almost undetectable level of activity in the absence of G6P and the mobility shift upon phosphatase treatment that our hGYS1 protein is highly phosphorylated, possibly at several sites. Because of the complex pattern of phosphorylation that is hierarchal in nature[4], and because multiple kinases are involved in the phosphorylation of the nine known sites in hGYS1 (Fig. 1), it is not possible to determine the exact level of phosphorylation or which sites are phosphorylated without detailed analysis by mass spectrometry.
Analysis of hGYS1 by Mass Spectrometry
To investigate the sites at which hGYS1 may be phosphorylated, we undertook a mass spectrometric analysis of our recombinant glycogen synthase. Following digestion of the protein at lysine and arginine residues and tandem mass spectrometry combined with phosphoRS analysis, the probable sites of modifications were revealed. Following post-translational modification mapping of serine, threonine, and tyrosine phosphorylation; lysine acetylation; and methionine oxidation using SEQUEST and X!Tandem, we were able to obtain 65% sequence coverage for hGYS1 and 42% sequence coverage for rGYG1 (with a false discovery rate of 0.23% and a mimimum peptide confidence of 90%). This result is consistent with the presence of glycogenin as the 40 kDa contaminant comprising the second most abundant protein in our hGSY1 purification. Muscle glycogen synthase from higher eukaryotes is subject to phosphorylation at nine distinct sites (Fig. 1). Several Ser/Thr-directed protein kinases are responsible for phosphorylation of these residues[3]. Fig. 5 and Table 1 highlight the peptides and relative probability scores from our analysis of recombinant hGYS1 phosphorylation. Fig. 5 highlights the different peptides as they were identified by SEQUEST/X!TANDEM (highlighted in green) and the site probabilities of phosphorylation as identified by PhosphoRS. We identified multiple peptides in different regions of the protein which could be subjected to phosphorylation but found no evidence of acetylation. We detected phosphorylation at the known sites 3a (S641), 3b (S645), 3c (S649), 4 (S652), 5 (S657) and 1b (S710) (Fig. 5 and Table 1). In addition to these well-characterized sites in glycogen synthase, there may be additional sites of phosphorylation for hGYS1 purified from insect cells; including T278, S412, S593, S647, S653 and S727 as surmised from their high probability scores (Table 1).
Fig. 5.

Post-translational modification mapping by LC/MS/MS for hGYS1. The complete amino acid sequence for hgys1 is shown with all peptides detected through database searches shaded as indicated in the peptide identification legend to the right. Phosphorylation sites are indicated with a ‘P’ above the modified residue(s) with color coded boxes shown to indicate the phosphorylation site probability range as determined by analysis with phosphoRS within Proteome Discoverer 1.3 (legend to the right). In addition, the phosphorylated residues which were detected by high confidence spectral mapping are listed to the left.
Table 1.
Sites of phosphorylation as detected by Mass spectrometry analysis of digested hGYS1 with known sites of phosphorylation indicated.
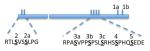
| Sites of phosphorylation |
Protein Position |
|---|---|
| 2a | 7 |
| 2b | 10 |
| 3a | 641 |
| 3b | 645 |
| 3c | 649 |
| 4 | 653 |
| 5 | 657 |
| 1a | 697 |
| 1b | 710 |
Phosphorylation of serine 412 was found to occur rarely (44/698 spectra phosphorylated) on three distinct peptides spanning amino acids 403-418. A peptide spanning amino acids 636-650 was found to be highly modified with 6.7% of all peptides being mono-phosphorylated, 61.7% di-phosphorylated, 8.7% tri-phosphorylated, and 16.7% tetra-phosphorylated. This indicates that serine residues 641, 645, 647, and 649 can all be phosphorylated in Hi5 cells.
We identified a peptide covering amino acids 651-674 which was found to be tri-phosphorylated each time it was identified (6/6) for sites 4 and 5 (amino acids 652 and 657). This +3 peptide fragmented poorly by collision induced dissociation (CID) leading to relatively low SEQUEST XCorr values (2.65-3.75) for these peptides. However, this peptide demonstrated both single and double neutral loss values with fragment ions covering phospho-S657 further supporting that this peptide is truly phosphorylated. The unmodified version of this peptide spanned amino acids 651-664 and was identified twice.
We identified 4 different peptides covering the C-terminus of hGYS1 spanning residues 708-736, 708-737, 709-736, and 709-737. We found that 59.3% of these peptides were phosphorylated. Specifically 33.4% of the C-terminal peptides were mono-phosphorylated, 14.9% were di-phosphorylated, 7.3% were tri-phosphorylated, and 3.7% were tetra-phosphorylated. Using the phosphoRS site prediction in conjunction with manual spectrum validation the most likely serine residues for phosphorylation in this sequence were S710, S723, S727, and S731 (with phosphoRS scores of 99, 95.1, 86.5, and 86.3 respectively). Although additional serine residues were assigned as phosphorylated by both SEQUEST and X!Tandem many of those identifications were ambiguous, lacking specific fragment ion coverage, and could therefore also be assigned to S710, S723, S727, and/or S731.
We did not detect the peptide covering site 1a (S698) nor the peptide containing the N-terminal sites of phosphorylation (sites 2 and 2a, S8 and S11) in either phosphorylated or unmodified form. The lack of detection for the peptide containing sites 2 and 2a is likely due to the large size of this tryptic peptide (amino acids 6-39).
The heavy and non-canonical phosphorylation of our recombinant hGYS1 is likely due to the combined action of both insect homologs to the specific protein kinases that regulate human glycogen synthase as well as non-specific action of other protein kinases due to the high levels of expression in these cells.
Conclusions
Numerous attempts were made to produce active recombinant human glycogen synthase. We show here that the only condition that reproducibly yielded soluble active protein was co-expression in insect cells with glycogenin. We propose that glycogenin allows hGYS1 to remain soluble and bound to the glycogen particle similar to what was previously observed in COS cells[9]. The ability to express substantial amounts of stable glycogen synthase will permit detailed structural and functional characterization of this complex and highly regulated enzyme, as well as the construction of various forms of the enzyme with different sites of phosphorylation through the use of intein-mediated peptide fusions and site-directed mutagenesis, leading towards an in-depth understanding of the mechanism of regulation by hGYS1.
Highlights.
Human Glycogen Synthase 1 (hGYS1) is purified by co-expression with Glycogenin
Recombinant hGYS1 produced in insect Hi5 cells is functional
Recombinant hGYS1 is highly phosphorylated
Acknowledgements
We would like to thank Dr. Imre Berger (EMBL Grenoble) for providing the reagents for MultiBac system. T.D.H. was supported by NIH grants R01-DK079887 and R37-DK027221. Y.T. was supported by US National Science Foundation grant (MCB 0843026 and MCB 1157688), an American Heart Association Scientist Development Award 0735395 and the Showalter Charitable Trust. TI was supported through a Human Frontier Science Program long-term fellowship and V.M.C and S.P-M. were supported by NIH T32-DK064466.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers hat apply to the journal pertain.
References
- [1].YOUNG FG. Claude Bernard and the Discovery of Glycogen | BMJ. British Medical Journal. 1957 doi: 10.1136/bmj.1.5033.1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Preiss J, Walsh DA. [CITATION][C] Biology of Carbohydrates. 1981 [Google Scholar]
- [3].Roach PJ, Depaoli-Roach AA, Hurley TD, Tagliabracci VS. Glycogen and its metabolism: some new developments and old themes. Biochem. J. 2012;441:763–787. doi: 10.1042/BJ20111416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Roach PJ. Control of glycogen synthase by hierarchal protein phosphorylation. Faseb J. 1990;4:2961–2968. [PubMed] [Google Scholar]
- [5].Roach PJ. Multisite and hierarchal protein phosphorylation. J. Biol. Chem. 1991;266:14139–14142. [PubMed] [Google Scholar]
- [6].Lomako J, Lomako WM, Whelan WJ. A self-glucosylating protein is the primer for rabbit muscle glycogen biosynthesis. The FASEB Journal. 1988 doi: 10.1096/fasebj.2.15.2973423. [DOI] [PubMed] [Google Scholar]
- [7].SMYTHE C, COHEN P. The discovery of glycogenin and the priming mechanism for glycogen biogenesis. Eur. J. Biochem. 1991 doi: 10.1111/j.1432-1033.1991.tb16225.x. [DOI] [PubMed] [Google Scholar]
- [8].Skurat AV, Dietrich AD, Roach PJ. Interaction between glycogenin and glycogen synthase. Arch. Biochem. Biophys. 2006;456:93–97. doi: 10.1016/j.abb.2006.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Skurat AV, Cao Y, Roach PJ. Glucose control of rabbit skeletal muscle glycogenin expressed in COS cells. J. Biol. Chem. 1993;268:14701–14707. [PubMed] [Google Scholar]
- [10].Pederson BA, Cheng C, Wilson WA, Roach PJ. Regulation of glycogen synthase. Identification of residues involved in regulation by the allosteric ligand glucose-6-P and by phosphorylation. J. Biol. Chem. 2000;275:27753–27761. doi: 10.1074/jbc.M003342200. [DOI] [PubMed] [Google Scholar]
- [11].Baskaran S, Roach PJ, Depaoli-Roach AA, Hurley TD. Structural basis for glucose-6-phosphate activation of glycogen synthase. Proc. Natl. Acad. Sci. U.S.a. 2010;107:17563–17568. doi: 10.1073/pnas.1006340107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Fitzgerald DJ, Berger P, Schaffitzel C, Yamada K, Richmond TJ, Berger I. Protein complex expression by using multigene baculoviral vectors. Nat. Methods. 2006;3:1021–1032. doi: 10.1038/nmeth983. [DOI] [PubMed] [Google Scholar]
- [13].Imasaki T, Calero G, Cai G, Tsai K-L, Yamada K, Cardelli F, et al. Architecture of the Mediator head module. Nature. 2011;475:240–243. doi: 10.1038/nature10162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].CAMICI M, DePaoli-Roach AA, Roach PJ. Rabbit liver glycogen synthase. Purification and comparison of the properties of glucose-6-P-dependent and glucose-6-P-independent forms of the enzyme. J. Biol. Chem. 1984;259:3429–3434. [PubMed] [Google Scholar]
- [15].Thomas JA, Schlender KK, Larner J. A rapid filter paper assay for UDPglucose-glycogen glucosyltransferase, including an improved biosynthesis of UDP-14C-glucose. Anal. Biochem. 1968;25:486–499. doi: 10.1016/0003-2697(68)90127-9. [DOI] [PubMed] [Google Scholar]
- [16].Washburn MP, Wolters D, Yates JR. Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nat. Biotechnol. 2001;19:242–247. doi: 10.1038/85686. [DOI] [PubMed] [Google Scholar]
- [17].Wolters DA, Washburn MP, Yates JR. An automated multidimensional protein identification technology for shotgun proteomics. Anal. Chem. 2001;73:5683–5690. doi: 10.1021/ac010617e. [DOI] [PubMed] [Google Scholar]
- [18].Mosley AL, Sardiu ME, Pattenden SG, Workman JL, Florens L, Washburn MP. Highly reproducible label free quantitative proteomic analysis of RNA polymerase complexes. Mol. Cell Proteomics. 2011;10:M110.000687. doi: 10.1074/mcp.M110.000687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Craig R, Beavis RC. TANDEM: matching proteins with tandem mass spectra. Bioinformatics. 2004;20:1466–1467. doi: 10.1093/bioinformatics/bth092. [DOI] [PubMed] [Google Scholar]
- [20].Taus T, Köcher T, Pichler P, Paschke C, Schmidt A, Henrich C, et al. Universal and confident phosphorylation site localization using phosphoRS. J. Proteome Res. 2011;10:5354–5362. doi: 10.1021/pr200611n. [DOI] [PubMed] [Google Scholar]
- [21].Bieniossek C, Imasaki T, Takagi Y, Berger I. MultiBac: expanding the research toolbox for multiprotein complexes. Trends Biochem. Sci. 2012;37:49–57. doi: 10.1016/j.tibs.2011.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Bieniossek C, Richmond TJ, Berger I. MultiBac: multigene baculovirus-based eukaryotic protein complex production. Curr Protoc Protein Sci. 2008 doi: 10.1002/0471140864.ps0520s51. Chapter 5. Unit 5.20. [DOI] [PubMed] [Google Scholar]
- [23].DePaoli-Roach AA, Vilardo PG, Kim JH, Mavila N, Vemuri B, Roach PJ. Determination of mammalian glycogen synthase phosphatase activity. Meth. Enzymol. 2003;366:17–34. doi: 10.1016/s0076-6879(03)66002-7. [DOI] [PubMed] [Google Scholar]
- [24].Skurat AV, Roach PJ. Phosphorylation of Sites 3a and 3b (Ser(640) Ans Ser(644)) in the Control of Rabbit Muscle Glycogen-Synthase. J. Biol. Chem. 1995;270:12491–12497. doi: 10.1074/jbc.270.21.12491. [DOI] [PubMed] [Google Scholar]
- [25].Skurat AV, Wang Y, Roach PJ. Rabbit skeletal muscle glycogen synthase expressed in COS cells. Identification of regulatory phosphorylation sites. J. Biol. Chem. 1994;269:25534–25542. [PubMed] [Google Scholar]
- [26].Skurat AV, Dietrich AD, Roach PJ. Glycogen synthase sensitivity to insulin and glucose-6-phosphate is mediated by both NH2- and COOH-terminal phosphorylation sites. Diabetes. 2000;49:1096–1100. doi: 10.2337/diabetes.49.7.1096. [DOI] [PubMed] [Google Scholar]