

Abstract
Shuttle plasmids are among the few routinely utilized tools in the Streptococcus mutans genetic system that still require the use of classical cloning methodologies and intermediate hosts for genetic manipulation. Accordingly, it typically requires considerably less time and effort to introduce mutations onto the S. mutans chromosome than it does to construct shuttle vectors for expressing genes in trans. Occasionally, shuttle vector constructs also exhibit toxicity in E. coli, which prevents their proper assembly. To circumvent these limitations, we modified a prolonged overlap extension PCR (POE-PCR) protocol to facilitate direct plasmid assembly in S. mutans. Using solely PCR, we created the reporter vector pZX7, which contains a single minimal streptococcal replication origin and harbors a spectinomycin resistance cassette and the gusA gene encoding β-glucuronidase. We compared the efficiency of pZX7 assembly using multiple strains of S. mutans and were able to obtain from 5×103 – 2×105 CFU/μg PCR product. Likewise, we used pZX7 to further demonstrate that Streptococcus sanguinis and Streptococcus gordonii are also excellent hosts for cloning-independent plasmid assembly, which suggests that this system is likely to function in numerous other streptococci. Consequently, it should be possible to completely forgo the use of E. coli – Streptococcus shuttle vectors in many streptococcal species, thereby decreasing the time and effort required to assemble constructs and eliminating any toxicity issues associated with intermediate hosts.
Keywords: Streptococcus mutans, Streptococcus sanguinis, Streptococcus gordonii, shuttle vector, overlap extension PCR
Introduction
Genetic research of streptococci has benefited tremendously from the efficient natural competence ability of many of these species coupled with the increasingly diverse palette of genetic tools that have been adapted for use in these organisms. Among the human oral streptococci, Streptococcus mutans has the most sophisticated genetic system. Various types of gene mutations can be engineered in a surprisingly short time frame using a variety of antibiotic resistance cassettes (Kuramitsu, 1993, Kuramitsu, 2003, Russell, 1994) or via several markerless mutagenesis systems (Atlagic et al., 2006, Banerjee and Biswas, 2008, Merritt et al., 2007, Xie et al., 2011). Multiple shuttle vectors are also available for complementation (Biswas et al., 2008, Chen and LeBlanc, 1992, Chen et al., 2011, Macrina et al., 1982). Likewise, these same tools are routinely employed to introduce numerous types of reporter genes to assay gene expression or for in situ imaging applications (Goodman and Gao, 1999, Honeyman et al., 2002, Hudson and Curtiss, 1990, Kreth et al., 2004, Yoshida and Kuramitsu, 2002). As is, the S. mutans genetic toolbox is already more akin to that of a model organism (Lemos et al., 2013). However, unlike E. coli genetic research, genetic work in S. mutans still requires intermediate hosts to clone constructs. Classical cloning methodologies utilizing restriction enzymes and ligases are also frequently the most frustrating and labor intensive steps during strain construction and can form significant obstacles to progress when the cloned DNA fragments exhibit toxicity in E. coli. Consequently, it has been one of our longstanding goals to improve the S. mutans genetic system to the point where we no longer require such cloning methodologies or intermediate hosts. We have recently made significant progress toward this goal by creating a cloning-independent markerless mutagenesis system for S. mutans (Xie, Okinaga, Qi, Zhang and Merritt, 2011). Using only overlap extension PCR, we are now able to create unlimited chromosomal deletions, point mutations, truncations, reporter gene insertions, etc. all within a wild-type parent background and free of antibiotic resistance cassettes or scars on the chromosome. This single system has already greatly diminished the need for E. coli in the majority of our S. mutans genetic work. A prominent exception is plasmid construction. Often times it is necessary to express a gene in trans, which necessitates cloning constructs onto E. coli – Streptococcus shuttle vectors. Since streptococcal promoters are typically active in E. coli, cloned genes tend to be highly expressed due to the plasmid copy number and frequently exhibit toxicity (Warren et al., 2007). Consequently, certain constructs may be difficult or impossible to assemble.
Recently, two separate cloning-independent plasmid construction methodologies have been reported for use in E. coli and B. subtilis (van den Ent and Lowe, 2006, You et al., 2011). These systems both utilize similar prolonged overlap extension PCR (POE-PCR) approaches to introduce inserts into target vectors. The insert is first PCR amplified with primers that add stretches of vector complementarity to the insert amplicon. Next, the insert amplicon is mixed in an equimolar concentration with either a circular plasmid or PCR amplified plasmid in a final primerless PCR reaction. For this reaction, the complementary sequences engineered into the insert amplicon serve as the primers for the final PCR step. Thus, additional primers are not required for polymerization. Each round of denaturation and annealing in the PCR reaction allows the terminal ends of the insert amplicon to hybridize to the target vector, which then facilitates ligation and amplification by the polymerase during the extension step. (Bryksin and Matsumura, 2010, You, Zhang and Zhang, 2011). The continual cycling in the PCR reaction ultimately generates a series of linear concatemers consisting of variable numbers of repeated subunits of vector + insert and will appear as high molecular weight smears in agarose gels after electrophoresis. Interestingly, if these linear concatemers are transformed, they are actually recircularized intracellularly into stably replicating plasmids, presumably due to recombination between the numerous repeats of vector + insert within the molecules (You, Zhang and Zhang, 2011). Given our previous success with overlap extension PCR for mutagenesis, we were curious whether PCR generated concatemers would function for plasmid assembly in S. mutans. Here, we demonstrate that this approach is highly efficient in multiple strains of S. mutans as well as in S. sanguinis and S. gordonii. These results suggest that numerous streptococcal species should be able to directly serve as hosts for vector construction, which eliminates the requirement for classical cloning methodologies, shuttle vectors, and intermediate hosts during plasmid assembly.
Materials and Methods
Bacterial strains and culture conditions
Bacterial strains and plasmids used in this study are listed in Table 1. All Streptococcus strains were grown anaerobically (in an atmosphere consisting of 85% N2, 10% CO2, and 5% H2) at 37°C. For natural transformation experiments, cells were maintained in Todd -Hewitt medium (Difco) supplemented with 0.3% (wt/vol) yeast extract (THYE). For the selection of antibiotic-resistant colonies in Streptococcus mutans, BHI plates were supplemented with spectinomycin (Sigma) (1000 μg ml−1). For the selection of antibiotic-resistant colonies in Streptococcus sanguinis or Streptococcus gordonii, BHI plates were supplemented with spectinomycin (500 μg ml−1). To visualize β-glucuronidase activity in transformants, BHI plates were supplemented with 0.75 mM 5-Bromo-4-chloro-3-indolyl-β-d-glucuronide (X-Glu) (Biosynth International Inc). The competence-stimulating peptide used for S. mutans transformation (CSP-Sm) was custom-synthesized by Anaspec, while the competence-stimulating peptide used for S. sanguinis transformation (CSP-Ss) was kindly provided by Dr. J. Kreth.
Table 1.
Bacterial strains and plasmids used in this study
| Strain or plasmid | Characteristics | Reference |
|---|---|---|
| Strains | ||
| UA140 | Wild-type Streptococcus mutans | (Qi et al., 2001) |
| UA159 | Wild-type Streptococcus mutans reference strain | (Ajdic et al., 2002) |
| GS5 | Wild-type Streptococcus mutans | (Biswas and Biswas, 2012) |
| ATCC 25175 | Wild-type Streptococcus mutans | |
| SK36 | Wild-type Streptococcus sanguinis | (Xu et al., 2007) |
| DL1 | Wild-type Streptococcus gordonii | (Vickerman et al., 2007) |
| PRgusA | UA140 hdrRp::gusA,ΔhdrRM, markerless | Unpublished data |
| Plasmids | ||
| pZX7 | pVA380:: hdrRP::gusA, specr | This study |
| pDL278 | E. coli – Streptococcus shuttle vector, specr | This study |
| pFW5 | Streptococcal suicide vector, specr | (Podbielski et al., 1996) |
| pET44b | E. coli expression vector, ampr | |
Cloning-independent plasmid construction in Streptococcus strains
A similar protocol was employed here as has been described in detail for use in E. coli and B. subtilis (You, Zhang and Zhang, 2011) and is briefly summarized. Firstly, linear DNA fragments of vector and insert were generated by PCR with prim ers adding >40 bp of complementarity the amplicon termini. Secondly, concatemers were generated by prolonged overlap extension PCR (POE-PCR) using the amplicons generated in the first PCR reaction as the template and primers. Finally, the POE-PCR products (concatemers) were directly transformed into Streptococcus strains via natural transformation. The primers used in this study are shown in Table 2. Phusion DNA polymerase (New England BioLabs) was used for the PCR steps in this study. Generation of the vector and insert amplicons in the first step PCR was performed according to the manufacturers’ instructions as follows: 10 μL 5X Phusion buffer, 1 μL 10mM dNTP mix, 1 μL of 10 μM forward and reverse primer, <10 ng template, and 1 unit of enzyme were mixed in 50 μL total volume. The reaction was cycled using an initial denaturation of 98°C for 30 sec. followed by 30 cycles of 98°C denaturation for 10 sec. + annealing at primer Tm for 15 sec. + 72°C extension for 2 Kb/min. and a final 72°C extension for 5 min. The PCR amplicons were subsequently purified using the Qiagen PCR purification kit. The second step PCR generating the concatemers was assembled as follows. Each 50 μL reaction contained: 100 ng insert amplicon, equimolar concentration of vector amplicon, 10 μL 5X Phusion reaction buffer, 1 μL 10mM dNTP mix, and 1 unit of Phusion DNA polymerase. The PCR program was set according to the following: 98°C initial denaturation for 30 seconds, 25 cycles of denaturation at 98°C for 10 seconds + 65°C annealing for 15 seconds + 72°C extension for a period based on the size of the final plasmid (2Kb/min.), and a final extension at 72°C for 5 minutes.
Table 2.
Primers used in this study
| Primer | Sequence (5′ → 3′) | Purpose |
|---|---|---|
| pRF-pDL TCTTAGAC | CACGAACGAAAATCGATCTTCTCACTAATGGTTACC | pZX7 construction and confirmation |
| gusAR-pDL | ACGAGGCTAGTTTACCGTATCTTCATTGTTTGCCTCCCTGCTGC | pZX7 construction and confirmation |
| pDLF-PR | GTCTAAGAGGTAACCATTAGTGAGAAGATCGATTTTCGTTCGTG | pZX7 construction |
| pDLR-gusA | GCAGCAGGGAGGCAAACAATGAAGATACGGTAAACTAGCCTCGT | pZX7 construction |
Natural transformation of S. mutans UA140, UA159, GS5, 25175
S. mutans strains were cultivated overnight in THYE. The following day, stationary phase cultures were diluted 1:20 in THYE and incubated at 37°C until the OD600 reached 0.2 – 0.3 (about 2 – 3 hours). 1 μg of transforming DNA (concatemer PCR) and 1 μg ml−1 final concentration CSP-Sm were added into each 500 μl cell culture and incubated for another 2 hours. After the incubation period, cells were diluted appropriately before plating a portion on selective BHI plates and incubating at 37°C until colonies were visible. Depending upon the efficiency of plasmid con struction in a given strain, it was necessary to plate varying dilutions of the transformation reactions to obtain the desired range of 200 – 500 CFU/plate.
Visualization of plasmids extracted from S. mutans
Six UA159 POE-PCR transformants were randomly selected and inoculated into separate tubes containing 3 mL BHI + spectinomycin. Stationary phase cultures were resuspended in 500 μL buffer (10 mM pH 7.5 Tris-Cl + 1 mM EDTA + 10 mg/mL lysozyme). The cells were incubated for 4hr at 37°C. Following the incubation period, the plasmids were extracted using the Qiagen Plasmid Miniprep kit beginning with the lysis step in the manufacturer’s protocol. 500 μL buffer P2 and 700 μL buffer N3 were used for alkaline lysis and neutralization, respectively. Clarified lysates from each culture were loaded into six spin columns and washed according to the manufacturer’s protocol. The plasmids were each eluted in a 50 μL total volume and then combined. The combined pool of 300 μL plasmid was further concentrated to a total volume of 50 μL using a Zymo DNA Clean and Concentrator-5 kit. As a negative control, the same plasmid preparation protocol was performed as described using a total of 18 mL culture from wild-type UA159. Concentrated plasmid preparations were finally loaded into an agarose gel for direct visualization.
Natural transformation of S. sanguinis SK36 and S. gordonii DL1
Bacteria were cultivated overnight in THYE. The next day, stationary phase cultures were diluted 1:20 in THYE and incubated at 37°C until the OD600 reached 0.1 (about 1 hour). 1 μg DNA (concatemer PCR) was added into each 500 μl culture and the cells were incubated for another 3 hours. For S. sanguinis transformation, a final concentration of 1 μg ml−1 CSP-Ss was added together with the transforming DNA. After the final incubation, cells were plated on selective BHI plates and incubating at 37°C until colonies were visible.
Construction of the gusA reporter plasmid
In order to test the efficiency of cloning-independent plasmid assembly in different Streptococcus species, the plasmid pZX7 (streptococcal replicon with gusA reporter insertion) was constructed. Firstly, the gusA open reading frame (ORF) + hdrR promoter fusion (~1.9 Kb) was amplified using primer pair pRF-pDL and gusAR-pDL and the genomic DNA of reporter strain PRgusA. This strain has both open reading frames of the S. mutans hdrRM operon markerlessly replaced with the E. coli gusA ORF (unpublished strain). The streptococcal plasmid replicon (~3.7 Kb) was generated by PCR with the primer pair pDLF-PR and pDLR-gusA using the E. coli – Streptococcus shuttle vector pDL278 as a template. Complementary regions between the insert and vector amplicons allowed for the generation of concatamers by POE-PCR. 5 μl of the resulting POE-PCR products (about 1 μg concatamer DNA) was used for transformation in Streptococcus species using the protocols described above. The final transformation culture was diluted before plating on BHI plates containing spectinomycin and X-Glu. Due to the excessive numbers of transformants, it was necessary to dilute the S. mutans, S. sanguinis, and S gordonii transformation reactions 1000-fold, 100-fold, and 10-fold, respectively in order to obtain the desired number of CFU per plate. After the colonies formed, the plates were kept at room temperature in an aerobic atmosphere for an additional overnight to complete color development from X-Glu hydrolysis. Five colonies were randomly selected for plasmid extraction. Using the plasmid as a template, PCR was performed to confirm the construct with the primer pair pRF-pDL and gusAR-pDL.
Results
Linear concatemers form replicating plasmids after transformation in S. mutans
In order to test the feasibility of cloning-independent plasmid construction in S. mutans, we designed a POE-PCR strategy that would result in the production of a reporter plasmid pZX7 expressing β-glucuronidase, which we could easily detect with the chromogenic substrate X-Glu (Fig. 1). We began by PCR amplifying the streptococcal origin of replication and a spectinomycin resistance cassette from the E. coli – Streptococcus shuttle vector pDL278. Based upon previous predictions (LeBlanc et al., 1992), we confirmed the minimal streptococcal plasmid replicon from this vector using a variety of primer combinations (data not shown). Our optimal primer combination for both amplicon size and quality is illustrated in figure 1A. For the vector insert, we PCR amplified a transcription fusion between the S. mutans hdrRM operon promoter and gusA open reading frame (ORF) (Fig. 2A) using a previously constructed reporter strain that we had already determined to hydrolyze X-Glu (unpublished). As expected, the POE-PCR reaction generated a high molecular weight smear when separated by electrophoresis (Fig. 2A). A portion of the PCR reaction contained concatemers too large to enter the gel, and therefore remained in the well throughout the entire electrophoresis ru n. These extremely high molecular weight concatemers were observed to correlate with the total number of PCR cycles. For PCR reactions containing >25 cycles, a much higher percentage of the final PCR product contained concatemers too large to separate by typical agarose gel electrophoresis (data not shown). A similar result was also reported to occur in E. coli POE-PCR plasmid constructions (Bryksin and Matsumura, 2010). We next transformed 5 μl of the PCR reaction directly into S. mutans wild-type strain UA140 using natural transformation and were able to obtain a tremendous number of β-glucuronidase producing transformants (Fig. 2B). For example, to generate the image shown in figure 2B, it was necessary to dilute the transformation reaction 1000-fold before plating. Five blue CFU were randomly selected from the transformation plate, inoculated into broth, and used for plasmid preparation. As expected, we were able to amplify the gusA transcription fusion construct from each plasmid (Fig. 2C). We were also interested to determine whether transformation of the concatemer mixture results in the creation of plasmid multimers in vivo. For this, it was necessary to transform strain UA159 because strain UA140 naturally harbors a cryptic plasmid of nearly identical size to pZX7 (Zou et al., 2001). We extracted plasmids from cultures of six separate POE-PCR transformants, combined the preparations, and then concentrated the samples. As shown in figure 2D, most, if not all, of the isolated plasmids were uniform in size and migrated at the expected size range for an uncut vector of 5.6 Kb. As a control, the plasmid extract was also linearized using BamHI, which only cuts within the gusA gene to yield a single 5.6 Kb fragment. Thus, we found no evidence to suggest that plasmid multimers were a common byproduct from transformation of linear concatemers. Next, we repeated the POE-PCR procedure using several naturally transformable S. mutans strains and found that all were highly efficient at recircularizing PCR generated concatemers. Strain UA140 yielded the greatest number of transformants, whereas strain ATCC 25175 yielded considerably fewer (Fig. 3). Though, this strain still yielded >5000 CFU/μg of PCR product. From these results, we conclude that the POE-PCR procedure is likely to function extremely well in naturally transformable strains of S. mutans.
Fig. 1. Protocol for creating replicating plasmids in S. mutans using POE-PCR.

(A) The E. coli – Streptococcus shuttle vector pDL278 is shown. The indicated primers were used to PCR amplify the streptococcal origin of replication together with the spectinomycin resistance cassette. (B) Illustration of the POE-PCR procedure used to create β-glucuronidase expressing plasmids. The target vector was amplified from the shuttle vector pDL278, while the transcription fusion to the gusA gene was PCR amplified from an existing S. mutans gusA reporter strain. The primers used to amplify the gusA transcription fusion also contained complementary regions to the intended insertion site of the target vector (represented in red and blue). The two resulting PCR amplicons were subsequently mixed in a similar molar ratio and then assembled into linear concatemers through a second round of PCR. The regions of complementarity between the insert and vector amplicons served as the primers in the final PCR. Thus, no additional primers were added to this reaction. The resulting mixture of PCR generated concatemers was transformed directly into S. mutans, where the concatemers were recircularized into replicating plasmids containing the desired insert.
Fig. 2. Verification of the POE-PCR approach to plasmid construction in S. mut ans.
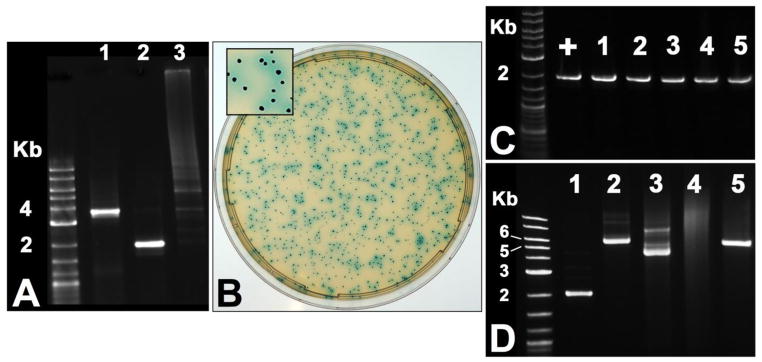
(A) In the first round of PCR, the target vector (Lane 1) and gusA insert (Lane 2) were amplified using typical PCR conditions. The gusA insert was amplified with primers containing 5′ terminal regions of complementarity to the target vector. The two amplicons were mixed in equimolar concentrations and subjected to a second round of primerless PCR. A successful amplification resulted in the generation of high molecular weight concatemers that appeared as a smear when separated by agarose gel electrophoresis (Lane 3). (B) The resulting PCR generated concatemers were transformed directly into S. mutans and plated on medium containing spectinomycin and X-Glu. In order to obtain well-separated colonies on the plate, it was necessary to dilute the transformation reaction 1000-fold prior to plating. (C) 5 blue colonies from the transformation plate were inoculated into liquid medium for plasmid preparation and then tested for the presence of the expected gusA insert. The sample denoted with a “+” indicates a positive control. (D) Plasmids generated by POE-PCR were also separated by electrophoresis to determine whether the resulting plasmids are of the expected size (5.6 Kb). Lane 1: uncut pFW5 (2.7 Kb); Lane 2: uncut pET44b (7.3 Kb); Lane 3: uncut pZX7 (5.6 Kb, combined plasmid preparation from 18 mL of 6 separate POE-PCR transformants); Lane 4: untransformed wild -type (plasmid preparation from 18 mL culture); Lane 5: linearized pZX7 (5.6 Kb, the same plasmid preparation as in Lane 3, but digested with BamHI)
Fig. 3. Assessment of cloning-independent plasmid construction in multiple strains of S. mutans.

PCR generated concatemers of pZX7 were transformed into four separate wild -type strains of S. mutans. The data are presented as the number of transformants per μg of concatemer DNA. Shown here are the averages of three independent experiments.
Cloning-independent assembly of pZX7 in other streptococci
Since numerous streptococci contain efficient natural transformation systems, we suspected that linear concatemers would generate replicating plasmids in a variety of streptococcal species other than S. mutans. To test this, we assayed the ability of Streptococcus sanguinis and Streptococcus gordonii to recircularize linear concatemers of the β-glucuronidase reporter plasmid pZX7. Both species were found to be highly amenable to cloning-independent plasmid assembly with each producing >2000 CFU/μg of PCR product (Fig. 4A). For S. gordonii, transformations were performed without added CSP, which suggests that the efficiency of the procedure could be further increased in this organism. It was also apparent that virtually every transformant of both species was able to hydrolyze X-Glu, indicating that the vast majority of the clones harbored the expected plasmid constructs (Fig. 4B & C). These results suggest that numerous species of Streptococcus are likely to serve as suitable hosts for assembling replicating plasmids from POE-PCR amplicons.
Fig. 4. Cloning-independent construction of pZX7 in other streptococci.
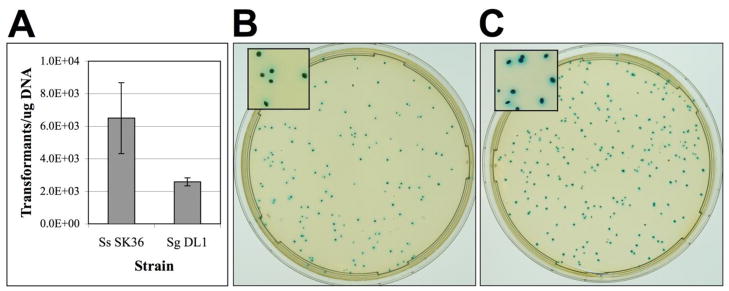
(A) POE-PCR was used to generate concatemers of the pDL278 streptococcal replicon and a gusA reporter fusion. The concatemers were transformed into S. sanguinis SK36 and S. gordonii DL1 and then assayed for the number of pZX7 transformants per μg of concatemer DNA. The averages of three independent experiments are shown. (B) The concatemer PCR reaction was transformed into S. sanguinis, diluted 100-fold, and plated on agar plates containing spectinomycin and X-Glu. (C) The concatemer PCR reaction was transformed into S. gordonii, diluted 10-fold, and plated on agar plates containing spectinomycin and X-Glu.
Discussion
In the present study, we provide the first demonstration of cloning-independent plasmid assembly in streptococci. This approach was both highly efficient and reliable, even for constructions requiring multiple inserts. For example, we recently constructed a plasmid containing three inserts using POE-PCR with no additional modifications to the protocol (data not shown). In our hands, the first round of PCR has been the primary determinant for successful concatemer formation. The individual vector and insert PCR reactions should all yield abundant, highly specific amplicons. Even weakly amplified nonspecific PCR products could potentially reduce the efficiency of the POE-PCR step or inhibit the reaction altogether. In our experience, gel purification of nonspecific PCR reactions also provides little improvement in the final results. Thus, if the first round PCR products contain nonspecific amplicons, the best approach is simply to synthesize alternative primers to improve the quality of the PCR reaction. In fact, this has been true of all of our overlap extension PCR constructions and is not exclusive to POE-PCR. It has also been reported that POE-PCR is highly influenced by the choice of polymerase with Phusion polymerase from NEB being among the best options (You, Zhang and Zhang, 2011). We used Phusion in this study and have not compared whether other polymerases function as well in POE-PCR reactions. Though, we have had previous success with polymerases other than Phusion for our typical overlap extension PCR reactions.
It should be noted that in this study, natural competence was the only approach used to transform linear concatemers into S. mutans, S. sanguinis, and S. gordonii. Since the natural competence state also primes the cells for recombination, it is conceivable that linear concatemers entering the cell via natural competence are much more apt to be recirularized than if introduced artificially. Consistent with this notion, transformation of linear concatem ers into naturally competent B. subtilis yielded several orders of magnitude greater CFU than with commercially prepared chemically competent E. coli (You, Zhang and Zhang, 2011). It is still unknown how the efficiency of recirularization would have been affected if the DNA were introduced via electroporation. This could be a potentially limiting factor for streptococci lacking efficient natural competence systems like S. pyogenes. Lastly, it is worth noting that β-glucuronidase has not been previously reported for use as a reporter system in S. sanguinis or S. gordonii genetic studies. Our results indicate that the gusA gene would likely serve as a useful genetic tool in these species and probably other streptococci as well.
Conclusions
In this study, we demonstrate the utility of cloning-independent plasmid construction in naturally competent streptococci. A variety of commonly used laboratory strains of S. mutans, S. sanguinis, and S. gordonii all had the ability to assemble the β-glucuronidase reporter plasmid pZX7 directly from linear PCR products. Thus, our results demonstrate the feasibility of assembling streptococcal plasmids without using intermediate hosts. This approach greatly decreases the time and effort required for plasmid construction and circumvents the problems of toxicity that are often encountered when cloning into E. coli – Streptococcus shuttle vectors.
Highlights.
A new reporter vector is constructed directly in species of Streptococcus using only PCR
Plasmid assembly is highly efficient and ranges anywhere from 2×103 – 2×105 CFU/μg DNA
Approach can negate the requirement for E. coli and shuttle vectors in genetic studies
Demonstrate the utility of β-glucuronidase as a reporter in 2 additional species of Streptococcus
Acknowledgments
This work was supported by an NIDCR DE018893 grant to J.M. We would like to thank Dr. J. Kreth for supplying strains of S. sanguinis and S. gordonii as well as Dr. Lanyan Zheng for helpful advice on the transformation of these organisms.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ajdic D, McShan WM, McLaughlin RE, Savic G, Chang J, Carson MB, Primeaux C, Tian R, Kenton S, Jia H, Lin S, Qian Y, Li S, Zhu H, Najar F, Lai H, White J, Roe BA, Ferretti JJ. Genome sequence of Streptococcus mutans UA159, a cariogenic dental pathogen. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:14434–14439. doi: 10.1073/pnas.172501299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atlagic D, Kilic AO, Tao L. Unmarked gene deletion mutagenesis of gtfB and gtfC in Streptococcus mutans using a targeted hit-and-run strategy with a thermosensitive plasmid. Oral microbiology and immunology. 2006;21:132–135. doi: 10.1111/j.1399-302X.2006.00267.x. [DOI] [PubMed] [Google Scholar]
- Banerjee A, Biswas I. Markerless multiple-gene-deletion system for Streptococcus mutans. Applied and environmental microbiology. 2008;74:2037–2042. doi: 10.1128/AEM.02346-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswas I, Jha JK, Fromm N. Shuttle expression plasmids for genetic studies in Streptococcus mutans. Microbiology. 2008;154:2275–2282. doi: 10.1099/mic.0.2008/019265-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswas S, Biswas I. Complete genome sequence of Streptococcus mutans GS-5, a serotype c strain. Journal of bacteriology. 2012;194:4787–4788. doi: 10.1128/JB.01106-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryksin AV, Matsumura I. Overlap extension PCR cloning: a simple and reliable way to create recombinant plasmids. BioTechniques. 2010;48:463–465. doi: 10.2144/000113418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YY, LeBlanc DJ. Genetic analysis of scrA and scrB from Streptococcus sobrinus 6715. Infection and immunity. 1992;60:3739–3746. doi: 10.1128/iai.60.9.3739-3746.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YY, Shieh HR, Lin CT, Liang SY. Properties and construction of plasmid pFW213, a shuttle vector with the oral Streptococcus origin of replication. Applied and environmental microbiology. 2011;77:3967–3974. doi: 10.1128/AEM.02828-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman SD, Gao Q. Firefly luciferase as a reporter to study gene expression in Streptococcus mutans. Plasmid. 1999;42:154–157. doi: 10.1006/plas.1999.1416. [DOI] [PubMed] [Google Scholar]
- Honeyman AL, Cote CK, Curtiss R., 3rd Construction of transcriptional and translational lacZ gene reporter plasmids for use in Streptococcus mutans. J Microbiol Methods. 2002;49:163–171. doi: 10.1016/s0167-7012(01)00368-2. [DOI] [PubMed] [Google Scholar]
- Hudson MC, Curtiss R., 3rd Regulation of expression of Streptococcus mutans genes important to virulence. Infection and immunity. 1990;58:464–470. doi: 10.1128/iai.58.2.464-470.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreth J, Merritt J, Bordador C, Shi W, Qi F. Transcriptional analysis of mutacin I (mutA) gene expression in planktonic and biofilm cells of Streptococcus mutans using fluorescent protein and glucuronidase reporters. Oral microbiology and immunology. 2004;19:252–256. doi: 10.1111/j.1399-302X.2004.00148.x. [DOI] [PubMed] [Google Scholar]
- Kuramitsu HK. Virulence factors of mutans streptococci: role of molecular genetics. Crit Rev Oral Biol Med. 1993;4:159–176. doi: 10.1177/10454411930040020201. [DOI] [PubMed] [Google Scholar]
- Kuramitsu HK. Molecular genetic analysis of the virulence of oral bacterial pathogens: an historical perspective. Crit Rev Oral Biol Med. 2003;14:331–344. doi: 10.1177/154411130301400504. [DOI] [PubMed] [Google Scholar]
- LeBlanc DJ, Lee LN, Abu-Al-Jaibat A. Molecular, genetic, and functional analysis of the basic replicon of pVA380–1, a plasmid of oral streptococcal origin. Plasmid. 1992;28:130–145. doi: 10.1016/0147-619x(92)90044-b. [DOI] [PubMed] [Google Scholar]
- Lemos JA, Quivey RG, Koo H, Abranches J. Streptococcus mutans: a new Gram -positive paradigm? Microbiology. 2013 doi: 10.1099/mic.0.066134-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macrina FL, Tobian JA, Jones KR, Evans RP, Clewell DB. A cloning vector able to replicate in Escherichia coli and Streptococcus sanguis. Gene. 1982;19:345–353. doi: 10.1016/0378-1119(82)90025-7. [DOI] [PubMed] [Google Scholar]
- Merritt J, Tsang P, Zheng L, Shi W, Qi F. Construction of a counterselection -based in-frame deletion system for genetic studies of Streptococcus mutans. Oral microbiology and immunology. 2007;22:95–102. doi: 10.1111/j.1399-302X.2007.00329.x. [DOI] [PubMed] [Google Scholar]
- Podbielski A, Spellerberg B, Woischnik M, Pohl B, Lutticken R. Novel series of plasmid vectors for gene inactivation and expression analysis in group A streptococci (GAS) Gene. 1996;177:137–147. doi: 10.1016/0378-1119(96)84178-3. [DOI] [PubMed] [Google Scholar]
- Qi F, Chen P, Caufield PW. The group I strain of Streptococcus mutans, UA140, produces both the lantibiotic mutacin I and a nonlantibiotic bacteriocin, mutacin IV. Applied and environmental microbiology. 2001;67:15–21. doi: 10.1128/AEM.67.1.15-21.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell RR. The application of molecular genetics to the microbiology of dental caries. Caries research. 1994;28:69–82. doi: 10.1159/000261625. [DOI] [PubMed] [Google Scholar]
- van den Ent F, Lowe J. RF cloning: a restriction-free method for inserting target genes into plasmids. Journal of biochemical and biophysical methods. 2006;67:67–74. doi: 10.1016/j.jbbm.2005.12.008. [DOI] [PubMed] [Google Scholar]
- Vickerman MM, Iobst S, Jesionowski AM, Gill SR. Genome-wide transcriptional changes in Streptococcus gordonii in response to competence signaling peptide. Journal of bacteriology. 2007;189:7799–7807. doi: 10.1128/JB.01023-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren TK, Lund SA, Jones KF, Hruby DE. Comparison of transformation protocols in Streptococcus gordonii and evaluation of native promoter strength using a multiple-copy plasmid. Canadian journal of microbiology. 2007;53:417–426. doi: 10.1139/W07-004. [DOI] [PubMed] [Google Scholar]
- Xie Z, Okinaga T, Qi F, Zhang Z, Merritt J. Cloning-independent and counterselectable markerless mutagenesis system in Streptococcus mutans. Applied and environmental microbiology. 2011;77:8025–8033. doi: 10.1128/AEM.06362-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu P, Alves JM, Kitten T, Brown A, Chen Z, Ozaki LS, Manque P, Ge X, Serrano MG, Puiu D, Hendricks S, Wang Y, Chaplin MD, Akan D, Paik S, Peterson DL, Macrina FL, Buck GA. Genome of the opportunistic pathogen Streptococcus sanguinis. Journal of bacteriology. 2007;189:3166–3175. doi: 10.1128/JB.01808-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida A, Kuramitsu HK. Streptococcus mutans biofilm formation: utilization of a gtfB promoter-green fluorescent protein (PgtfB::gfp) construct to monitor development. Microbiology. 2002;148:3385–3394. doi: 10.1099/00221287-148-11-3385. [DOI] [PubMed] [Google Scholar]
- You C, Zhang XZ, Zhang YH. Simple cloning via direct transformation of PCR product (DNA Multimer) to Escherichia coli and Bacillus subtilis. Applied and environmental microbiology. 2011;78:1593–1595. doi: 10.1128/AEM.07105-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou X, Caufield PW, Li Y, Qi F. Complete nucleotide sequence and characterization of pUA140, a cryptic plasmid from Streptococcus mutans. Plasmid. 2001;46:77–85. doi: 10.1006/plas.2001.1539. [DOI] [PubMed] [Google Scholar]