

Abstract
The promise of genomic medicine has received great attention over the past decade, projecting how genomics will soon guide the prevention, diagnosis, and treatment of human diseases. However, this evolution has been slower than forecast, even where evidence is often strong (e.g., pharmacogenomics). Reasons include the requirement for institutional resources and the need for the will to push beyond barriers impeding health-care changes. Here, we illustrate how genomics has been deployed to advance the treatment of childhood leukemia.
Keywords: pharmacogenomics, genetic testing, personalized medicine, cancers, pharmacokinetics
Health care is in the early stages of translating the promise of “genomic medicine” into evidence-based strategies to enhance the use of medications and the treatment of human diseases. This represents an evolution from the current strategy of selecting medications and their dosages on the basis of population data (average dose) and “trial and error” (if not marketing propaganda) to an approach that uses genomic criteria (among others) to make decisions about what drug and what dosage are best for the individual patient. Unfortunately, today there are many examples of genomic diagnostics that are well established, yet rarely used, including many such examples in pharmacogenomics. Why are diagnostics that are clearly helpful and relatively simple to perform still not routinely used outside of academic medical centers? And why are there still many patients treated within academic medical centers who do not routinely benefit from genomic medicine? Lack of money, time, expertise, and evidence are some of the common explanations.1 Our fractionated health-care system in the United States, the lack of robust decision-support tools, and professional inertia may also be major contributing factors.2
When things are complicated and expensive, taking a stepwise approach can be the best way forward. At our institution, we began to use genomics to individualize the treatment of acute lymphoblastic leukemia (ALL) in the 1980s; we have added many new features in the decades since (Figure 1), and the list continues to expand today. The first steps can be the hardest in a journey that has no end in sight, but when the benefits to patients are clear, the way forward becomes easier. Here, we use the treatment of ALL to illustrate the often stepwise process of translating genomics into diagnostics that guide treatment decisions and to exemplify the potential it holds for improving the outcome of serious human diseases. This review reflects our experiences as collaborators for the past 35 years and emanates from our perspectives as principal investigators, as clinicians, as department chairs, and as an institutional chief executive officer, each offering a slightly different view of how things happened—or didn’t happen. We have had the advantage of doing all this at an institution that encouraged treatment protocols to be conceptually driven and not compromised by short-term medical economics. We acknowledge that working at such a place may not be the “real world,” but we would argue that if this model represents a better way, then the real world (e.g., “payers”) will find a way to embrace it.
Figure 1.
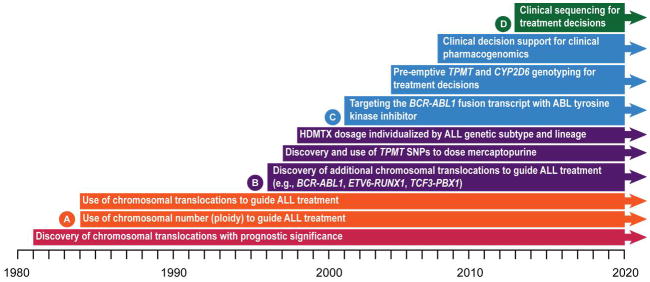
A time line of discovery and clinical implementation of genomics to advance treatment of childhood ALL. A (orange): corresponds to time line for studies Total XI–XII in Figure 2; B (purple): corresponds to time line for studies Total XIII–XIV in Figure 2; C (blue): corresponds to time line for study Total XV in Figure 2; D (green): denotes future clinical use of genomics. ALL, acute lymphoblastic leukemia; SNP, single-nucleotide polymorphism; TPMT, thiopurine methyltransferase.
As depicted in Figure 1, we began using genomics to guide treatment of childhood ALL more than 30 years ago, shortly after it was discovered that specific chromosomal abnormalities in leukemia cells have prognostic importance. In the 1990s, we began to also use inherited genome variations to determine the optimal dosage of medications for individual patients. Today, we continue to expand the discovery and clinical use of both somatic and germline DNA variations that are emerging from a broad spectrum of genomics research and clinical trials.3,4 We hope there is helpful information to be gleaned from our “work in progress” that will prove useful to those working to translate genomics into clinical diagnostics to improve the treatment of human diseases.
Identification of Non-random Chromosomal Abnormalitiesin Leukemia Cells
In 1978, Secker-Walker et al.5 first showed that chromosome number (ploidy) of leukemic cells at diagnosis had prognostic significance in childhood ALL, with hyperdiploidy associated with the most durable response to treatment.By 1981, four chromosomal translocations, including the Philadelphia chromosome, were described in adult ALL,6 and in 1984 the first two phenotype-specific chromosomal translocations in childhood ALL—t(1;19)(q23;p13.3) in pre-B ALL and t(11;14)(p13;q13) in T-cell ALL—were identified at our center.7 Soon thereafter, specific chromosomal translocations were associated with an increased risk of treatment failure.8 During that period, it was shown at our center that flow cytometry could be used to identify hyperdiploid ALL, making the diagnosis of this favorable genetic subtype relatively easy.9
RiskStratification and Treatment Based on Leukemia Cell Genotypes
On the basis of the aforementioned discoveries, the St. Jude ALL Study “Total Therapy XI” (1984–1988) became the first treatment protocol in which genetic features (the presence of chromosomal translocations or hyperdiploidy) were used to assign patients to risk-directed treatments (higher-risk patients received more aggressive therapy).10
Recognizing the favorable prognostic significance of the ETV6-RUNX1 fusion (also known as TEL-AML1),11 we used this genetic abnormality in our subsequent Total Therapy XVprotocol (2000–2007) to identify patients with a low risk of relapse to receive standard (less aggressive) therapy, with subsequent treatment intensified in a small subset that continued to have minimal residual disease (submicroscopic leukemia in the bone marrow, detected by flow cytometry or PCR) at the end of 6 weeks of remission induction therapy.12 In this same protocol (Total XV), the presence of a Philadelphia chromosome containing the BCR-ABL1 fusion, once associated with a dire prognosis, was used together with poor treatment response to remission induction as determined by minimal residual disease level to identify patients to receive allogeneic hematopoietic stem cell transplantation, and more recently to receive an ABL tyrosine kinase inhibitor (e.g., imatinib).13
Optimal use of existing antileukemic agents, more precise risk classification, and improved supportive care have brought the 5-year survival rate of childhood leukemia to >85%. The majority of US children with ALL receive contemporary therapy according to frontline studies at St. Jude or the Children’s Oncology Group, which largely parallel the risk stratification and treatment parameters of the St. Jude protocols. Only St. Jude protocols use preemptive thiopurine methyltransferase (TPMT) genotyping for all patients, but the Children’s Oncology Group studies include guidelines for using TPMT status for dosing mercaptopurine (MP), although this is currently at the clinician’s discretion. By individualizing therapy based on prognostic factors, the 5-year overall survival rate observed in the St. Jude XV study (93.5%) compares favorably with those achieved in other contemporary pediatric ALL trials worldwide (Table 1). This advancement in survival rate has occurred despite the fact that there have been no new frontline antileukemic drugs approved in the past 50 years, except for tyrosine kinase inhibitors, which are used in a small minority of patients as frontline therapy (~2–3% of patients)13, and clofarabine, which is used to treat infants with ALL (~1% of patients).14
Table 1.
| Study group | Years of study | No. of patients | Age (years), range | T-cell ALL, % | 5-Year outcome, % | Data source | ||
|---|---|---|---|---|---|---|---|---|
| Cumulative CNS relapse rate | EFS | Survival | ||||||
| SJCRH XV | 2000–2007 | 498 | 1–18 | 15 | 2.7±0.8 | 85.6±2.9 | 93.5±1.9 | Pui et al. 12 |
| AIEOP-95 | 1995–2000 | 1,743 | 0–18 | 11 | 1.2±0.3 | 75.9±1.0 | 85.5±0.8 | Conter et al.47 |
| BFM-95 | 1995–1999 | 2,169 | 0–18 | 13 | 4.0±0.4 | 79.6±0.9 | 87.0±0.7 | Möricke et al.48 |
| COG | 2000–2005 | 7,153 | 0–21 | 7 | NA | NA | 90.4±0.5 | Hunger et al.49 |
| DCOG-9 | 1997–2004 | 859 | 1–18 | 11 | 2.6±0.6 | 80.6±1.4 | 86.4±1.2 | Veerman et al.50 |
| DFCI00-01 | 2000–2004 | 492 | 1–18 | 11 | NA | 80.0±2 | 91±1 | Vrooman et al.51 |
| NOPHO-2000 | 2002–2007 | 1,023 | 1–15 | 11 | 2.7±0.6 | 79.4±1.5 | 89.1±11 | Schmiegelow et al.52 |
| UKALL 97/99 | 1999–2002 | 938 | 1–18 | 11 | 3.0±0.6 | 80.0±1.3 | 88.0±1.1 | Mitchell et al.53 |
AIEOP, Associazione Italiana di Ematologia ed Oncologia Pediatrica; ALL. acutelymphoblastic leukemia; BFM, Berlin-Frankfurt-Münster; CNS, central nervous system; COG. Children’s Oncology Group; DCOG, Dutch Childhood Oncology Group; DFCI, Dana-Farber Cancer Institute consortium; EPS, event-free survival; NA, not available; NOPHO, Nordic Society of Pediatric Hematology and Oncology; SJCRH, St Jude Children’s Research Hospital; UKALL, United Kingdom Medical Research Council Working Party on Childhood Leukaemia.
Adapted from ref. 14.
Acquired (somatic)and inherited (germline)DNA variation can alterthe pharmacokinetics and pharmacodynamics of antileukemic agents
From early studies showing that pharmacokinetic variability could influence treatment outcome in ALL,15 we sought to understand why there were such large differences (greater than 5-to 10-fold) in both the systemic and intracellular (leukemia cell) pharmacokinetics and pharmacodynamics of antileukemic agents in children. We and others found many sources of variation, some environmental (e.g., hydration, drug interactions) and some genetic.16 Here, we focus on two examples of the latter, using two medications that every child with ALL receives (methotrexate (MTX) and mercaptopurine (MP)).
The intracellular levels of the active form of MTX (MTX polyglutamates) were found to vary 10-fold in leukemia cells across a population of children given the same intravenous dosage of MTX (1μg/m2), with significant differences among specific genetic subtypes of ALL; genome-wide analyses revealed distinct genomic mechanisms for these subtype differences.17 We now use higher doses of MTX in patients with T-lineage ALL and B-lineage ALL with the t(1;19) and TCF3-PBX1 fusion, and lower doses in patients with hyperdiploid ALL who avidly accumulate MTX-polyglutamates in their leukemia cells.17,18 We have more recently identified germline polymorphisms in SLCO1B1 that significantly influence MTX systemic clearance and alter the risk of gastrointestinal toxicity.19,20
The relation between inherited polymorphisms in TPMT and the metabolism and hematopoietic toxicity of MP became a focus of our work after we encountered patients who experienced severe hematopoietic toxicity whenever treated with MP.21,22 Building on early work from the Weinshilboum lab,23 we identified three inherited single-nucleotide polymorphisms that define the major variant alleles associated with inherited TPMT enzyme deficiency.24,25,26 Patients who inherit one or two of these variant alleles are at significantly higher risk of hematopoietic toxicity27 but could be safely treated with reduced doses of MP.16 On the basis of this strong association we now preemptively genotype all ALLg patients for TPMT before their first dose of MP, and adjust their MP dosages accordingly (e.g., a 10-fold dose reduction for patients inheriting two variant TPMT alleles).
We have more recently found that de novo sensitivity of ALL cells to prednisolone, vincristine, asparaginase, or doxorubicin is related to the expression of 20–40 genes (per drug) in ALL cells, and that their expression pattern is drug specific and predictive of treatment outcome.28 Moreover, multidrug cross-resistance (two or more drugs) was related to the expression of a different set of genes and identified patients at the highest risk of relapse.29 Ongoing studies aim to elucidate the mechanisms responsible for these differences in gene expression (e.g., DNA methylation, microRNA expression, DNA copy-number variations, single-nucleotide polymorphisms) and to develop strategies for overcoming resistance by targeting one or more of these genes.
In 2007 we implemented a routine clinical genotyping test for cytochrome P450 2D6 (CYP2D6), a polymorphic gene involved in the metabolism of codeine (a prodrug) to morphine; hence, the efficacy and safety of codeine have been shown to be influenced by CYP2D6 polymorphisms.30,31 We genotype patients to identify CYP2D6 poor metabolizers because these patients are at high risk for a lack of response to codeine, and many of our patients require codeine during the course of their ALL therapy.32 Likewise, we identify patients with a duplication of functional CYP2D6 alleles and alert clinicians at the time of ordering that such patients may be “ultrarapid metabolizers” of drugs metabolized by CYP2D6 and that these patients are at high risk for toxicity with “normal” doses of codeine.
For many years, our group has used single-gene tests for TPMT and CYP2D6 to preemptively guide clinical prescribing decisions for thiopurines and codeine, respectively.33,34 Because there are some inherent disadvantages (e.g., high expense and long turnaround time) associated with determining genotypes one gene at a time, more recently we have begun preemptive array-based pharmacogenomic testing of >200 genes on a single array.4 Unlike single-gene testing, array-based preemptive testing can include a large number of relevant genes that cover many “high-risk” drugs that may be prescribed to a patient over the course of his or her lifetime, and its relatively low cost makes it feasible for every patient entering the health-care system. Making these genotypes available prior to any prescribing decision is consistent with our vision that patient genomes will be considered in every prescribing decision as an inherent patient characteristic, as are gross patient characteristics such as renal and liver function.
Genome-wide Analyses to Identify New Genotypes and Novel Treatment Targets
With the advent of high-resolution genome-wide analyses, we performed some of the first studies of gene expression,35 DNA copy-number alterations,36 and next-generation whole-genome and whole-transcriptome sequencing3 in childhood ALL, providing new insights into leukemogenesis, drug resistance, and potential novel treatment targets. We and others observed that IKZF1 alteration is a hallmark of two high-risk ALL subtypes: Philadelphia chromosome (BCR-ABL1)-positive ALL37 and a new subtype termed “BCR-ABL1-like” ALL.38,39 Among genetic abnormalities identified in BCR-ABL1-like cases, EBF1-PDGFB or NUP214-ABL1 fusions responded to ABL tyrosine kinase inhibitors (which also inhibit PDGFB), and BCR-JAK2 or mutated IL7R responded to a JAK2 inhibitor in preclinical studies.40 In separate analyses, we and others found rearrangement of CRLF2 in up to 8% of childhood ALL and more interestingly in ALL cells of 50–60% of patients with Down syndrome.41,42 In patients with or without Down syndrome, approximately 50% of cases with CRLF2 rearrangements harbor concomitant activating mutations in the Janus kinase genes JAK1 or JAK2, findings leading to a Children’s Oncology Group phase I clinical trial of a JAK inhibitor (ruxolitinib). Whole-genome analysis has also disclosed the mutational spectrum and global transcriptional profile in early T-cell precursor ALL, a recently discovered subtype of T-cell ALL, similar to those of myeloid leukemia.43 To this end, the identification of histone-modifying genes in early T-cell precursor ALL43 and relapsed ALL cases44 suggested that epigenetic therapy may also play a role in the treatment of ALL in future clinical trials. We expect that future studies will further improve the ALL cure rate by discovery of new molecular lesions, coupled with the development of novel targeted treatment through high-throughput genomics and contemporary drug-screening systems.
Therefore, what was once considered a single disease, “ALL”, is now known to comprise numerous subtypes when defined at the genetic level. Of note, every major treatment center for childhood ALL now uses these somatic DNA alterations in ALL cells as diagnostics to determinethe treatment regimen for a child with ALL. These genetic diagnostic tests will become ever more comprehensive as new primary and cooperative genetic abnormalities with prognostic and therapeutic relevance are discovered. Furthermore, ongoing genomic studies are certain to lead to additional novel targeted therapies that are more effective and less toxic than conventional chemotherapy.
Developing decision support in the Electronic Health Record
With the burgeoning amount of genomic data and the complexities of translating the data in specific clinical situations, it will be difficult (if not impossible) for clinicians to remain cognizant of all the genotype–phenotype associations that alter drug effects. In addition, germline pharmacogenomic test results differ from other test results because they have lifelong implications. It is plausible that a pharmacogenomic genotype could be diagnosed many decades before a person is prescribed a medication that is affected by this trait. For this reason, it is essential to deliver genomic information and guidance to clinicians at the point and time of care.45 As implementation of genomics into routine clinical practice progresses, results must be both available statically in the medical record and provided actively as alerts to clinicians at the point of care. To this end, we have instituted both passive clinical decision support such as result interpretations in our electronic health record, and active rules and alerts that alert clinicians only when a high-priority genotype and a prescription for a high-risk medication are both present.4,46
Conclusions
Today, approximately 90% of children with ALL can be cured with current therapy (Figure 2). Yet better treatment is needed for all children, the 10% who are not cured and the 90% who are cured with cytotoxic drugs that are associated with substantial toxicities. Genomics has played an important role in advancing ALL cure rates over the past 25 years and likewise holds promise to radically change the nature of ALL treatment over the coming decade by leading to more targeted medications and by providing diagnostics that will guide the dosage and schedule of medications for each patient. We envisage the day when the entire cancer and germline genomes of every ALL patient will be sequenced at the time of diagnosis and used along with epigenetic variation to select the optimal treatment for each child (until germline genomes are routinely sequenced early in life, we will have to sequence both genomes in each cancer patient).3 Because most cancers have various subclones, the ultimate genomic interrogation would be single-cell DNA sequencing. Interrogation of epigenetic variations will also be increasingly important. Continuing advances in technologies for assessing genome variation (e.g., DNA sequence, epigenetic changes, messenger RNA and microRNA expression) coupled with lower cost and greater availability will eventually make assessment of genome variation the principal diagnostic workup of cancer patients. This, coupled with the discovery and development of additional agents that target mutations underlying human cancers, will lead to greater individualization of cancer treatment. Medications will no longer be selected primarily based on the tissue of origin of the cancer (lung, liver, leukemia); instead, they will be based on the major mutations found in each patient’s cancer cells, regardless of tissue of origin. For this to become a reality, we need better and cheaper technology for genomic characterization, better tools for analyzing genomic data and translating it into actionable findings, and more medications that are targeted to the mutations driving a larger spectrum of human cancers.
Figure 2.
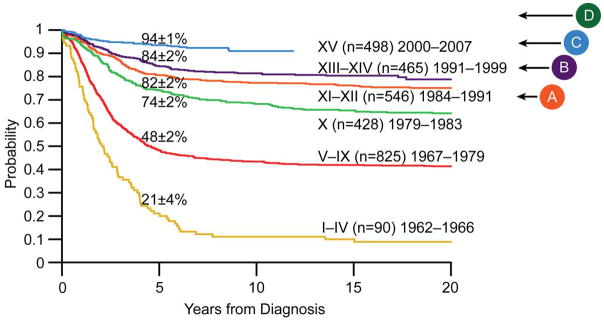
Kaplan–Meier analysis of treatment outcome (survival) for children enrolled in successive clinical trials at St Jude Children’s Research Hospital between 1962 and 2007. Advances in the cure rate of childhood ALL have occurred over this time period despite the introduction of only one new antileukemic agent (imatinib was developed for Philadelphia chromosome (BCR-ABL1)-positive ALL, which constitutes only 3% of childhood ALL cases). Better supportive care has been important (e.g., new antibiotics, new antifungals, and better diagnostics). However, genomics has played an important role in determining the optimal treatment based on both somatic and germline DNA variation. A: institution of use of chromosomal number (or DNA content) and translocations to guide therapy;B: implementation of TPMT genotype to dose mercaptopurine; C: institution of the use of the presence of Philadelphia chromosome (BCR-ABL1) to guide treatment, preemptive genotyping, and clinical decision support; D: use of next-generation sequencing for discovery (2010), clinical sequencing begins (2013). Numbers on the curves denote overall survival rates at 5 years. ALL, acute lymphoblastic leukemia; TPMT, thiopurine methyltransferase.
Our institution is fully committed to both the discovery and translation of genomics as a way to improve treatment of all childhood cancers, seeing the additional short-term expenses as merely a down payment on more cost-effective treatments of the future. For those who think that one day the switch will be flipped and genomic medicine will be a reality, it is not going to happen that way. Rather, this will occur in a stepwise fashion over time (as illustrated here for ALL); those who sit and wait run the risk of looking back one day and seeing that the genomic medicine “train” left the station years ago, without them. Now is the time to come aboard.
Acknowledgments
This work is supported in part by grants from the National Institutes of Health (CA21765, CA36401, and GM92666) and by the American Lebanese Syrian Associated Charities.
Footnotes
Conflict of Interest: W.E.E. and St. Jude Children’s Research Hospital have received patent royalties from TPMT genotyping tests. The other authors declared no conflict of interest.
References
- 1.Stanek EJ, et al. Adoption of pharmacogenomic testing by US physicians: results of a nationwide survey. Clin Pharmacol Ther. 2012;91:450–458. doi: 10.1038/clpt.2011.306. [DOI] [PubMed] [Google Scholar]
- 2.Scott SA. Personalizing medicine with clinical pharmacogenetics. Genet Med. 2011;13:987–995. doi: 10.1097/GIM.0b013e318238b38c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Downing JR, et al. The Pediatric Cancer Genome Project. Nat Genet. 2012;44:619–622. doi: 10.1038/ng.2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Crews KR, Hicks JK, Pui CH, Relling MV, Evans WE. Pharmacogenomics and individualized medicine: translating science into practice. Clin Pharmacol Ther. 2012;92:467–475. doi: 10.1038/clpt.2012.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Secker-Walker LM, Lawler SD, Hardisty RM. Prognostic implications of chromosomal findings in acute lymphoblastic leukaemia at diagnosis. Br Med J. 1978;2:1529–1530. doi: 10.1136/bmj.2.6151.1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bloomfield CD, et al. Chromosomal abnormalities in acute lymphoblastic leukemia. Cancer Res. 1981;41:4838–4843. [PubMed] [Google Scholar]
- 7.Williams DL, et al. New chromosomal translocations correlate with specific immunophenotypes of childhood acute lymphoblastic leukemia. Cell. 1984;36:101–109. doi: 10.1016/0092-8674(84)90078-3. [DOI] [PubMed] [Google Scholar]
- 8.Williams DL, et al. Chromosomal translocations play a unique role in influencing prognosis in childhood acute lymphoblastic leukemia. Blood. 1986;68:205–212. [PubMed] [Google Scholar]
- 9.Look AT, et al. Prognostic importance of blast cell DNA content in childhood acute lymphoblastic leukemia. Blood. 1985;65:1079–1086. [PubMed] [Google Scholar]
- 10.Rivera GK, et al. Improved outcome in childhood acute lymphoblastic leukaemia with reinforced early treatment and rotational combination chemotherapy. Lancet. 1991;337:61–66. doi: 10.1016/0140-6736(91)90733-6. [DOI] [PubMed] [Google Scholar]
- 11.Shurtleff SA, et al. TEL/AML1 fusion resulting from a cryptic t(12;21) is the most common genetic lesion in pediatric ALL and defines a subgroup of patients with an excellent prognosis. Leukemia. 1995;9:1985–1989. [PubMed] [Google Scholar]
- 12.Pui CH, et al. Treating childhood acute lymphoblastic leukemia without cranial irradiation. N Engl J Med. 2009;360:2730–2741. doi: 10.1056/NEJMoa0900386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schultz KR, et al. Improved early event-free survival with imatinib in Philadelphia chromosome-positive acute lymphoblastic leukemia: a children’s oncology group study. J Clin Oncol. 2009;27:5175–5181. doi: 10.1200/JCO.2008.21.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pui CH, et al. Biology, risk stratification, and therapy of pediatric acute leukemias: an update. J Clin Oncol. 2011;29:551–565. doi: 10.1200/JCO.2010.30.7405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Evans WE, Relling MV, Rodman JH, Crom WR, Boyett JM, Pui CH. Conventional compared with individualized chemotherapy for childhood acute lymphoblastic leukemia. N Engl J Med. 1998;338:499–505. doi: 10.1056/NEJM199802193380803. [DOI] [PubMed] [Google Scholar]
- 16.Cheok MH, Evans WE. Acute lymphoblastic leukaemia: a model for the pharmacogenomics of cancer therapy. Nat Rev Cancer. 2006;6:117–129. doi: 10.1038/nrc1800. [DOI] [PubMed] [Google Scholar]
- 17.Kager L, et al. Folate pathway gene expression differs in subtypes of acute lymphoblastic leukemia and influences methotrexate pharmacodynamics. J Clin Invest. 2005;115:110–117. doi: 10.1172/JCI22477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Panetta JC, Sparreboom A, Pui CH, Relling MV, Evans WE. Modeling mechanisms of in vivo variability in methotrexate accumulation and folate pathway inhibition in acute lymphoblastic leukemia cells. PLoS Comput Biol. 2010;6:e1001019. doi: 10.1371/journal.pcbi.1001019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Treviño LR, et al. Germline geneticvariation in an organic anion transporter polypeptide associated with methotrexate pharmacokinetics and clinical effects. J Clin Oncol. 2009;27:5972–5978. doi: 10.1200/JCO.2008.20.4156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ramsey LB, et al. Rare versus common variants in pharmacogenetics: SLCO1B1 variation and methotrexate disposition. Genome Res. 2012;22:1–8. doi: 10.1101/gr.129668.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Evans WE, Horner M, Chu YQ, Kalwinsky D, Roberts WM. Altered mercaptopurine metabolism, toxic effects, and dosage requirement in a thiopurine methyltransferase-deficient child with acute lymphocytic leukemia. J Pediatr. 1991;119:985–989. doi: 10.1016/s0022-3476(05)83063-x. [DOI] [PubMed] [Google Scholar]
- 22.Evans WE, et al. Preponderance of thiopurine S-methyltransferase deficiency and heterozygosity among patients intolerant to mercaptopurine or azathioprine. J Clin Oncol. 2001;19:2293–2301. doi: 10.1200/JCO.2001.19.8.2293. [DOI] [PubMed] [Google Scholar]
- 23.Lee D, et al. Thiopurine methyltransferase pharmacogenetics. Cloning of human liver cDNA and a processed pseudogene on human chromosome 18q21.1. Drug Metab Dispos. 1995;23:398–405. [PubMed] [Google Scholar]
- 24.Krynetski EY, Schuetz JD, Galpin AJ, Pui CH, Relling MV, Evans WE. A single point mutation leading to loss of catalytic activity in human thiopurine S-methyltransferase. Proc Natl Acad Sci USA. 1995;92:949–953. doi: 10.1073/pnas.92.4.949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tai HL, et al. Thiopurine S-methyltransferase deficiency: two nucleotide transitions definethe most prevalent mutant allele associated with loss of catalytic activity in Caucasians. Am J Hum Genet. 1996;58:694–702. [PMC free article] [PubMed] [Google Scholar]
- 26.Yates CR, et al. Molecular diagnosis of thiopurine S-methyltransferase deficiency: genetic basis for azathioprine and mercaptopurine intolerance. Ann Intern Med. 1997;126:608–614. doi: 10.7326/0003-4819-126-8-199704150-00003. [DOI] [PubMed] [Google Scholar]
- 27.Relling MV, et al. Mercaptopurine therapy intolerance and heterozygosity at the thiopurine S-methyltransferase gene locus. J Natl Cancer Inst. 1999;91:2001–2008. doi: 10.1093/jnci/91.23.2001. [DOI] [PubMed] [Google Scholar]
- 28.Holleman A, et al. Gene-expression patterns in drug-resistant acute lymphoblastic leukemia cells and response to treatment. N Engl J Med. 2004;351:533–542. doi: 10.1056/NEJMoa033513. [DOI] [PubMed] [Google Scholar]
- 29.Lugthart S, et al. Identification of genes associated with chemotherapy crossresistance and treatment response in childhood acute lymphoblastic leukemia. Cancer Cell. 2005;7:375–386. doi: 10.1016/j.ccr.2005.03.002. [DOI] [PubMed] [Google Scholar]
- 30.Kirchheiner J, et al. Pharmacokinetics of codeine and its metabolite morphine in ultra-rapid metabolizers due to CYP2D6 duplication. Pharmacogenomics J. 2007;7:257–265. doi: 10.1038/sj.tpj.6500406. [DOI] [PubMed] [Google Scholar]
- 31.Lötsch J, Rohrbacher M, Schmidt H, Doehring A, Brockmöller J, Geisslinger G. Can extremely low or high morphine formation from codeine be predicted prior to therapy initiation? Pain. 2009;144:119–124. doi: 10.1016/j.pain.2009.03.023. [DOI] [PubMed] [Google Scholar]
- 32.Crews KR, et al. Clinical Pharmacogenetics Implementation Consortium. Clinical Pharmacogenetics Implementation Consortium (CPIC) guidelines for codeine therapy in the context of cytochrome P450 2D6 (CYP2D6) genotype. Clin Pharmacol Ther. 2012;91:321–326. doi: 10.1038/clpt.2011.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Relling MV, Pui CH, Cheng C, Evans WE. Thiopurine methyltransferase in acute lymphoblastic leukemia. Blood. 2006;107:843–844. doi: 10.1182/blood-2005-08-3379. [DOI] [PubMed] [Google Scholar]
- 34.Crews KR, et al. Development and implementation of a pharmacist-managed clinical pharmacogenetics service. Am J Health Syst Pharm. 2011;68:143–150. doi: 10.2146/ajhp100113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yeoh EJ, et al. Classification, subtype discovery, and prediction of outcome in pediatric acute lymphoblastic leukemia by gene expression profiling. Cancer Cell. 2002;1:133–143. doi: 10.1016/s1535-6108(02)00032-6. [DOI] [PubMed] [Google Scholar]
- 36.Mullighan CG, et al. Genome-wide analysis of genetic alterations in acute lymphoblastic leukaemia. Nature. 2007;446:758–764. doi: 10.1038/nature05690. [DOI] [PubMed] [Google Scholar]
- 37.Mullighan CG, et al. BCR-ABL1 lymphoblastic leukaemia is characterized by the deletion of Ikaros. Nature. 2008;453:110–114. doi: 10.1038/nature06866. [DOI] [PubMed] [Google Scholar]
- 38.Mullighan CG, et al. Children’s Oncology Group. Deletion of IKZF1and prognosis in acute lymphoblastic leukemia. N Engl J Med. 2009;360:470–480. doi: 10.1056/NEJMoa0808253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Den Boer ML, et al. A subtype of childhood acute lymphoblastic leukaemia with poor treatment outcome: a genome-wide classification study. Lancet Oncol. 2009;10:125–134. doi: 10.1016/S1470-2045(08)70339-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Roberts KG, et al. Genetic alterations activating kinase and cytokine receptor signaling in high-risk acute lymphoblastic leukemia. Cancer Cell. 2012;22:153–166. doi: 10.1016/j.ccr.2012.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mullighan CG, et al. Rearrangement of CRLF2 in B-progenitor-and Down syndrome-associated acute lymphoblastic leukemia. Nat Genet. 2009;41:1243–1246. doi: 10.1038/ng.469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mullighan CG, et al. JAK mutations in high-risk childhood acute lymphoblastic leukemia. Proc Natl Acad Sci USA. 2009;106:9414–9418. doi: 10.1073/pnas.0811761106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang J, et al. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature. 2012;481:157–163. doi: 10.1038/nature10725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mullighan CG, et al. CREBBP mutations in relapsed acute lymphoblastic leukaemia. Nature. 2011;471:235–239. doi: 10.1038/nature09727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pulley JM, et al. Operational implementation of prospectivegenotyping for personalized medicine: the design of the Vanderbilt PREDICT project. Clin Pharmacol Ther. 2012;92:87–95. doi: 10.1038/clpt.2011.371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hicks JK, et al. A clinician-driven automated system for integration of pharmacogenetic interpretations into an electronic medical record. Clin Pharmacol Ther. 2012;92:563–566. doi: 10.1038/clpt.2012.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Conter V, et al. Associazione Italiana di Ematologia ed Oncologia Pediatrica. Long-term results of the Italian Association of Pediatric Hematology and Oncology (AIEOP) Studies 82, 87, 88, 91 and 95 for childhood acute lymphoblastic leukemia. Leukemia. 2010;24:255–264. doi: 10.1038/leu.2009.250. [DOI] [PubMed] [Google Scholar]
- 48.Möricke A, et al. Long-term results of five consecutive trials in childhood acute lymphoblastic leukemia performed by the ALL-BFM study group from 1981 to 2000. Leukemia. 2010;24:265–284. doi: 10.1038/leu.2009.257. [DOI] [PubMed] [Google Scholar]
- 49.Hunger SP, et al. Improved survival for children and adolescents with acute lymphoblastic leukemia between 1990 and 2005: a report from the children’s oncology group. J Clin Oncol. 2012;30:1663–1669. doi: 10.1200/JCO.2011.37.8018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Veerman AJ, et al. Dutch Childhood Oncology Group. Dexamethasone-based therapy for childhood acute lymphoblastic leukaemia: results of the prospective Dutch Childhood Oncology Group (DCOG) protocol ALL-9 (1997–2004) Lancet Oncol. 2009;10:957–966. doi: 10.1016/S1470-2045(09)70228-1. [DOI] [PubMed] [Google Scholar]
- 51.Vrooman LM, et al. Post induction dexamethasone and individualized dosing of Escherichia coli L-asparaginase each improve outcome of children and adolescents with newly diagnosed acute lymphoblastic leukemia: results from a randomized study, Dana-Farber Cancer Institute ALL Consortium Protocol 00–01. J Clin Oncol. 2013 doi: 10.1200/JCO.2012.43.2070. e-pub ahead of print 28 January 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schmiegelow K, et al. Nordic Society of Paediatric Haematology and Oncology. Long-term results of NOPHO ALL-92 and ALL-2000 studies of childhood acute lymphoblastic leukemia. Leukemia. 2010;24:345–354. doi: 10.1038/leu.2009.251. [DOI] [PubMed] [Google Scholar]
- 53.Mitchell C, Richards S, Harrison CJ, Eden T. Long-term follow-up of the United Kingdom medical research council protocols for childhood acute lymphoblastic leukaemia, 1980–2001. Leukemia. 2010;24:406–418. doi: 10.1038/leu.2009.256. [DOI] [PMC free article] [PubMed] [Google Scholar]