

Abstract
Abnormal activation of Mer kinase has been implicated in the oncogenesis of many human cancers including acute lymphoblastic and myeloid leukemia, non-small cell lung cancer, and glioblastoma. We have discovered a new family of small molecule Mer inhibitors, pyrazolopyrimidine sulfonamides, that potently inhibit the kinase activity of Mer. Importantly, these compounds do not demonstrate significant hERG activity in the PatchXpress assay. Through structure-activity relationship studies, 35 (UNC1062) was identified as a potent (IC50 = 1.1 nM) and selective Mer inhibitor. When applied to live tumor cells, UNC1062 inhibited Mer phosphorylation and colony formation in soft agar. Given the potential of Mer as a therapeutic target, UNC1062 is a promising candidate for further drug development.
Keywords: Mer inhibitors, pyrazolopyrimidines, sulfonamides, leukemia, non-small cell lung cancer, glioblastoma
INTRODUCTION
Mer belongs to the TAM (Tyro3, Axl, and Mer) family of receptor tyrosine kinases (RTKs).1 Under normal physiological conditions, Mer kinase promotes cell proliferation and survival, platelet aggregation, macrophage clearance of apoptotic cells, and cytokine release.2 Abnormal activation of Mer has been implicated in the oncogenesis of many human cancers and is often associated with poor prognostic indicators.2–3 For example, Mer is ectopically expressed in both B- and T-cell acute lymphoblastic leukemias (ALL)4 but not in normal mouse and human T- and B-lymphocytes at any stage of development. Similarly, Mer is expressed in the majority of acute myeloid leukemia (AML) cell lines and patient samples but not in normal bone marrow myeloid cells.5 Finally, Mer is frequently overexpressed in solid tumors, including non-small cell lung cancer (NSCLC) cell lines6 and patient samples, even though normal human bronchial epithelial (NHBE) cells and lung tissue adjacent to tumors do not express Mer protein.6 Similar findings have been reported in other solid tumors, including glioblastoma (GBM)3b, 7 and metastatic melanoma.8 This tumor specific expression pattern may confer a large therapeutic window - at least in these tumor types. Expression of a Mer transgene in hematopoietic cells leads to development of leukemia and/or lymphoma in mice.9 In addition, when Mer is inhibited by sh-RNA knockdown in leukemia,4c NSCLC,6 or GBM cells,7a they are more susceptible to apoptotic death and exhibit reduced colony formation in soft agar. Moreover, in orthotropic ALL and AML xenograft mouse models, onset and progression of disease were delayed and survival was significantly increased in mice transplanted with Mer knockdown leukemia cell lines relative to mice transplanted with wild-type cell lines.4c, 5 Similarly, in a NSCLC xenograft mouse model growth of tumors is markedly slowed by Mer knockdown. In addition, the combination of standard cytotoxic chemotherapies with shRNA-mediated Mer knockdown results in synergistic tumor cell killing. Taken together, these data demonstrate important roles of Mer for oncogenesis and anti-apoptotic activity in multiple tumor types. Therefore, Mer provides a novel therapeutic target for the treatment of ALL, AML, NSCLC, and other Mer-related diseases. Consequently, Mer inhibitors are expected to both mediate direct anti-tumor, proapoptotic effects and augment chemosensitivity in combination with standard therapies.
Few small molecule Mer inhibitors have been reported,10 among them compound 1 (UNC569) (Figure 1) is the most promising one demonstrating inhibition of Mer at sub-nanomolar concentrations.11 However, UNC569 has undesirable activity toward the human ether-a-go-go-related gene potassium channel (hERG) in the conventional PatchXpress assay (EC50 = 1.7 μM). 12 Inhibition of hERG could lead to long QT syndrome and sudden death and is the reason for the withdrawal of several FDA-approved drugs.13 In an effort to decrease the hERG activity of UNC569 through analog synthesis, we discovered that sulfonamide 2 (Figure 1) had similar activity against Mer kinase with better physical properties. Consequently, compound 2 is a better starting point than UNC569 to develop potent Mer inhibitors as potential therapeutics. Herein we report the structure–activity relationship (SAR) of a series of compounds related to 2 on inhibiting the TAM family kinases and the discovery of potent Mer inhibitors with minimal hERG activity and excellent cellular activity.
Figure 1.
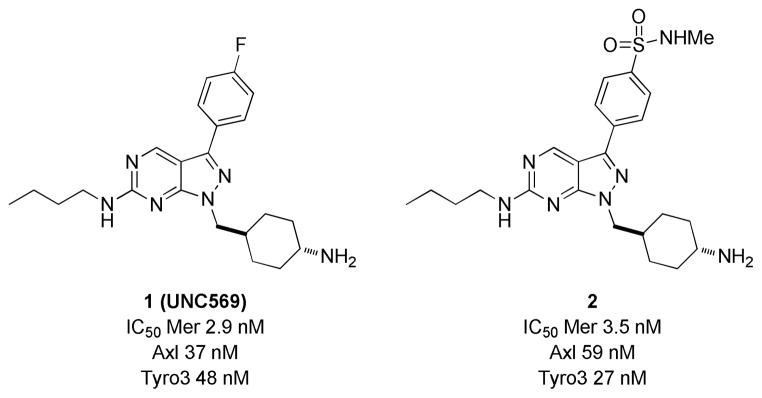
The structures and enzymatic activities of 1 and 2.
SYNTHESIS
The synthetic routes to sulfonamide analogues (Scheme 1) are similar to what we have previously developed for the synthesis of analogues of UNC569.11 Briefly, 3-chlorobenzoperoxoic acid (mCPBA) oxidation of the known compound 314 followed by the displacement of the methyl sulfinyl/sulfonyl group by amines provided the intermediate 4, which was subsequently alkylated at the N1 position of the pyrazole ring to yield 5. Suzuki coupling between 5 and a boronic acid provided the final product 6.
Scheme 1.

The synthetic routes for sulfonamide analogues
RESULTS AND DISCUSSION
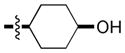
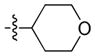
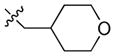
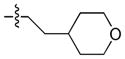
Most hERG inhibitors contain a basic amine group and we thus speculate that the hERG activity of 1 may be due to the presence of a primary amine (NH2) at the R2 position (Table 1). Consequently, we first synthesized a focused library of compounds with different R2 groups while keeping R1 fixed as N-methyl benzenesulfonamide. Inhibition of Mer kinase activity by analogues was tested at the Km for ATP using a microfluidic capillary electrophoresis (MCE) assay15 in which phosphorylated and unphosphorylated substrate peptides were separated and analyzed using a LabChip EZ Reader. For comparison, inhibition of other members in the TAM RTK family, Tyro3 and Axl, was also measured. As shown in Table 1, when the NH2 group at the R2 position (2) was replaced by a hydroxyl (OH) group (7), the activity of the analogue 7 was decreased 6-fold. Increasing the length of the linker between the pyrazole ring and the cyclohexyl group also decreased the activity of compounds (8, 9 vs 2). In contrast, when the length of the linker was shortened and R2 was replaced with a trans-4-hydroxycyclohexyl group, the corresponding analogue 10 had enhanced activity compared to the parent 7. More specifically, the IC50 value was changed from 18 nM for 7 to 2.4 nM for 10, suggesting that the conformational rigidity and the size of R2 was important for Mer inhibition. Consistent with this notion, analogs 12 and 13, which featured open chain versions of cyclohexanol at the R2 position, had decreased Mer inhibition. Using a tetrahydropyran to replace the 4-hydroxycyclohexyl group provided analogues that were less active (14, 15 & 16). Furthermore, a cyclohexyl group alone at the R2 position resulted in a 28-fold weaker inhibitor 17, which further confirmed our previous observation: a hydrogen bond between the polar substituent at the R2 position with the carbonyl of Arg727 of Mer protein was important.11 The configuration of the hydroxyl group was not critical, as the cis-analogue 11 had similar activity as trans-10.
Table 1.
Preliminary SAR at the R2 position
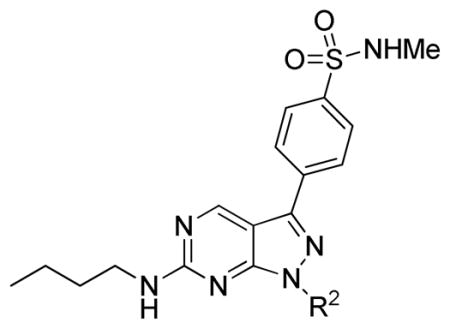
| ||||
|---|---|---|---|---|
| Compound | R2 | IC50 (nM)a
|
||
| Mer | Axl | Tyro3 | ||
| 2 |
|
3.5 | 59 | 27 |
| 7 |
|
18 | 140 | 130 |
| 8 |
|
35 | 120 | 200 |
| 9 |
|
50 | 190 | 82 |
| 10 |
|
2.4 | 74 | 47 |
| 11 |
|
4.4 | 70 | 44 |
| 12 |
|
250 | 2400 | 860 |
| 13 |
|
130 | 5000 | 730 |
| 14 |
|
23 | 450 | 280 |
| 15 |
|
47 | 620 | 810 |
| 16 |
|
54 | 360 | 430 |
| 17 |
|
100 | 26000 | 720 |
Values are the mean of two or more independent assays performed at the ATP Km.
We also tested the activity of the synthetic analogues against Axl and Tyro3 to monitor their selectivity for TAM family members and develop a pan-TAM SAR. In general, these analogues showed modest selectivity within the TAM family and are generally less potent against Axl and Tyro3. In addition, to test our hypothesis regarding on the cause of the hERG activity, compounds 2 & 10 were characterized in the hERG binding assay. Indeed, 10 had no hERG activity in the PatchXpress assay while 2 was active with an EC50 of 3.2 μM. Removal of the primary amine at R2 position eliminated the binding of our compounds to the hERG channel.
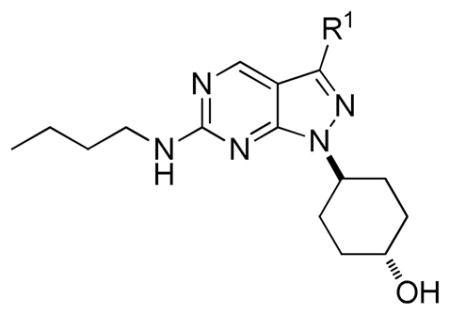
Next, the SAR at the R1 site was explored (Table 2). In general, these analogues are very potent against Mer (at sub-nanomolar concentrations) and have good selectivity for Mer over Axl (> 16 fold) and Tyro3 (> 9 fold). A reversed sulfonamide functional group (18), sulfonic acid (19) and unsubstituted sulfonamide (20) at the R1 site were tolerated. When one of the acidic hydrogens in sulfonamide was substituted with a small alkyl group such as propyl (21) or iso-propyl (22), the activity of the analogues remained the same. In contrast, a cycloalkyl group such as cyclopropyl (23), cyclopentyl (24) and cyclohexyl (25) at this position yielded a more active analogue. A tetrahydropyran (26) or 4-piperidinylmethyl (28) group was also well tolerated at this position while a para-fluorophenyl ring (27) was less suitable. Interestingly, 29, the precursor of 28 with a Boc protection group at piperidinyl nitrogen, was equally potent, which could be explained by the fact that the R1 position was exposed to solvents as observed in the X-ray structure of UNC569 complexed with Mer.11 To further confirm this explanation, we introduced a large side chain at this position and found that the corresponding derivative 30 was still active. Finally, when the acidic hydrogen on the sulfonamide functional group was replaced by a variety of alkyl groups, the activity and TAM RTK-selectivity of the resulting analogues 31–36 were maintained.16
Table 2.
SAR Study of R1
| ||||
|---|---|---|---|---|
| Compound | R1 | IC50 (nM)a
|
||
| Mer | Axl | Tyro3 | ||
| 18 |
|
4.0 | 110 | 35 |
| 19 |
|
1.8 | 130 | 55 |
| 20 |
|
1.4 | 29 | 54 |
| 21 |
|
1.8 | 62 | 35 |
| 22 |
|
3.2 | 120 | 68 |
| 23 |
|
1.1 | 75 | 38 |
| 24 |
|
0.80 | 67 | 31 |
| 25 |
|
1.1 | 350 | 45 |
| 26 |
|
1.2 | 69 | 28 |
| 27 |
|
9.7 | 6500 | 290 |
| 28 |
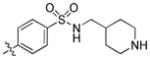
|
1.1 | 55 | 47 |
| 29 |
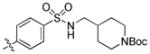
|
1.4 | 73 | 150 |
| 30 |
|
18 | 780 | 320 |
| 31 |
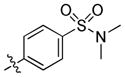
|
1.8 | 71 | 47 |
| 32 |
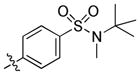
|
1.2 | 110 | 160 |
| 33 |
|
1.0 | 42 | 27 |
| 34 |
|
0.65 | 46 | 22 |
| 35 |
|
1.1 | 85 | 60 |
| 36 |
|
1.4 | 23 | 76 |
Values are the mean of two or more independent assays performed at the ATP Km.
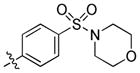
Compared to UNC569, 35 (UNC1062) had improved selectivity and activity against Mer relative to other TAM family RTKs (78-fold selectivity over Axl and 36-fold over Tyro3). Morrison tight-binding inhibition studies revealed that UNC1062 had a Ki of 0.33 nM against Mer.17 These outstanding inhibitory profiles of UNC1062 prompted us to further investigate its activity in intact cell systems.
The ability of UNC1062 to inhibit Mer auto-phosphorylation in different cell lines was first tested. Briefly, human pre-B leukemia 697 cells were grown in suspension and treated with various concentrations of UNC1062 for 1 h prior to addition of a phosphatase inhibitor to stabilize phospho-Mer. Mer protein was immunoprecipitated from lysates and phospho-Mer and total Mer proteins were detected by western blot. In this cell-based assay, the IC50 value for inhibition of phospho-Mer by UNC1062 was 6.4 nM (Figure 2). In comparison, the IC50 of UNC569 in the same assay was 141 nM,11 suggesting that UNC1062 was significantly more potent than UNC569 both with purified Mer kinase and in intact cells.
Figure 2. UNC1062 inhibits accumulation of activated Mer protein in acute leukemia cells.
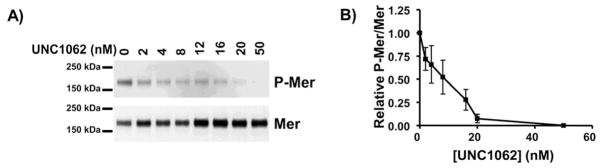
697 cell cultures were treated with the indicated concentrations of UNC1062 for 1 h. Pervanadate was added to cultures for 3 min to stabilize the phosphorylated form of Mer. Mer was immunoprecipitated from cell lysates and total Mer protein and Mer phosphoprotein were detected by immunoblot. A) Representative western blots. B) Relative levels of phospho-Mer and Mer proteins were determined. Mean values +/− standard error derived from 4 independent experiments are shown. IC50 = 6.4 nM with a 95% confidence interval of 3.8 nM to 10.7 nM.
To determine if UNC1062 inhibits Mer phosphorylation in adherent cells, similar studies were performed using a human, adherent, pediatric rhabdoid brain tumor cell line (BT-12) and two NSCLC cell lines (A549 and Colo699). Inhibition of Mer phosphorylation was evident in cells when the concentration of UNC1062 was above 300 nM (Figure 3A). Similarly, phosphorylation of Mer was stably inhibited over 72 h of culture in A549 and Colo699 NSCLC cells treated with 250 nM or 500 nM UNC1062 (Figure 3B). Thus, while solid tumor cells require significantly higher concentrations of UNC1062 compared to non-adherent leukemia cells, possibly as a result of decreased exposure across adherent cell membranes, treatment with UNC1062 at sub-micromolar concentrations was sufficient to inhibit Mer phosphorylation in adherent cell lines.
Figure 3. UNC1062 inhibits Mer activation in adherent tumor cell lines.
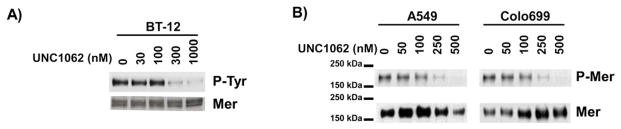
A) BT-12 pediatric rhabdoid tumor cells were treated with UNC1062 or vehicle only for 1 h prior to the addition of pervanadate for 15 min. Cell lysates were prepared, Mer protein was immunoprecipitated, and phospho-tyrosine and Mer protein were detected by western blot. B) The indicated NSCLC cell lines were treated with UNC1062 or an equivalent volume of vehicle only for 72 h prior to treatment with pervanadate for 1.0 (Colo699) or 5.0 (A549) min. Whole cell lysates were prepared, Mer protein was immunoprecipitated, and phosphorylated and total Mer proteins were detected by western blot. The results shown are representative of 3 independent experiments.
To determine if UNC1062 can mediate functional anti-tumor effects, BT-12, A549, and Colo699 cells were cultured in soft agar overlaid with medium containing UNC1062 or vehicle only. Treatment with 1.0 μM UNC1062 was sufficient to completely abrogate growth of BT-12 rhabdoid tumor colonies in soft agar (Figure 4A). Similarly, treatment with UNC1062 inhibited colony formation in both A549 and Colo699 NSCLC cultures. Moreover, inhibition of colony formation in long-term soft agar assays and inhibition of Mer phosphorylation in short-term assays occurred at similar concentrations of UNC1062 in these cell lines, consistent with a causal relationship.
Figure 4. UNC1062 inhibits colony formation in solid tumor cell lines.
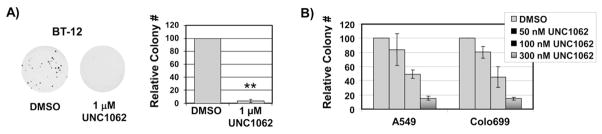
A) BT-12 rhabdoid tumor cells were cultured in soft agar containing 1.0 μM UNC1062 or vehicle only and overlaid with medium containing UNC1062 or vehicle only. Medium and UNC1062 were refreshed twice weekly. Colonies were stained and counted. Representative plates are shown. B) A549 and Colo699 NSCLC cells were cultured in 0.35% soft agar overlaid with medium containing UNC1062 or vehicle only. Medium and UNC1062 were refreshed 3 times per week. Colonies were stained and counted. Mean values ± SEM derived from 3 independent experiments are shown. *Statistically significant results were determined using the student’s paired t test (* p < 0.02, ** p < 0.001 relative to vehicle only).
CONCLUSIONS
In summary, we have identified and synthesized a new family of small molecule Mer inhibitors, pyrazolopyrimidine sulfonamides, which potently inhibit the kinase activity of Mer. Importantly, this new generation of inhibitors do not show significant hERG activity in the PatchXpress assay. Through structure-activity relationship studies, 35 (UNC1062) was identified as both a potent (Ki = 0.33 nM) and selective Mer inhibitor. When applied to live cells, UNC1062 inhibited Mer phosphorylation and colony formation in soft agar. These results provide the first evidence of anti-tumor activity mediated by a member of this novel class of inhibitors, thus further validate Mer as a drug target for cancer.
EXPERIMENTAL
General
Microwave reaction was carried out using a Discover-S reactor with a vertically-focused IR external temperature sensor and an Explorer 72 autosampler. The dynamic mode was used to set up the desired temperature and hold time with the following fixed parameters: PreStirring, 1 min; Pressure, 200 psi; Power, 200 W; PowerMax, off; Stirring, high. Flash chromatography was carried out on pre-packed silica gel disposable columns. Analytical thin-layer chromatography (TLC) was performed with silica gel 60 F254, 0.25 mm pre-coated TLC plates. TLC plates were visualized using UV254 or phosphomolybdic acid with charring. All 1H NMR spectra were obtained with a 400 MHz spectrometer and 13C NMR spectra were obtained with a 100 MHz spectrometer. Preparative HPLC was performed with the UV detection at 220 or 254 nm. LC-MS was performed with the UV detection at 220 nm, 254 nm, and 280 nm, and a single quadrupole mass spectrometer using electrospray ionization (ESI) source. High-resolution (positive ion) mass spectra (HRMS) were acquired using a LCMS-TOF mass spectrometer.
Synthesis
3-Bromo-N-butyl-1H-pyrazolo[3,4-d]pyrimidin-6-amine (4)
To a mixture of 3-bromo-6-(methylthio)-1H-pyrazolo[3,4-d] pyrimidine (24.5 g, 100 mmol) in THF (100 mL) was added meta-chloroperoxybenzoic acid (33.7 g, 77%, 150 mmol) in portions at room temperature. The white mixture was stirred for 2 h and transferred into a THF (50 mL) solution of n-butylamine (49.4 mL, 500 mmol) at 0 °C. The resulting solution was heated at 60 °C for 2.0 h. After removal of the solvent under reduced pressure, MeOH was added and the mixture was filtered. The white solid was washed with MeOH (3x) and dried to afford the title compound 4 (23.6 g, 87%). 1H NMR (400 MHz, DMSO-d6) δ 8.59 (s, 1H), 7.63 (s, 1H), 3.37–3.11 (m, 3H), 1.54–1.41 (m, 2H), 1.29 (dq, J = 14.4, 7.3 Hz, 2H), 0.84 (t, J = 7.3 Hz, 3H); 13C NMR (100 MHz, DMSO-d6) δ 162.0, 157.4, 153.1, 120.5, 107.1, 41.0, 31.0, 20.1, 14.2; MS m/z 270.1 [M+H]+.
4-(1-((trans-4-Aminocyclohexyl)methyl)-6-(butylamino)-1H-pyrazolo[3,4-d]pyrimidin-3-yl)-N-methylbenzenesulfonamide (2)
A mixture of 4 (0.054 g, 0.20 mmol), K2CO3 (0.11 g, 0.8 mmol), DMF (2.0 mL), and tert-butyl (trans-4-(chloromethyl)cyclohexyl)carbamate (0.062 g, 0.25 mmol) in a 10 mL microwave tube was heated under microwave irradiation at 150 °C for 10 min. After cooling to room temperature, (4-(N-methylsulfamoyl)phenyl)boronic acid (0.065 g, 0.30 mmol), Pd(PPh3)4 (0.023 g, 0.020 mmol), and H2O (1.0 mL) were added sequentially. The resulting mixture was stirred at room temperature for 1.0 min and then heated under microwave irradiation at 150 °C for 15 min. After cooling to room temperature, the mixture was quenched with H2O and extracted with EtOAc (3x). The combined organic layer was dried (Na2SO4) and concentrated. The residue was purified by an ISCO silica gel column to provide tert-butyl (trans-4-((3-(4-(N-methylsulfamoyl)phenyl)-6-(butyllamino)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)methyl) cyclohexyl)carbamate as a white solid. This white solid was dissolved in CH2Cl2 (2.0 mL), then was added TFA (0.5 mL) at room temperature. The resulting solution was stirred for 1.0 h and was concentrated. The residue was purified by prep-HPLC to provide the title compound 2 as a white solid (0.053 g, 48% over 3 steps). 1H NMR (400 MHz, CDCl3) δ 1H NMR (400 MHz, CD3OD) δ 9.17 (s, 1H), 8.20–8.14 (m, 2H), 8.01–7.95 (m, 2H), 4.26 (d, J = 6.8 Hz, 2H), 3.52 (t, J = 7.1 Hz, 2H), 3.13–3.00 (m, 1H), 2.81 (s, 3H), 2.15 –2.01 (m, 3H), 1.86 (d, J = 12.0 Hz, 2H), 1.68 (dt, J = 12.7, 7.4 Hz, 2H), 1.48 (dt, J = 14.8, 7.3 Hz, 2H), 1.41–1.21 (m, 4H), 1.00 (t, J = 7.4 Hz, 3H); LC-MS: >97% purity, tR = 4.379 min; MS m/z 472.3 [M+1]+.
4-(1-((trans-4-Hydroxycyclohexyl)methyl)-6-(butylamino)-1H-pyrazolo[3,4-d]pyrimidin-3-yl)-N-methylbenzenesulfonamide (7)
(General Procedure A) A mixture of 4 (0.054 g, 0.20 mmol), K2CO3 (0.11 g, 0.8 mmol), DMF (2.0 mL), and tert-butyl((trans-4-(chloromethyl)cyclohexyl)oxy)dimethylsilane (0.066 g, 0.25 mmol) in a 10 mL microwave tube was heated under microwave irradiation at 150 °C for 10 min. After the reaction was cooled to room temperature, (4-(N-methylsulfamoyl)phenyl)boronic acid (0.065 g, 0.30 mmol), Pd(PPh3)4 (0.023 g, 0.020 mmol), and H2O (1.0 mL) were added sequentially. The mixture was stirred at room temperature for 1.0 min and then heated under microwave irradiation at 150 °C for 15 min. After cooling to room temperature, the mixture was quenched with H2O and extracted with EtOAc (3x). The combined organic layers were dried (Na2SO4) and concentrated. The residue was purified by an ISCO silica gel column to provide 4-(1-((trans-4-((tert-butyldimethylsilyl)oxy)cyclohexyl)methyl)-6-(butylamino)-1H-pyrazolo[3,4-d]pyrimidin-3-yl)-N-methylbenzenesulfonamide as a white solid. This white solid was dissolved in THF (2.0 mL) and was added TBAF (1.0 M in THF, 1 mL) at room temperature. The resulting solution was refluxed for 2.0 h and was concentrated. The residue was purified by prep-HPLC to provide the title compound 7 (0.040 g, 44% over 3 steps) as a white solid. 1H NMR (400 MHz, CDCl3) δ 9.30 (s, 1H), 8.20 (d, J = 8.0 Hz, 2H), 8.01 (d, J = 8.0 Hz, 2H), 4.26 (d, J = 7.0 Hz, 2H), 3.58 (t, J = 7.2 Hz, 2H), 3.55–3.45 (m, 1H), 2.58 (s, 3H), 2.11–1.93 (m, 3H), 1.73 (td, J = 14.9, 7.9 Hz, 4H), 1.49 (dq, J = 14.8, 7.5 Hz, 2H), 1.32–1.14 (m, 4H), 1.02 (t, J = 7.7Hz, 3H); LC-MS: >97% purity, tR = 6.629 min; MS m/z 473.3 [M+1]+.
4-(1-(2-(trans-4-Hydroxycyclohexyl)ethyl)-6-(propylamino)-1H-pyrazolo[3,4-d]pyrimidin-3-yl)-N-methylbenzenesulfonamide (8)
The title compound 8 (0.040 g, 42%) was prepared according to general procedure A from tert-butyl((trans-4-(2-chloroethyl)cyclohexyl)oxy)dimethylsilane (0.069 g, 0.25 mmol) as a white solid. 1H NMR (400 MHz, CDCl3) δ 8.79 (s, 1H), 7.92 (d, J = 8.6 Hz, 2H), 7.83 (d, J = 8.6 Hz, 2H), 4.24 (t, J = 7.0 Hz, 2H), 3.45–3.30 (m, 3H), 2.50 (s, 3H), 1.81 (t, J = 11.5 Hz, 3H), 1.71 (dd, J = 13.7, 6.8 Hz, 2H), 1.53 (dt, J = 14.9, 7.3 Hz, 2H), 1.40–1.27 (m, 2H), 1.08 (dd, J = 23.1, 12.5 Hz, 4H), 1.00–0.89 (m, 2H), 0.86 (t, J = 7.9 Hz, 3H); LC-MS: >97% purity, tR = 5.759 min; MS m/z 487.3 [M+1]+.
4-(1-(3-(trans-4-Hydroxycyclohexyl)propyl)-6-(propylamino)-1H-pyrazolo[3,4-d]pyrimidin-3-yl)-N-methylbenzenesulfonamide (9)
The title compound 9 (0.055 g, 56%) was prepared according to general procedure B from tert-butyl((trans-4-(3-chloropropyl)cyclohexyl)oxy)dimethylsilane (0.069 g, 0.25 mmol) as a white solid. 1H NMR (400 MHz, CDCl3) δ 9.03 (s, 1H), 8.17–8.09 (m, 2H), 7.99–7.92 (m, 2H), 4.32 (t, J = 6.9 Hz, 2H), 3.48 (t, J = 7.0 Hz, 2H), 3.42 (dt, J = 10.8, 4.2 Hz, 1H), 2.58 (s, 3H), 2.02–1.84 (m, 4H), 1.76 (d, J = 11.6 Hz, 2H), 1.66 (dt, J = 14.8, 7.3 Hz, 2H), 1.52–1.39 (m, 2H), 1.29–1.13 (m, 5H), 0.99 (t, J = 7.4 Hz, 3H), 0.96–0.86 (m, 2H); LC-MS: >97% purity, tR = 5.856 min; MS m/z 501.3 [M+1]+.
4-(1-(trans-4-Hydroxycyclohexyl)-6-(propylamino)-1H-pyrazolo[3,4-d]pyrimidin-3-yl)-N-methylbenzenesulfonamide (10) and 4-(1-(cis-4-Hydroxycyclohexyl)-6-(propylamino)-1H-pyrazolo[3,4-d]pyrimidin-3-yl)-N-methylbenzenesulfonamide (11)
(General Procedure B) A mixture of 4 (0.054 g, 0.20 mmol), K2CO3 (0.11 g, 0.8 mmol), DMF (2.0 mL), and 4-chlorocyclohexanol (0.081 g, 0.6 mmol) in a 10 mL microwave tube was heated under microwave irradiation at 200 °C for 30 min. After cooling to room temperature, (4-(N-methylsulfamoyl)phenyl)boronic acid (0.065 g, 0.30 mmol), Pd(PPh3)4 (0.023 g, 0.020 mmol), and H2O (1.0 mL) were added sequentially. The resulting mixture was stirred at room temperature for 1.0 min and then heated under microwave irradiation at 150 °C for 15 min. After cooling to room temperature, the mixture was quenched with H2O and extracted with EtOAc (3x). The combined organic layer was dried (Na2SO4) and concentrated. The residue was purified by prep-HPLC to provide the title compounds 10 (0.011 g, 13% over 3 steps) and 11 (0.020 g, 23% over 3 steps) as white solids. 10: 1H NMR (400 MHz, CDCl3) δ 8.78 (s, 1H), 7.92 (d, J = 8.4 Hz, 2H), 7.82 (d, J = 8.3 Hz, 2H), 4.51 (s, 1H), 3.64 (s, 1H), 3.37 (t, J = 7.0 Hz, 2H), 2.50 (s, 3H), 2.17–1.86 (m, 6H), 1.61–1.29 (m, 6H), 0.87 (t, J = 7.3 Hz, 3H); LC-MS: >97% purity, tR = 5.402 min; HRMS (TOF, ESI+) m/z: [M+H]+ calculated for C22H31N6O3S, 459.2178; found 459.2155. 11: 1H NMR (400 MHz, CDCl3 + CD3OD) δ 8.82 (s, 1H), 7.95 (dd, J = 8.2, 6.2 Hz, 2H), 7.84 (dd, J = 8.5, 1.8 Hz, 2H), 4.66–4.48 (m, 1H), 3.44–3.33 (m, 3H), 2.53 (s, 3H), 2.46–2.32 (m, 1H), 1.99–1.81 (m, 3H), 1.79– 1.61 (m, 3H), 1.55 (dt, J = 14.8, 7.4 Hz, 2H), 1.36 (td, J = 14.8, 7.4 Hz, 3H), 0.88 (t, J = 7.3 Hz, 3H); LC-MS: >97% purity, tR = 5.590 min; MS m/z 459.2 [M+1]+.
4-(1-(2-Hydroxyethyl)-6-(propylamino)-1H-pyrazolo[3,4-d]pyrimidin-3-yl)-N-methylbenzenesulfonamide (12)
(General Procedure C) A mixture of 4 (0.054 g, 0.20 mmol), K2CO3 (0.11 g, 0.8 mmol), DMF (2.0 mL), and 2-bromoethanol (0.038 g, 0.30 mmol) in a 10 mL microwave tube was heated under microwave irradiation at 150 °C for 10 min. After cooling to room temperature, (4-(N-methylsulfamoyl)phenyl)boronic acid (0.065 g, 0.30 mmol), Pd(PPh3)4 (0.023 g, 0.020 mmol), and H2O (1.0 mL) were added sequentially. The resulting mixture was stirred at room temperature for 1.0 min and then heated under microwave irradiation at 150 °C for 15 min. After cooling to room temperature, the mixture was quenched with H2O and extracted with EtOAc (3x). The combined organic layers were dried (Na2SO4) and concentrated. The residue was purified by an ISCO silica gel column to provide the title compound 12 (0.044 g, 57%) as a white solid. 1 H NMR (400 MHz, CDCl3) δ 8.86 (s, 1H), 8.00–7.92 (m, 2H), 7.91–7.83 (m, 2H), 4.42 (t, J = 5.0 Hz, 2H), 4.01 (t, J = 5.0 Hz, 2H), 3.38 (t, J = 7.1 Hz, 3H), 2.56 (s, 3H), 1.63–1.50 (m, 2H), 1.44–1.30 (m, 2H), 0.90 (t, J = 7.3 Hz, 3H); LC-MS: >97% purity, tR = 5.244 min; MS m/z 405.2 [M+1]+.
4-(1-(3-Hydroxypropyl)-6-(propylamino)-1H-pyrazolo[3,4-d]pyrimidin-3-yl)-N-methylbenzenesulfonamide (13)
The title compound 13 (0.045 g, 56%) was prepared according to general procedure C from 3-chloropropan-1-ol (0.029 g, 0.30 mmol) as a white solid. 1H NMR (400 MHz, CDCl3) δ 8.88 (s, 1H), 7.98 (d, J = 8.6 Hz, 2H), 7.90 (d, J = 8.6 Hz, 2H), 4.43 (t, J = 6.2 Hz, 2H), 3.50 (t, J = 5.8 Hz, 2H), 3.40 (t, J = 7.1 Hz, 2H), 2.59 (s, 3H), 2.10–2.00 (m, 2H), 1.60 (dt, J = 14.9, 7.4 Hz, 2H), 1.40 (dq, J = 14.5, 7.3 Hz, 2H), 0.92 (t, J = 7.3 Hz, 3H); LC-MS: >97% purity, tR = 5.336 min; MS m/z 419.2 [M+1]+.
N-Methyl-4-(6-(propylamino)-1-(tetrahydro-2H-pyran-4-yl)-1H-pyrazolo[3,4-d]pyrimidin-3-yl)benzenesulfonamide (14)
(General Procedure D) A mixture of 4 (0.054 g, 0.20 mmol), K2CO3 (0.11 g, 0.8 mmol), DMF (2.0 mL), and 4-chlorotetrahydro-2H-pyran (0.072 g, 0.60 mmol) in a 10 mL microwave tube was heated under microwave irradiation at 200 °C for 30 min. After cooling to room temperature, (4-(N-methylsulfamoyl)phenyl)boronic acid (0.065 g, 0.30 mmol), Pd(PPh3)4 (0.023 g, 0.020 mmol), and H2O (1.0 mL) were added sequentially. The resulting mixture was stirred at room temperature for 1.0 min and then heated under microwave irradiation at 150 °C for 15 min. After cooling to room temperature, the mixture was quenched with H2O and extracted with EtOAc (3x). The combined organic layers were dried (Na2SO4) and concentrated. The combined organic layer was dried (Na2SO4) and concentrated under reduced pressure. The residue was purified by an ISCO silica gel column to provide the title compound 14 as a white solid (0.052 g, 61% over 2 steps). 1H NMR (400 MHz, CDCl3) δ 8.96 (s, 1H), 8.07 (d, J = 8.5 Hz, 2H), 7.97 (d, J = 8.5 Hz, 2H), 5.33 (s, 1H), 4.92–4.79 (m, 1H), 4.32 (dd, J = 10.8, 5.4 Hz, 1H), 4.17 (dd, J = 11.7, 3.3 Hz, 2H), 3.64 (dd, J = 11.9, 10.2 Hz, 2H), 3.52 (dd, J = 12.9, 6.9 Hz, 2H), 2.71 (d, J = 5.4 Hz, 3H), 2.48 (ddd, J = 25.0, 12.5, 4.7 Hz, 2H), 1.97 (d, J = 10.8 Hz, 2H), 1.66 (dt, J = 14.8, 7.2 Hz, 2H), 1.47 (dq, J = 14.5, 7.3 Hz, 2H), 0.99 (t, J = 7.3 Hz, 3H); LC-MS: 96% purity, tR = 5.689 min; MS m/z 445.2 [M+1]+.
N-Methyl-4-(6-(propylamino)-1-((tetrahydro-2H-pyran-4-yl)methyl)-1H-pyrazolo[3,4-d]pyrimidin-3-yl)benzenesulfonamide (15)
The title compound 15 (0.062 g, 70% over 2 steps) was prepared according to general procedure C from 4-(bromomethyl)tetrahydro-2H-pyran (0.045 g, 0.25 mmol) as a white solid. 1H NMR (400 MHz, CDCl3) δ 8.95 (s, 1H), 8.10–8.03 (m, 2H), 8.00–7.93 (m, 2H), 5.37 (bs, 1H), 4.41–4.30 (m, 1H), 4.24 (d, J = 7.1 Hz, 2H), 4.03–3.91 (m, 2H), 3.51 (dd, J = 13.0, 6.9 Hz, 2H), 3.38 (td, J = 11.5, 2.6 Hz, 2H), 2.71 (d, J = 5.4 Hz, 3H), 2.40 – 2.24 (m, 1H), 1.71–1.61 (m, 2H), 1.60–1.56 (m, 1H), 1.54–1.38 (m, 5H), 0.99 (t, J = 7.3 Hz, 3H); LC-MS: >97% purity, tR = 5.714 min; MS m/z 459.3 [M+1]+.
N-Methyl-4-(6-(propylamino)-1-(2-(tetrahydro-2H-pyran-4-yl)ethyl)-1H-pyrazolo[3,4-d]pyrimidin-3-yl)benzenesulfonamide (16)
The title compound 16 (0.058 g, 63% over 2 steps) was prepared according to general procedure C from 4-(2-bromoethyl)tetrahydro-2H-pyran (0.048 g, 0.25 mmol) as a white solid. 1 H NMR (400 MHz, CDCl3) δ 8.95 (s, 1H), 8.10–8.03 (m, 2H), 7.99–7.93 (m, 2H), 5.38 (bs, 1H), 4.47–4.33 (m, 3H), 3.95 (dd, J = 11.5, 3.5 Hz, 2H), 3.49 (dd, J = 13.1, 6.8 Hz, 2H), 3.33 (td, J = 11.7, 1.8 Hz, 2H), 2.71 (d, J = 5.4 Hz, 3H), 1.97–1.86 (m, 2H), 1.76 (d, J = 12.7 Hz, 2H), 1.65 (dt, J = 12.7, 7.4 Hz, 2H), 1.55–1.30 (m, 5H), 0.98 (t, J = 7.3 Hz, 3H); LC-MS: 97% purity, tR = 5.835 min; MS m/z 473.3 [M+1]+.
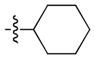
4-(1-Cyclohexyl-6-(propylamino)-1H-pyrazolo[3,4-d]pyrimidin-3-yl)-N-methylbenzene sulfonamide (17)
The title compound 17 (0.035 g, 40% over 2 steps) was prepared according to general procedure D from chlorocyclohexane (0.071 g, 0.60 mmol) as a white solid. 1H NMR (400 MHz, CDCl3) δ 8.94 (s, 1H), 8.10–8.03 (m, 2H), 7.99–7.92 (m, 2H), 5.30 (bs, 1H), 4.68–4.56 (m, 1H), 4.33 (q, J = 5.4 Hz, 1H), 3.52 (dd, J = 12.9, 7.0 Hz, 2H), 2.70 (d, J = 5.4 Hz, 3H), 2.16–1.89 (m, 6H), 1.77 (d, J = 12.7 Hz, 1H), 1.66 (dt, J = 14.8, 7.3 Hz, 2H), 1.54–1.28 (m, 5H), 0.99 (t, J = 7.3 Hz, 3H); LC-MS: >97% purity, tR = 6.358 min; MS m/z 443.2 [M+1]+.
trans-4-(3-Bromo-6-(propylamino)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)cyclohexanol
A solution of 4 (2.70 g, 10 mmol) and cis-4-chlorocyclohexanol (4.04 g, 30 mmol) in DMF (50 mL) was added K2CO3 (5.52 g, 40 mmol). The reaction mixture was heated at 150 °C for overnight, quenched with water at room temperature and extracted with EtOAc (3x). The combined organic layer was dried (Na2SO4) and concentrated. The residue was purified by an Isco silica gel column to afford the title compound (1.83 g, 52%) as a white solid. 1H NMR (400 MHz, CD3OD) δ 8.49 (s, 1H), 4.59–4.41 (m, 1H), 3.69–3.56 (m, 1H), 3.42 (t, J = 7.1 Hz, 2H), 2.13–2.00 (m, 4H), 1.98–1.89 (m, 2H), 1.66–1.56 (m, 2H), 1.52–1.36 (m, 4H), 0.96 (t, J = 7.4 Hz, 3H); 13C NMR (100 MHz, dmso-d6) δ 161.6, 155.0, 153.4, 119.1, 110.3, 107.7, 68.3, 55.5, 34.6, 30.9, 29.8, 20.0, 14.1; MS m/z 369.1 [M+H]+.
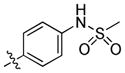
trans-N-(4-(6-(Butylamino)-1-(4-hydroxycyclohexyl)-1H-pyrazolo[3,4-d]pyrimidin-3-yl)phenyl)methanesulfonamide (18)
The title compound 18 (0.008 g, 18%) was prepared according to general procedure B from (4-(methylsulfonamido)phenyl)boronic acid (0.033 g, 0.15 mmol) as a white solid. 1H NMR (400MHz, CDCl3) δ 8.76 (s, 1H), 7.76–7.72 (m, 2H), 7.28–7.24 (m, 2H), 4.56–4.48 (m, 1H), 3.71–3.61 (m, 1H), 3.42–3.39 (m, 1H), 3.29–3.27 (m, 2H), 2.92 (s, 3H), 2.14–2.02 (m, 4H), 1.97–1.90 (m, 2H), 1.61–1.53 (m, 2H), 1.47–1.32 (m, 6H), 0.90 (t, J = 7.36 Hz, 3H); LC-MS: >97% purity, tR = 5.538 min; MS m/z 459.3 [M+1]+.
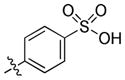
4-(1-(trans-4-Hydroxycyclohexyl)-6-(propylamino)-1H-pyrazolo[3,4-d]pyrimidin-3-yl)benzenesulfonic acid (19)
(General Procedure E) A mixture of trans-4-(3-bromo-6-(propylamino)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)cyclohexanol (0.055 g, 0.15 mmol), K3PO4 (0.063 g, 0.30 mmol), 4-boronobenzenesulfonic acid (0.045 g, 0.225 mmol), Pd(PPh3)4 (0.017 g, 0.015 mmol), dioxane (2.0 mL) and H2O (0.5 mL) in a 10 mL microwave tube was heated under microwave irradiation at 150 °C for 10 min. After cooling to room temperature, the mixture was quenched with H2O and extracted with EtOAc (3x). The combined organic layer was dried (Na2SO4) and concentrated. The residue was filtered through a plug of Celite and then purified by prep-HPLC to afford the title compound 19 (0.027 g, 30%) as a white solid. 1H NMR (400 MHz, CD3OD) δ 9.19 (s, 1H), 8.05–7.93 (m, 4H), 4.77–4.61 (m, 1H), 3.73 (tt, J = 10.7, 4.1 Hz, 1H), 3.57 (t, J = 7.1 Hz, 2H), 2.31–2.02 (m, 6H), 1.77–1.67 (m, 2H), 1.61–1.44 (m, 4H), 1.03 (t, J = 7.4 Hz, 3H); LC-MS: >97% purity, tR = 4.890 min; MS m/z 444.2 [M-1]−.
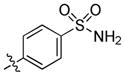
trans-4-(6-(Butylamino)-1-(4-hydroxycyclohexyl)-1H-pyrazolo[3,4-d]pyrimidin-3-yl)benzenesulfonamide (20)
The title compound 20 (0.064 g, 71%) was prepared according to general procedure E from 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzenesulfonamide (0.086 g, 0.306 mmol) as a white solid. 1H NMR (400 MHz, CD3OD+CDCl3) δ 8.74 (s, 1H), 7.87–7.78 (m, 4H), 4.46–4.43 (m, 1H), 3.59–3.55 (m, 1H), 3.33 – 3.28 (m, 2H), 2.03 – 1.93 (m, 4H), 1.90 – 1.85 (m, 2H), 1.50 – 1.45 (m, 2H), 1.39–1.33 (m, 2H), 1.30–1.25 (m, 2H), 1.08–1.06 (m, 3H), 0.80 (t, J = 7.4 Hz, 3H); LC-MS: 97% purity, tR = 5.147 min; MS m/z 445.2 [M+1]+.
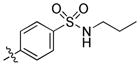
trans-4-(6-(Butylamino)-1-(4-hydroxycyclohexyl)-1H-pyrazolo[3,4-d]pyrimidin-3-yl)-N-propylbenzenesulfonamide (21)
The title compound 21 (0.019 g, 13%) was prepared according to general procedure B from (4-(N-propylsulfamoyl)phenyl)boronic acid (0.109 g, 0.45 mmol) as a white solid. 1H NMR (400MHz, CDCl3) δ 8.83 (s, 1H), 7.98 (s, 4H), 4.68–4.62 (m, 1H), 4.42 (t,, J = 6.16 Hz 1H), 3.87–3.81 (m, 1H), 3.57–3.51 (m, 2H), 2.98 (dd, J = 6.95, 13.46 Hz, 2H), 2.25–2.16 (m, 4H), 2.10–2.07 (m, 2H), 1.71 (dt, J = 7.43, 14.83 Hz, 2H), 1.60–1.42 (m, 6H), 1.00 (t, J = 7.36 Hz, 3H), 0.89 (t, J = 7.40 Hz, 3H); LC-MS: >97% purity, tR = 6.217 min; MS m/z 487.3 [M+1]+.
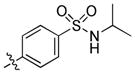
trans-4-(6-(Butylamino)-1-(4-hydroxycyclohexyl)-1H-pyrazolo[3,4-d]pyrimidin-3-yl)-N-isopropylbenzenesulfonamide (22)
The title compound 22 (0.016 g, 13%) was prepared according to general procedure B from (4-(N-isopropylsulfamoyl)phenyl)boronic acid (0.095 g, 0.39 mmol) as a white solid. 1H NMR (400MHz, CDCl3) δ 8.91 (s, 1H), 8.00 (dd, J = 8.62, 20.77 Hz, 4H), 4.69–4.61 (m, 1H), 4.26 (d, J = 7.61 Hz, 1H), 3.88–3.80 (m, 1H), 3.57–3.48 (m, 2H ), 2.27–2.18 (m, 4H), 2.07 (d, J = 10.85 Hz, 2H), 1.71–1.53 (m, 6H), 1.47 (dd, J = 7.36, 14.90 Hz, 2H), 1.11 (d, J = 6.52 Hz, 6H), 0.99 (t, J = 7.34 Hz, 3H); LC-MS: >97% purity, tR = 5.826 min; MS m/z 487.3 [M+1]+.
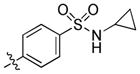
4-(6-(Butylamino)-1-(trans-4-hydroxycyclohexyl)-1H-pyrazolo[3,4-d]pyrimidin-3-yl)-N-cyclopropylbenzenesulfonamide (23)
The title compound 23 (0.003 g, 4%) was prepared according to general procedure E from (4-(N-cyclopropylsulfamoyl)phenyl)boronic acid (0.090 g, 0.375 mmol) as a white solid. 1H NMR (400 MHz, CD3OD). δ 9.11 (s, 1H), 8.17 (d, J = 8.6 Hz, 2H), 8.00 (d, J = 8.6 Hz, 2H), 4.73–4.63 (m, 1H), 3.79–3.67 (m, 1H), 3.51 (t, J = 7.1 Hz, 2H), 2.30–2.01 (m, 7H), 1.69 (td, J = 14.7, 7.4 Hz, 2H), 1.61–1.42 (m, 4H), 1.01 (t, J = 7.4 Hz, 3H), 0.62–0.47 (m, 4H); LC-MS: 95% purity, tR = 5.957 min; MS m/z 485.0 [M+1]+.
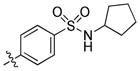
trans-4-(6-(Butylamino)-1-(4-hydroxycyclohexyl)-1H-pyrazolo[3,4-d]pyrimidin-3-yl)-N-cyclopentylbenzenesulfonamide (24)
The title compound 24 (0.015 g, 10%) was prepared according to general procedure B from (4-(N-cyclopentylsulfamoyl)phenyl)boronic acid (0.121 g, 0.45 mmol) as a white solid. 1H NMR (400MHz, CDCl3) δ 8.82 (s, 1H), 7.99 (q, J = 8.67 Hz, 4H), 4.68–4.64 (m, 1H), 4.44 (d, J = 7.34Hz, 1H), 3.86–3.82 (m, 1H), 3.67 (dd, J = 6.82, 13.53 Hz, 1H), 3.55–3.50 (m, 2H), 2.22–2.16 (m, 4H), 2.10–2.07 (m, 2H), 1.84 (dd, J = 5.70, 12.36 Hz, 2H), 1.71 (dd, J = 7.63, 14.79 Hz, 2H), 1.63–1.57 (m, 4H), 1.54–1.48 (m, 4H), 1.40–1.35 (m, 2H), 1.00 (t, J = 7.35 Hz, 3H); LC-MS: 95 % purity, tR = 6.186 min; MS m/z 513.3 [M+1]+.
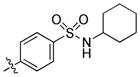
trans-4-(6-(Butylamino)-1-(4-hydroxycyclohexyl)-1H-pyrazolo[3,4-d]pyrimidin-3-yl)-N-cyclohexylbenzenesulfonamide (25)
The title compound 25 (0.015 g, 11%) was prepared according to general procedure B from (4-(N-cyclohexylsulfamoyl)phenyl)boronic acid (0.110 g, 0.39 mmol) as a white solid. 1H NMR (400MHz, CDCl3) δ 8.95 (s, 1H), 8.04 (d, J = 8.61 Hz, 2H), 7.97 (d, J = 8.59 Hz, 2H), 5.36 (brs, 1H), 4.67–4.64 (m, 1H), 4.34 (d, J = 7.55 Hz, 1H), 3.87–3.80 (m, 1H), 3.52 (dd, J = 7.03, 12.98 Hz, 2H), 3.24–3.15 (m, 1H), 2.28–2.18 (m, 4H), 2.09–2.06 (m, 2H), 1.82–1.79 (m, 2H), 1.67–1.56 (m, 6H), 1.52–1.46 (m, 4H), 1.27–1.14 (m, 6H), 1.00 (t, J = 7.35 Hz, 3H); LC-MS: >97% purity, tR = 6.171 min; MS m/z 527.0 [M+1]+.
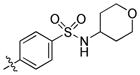
trans-4-(6-(Butylamino)-1-(4-hydroxycyclohexyl)-1H-pyrazolo[3,4-d]pyrimidin-3-yl)-N-(tetrahydro-2H-pyran-4-yl)benzenesulfonamide (26)
The title compound 26 (0.016 g, 10%) was prepared according to general procedure B from (4-(N-(tetrahydro-2H-pyran-4-yl)sulfamoyl)phenyl)boronic acid (0.128 g, 0.45 mmol) as a white solid. 1H NMR (400MHz, CDCl3) δ 9.44 (brs, 1H), 8.92 (s, 1H), 8.00 (q, J = 8.67 Hz, 4H), 4.69–4.61 (m, 2H), 3.90–3.84 (m, 4H), 3.55 (t, J = 7.04 Hz, 2H), 3.43–3.33 (m, 2H), 2.21–2.07 (m, 6H), 1.79–1.68 (m, 4H), 1.60–1.44 (m, 6H), 1.00 (t, J = 7.36 Hz, 3H); LC-MS: 95% purity, tR = 5.747 min; MS m/z 529.3 [M+1]+.
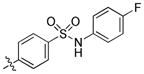
trans-4-(6-(Butylamino)-1-(4-hydroxycyclohexyl)-1H-pyrazolo[3,4-d]pyrimidin-3-yl)-N-(4-fluorophenyl)benzenesulfonamide (27)
The title compound 27 (0.009 g, 6%) was prepared according to general procedure B from 4-(N-(4-fluorophenyl)sulfamoyl)phenyl)boronic acid (0.133 g, 0.45 mmol) as a white solid. 1H NMR (400MHz, CDCl3) δ 9.38 (s, 1H), 8.82 (s, 1H), 7.94–7.89 (m, 2H), 7.85–7.81 (m, 2H), 7.09–7.05 (m, 2H), 6.98–6.94 (m, 2H), 6.57 (s, 1H), 4.67–4.61 (m, 1H), 3.98–3.80 (m, 1H), 3.57–3.51 (m, 2H), 2.22–2.08 (m, 6H), 1.71 (dt, J = 7.29, 14.74 Hz, 2H), 1.62–1.57 (m, 2H), 1.45 (dt, J = 7.37, 14.45 Hz, 2H), 0.99 (t, J = 7.38 Hz, 3H); LC-MS: >97% purity, tR = 6.198 min; MS m/z 539.3 [M+1]+.
trans-tert-Butyl 4-((4-(6-(butylamino)-1-(4-hydroxycyclohexyl)-1H-pyrazolo[3,4-d]pyrimidin-3-yl)phenylsulfonamido)methyl)piperidine-1-carboxylate (29)
The title compound 29 (0.011 g, 9%) was prepared according to general procedure B from (4-(N-((1-(tert-butoxycarbonyl)piperidin-4-yl)methyl)sulfamoyl)phenyl)boronic acid (0.119 g, 0.30 mmol, 1.5 eq) as a white solid. 1H NMR (400MHz, CDCl3+CD3OD) δ 8.78 (s, 1H), 7.90 (d, J = 8.44 Hz, 2H), 7.80 (d, J = 7.34 Hz, 2H), 4.46–4.43 (m, 1H), 3.91 (d, J = 12.95 Hz, 2H), 3.63–3.61 (m, 1H), 2.36 (t, J = 7.08 Hz, 2H), 3.22 (dt, J = 1.61, 3.24 Hz, 3H), 2.65 (d, J = 6.62 Hz, 2H), 2.54–2.51 (m, 2H), 2.08–2.00 (m, 4H), 1.93–1.90 (m, 2H), 1.58–1.46 (m, 4H), 1.31 (s, 12H), 0.97–0.91 (m, 2H), 0.89 (t, J = 7.35 Hz, 3H); LC-MS: 96% purity, tR = 6.305 min; MS m/z 642.4 [M+1]+.
trans-4-(6-(Butylamino)-1-(4-hydroxycyclohexyl)-1H-pyrazolo[3,4-d]pyrimidin-3-yl)-N-(piperidin-4-ylmethyl)benzenesulfonamide (28)
A mixture 29 (20 mg, 0.03 mmol) and TFA (2.3 mL) in dichloromethane (4.7 mL) were stirred at room temperature for 2.0 h, then diluted with dichloromethane (20 mL) and washed with a saturated aqueous NaHCO3 solution, water and brine. The organic layer was dried (Na2SO4) and concentrated to afford the title compound 28 (12.7 mg, 75%) as a white solid. 1H NMR (400MHz, CDCl3+CD3OD) δ 8.25 (s, 1H), 7.23 (d, J = 8.45 Hz, 2H), 7.14 (d, J = 8.20 Hz, 2H), 3.85–3.80 (m, 1H), 2.96–2.91 (m, 1H), 2.72 (t, J = 7.15 Hz, 2H), 2.54–2.52 (m, 3H), 2.07–1.99 (m, 4H), 1.41–1.32 (m, 4H), 1.26–1.23 (m, 2H), 1.12 (d, J = 13.63 Hz, 2H), 0.98–0.94 (m, 1H), 0.91–0.84 (m, 2H), 0.79–0.75 (m, 2H), 0.67–0.56 (m, 4H), 0.17 (t, J = 7.35 Hz, 3H); LC-MS: >97% purity, tR = 4.975 min; MS m/z 542.3 [M+1]+.
trans-1-(4-(6-(Butylamino)-1-(4-hydroxycyclohexyl)-1H-pyrazolo[3,4-d]pyrimidin-3-yl)phenylsulfonamido)-N-(4-fluorophenyl)cyclopropanecarboxamide (30)
The title compound 30 (0.009 g, 7%) was prepared according to general procedure B from (4-(N-(1-((4-fluorophenyl)carbamoyl)cyclopropyl)sulfamoyl)phenyl)boronic acid (0.160 g, 0.42 mmol, 2.0 eq) as a white solid. 1H NMR (400MHz, CD3OD) δ 9.17 (s, 1H), 8.95 (s, 1H), 8.06 (d, J = 8.54 Hz, 2H), 7.96 (d, J = 8.33 Hz, 2H), 7.34–7.31 (m, 2H), 6.90–6.86 (m, 2H), 4.68–4.62 (m, 1H), 3.75–3.70 (m, 1H), 3.50 (t, J = 7.90 Hz, 2H), 2.24–2.13 (m, 4H), 2.06–2.03 (m, 2H), 1.71–1.64 (m, 2H), 1.58–1.38 (m, 6H), 1.10 (dd, J = 4.67, 7.89 Hz, 2H), 1.00 (t, J = 7.36 Hz, 3H); LC-MS: 96% purity, tR = 6.051 min; MS m/z 622.3 [M+1]+.
4-(6-(Butylamino)-1-(trans-4-hydroxycyclohexyl)-1H-pyrazolo[3,4-d]pyrimidin-3-yl)-N,N-dimethylbenzenesulfonamide (31)
The title compound 31 (0.052 g, 74%) was prepared according to general procedure E from (4-(N,N-dimethylsulfamoyl)phenyl)boronic acid (0.052 g, 0.225 mmol) as a white solid. 1H NMR (400 MHz, CDCl3) δ 8.94 (s, 1H), 8.07 (d, J = 8.4 Hz, 2H), 7.88 (d, J = 8.3 Hz, 2H), 5.34 (bs, 1H), 4.73–4.59 (m, 1H), 3.90–3.76 (m, 1H), 3.52 (dd, J = 13.0, 6.8 Hz, 2H), 2.75 (s, 6H), 2.29–2.15 (m, 4H), 2.13–2.02 (m, 2H), 1.66 (dt, J = 14.9, 7.3 Hz, 2H), 1.59–1.52 (t, J = 10.2 Hz, 3H), 1.47 (dd, J = 15.0, 7.4 Hz, 2H), 1.00 (t, J = 7.3 Hz, 3H); LC-MS: 97% purity, tR = 6.011 min; MS m/z 473.3 [M+1]+.
N-(tert-Butyl)-4-(6-(butylamino)-1-(trans-4-hydroxycyclohexyl)-1H-pyrazolo[3,4-d]pyrimidin-3-yl)-N-methylbenzenesulfonamide (32)
The title compound 32 (0.058 g, 76%) was prepared according to general procedure E from ((4-(N-(tert-butyl)-N-methylsulfamoyl)phenyl)boronic acid (0.061 g, 0.225 mmol) as a white solid. 1H NMR (400 MHz, CDCl3) δ 8.93 (s, 1H), 8.07–7.97 (m, 2H), 7.96–7.89 (m, 2H), 5.40 (bs, 1H), 4.73–4.58 (m, 1H), 3.91–3.76 (m, 1H), 3.52 (dd, J = 12.9, 6.9 Hz, 2H), 3.01 (s, 3H), 2.30–2.14 (m, 4H), 2.07 (d, J = 10.8 Hz, 2H), 1.68–1.44 (m, 7H), 1.38 (d, J = 4.8 Hz, 9H), 0.99 (t, J = 7.3 Hz, 3H); LC-MS: 96% purity, tR = 6.328 min; MS m/z 515.3 [M+1]+.
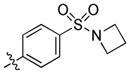
trans-4-(3-(4-(Azetidin-1-ylsulfonyl)phenyl)-6-(butylamino)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)cyclohexanol (33)
The title compound 33 (0.054 g, 74%) was prepared according to general procedure E from (4-(azetidin-1-ylsulfonyl)phenyl)boronic acid (0.054 g, 0.225 mmol) as a white solid. 1H NMR (400 MHz, CDCl3) δ 8.96 (s, 1H), 8.11 (d, J = 8.3 Hz, 2H), 7.95 (d, J = 8.2 Hz, 2H), 5.33 (s, 1H), 4.75–4.57 (m, 1H), 3.92–3.74 (m, 5H), 3.53 (dd, J = 13.1, 6.7 Hz, 2H), 2.32–2.16 (m, 4H), 2.15–2.03 (m, 4H), 1.66 (dt, J = 14.8, 7.2 Hz, 2H), 1.55–1.50 (m, 3H), 1.45 (dd, J = 14.6, 7.3 Hz, 2H), 1.00 (t, J = 7.3 Hz, 3H); LC-MS: >97% purity, tR = 5.992 min; MS m/z 485.3 [M+1]+.
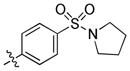
trans-4-(6-(Butylamino)-3-(4-(pyrrolidin-1-ylsulfonyl)phenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)cyclohexanol (34)
The title compound 34 (0.020 g, 27%) was prepared according to general procedure E from (4-(pyrrolidin-1-ylsulfonyl)phenyl)boronic acid (0.096 g, 0.375 mmol) as a white solid. 1H NMR (400 MHz, CD3OD). δ 9.20 (s, 1H), 8.20 (d, J = 8.5 Hz, 2H), 7.98 (d, J = 8.5 Hz, 2H), 4.75–4.64 (m, 1H), 3.78–3.67 (m, 1H), 3.55 (t, J = 7.1 Hz, 2H), 3.32–3.23 (m, 4H), 2.30–2.02 (m, 6H), 1.85–1.64 (m, 6H), 1.62–1.42 (m, 4H), 1.02 (t, J = 7.4 Hz, 3H); LC-MS: >97% purity, tR = 6.100 min; MS m/z 499.0 [M+1]+.
trans-4-(6-(Butylamino)-3-(4-(morpholinosulfonyl)phenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)cyclohexanol (35, UNC1062)
The title compound 35 (0.063 g, 82%) was prepared according to general procedure E from 4-((4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)sulfonyl)morpholine (0.079 g, 0.225 mmol) as a white solid. 1H NMR (400 MHz, CD3OD). δ 9.20 (s, 1H), 8.26–8.20 (m, 2H), 7.95–7.89 (m, 2H), 4.75–4.64 (m, 1H), 3.79–3.68 (m, 1H), 3.73 (dd, J = 10.2, 5.7 Hz, 4H), 3.55 (t, J = 7.1 Hz, 2H), 3.08–2.97 (m, 4H), 2.36–2.01 (m, 6H), 1.76–1.65 (m, 2H), 1.62–1.42 (m, 4H), 1.02 (t, J = 7.4 Hz, 3H); 13C NMR (100 MHz, DMSO-d6) δ 160.6, 155.2, 154.0, 140.6, 136.9, 133.7, 128.3, 127.0, 105.1, 68.0, 65.3, 55.0, 45.9, 40.3, 34.2, 30.6, 29.4, 19.6, 13.7; LC-MS: >97% purity, tR = 5.657 min; HRMS (TOF, ESI+) m/z: [M+H]+ calculated for C25H35N6O4S, 515.2440; found 515.2421.
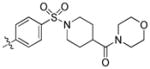
trans-(1-((4-(6-(Butylamino)-1-(4-hydroxycyclohexyl)-1H-pyrazolo[3,4-d]pyrimidin-3-yl)phenyl)sulfonyl)piperidin-4-yl)(morpholino)methanone (36)
The title compound 36 (0.065 g, 66%) was prepared according to general procedure E from (4-((4-(morpholine-4-carbonyl)piperidin-1-yl)sulfonyl)phenyl)boronic acid (0.091 g, 0.24 mmol) as a white solid. 1H NMR (400 MHz, CD3OD+CDCl3) δ 9.21 (s, 1H), 8.37–8.28 (m, 2H), 8.24–8.16 (m, 2H), 5.04–4.94 (m, 1H), 4.60–4.38 (m, 2H), 4.23–4.07 (m, 3H), 3.99–3.94 (m, 3H), 3.93–3.82 (m, 4H), 3.78–3.68 (m, 2H), 2.91–2.82 (m, 2H), 2.76 (ddd, J = 10.4, 7.2, 3.9 Hz, 1H), 2.61–2.46 (m, 4H), 2.46–2.36 (m, 2H), 2.30–2.17 (m, 2H), 2.15–2.06 (m, 2H), 2.06–1.98 (m, 2H), 1.98–1.86 (m, 2H), 1.85–1.74 (m, 2H), 1.33 (t, J = 7.4 Hz, 3H); LC-MS: 96% purity, tR = 5.468 min; MS m/z 626.3 [M+1]+.
Microfluidic Capillary Electrophoresis (MCE) Assay
Activity assays were performed in a 384 well, polypropylene microplate with a final volume of 50 μL in 50 mM Hepes, Ph 7.4 containing 10 mM MgCl2, 1.0 mM DTT, 0.01% Triton X-100, 0.1% Bovine Serum Albumin (BSA), using 1.0 μM fluorescent substrate (table 3) and ATP at Km for each enzyme (Table 3). All reactions were terminated by addition of 20 μL of 70 mM EDTA. Phosphorylated and unphosphorylated substrate peptides (Table 3) were separated following a 180 min incubation on a LabChip EZ Reader equipped with a 12-sipper chip in separation buffer supplemented with 1 x CR-8 and analyzed using EZ Reader software.
Table 3.
Assay conditions for MCE assays currently used
| Kinase | Peptide Substrate | Kinase (nM) | ATP (uM) |
|---|---|---|---|
| Mer | 5-FAM-EFPIYDFLPAKKK-CONH2 | 2.0 | 5.0 |
| Axl | 5-FAM-KKKKEEIYFFF-CONH2 | 120 | 65 |
| Tyro | 5-FAM-EFPIYDFLPAKKK-CONH2 | 10 | 21 |
Morrison Ki Measurement
Inhibition of Mer kinase by UNC1062 was measured in the MCE assay using the Morrison Tight-Binding Method17b. Reactions were performed in a 384 well, polypropylene microplate in a final volume of 80 μL in 50 mM Hepes, Ph 7.4 containing 10 mM MgCl2, 1.0 mM DTT, 0.01% Triton X-100, 0.1% Bovine Serum Albumin (BSA), and 5.0 μM ATP. Compounds were tested in 20 dilution (1.5 fold) dose curves spanning a concentration range of 60 nM to 0.027 nM, 2.0 nM Mer, 1.0 μM Peptide substrate, and 5.0 μM ATP, final. To 40 μL of enzyme (4 nM Mer), 8.0 μL of compound in 10% DMSO was added and allowed to incubate for 10 min followed by addition of 32 μL of substrate mix (containing 2.5 μM peptide substrate and 12.5 μM ATP).
Kinetic reads were taken from 7.0 to 209 min, 12 reads total, on an EZReader, using upstream voltage = −2250 V, downstream voltage = −500 V and pressure = −1.0 psi (sip time 0.2 sec, buffer 30 sec). The steady-state velocity was analyzed by linear regression of the Peptide as percent-conversion/min, and a plot of nM compound (UNC1062) vs. velocity was fit to the Morison equation using GraphPad Prism 5.0., Y=Vo*(1-((((Et+X+(Ki*(1+(S/Km))))-(((Et+X+(Ki*(1+(S/Km))))^2)-4*Et*X)^0.5))/(2*Et))) (Figure 5). The total enzyme concentration (Et) was fixed to 0.855 nM as determined from previous titration studies (data not shown).17b (Y = velocity, X = inhibitor concentration).
For UNC1062, the Morrison Ki was 0.33 ± 0.083 nM (n=3, error reported as S.D.).
hERG Binding Assay
Assay Buffer: Hank’s Balanced Salt Solution, 20 mM HEPES, 2.5 mM probenecid, pH 7.4, membrane potential sensitive dye (Molecular Devices).
To screen for drugs that block or open HERG K+ channels, we employ a fluorescence-based membrane potential assay (Molecular Devices). HEK293 cells stably expressing recombinant human HERG (provided by Dr. J. Overholt, Case Western Reserve Univeristy, Cleveland, OH; cells originally from Drs. A. Brown and E. Ficker, MetroHealth Medical Center, Cleveland, OH) are seeded in poly-L-lysine-coated 96-well plates (45,000 cells/100 μL DMEM supplemented with 10% fetal bovine serum/well) one day prior to assay. The next day, the medium is removed and replaced with 30 μL/well of assay buffer containing membrane potential dye (Molecular Devices) (the lyophilized dye is reconstituted with 15 mL of assay buffer). After a 15 min incubation at 37 degrees centigrade, 30 μL/well of 2X dilutions of terfenadine (a known HERG blocker used as a reference compound) or test compound (final assay concentration ranging from 0.1 nM to 10 μM) are added to the cells (each concentration assayed in triplicate). Baseline fluorescence (excitation 530 nM, emission 565 nM) is measured over 15 min, then 140 μL of depolarization solution (143 mM KCl in distilled water) containing test or reference compound (1x) are added to the cells and fluorescence is recorded for 3 min. Raw fluorescence data are analyzed by GraphPad Prism 4.0.
Cell Based Assays for Mer Kinase Inhibition
697, BT-12, A549, or Colo699 cells were cultured in the presence of UNC1062 or vehicle only for 1.0 h (697, BT-12) or 72 h (A549, Colo699). Pervanadate solution was prepared fresh by combining 20 mM sodium orthovanadate in 0.9x PBS in a 1:1 ratio with 0.3% (w/w) hydrogen peroxide in PBS for 15–20 min at room temperature. Cultures were treated with 120 μM pervanadate prior to collection for preparation of whole cell lysates, immunoprecipitation of Mer, and analysis by western blot.
697, A549, and Colo699 cells were treated with pervanadate for 3.0, 5.0, and 1.0 min respectively. Cell lysates were prepared in 50 mM HEPES pH 7.5, 150 mM NaCl, 10 mM EDTA, 10% glycerol, and 1% Triton X-100, supplemented with protease inhibitors (Roche Molecular Biochemicals, #11836153001). Mer protein was immunoprecipitated with a monoclonal anti-Mer antibody (R&D Systems, #MAB8912) and Protein G agarose beads (InVitrogen). Phospho-Mer was detected by western blot using a polyclonal anti-phospho-Mer antibody raised against a peptide derived from the tri-phosphorylated activation loop of Mer. Nitrocellulose membranes were stripped and total Mer protein was detected using a second anti-Mer antibody (Epitomics Inc., #1633-1). For 697 cells, relative phosphorylated and total Mer protein levels were determined by densitometry and IC50 values were determined by non-linear regression.
Confluent BT-12 cultures were treated with pervanadate for 15 min. Cell lysates were prepared in 20 mM HEPES pH 7.5, 500 mM NaCl, 5.0 mM EDTA, 10% glycerol, and 1% Triton X-100, supplemented with protease inhibitors (10 μg/mL leupeptin, 10 μg/mL phenylmethylsulfonyl fluoride, and 20 μg/ml aprotinin) and phosphatase inhibitors (50 mM sodium fluoride and 1.0 mM sodium orthovanadate) and Mer protein was immunoprecipitated using a custom polyclonal rabbit anti-Mer antisera raised against a peptide derived from the N-terminus of Mer and Protein A agarose beads (Santa Cruz Biotechnology). Phosphotyrosine-containing proteins were detected by western blot with a monoclonal HRP-conjugated anti-phosphotyrosine antibody (Santa Cruz Biotechnology, #sc-508). Antibodies were stripped from membranes and total Mer levels were determined using a custom polyclonal rabbit anti-Mer raised against a peptide derived from the catalytic domain of Mer.
Soft Agar Colony Formation Assays
BT-12 cells (10,000 cells) were cultured in 2.0 mL of 0.35% soft agar containing 0.5x RPMI medium, 7.5% FBS, and the indicated concentrations of UNC1062 or DMSO vehicle only and overlaid with 0.5 mL of 1x RPMI medium containing 15% FBS and UNC1062 or DMSO vehicle only. Medium and UNC1062 or vehicle were refreshed 2 times per week. Colonies were stained with thiazolyl blue tetrazolium bromide (Sigma Aldrich, #M5655) and counted after 3 weeks.
A549 or Colo699 cells (15,000 cells) were cultured in 1.5 mL of 0.35% soft agar containing 1x RPMI medium and 10% FBS and overlaid with 2.0 mL of 1x RPMI medium containing 10% FBS and the indicated concentrations of UNC1062 or DMSO vehicle only. Medium and UNC1062 or vehicle were refreshed 3 times per week. Colonies were stained with nitrotetrazolium blue chloride (Sigma Aldrich, #N6876) and counted after 2 weeks.
Supplementary Material
Highlights.
Discovery of pyazolopyrimidine sulfonamides as potent and selective Mer inhibitors.
UNC1062 selectively inhibits Mer both in purified enzymes and intact cell systems.
UNC1062 inhibits the colony formation of multiple tumor cell lines in soft agar.
The anti-tumor activity of UNC1062 further validates Mer as a therapeutic target.
Acknowledgments
HERG data was generously provided by the National Institute of Mental Health’s Psychoactive Drug Screening Program, Contract # HHSN-271-2008-025C(NIMH PDSP). The NIMH PDSP is directed by Bryan L. Roth MD, PhD at the University of North Carolina at Chapel Hill and Project Officer Jamie Driscol at NIMH, Bethesda MD, USA. We also thank Yingqiu Zhou for her help with MCE assays. This work was supported by the University of North Carolina Cancer Research Fund and Federal Funds from the National Cancer institute, National Institute of Health, under Contract No. HHSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Footnotes
SUPPORTING INFORMATION Experimental details and characterization of all compounds and biological methods. This material is available free of charge via the internet at http://pubs.acs.org.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Chen J, Carey K, Godowski PJ. Identification of Gas6 as a ligand for Mer, a neural cell adhesion molecule related receptor tyrosine kinase implicated in cellular transformation. Oncogene. 1997;14(17):2033–9. doi: 10.1038/sj.onc.1201039. [DOI] [PubMed] [Google Scholar]
- 2.Linger RMA, Keating AK, Earp HS, Graham DK. Advances in Cancer Research. Vol. 100. Academic Press; New York: 2008. TAM Receptor Tyrosine Kinases: Biologic Functions, Signaling, and Potential Therapeutic Targeting in Human Cancer; pp. 35–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.(a) Linger RMA, Keating AK, Earp HS, Graham DK. Taking aim at Mer and Axl receptor tyrosine kinases as novel therapeutic targets in solid tumors. Expert Opin Ther Targets. 2010;14(10):1073–1090. doi: 10.1517/14728222.2010.515980. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Brandao L, Migdall-Wilson J, Eisenman K, Graham DK. TAM receptors in leukemia: expression, signaling, and therapeutic implications. Critical reviews in oncogenesis. 2011;16(1–2):47–63. doi: 10.1615/critrevoncog.v16.i1-2.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.(a) Graham DK, Salzberg DB, Kurtzberg J, Sather S, Matsushima GK, Keating AK, Liang X, Lovell MA, Williams SA, Dawson TL, Schell MJ, Anwar AA, Snodgrass HR, Earp HS. Ectopic expression of the proto-oncogene Mer in pediatric T-cell acute lymphoblastic leukemia. Clin Cancer Res. 2006;12(9):2662–9. doi: 10.1158/1078-0432.CCR-05-2208. [DOI] [PubMed] [Google Scholar]; (b) Yeoh EJ, Ross ME, Shurtleff SA, Williams WK, Patel D, Mahfouz R, Behm FG, Raimondi SC, Relling MV, Patel A, Cheng C, Campana D, Wilkins D, Zhou X, Li J, Liu H, Pui CH, Evans WE, Naeve C, Wong L, Downing JR. Classification, subtype discovery, and prediction of outcome in pediatric acute lymphoblastic leukemia by gene expression profiling. Cancer cell. 2002;1(2):133–43. doi: 10.1016/s1535-6108(02)00032-6. [DOI] [PubMed] [Google Scholar]; (c) Brandao LN, Winges A, Christoph S, Sather S, McGranahan A, Liang X, Gao D, DeRyckere D, Graham DK. Inhibition of MerTK increases chemosensitivity and decreases oncogenic potential in T-cell acute lymphoblastic leukemia. Blood Cancer J. 2013;3:e101. doi: 10.1038/bcj.2012.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee-Sherick AB, Eisenman KM, Sather S, McGranahan A, Armistead PM, McGary CS, Hunsucker SA, Schlegel J, Martinson H, Cannon C, Keating AK, Earp HS, Liang X, DeRyckere D, Graham DK. Aberrant Mer receptor tyrosine kinase expression contributes to leukemogenesis in acute myeloid leukemia. Oncogene. 2013 doi: 10.1038/onc.2013.40. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Linger RM, Cohen RA, Cummings CT, Sather S, Migdall-Wilson J, Middleton DH, Lu X, Baron AE, Franklin WA, Merrick DT, Jedlicka P, Deryckere D, Heasley LE, Graham DK. Mer or Axl receptor tyrosine kinase inhibition promotes apoptosis, blocks growth and enhances chemosensitivity of human non-small cell lung cancer. Oncogene. 2012 doi: 10.1038/onc.2012.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Keating AK, Kim GK, Jones AE, Donson AM, Ware K, Mulcahy JM, Salzberg DB, Foreman NK, Liang X, Thorburn A, Graham DK. Inhibition of Mer and Axl Receptor Tyrosine Kinases in Astrocytoma Cells Leads to Increased Apoptosis and Improved Chemosensitivity. Mol Cancer Ther. 2010;9(5):1298–1307. doi: 10.1158/1535-7163.MCT-09-0707. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Rogers AE, Le JP, Sather S, Pernu BM, Graham DK, Pierce AM, Keating AK. Mer receptor tyrosine kinase inhibition impedes glioblastoma multiforme migration and alters cellular morphology. Oncogene. 2012;31(38):4171–81. doi: 10.1038/onc.2011.588. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Wang Y, Moncayo G, Morin P, Jr, Xue G, Grzmil M, Lino MM, Clement-Schatlo V, Frank S, Merlo A, Hemmings BA. Mer receptor tyrosine kinase promotes invasion and survival in glioblastoma multiforme. Oncogene. 2012:1–11. doi: 10.1038/onc.2012.104. [DOI] [PubMed] [Google Scholar]
- 8.(a) Schlegel JS, MJ, Sather S, Moschos SJ, Tan A-C, Winges A, DeRyckere D, Carson CC, Trembath DG, Tentler JJ, Eckhardt SG, Kuan P-F, Hamilton RL, Duncan LM, Alldredge V, Groben PA, Ollila DW, Thomas NE, Liu J, Zhang W, Yang C, Wang X, Frye SV, Earp HS, Shields JM, Graham DK. Mer receptor tyrosine kinase is a novel therapeutic target in melanoma. J Clin Invest. doi: 10.1172/JCI67816. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Tworkoski K, Singhal G, Szpakowski S, Zito CI, Bacchiocchi A, Muthusamy V, Bosenberg M, Krauthammer M, Halaban R, Stern DF. Phosphoproteomic screen identifies potential therapeutic targets in melanoma. Molecular cancer research : MCR. 2011;9(6):801–12. doi: 10.1158/1541-7786.MCR-10-0512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Keating AK, Salzberg DB, Sather S, Liang X, Nickoloff S, Anwar A, Deryckere D, Hill K, Joung D, Sawczyn KK, Park J, Curran-Everett D, McGavran L, Meltesen L, Gore L, Johnson GL, Graham DK. Lymphoblastic leukemia/lymphoma in mice overexpressing the Mer (MerTK) receptor tyrosine kinase. Oncogene. 2006;25(45):6092–100. doi: 10.1038/sj.onc.1209633. [DOI] [PubMed] [Google Scholar]
- 10.(a) Huang X, Finerty P, Jr, Walker JR, Butler-Cole C, Vedadi M, Schapira M, Parker SA, Turk BE, Thompson DA, Dhe-Paganon S. Structural insights into the inhibited states of the Mer receptor tyrosine kinase. J Struct Biol. 2009;165(2):88–96. doi: 10.1016/j.jsb.2008.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Greshock J, Bachman KE, Degenhardt YY, Jing J, Wen YH, Eastman S, McNeil E, Moy C, Wegrzyn R, Auger K, Hardwicke MA, Wooster R. Molecular Target Class Is Predictive of In vitro Response Profile. Cancer Res. 2010;70(9):3677–3686. doi: 10.1158/0008-5472.CAN-09-3788. [DOI] [PubMed] [Google Scholar]; (c) Howard S, Berdini V, Boulstridge JA, Carr MG, Cross DM, Curry J, Devine LA, Early TR, Fazal L, Gill AL, Heathcote M, Maman S, Matthews JE, McMenamin RL, Navarro EF, O’Brien MA, O’Reilly M, Rees DC, Reule M, Tisi D, Williams G, Vinkovic M, Wyatt PG. Fragment-based discovery of the pyrazol-4-yl urea (AT9283), a multitargeted kinase inhibitor with potent aurora kinase activity. J Med Chem. 2009;52(2):379–88. doi: 10.1021/jm800984v. [DOI] [PubMed] [Google Scholar]; (d) Schroeder GM, An Y, Cai ZW, Chen XT, Clark C, Cornelius LA, Dai J, Gullo-Brown J, Gupta A, Henley B, Hunt JT, Jeyaseelan R, Kamath A, Kim K, Lippy J, Lombardo LJ, Manne V, Oppenheimer S, Sack JS, Schmidt RJ, Shen G, Stefanski K, Tokarski JS, Trainor GL, Wautlet BS, Wei D, Williams DK, Zhang Y, Fargnoli J, Borzilleri RM. Discovery of N-(4-(2-amino-3-chloropyridin-4-yloxy)-3-fluorophenyl)-4-ethoxy-1-(4-fluorophenyl )-2-oxo-1,2-dihydropyridine-3-carboxamide (BMS-777607), a selective and orally efficacious inhibitor of the Met kinase superfamily. J Med Chem. 2009;52(5):1251–4. doi: 10.1021/jm801586s. [DOI] [PubMed] [Google Scholar]
- 11.Liu J, Yang C, Simpson C, DeRyckere D, Van DA, Miley MJ, Kireev D, Norris-Drouin J, Sather S, Hunter D, Korboukh VK, Patel HS, Janzen WP, Machius M, Johnson GL, Earp HS, Graham DK, Frye SV, Wang X. Discovery of Small Molecule Mer Kinase Inhibitors for the Treatment of Pediatric Acute Lymphoblastic Leukemia. ACS Med Chem Lett. 2012;3:129–134. doi: 10.1021/ml200239k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.(a) Dubin AE, Nasser N, Rohrbacher J, Hermans AN, Marrannes R, Grantham C, Van Rossem K, Cik M, Chaplan SR, Gallacher D, Xu J, Guia A, Byrne NG, Mathes C. Identifying modulators of hERG channel activity using the PatchXpress planar patch clamp. Journal of biomolecular screening. 2005;10(2):168–81. doi: 10.1177/1087057104272295. [DOI] [PubMed] [Google Scholar]; (b) Witchel HJ, Milnes JT, Mitcheson JS, Hancox JC. Troubleshooting problems with in vitro screening of drugs for QT interval prolongation using HERG K+ channels expressed in mammalian cell lines and Xenopus oocytes. Journal of pharmacological and toxicological methods. 2002;48(2):65–80. doi: 10.1016/S1056-8719(03)00041-8. [DOI] [PubMed] [Google Scholar]
- 13.(a) Priest B, Bell IM, Garcia M. Role of hERG potassium channel assays in drug development. Channels. 2008;2(2):87–93. doi: 10.4161/chan.2.2.6004. [DOI] [PubMed] [Google Scholar]; (b) Redfern WS, Carlsson L, Davis AS, Lynch WG, MacKenzie I, Palethorpe S, Siegl PK, Strang I, Sullivan AT, Wallis R, Camm AJ, Hammond TG. Relationships between preclinical cardiac electrophysiology, clinical QT interval prolongation and torsade de pointes for a broad range of drugs: evidence for a provisional safety margin in drug development. Cardiovascular research. 2003;58(1):32–45. doi: 10.1016/s0008-6363(02)00846-5. [DOI] [PubMed] [Google Scholar]; (c) Picard S, Lacroix P. QT interval prolongation and cardiac risk assessment for novel drugs. Curr Opin Investig Drugs. 2003;4(3):303–8. [PubMed] [Google Scholar]; (d) De Ponti F, Poluzzi E, Cavalli A, Recanatini M, Montanaro N. Safety of non-antiarrhythmic drugs that prolong the QT interval or induce torsade de pointes: an overview. Drug safety : an international journal of medical toxicology and drug experience. 2002;25(4):263–86. doi: 10.2165/00002018-200225040-00004. [DOI] [PubMed] [Google Scholar]
- 14.Liu J, Wang X. Microwave-Assisted, Divergent Solution-Phase Synthesis of 1,3,6-Trisubstituted Pyrazolo[3,4-d]pyrimidines. ACS Combinatorial Science. 2011;13(4):414–420. doi: 10.1021/co200039k. [DOI] [PubMed] [Google Scholar]
- 15.(a) Pommereau A, Pap E, Kannt A. Two simple and generic antibody-independent kinase assays: comparison of a bioluminescent and a microfluidic assay format. Journal of biomolecular screening. 2004;9(5):409–16. doi: 10.1177/1087057104264175. [DOI] [PubMed] [Google Scholar]; (b) Dunne J, Reardon H, Trinh V, Li E, Farinas J. Comparison of on-chip and off-chip microfluidic kinase assay formats. Assay Drug Dev Technol. 2004;2(2):121–9. doi: 10.1089/154065804323056468. [DOI] [PubMed] [Google Scholar]; (c) Bernasconi P, Chen M, Galasinski S, Popa-Burke I, Bobasheva A, Coudurier L, Birkos S, Hallam R, Janzen WP. A chemogenomic analysis of the human proteome: application to enzyme families. Journal of biomolecular screening. 2007;12(7):972–82. doi: 10.1177/1087057107306759. [DOI] [PubMed] [Google Scholar]
- 16.Wang X, Liu J, Yang C, Zhang W, Frye S, Kireev D. WO2011146313A1. Preparation of pyrazolopyrimidine compounds for the treatment of cancer. 2011
- 17.(a) Morrison JF. Kinetics of the reversible inhibition of enzyme-catalysed reactions by tight-binding inhibitors. Biochimica et biophysica acta. 1969;185(2):269–86. doi: 10.1016/0005-2744(69)90420-3. [DOI] [PubMed] [Google Scholar]; (b) Copeland RA. Evaluation of enzyme inhibitors in drug discovery. A guide for medicinal chemists and pharmacologists. Methods of biochemical analysis. 2005;46:1–265. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.