

Abstract
Chronic hypoxia causes pulmonary vasoconstriction and vascular remodeling, which lead to hypoxic pulmonary hypertension (HPH). HPH is associated with living at high altitudes and is a complication of many lung diseases, including chronic obstructive pulmonary disease, cystic fibrosis, and obstructive sleep apnea. Pulmonary vascular changes that occur with HPH include stiffening and narrowing of the pulmonary arteries that appear to involve all vascular cell types and sub-layers of the arterial wall. Right ventricular (RV) changes that occur with HPH include RV hypertrophy and RV fibrosis, often with preserved systolic and diastolic function and ventricular-vascular coupling efficiency. Both vascular stiffening and narrowing are important contributors to RV afterload via increases in oscillatory and steady ventricular work, respectively. The increased blood viscosity that occurs in HPH can be quite dramatic and is another important contributor to RV afterload. However, the viscosity, vascular mechanics and ventricular changes that occur with HPH are all reversible. Furthermore, even with continued hypoxia vascular remodeling does not progress to the obliterative, plexiform lesions that are seen clinically in severe pulmonary hypertension. In animal models, the RV changes appear adaptive, not maladaptive. In summary, HPH-induced vascular mechanical changes affect ventricular function but both are adaptive and reversible, which differentiates HPH from severe pulmonary hypertension. The mechanisms of adaptation and reversibility may provide useful insight into therapeutic targets for the clinical disease state.
Keywords: Hypoxia, pulmonary artery, ventricle
Introduction
Pulmonary hypertension (PH) is manifested as the elevation of pulmonary arterial pressure (Ppa) and is often hemodynamically defined as a mean resting Ppa greater than 25 mmHg. PH is a complex pulmonary disorder associated with a variety of causes; hypoxia-induced PH (HPH) is categorized in the third group of PH according to revised WHO classifications (McLaughlin et al., 2009). Clinically, HPH can be caused by living at high altitudes and is a complication of many lung diseases, including chronic obstructive pulmonary disease, cystic fibrosis, and obstructive sleep apnea. The preclinical animal model of HPH was introduced in 1970s (Zaiman et al., 2005) and is now widely used in rodents and calves to study the biological and functional changes in the pulmonary vasculature during PH progression.
While the pulmonary vascular changes in the most severe form of clinical PH – pulmonary arterial hypertension (PAH) – can be dramatic, the cause of death is typically right ventricular (RV) failure. Therefore, biological and functional changes in the RV with PH progression, and the interactions between pulmonary vascular and RV dysfunction have gained more attention recently. In this context, it is necessary to revisit the current understanding of HPH as well as its use as a model to study vascular and ventricular changes in PH progression.
Biological changes in pulmonary arteries during HPH
Hypoxia, the pathological condition in which one is deprived of adequate oxygen supply, can be achieved by exposing subjects to high altitude (which causes hypobaric hypoxia) or a low oxygen environment at normal barometric pressure (which causes normobaric hypoxia). Acutely, hypoxia leads to pulmonary vasoconstriction, mostly in the pre-capillary small pulmonary arteries (PAs) as evidenced by recent synchrotron radiation experiments (Schwenke et al., 2007). The cellular and molecular mechanisms of acute hypoxic pulmonary vasoconstriction have been extensively reviewed recently (Sylvester et al., 2012). Chronically, both continuous and intermittent hypoxia causes remodeling in large, proximal arteries and small, distal arterioles as well as RV hypertrophy (Stenmark et al., 2006). These arterial changes involve all vascular cell types (i.e., endothelial cells, smooth muscle cells and adventitial fibroblasts) and include altered cell proliferation and apoptosis, expression of growth factors, cytokines and receptors, as well as inflammatory responses (Humbert et al., 2004; Stenmark et al., 2006; Zhang et al., 2012).
While the HPH model continues to serve as a model of human PH, and is especially suitable for studying forms of PH associated with respiratory disorders, it is also well recognized that the HPH-induced remodeling in PAs lacks the marked distal luminal reduction by intimal growth and complex vascular lesions found in severe PAH (Zaiman et al., 2005; Nicolls et al., 2012). The absence of this biological signature of PAH in HPH may explain the mild to moderate and reversible functional changes in PAs and RV that we will discuss later.
Another major characteristic of PA remodeling during HPH is the accumulation of extracellular matrix (ECM) including elastin and collagen, especially in the proximal PAs (Poiani et al., 1990; Tozzi et al., 1994; Kobs et al., 2005; Drexler, 2008; Lammers et al., 2008; Estrada & Chesler, 2009; Ooi et al., 2010; Wang & Chesler, 2011b; Wang et al., 2013b). A recent study suggests elastin remodeling contributes to proximal PA stiffening in response to HPH in neonatal calves (Lammers et al., 2008), but discrepant observations are also reported in other species in adults. For example, there is no change in elastin content in rodent large PAs after chronic hypoxia (Merklinger et al., 2005; Drexler, 2008; Ooi et al., 2010). Our group has found that in mouse HPH, the ECM changes are dominated by collagen and in particular the type I isoform is elevated significantly (Ooi et al., 2010; Wang & Chesler, 2011b; Wang et al., 2013b). Changes in collagen cross-linking also occur with HPH progression and may affect blood flow dynamics (Wang & Chesler, 2011b; Wang et al., 2013b). Moreover, limiting collagen synthesis has been shown to limit HPH severity and RV dysfunction although the mechanism is unclear (Kerr et al., 1984; Kerr et al., 1987; Schreier et al., 2013). These studies suggest an important role of collagen in HPH severity and progression. The ECM remodeling that occurs with HPH has been postulated to be preceded by endothelial dysfunction, which in turn increases SMC-mediated proteolysis (Budhiraja et al., 2004; Rabinovitch, 2012). The proteolytic enzymes include matrix metalloproteinases (MMP) and their counteracting inhibitors (TIMP), which are elevated in experimental HPH as well as in clinical PAH (Hassoun, 2005). Therefore, the exact regulatory mechanisms of ECM remodeling in PAs may be essential keys to potential therapeutic targets in PH.
Mechanical changes in pulmonary arteries during HPH
Functionally, acute and chronic HPH cause distal pulmonary arteriolar narrowing that increases pulmonary vascular resistance (PVR), which is defined as (mPpa−Pla)/Q, where mPpa is the mean Ppa, Pla is the left atrial pressure, and Q is the mean pulmonary flow (or cardiac output). PVR is thus a useful parameter describing the degree of narrowing in the distal, small PAs and is markedly increased in HPH. From the mean pressure-flow relationship, one can also derive distal PA stiffness assuming a fully recruited and dilated pulmonary vasculature (Linehan et al., 1992); in particular, the distal PA distensibility, which the inverse of the distal PA stiffness, is:
where Pv is pulmonary venous pressure, CO is cardiac output, R0 is the vascular resistance of the unstressed lung (when vascular pressure approaches zero), Hct is the hematocrit, and α is the distal PA distensibility (mmHg−1), which is assumed to be constant throughout the pulmonary vascular bed. Since the equation above cannot be explicitly solved for α, typically the distensibility (α) is obtained by curve-fitting the experimental data to this equation. In vivo (Reeves et al., 2005; Blyth et al., 2007) and ex vivo (Chesler et al., 2009) studies have demonstrated that chronic PH decreases α. However, no change in α has been observed in acute HPH (Reeves et al., 2005).
In proximal arteries, there is no evidence that acute HPH has an effect but chronic HPH leads to remodeling and stiffening via increased ECM production and wall thickening. The stiffness of proximal, extralobar PAs is often measured by ex vivo or in vivo pressure-diameter relationships (Wang & Chesler, 2011a). In clinical settings, parameters that can be obtained non-invasively have been introduced, such as relative area change (RAC). RAC is not a stiffness but an area strain and is calculated as the relative cross-section area change (ΔA/A) of the proximal PA from systole to diastole; it is reduced significantly in PH patients (Gan et al., 2007). Another parameter frequently used in clinics to assess PA stiffening is the compliance (C), which is calculated as the ratio of stroke volume (SV) to pulse pressure (PP). Whereas in the systemic circulation, this metric reflects aortic stiffness because of the substantial length of the aorta before it branches into smaller arteries, in the pulmonary circulation it may depend on intermediate and distal PA stiffness as well as proximal PA stiffness (Saouti et al., 2010). Our recent data show that in mouse models of HPH, C does not always correlate with large, extralobar PA stiffness measured ex vivo (Fig. 1) (Wang et al., 2013a).
Figure 1.

Compliance measured in vivo for whole lung (CLUNG, by SV/PP) and ex vivo for extralobar left PAs (CLPA, by ΔV/ΔP) in mice with a collagen mutation (see (Wang et al., 2013b) for a description of this strain and the experimental protocol for CLPA measurements) exposed to normoxia, 10 days of hypoxia, and 10 days of hypoxia with beta-aminopropionitrile (BAPN). The hypoxia+BAPN group showed persistently low in vivo compliance compared to the hypoxia group, suggesting similar levels of overall pulmonary vascular stiffening. However, extralobar left PA compliance tended to increase in the hypoxia+BAPN group compared to the hypoxia group, suggesting more compliant proximal large PAs with BAPN treatment. Results are shown as mean ±SE. *p<0.05 vs. Normoxia.
Hemodynamic consequences of pulmonary arterial mechanical changes during HPH
The hemodynamic consequences of changes in both distal and proximal PAs as well as their interactions (e.g., pulse wave reflection) can be measured via the pulmonary vascular impedance (PVZ), which is derived from pulsatile pressure-flow relationships. Two approaches are commonly used to obtain PVZ – a frequency domain method and a time domain method (Wang & Chesler, 2011a). Both methods yield two important impedance metrics: the input impedance (Z0), which is either calculated as the impedance magnitude at 0 Hz (in the frequency domain) or total PVR (in the time domain), and the characteristic impedance (Zc), which is either calculated as the impedance magnitude at high frequencies (in the frequency domain) or the slope of the pressure-flow relationship in early systole (in the time domain).
Z0 is essentially as a measure of distal pulmonary constriction and increases with HPH progression; it is the PVZ in the absence of flow oscillations. Zc depends principally on the ratio of stiffness of the proximal arteries to fluid inertia in the proximal arteries; it is PVZ in the absence of wave reflections. In animal studies, Zc increases with PH in some species (Maggiorini et al., 1998; Wauthy et al., 2004) but not others (Ewalenko et al., 1997; Pagnamenta et al., 2003; Wauthy et al., 2004; Vanderpool et al., 2010b; Tabima et al., 2012), which likely reflects species-dependent differences in proximal PA stiffening vs. dilation in response to PH.
It is important to note also that PA stiffness itself is pressure-dependent. That is, as pressure increases, the PAs distend and more collagen fibers engage such that stiffness increases. Thus, it can be difficult to separate the effects of remodeling-induced stiffening from strain- or dilation-induced stiffening. Ideally, stiffness measurements should be made at multiple strain levels such that these two mechanisms of stiffening can be differentiated (Vanderpool et al., 2010a).
Changes in right ventricle during HPH
Proximal and distal arterial stiffening and narrowing are important contributors to RV afterload via increases in steady and oscillatory ventricular work. The steady work of the RV is typically calculated as the product of mPpa and stroke volume. Thus, it is the work required to overcome total PVR and to produce forward blood flow into pulmonary circulation; it largely depends on distal PA narrowing. The oscillatory work of the RV is calculated as the difference between the total work (stroke work) and the steady work. The oscillatory work is the work required to produce zero-mean oscillations in blood flow; it largely depends on pulmonary arterial compliance. Typically, oscillatory work is considered ‘wasted’ so an increase in the ratio of oscillatory to total work is considered a sign of decreased RV efficiency. See (Bellofiore & Chesler, 2013) for a recent review of this topic. It is currently not known whether the RV remodels differently in response to increased steady and oscillatory work demands.
Known consequences of increased RV afterload include RV hypertrophy and fibrosis. RV hypertrophy is typically quantified by the Fulton index, which is the weight ratio of right ventricle to the sum of left ventricle and septum (RV/(LV+S)). Increased Fulton index is universally observed in HPH. RV fibrosis, which is a hallmark of dysfunctional or failing RV, is seldom reported (or examined) in HPH. Our group has recently found a significant increase in collagen content in mouse RVs after 10 days of hypoxia (Schreier et al., 2013). But the structural and functional changes in the RV with chronic HPH have been less well studied than those in the pulmonary circulation until recently (Tabima et al., 2010; Walker et al., 2011; Schreier et al., 2013).
Our group has successfully established methods to measure RV function in mice in vivo (Tabima et al., 2010). With HPH progression, we have consistently observed increased right ventricular systolic pressure, significant RV hypertrophy, and increased effective arterial elastance (Ea), which is an index of RV afterload (Tabima et al., 2010; Schreier et al., 2013). Because of the pressure overload, RV contractility increases as measured by preload recruitable stroke work and ventricular end-systolic elastance (Ees). The ratio of Ees to Ea, which is an index of ventricular-vascular coupling efficiency, is typically maintained (Tabima et al., 2010; Schreier et al., 2013). To date we have seen no significant changes in cardiac output or ejection fraction in mice with chronic HPH. In large animals exposed to acute hypoxia, both Ees and Ea increase but Ees/Ea remains at control levels, which indicates preserved ventricular-vascular coupling (Wauthy et al., 2004). Therefore, RV functional changes during HPH seem to be adaptive and moderate, with preserved systolic and diastolic function.
Hematocrit and blood viscosity increases
A unique change associated with HPH but not other types of PH is the increased expression of erythropoietin, resulting in increased red blood cells, hematocrit (Hct) and hemoglobin levels. Hct can increase from ~45% at normal levels to up to 80% after chronic hypoxia exposure. This increases blood viscosity, which consequently increases pulmonary resistance. Whittaker and Winston found an power-law relationship between PVR and hematocrit (Whittaker & Winton, 1933):
where R0 is PVR at a Hct of 45% (normal) and ϕ is the measured Hct.
We recently measured PVZ in control mice and those exposed to 21 days of hypoxia at different hematocrits by partially replacing high viscosity blood with hydroxyethylstarch (unpublished data). By reducing the hemoglobin to normal levels (i.e. Hct~42%), Z0 (or PVR) decreased about 31% and Zc decreased about 47% (Fig. 2). These data suggest that the increase in hematocrit is a significant contributor to the increased RV afterload in chronic HPH.
Figure 2.
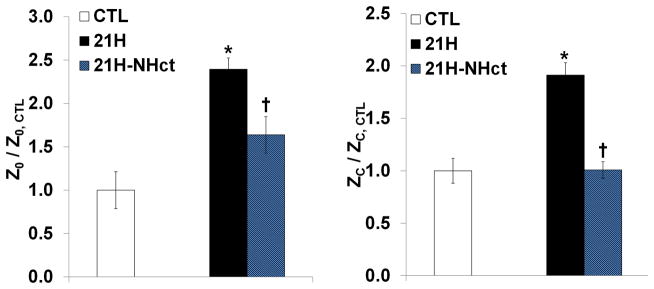
Pulmonary vascular Z0 and Zc in wild-type mice exposed to 21 days of hypoxia normalized to those measured in normoxic mice. Measurements were obtained in chronically hypoxic mice with elevated Hct (21H) as well as normal levels of Hct (21H-NHct) by blood dilution to show that decreased hematocrit led to significant reductions in Z0 and Zc. Results are shown as mean ±SE. *p<0.05 vs. Normoxia; †p<0.05 vs. 21H.
HPH: A moderate and reversible type of PH
It is well known that even with prolonged hypoxia exposure, distal vascular remodeling does not progress to the obliterative, plexiform lesions that are seen clinically in severe PH (Gomez-Arroyo et al., 2012; Nicolls et al., 2012), suggesting the vascular changes are only moderate in HPH. Another important characteristic of HPH that is different from severe PH is its reversibility. It has been shown in many studies that if allowed to recover in normoxic conditions, subjects will undergo a reverse remodeling in PAs and a decrease of Ppa, with a reduction in Hct as well (Rabinovitch et al., 1981; Liu, 1997; Tozzi et al., 1998; Riley et al., 2000; Li et al., 2004; Ooi et al., 2010; Tabima et al., 2012). In terms of vascular mechanics, the recovery process is accompanied by reduced proximal PA stiffening and reduced distal PA narrowing (Ooi et al., 2010; Tabima et al., 2012), which then significantly reduces the RV afterload. As a consequence, a regression in RV hypertrophy is often observed.
Because HPH is a moderate and reversible type of PH, it does not capture the key features of severe, clinical PH. Recently, the appropriate usage of this model has been reconsidered (Gomez-Arroyo et al., 2012; Nicolls et al., 2012). Instead of a limitation, however, the reversibility of HPH may in fact be an advantage; that is, the contrasts between HPH and severe PH may shed light on key factors that determine the reversibility of RV and PA remodeling and the critically important transition from RV adaptation to RV failure.
Summary and conclusions
In summary, HPH-induced vascular mechanical changes affect ventricular function but both are adaptive and reversible, which differentiates HPH from severe pulmonary hypertension. The mechanisms of adaptation and reversibility may provide useful insight into therapeutic targets for the clinical disease state.
New Findings.
This article reviews pulmonary vascular and right ventricular (RV) changes due to hypoxic pulmonary hypertension (HPH), which is a type of pulmonary hypertension (PH) found clinically and has been widely used to induce PH in animal models. As research into clinical PH progression broadens to include RV as well as pulmonary vascular remodeling, an improved understanding of the effects of HPH on the RV is required. This article highlights the moderate, adaptive and reversible nature of RV and pulmonary vascular remodeling in HPH. Moreover, we show that increased hematocrit in HPH contributes significantly to RV overload, which warrants additional attention.
Acknowledgments
We thank David A. Schreier for sharing data and for useful discussions. Preparation of this symposium report was supported by NIH R01 HL086939 (NCC).
References
- Bellofiore A, Chesler NC. Methods for Measuring Right Ventricular Function and Hemodynamic Coupling with the Pulmonary Vasculature. Ann Biomed Eng. 2013 doi: 10.1007/s10439-013-0752-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blyth KG, Syyed R, Chalmers J, Foster JE, Saba T, Naeije R, Melot C, Peacock AJ. Pulmonary arterial pulse pressure and mortality in pulmonary arterial hypertension. Respir Med. 2007;101:2495–2501. doi: 10.1016/j.rmed.2007.07.004. [DOI] [PubMed] [Google Scholar]
- Budhiraja R, Tuder RM, Hassoun PM. Endothelial dysfunction in pulmonary hypertension. Circulation. 2004;109:159–165. doi: 10.1161/01.CIR.0000102381.57477.50. [DOI] [PubMed] [Google Scholar]
- Chesler NC, Argiento P, Vanderpool R, D’Alto M, Naeije R. How to measure peripheral pulmonary vascular mechanics. Conf Proc IEEE Eng Med Biol Soc. 2009;2009:173–176. doi: 10.1109/IEMBS.2009.5333299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drexler ES, Bischoff JE, Slifka AJ, McCowan CN, Quinn TP, Shandas R, Ivy DD, Stenmark KR. Stiffening of the extrapulmonary arteries from rats in chronic hypoxic pulmonary hypertension. Journal of Research of the National Institute of Standards and Technology. 2008;113:239–249. doi: 10.6028/jres.113.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estrada KD, Chesler NC. Collagen-related gene and protein expression changes in the lung in response to chronic hypoxia. Biomech Model Mechanobiol. 2009;8:263–272. doi: 10.1007/s10237-008-0133-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewalenko P, Brimioulle S, Delcroix M, Lejeune P, Naeije R. Comparison of the effects of isoflurane with those of propofol on pulmonary vascular impedance in experimental embolic pulmonary hypertension. Br J Anaesth. 1997;79:625–630. doi: 10.1093/bja/79.5.625. [DOI] [PubMed] [Google Scholar]
- Gan CT, Lankhaar JW, Westerhof N, Marcus JT, Becker A, Twisk JW, Boonstra A, Postmus PE, Vonk-Noordegraaf A. Noninvasively assessed pulmonary artery stiffness predicts mortality in pulmonary arterial hypertension. Chest. 2007;132:1906–1912. doi: 10.1378/chest.07-1246. [DOI] [PubMed] [Google Scholar]
- Gomez-Arroyo J, Saleem SJ, Mizuno S, Syed AA, Bogaard HJ, Abbate A, Taraseviciene-Stewart L, Sung Y, Kraskauskas D, Farkas D, Conrad DH, Nicolls MR, Voelkel NF. A brief overview of mouse models of pulmonary arterial hypertension: problems and prospects. Am J Physiol Lung Cell Mol Physiol. 2012;302:L977–991. doi: 10.1152/ajplung.00362.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassoun PM. Deciphering the “matrix” in pulmonary vascular remodelling. European Respiratory Journal. 2005;25:778–779. doi: 10.1183/09031936.05.00027305. [DOI] [PubMed] [Google Scholar]
- Humbert M, Morrell NW, Archer SL, Stenmark KR, MacLean MR, Lang IM, Christman BW, Weir EK, Eickelberg O, Voelkel NF, Rabinovitch M. Cellular and molecular pathobiology of pulmonary arterial hypertension. J Am Coll Cardiol. 2004;43:13S–24S. doi: 10.1016/j.jacc.2004.02.029. [DOI] [PubMed] [Google Scholar]
- Kerr JS, Riley DJ, Frank MM, Trelstad RL, Frankel HM. Reduction of chronic hypoxic pulmonary hypertension in the rat by beta-aminopropionitrile. J Appl Physiol. 1984;57:1760–1766. doi: 10.1152/jappl.1984.57.6.1760. [DOI] [PubMed] [Google Scholar]
- Kerr JS, Ruppert CL, Tozzi CA, Neubauer JA, Frankel HM, Yu SY, Riley DJ. Reduction of chronic hypoxic pulmonary hypertension in the rat by an inhibitor of collagen production. Am Rev Respir Dis. 1987;135:300–306. doi: 10.1164/arrd.1987.135.2.300. [DOI] [PubMed] [Google Scholar]
- Kobs RW, Muvarak NE, Eickhoff JC, Chesler NC. Linked mechanical and biological aspects of remodeling in mouse pulmonary arteries with hypoxia-induced hypertension. Am J Physiol Heart Circ Physiol. 2005;288:H1209–1217. doi: 10.1152/ajpheart.01129.2003. [DOI] [PubMed] [Google Scholar]
- Lammers SR, Kao PH, Qi HJ, Hunter K, Lanning C, Albietz J, Hofmeister S, Mecham R, Stenmark KR, Shandas R. Changes in the structure-function relationship of elastin and its impact on the proximal pulmonary arterial mechanics of hypertensive calves. Am J Physiol Heart Circ Physiol. 2008;295:H1451–1459. doi: 10.1152/ajpheart.00127.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Huang W, Jiang ZL, Gregersen H, Fung YC. Tissue remodeling of rat pulmonary arteries in recovery from hypoxic hypertension. Proc Natl Acad Sci U S A. 2004;101:11488–11493. doi: 10.1073/pnas.0404084101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linehan JH, Haworth ST, Nelin LD, Krenz GS, Dawson CA. A simple distensible vessel model for interpreting pulmonary vascular pressure-flow curves. J Appl Physiol. 1992;73:987–994. doi: 10.1152/jappl.1992.73.3.987. [DOI] [PubMed] [Google Scholar]
- Liu SQ. Regression of hypoxic hypertension-induced changes in the elastic laminae of rat pulmonary arteries. J Appl Physiol. 1997;82:1677–1684. doi: 10.1152/jappl.1997.82.5.1677. [DOI] [PubMed] [Google Scholar]
- Maggiorini M, Brimioulle S, De Canniere D, Delcroix M, Naeije R. Effects of pulmonary embolism on pulmonary vascular impedance in dogs and minipigs. J Appl Physiol. 1998;84:815–821. doi: 10.1152/jappl.1998.84.3.815. [DOI] [PubMed] [Google Scholar]
- McLaughlin VV, Archer SL, Badesch DB, Barst RJ, Farber HW, Lindner JR, Mathier MA, McGoon MD, Park MH, Rosenson RS, Rubin LJ, Tapson VF, Varga J, Harrington RA, Anderson JL, Bates ER, Bridges CR, Eisenberg MJ, Ferrari VA, Grines CL, Hlatky MA, Jacobs AK, Kaul S, Lichtenberg RC, Moliterno DJ, Mukherjee D, Pohost GM, Schofield RS, Shubrooks SJ, Stein JH, Tracy CM, Weitz HH, Wesley DJ. ACCF/AHA 2009 expert consensus document on pulmonary hypertension: a report of the American College of Cardiology Foundation Task Force on Expert Consensus Documents and the American Heart Association: developed in collaboration with the American College of Chest Physicians, American Thoracic Society, Inc., and the Pulmonary Hypertension Association. Circulation. 2009;119:2250–2294. doi: 10.1161/CIRCULATIONAHA.109.192230. [DOI] [PubMed] [Google Scholar]
- Merklinger SL, Wagner RA, Spiekerkoetter E, Hinek A, Knutsen RH, Kabir MG, Desai K, Hacker S, Wang L, Cann GM, Ambartsumian NS, Lukanidin E, Bernstein D, Husain M, Mecham RP, Starcher B, Yanagisawa H, Rabinovitch M. Increased fibulin-5 and elastin in S100A4/Mts1 mice with pulmonary hypertension. Circ Res. 2005;97:596–604. doi: 10.1161/01.RES.0000182425.49768.8a. [DOI] [PubMed] [Google Scholar]
- Nicolls MR, Mizuno S, Taraseviciene-Stewart L, Farkas L, Drake JI, Al Husseini A, Gomez-Arroyo JG, Voelkel NF, Bogaard HJ. New models of pulmonary hypertension based on VEGF receptor blockade-induced endothelial cell apoptosis. Pulm Circ. 2012;2:434–442. doi: 10.4103/2045-8932.105031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ooi CY, Wang Z, Tabima DM, Eickhoff JC, Chesler NC. The role of collagen in extralobar pulmonary artery stiffening in response to hypoxia-induced pulmonary hypertension. Am J Physiol Heart Circ Physiol. 2010;299:H1823–1831. doi: 10.1152/ajpheart.00493.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagnamenta A, Fesler P, Vandinivit A, Brimioulle S, Naeije R. Pulmonary vascular effects of dobutamine in experimental pulmonary hypertension. Crit Care Med. 2003;31:1140–1146. doi: 10.1097/01.CCM.0000060126.75746.32. [DOI] [PubMed] [Google Scholar]
- Poiani GJ, Tozzi CA, Yohn SE, Pierce RA, Belsky SA, Berg RA, Yu SY, Deak SB, Riley DJ. Collagen and elastin metabolism in hypertensive pulmonary arteries of rats. Circ Res. 1990;66:968–978. doi: 10.1161/01.res.66.4.968. [DOI] [PubMed] [Google Scholar]
- Rabinovitch M. Molecular pathogenesis of pulmonary arterial hypertension. J Clin Invest. 2012;122:4306–4313. doi: 10.1172/JCI60658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinovitch M, Gamble WJ, Miettinen OS, Reid L. Age and sex influence on pulmonary hypertension of chronic hypoxia and on recovery. Am J Physiol. 1981;240:H62–72. doi: 10.1152/ajpheart.1981.240.1.H62. [DOI] [PubMed] [Google Scholar]
- Reeves JT, Linehan JH, Stenmark KR. Distensibility of the normal human lung circulation during exercise. Am J Physiol Lung Cell Mol Physiol. 2005;288:L419–425. doi: 10.1152/ajplung.00162.2004. [DOI] [PubMed] [Google Scholar]
- Riley DJ, Thakker-Varia S, Wilson FJ, Poiani GJ, Tozzi CA. Role of proteolysis and apoptosis in regression of pulmonary vascular remodeling. Physiol Res. 2000;49:577–585. [PubMed] [Google Scholar]
- Saouti N, Westerhof N, Postmus PE, Vonk-Noordegraaf A. The arterial load in pulmonary hypertension. Eur Respir Rev. 2010;19:197–203. doi: 10.1183/09059180.00002210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreier D, Hacker T, Song G, Chesler N. The role of collagen synthesis in ventricular and vascular adaptation to hypoxic pulmonary hypertension. J Biomech Eng. 2013;135:021018. doi: 10.1115/1.4023480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwenke DO, Pearson JT, Umetani K, Kangawa K, Shirai M. Imaging of the pulmonary circulation in the closed-chest rat using synchrotron radiation microangiography. Journal of Applied Physiology. 2007;102:787–793. doi: 10.1152/japplphysiol.00596.2006. [DOI] [PubMed] [Google Scholar]
- Stenmark KR, Fagan KA, Frid MG. Hypoxia-induced pulmonary vascular remodeling: cellular and molecular mechanisms. Circ Res. 2006;99:675–691. doi: 10.1161/01.RES.0000243584.45145.3f. [DOI] [PubMed] [Google Scholar]
- Sylvester JT, Shimoda LA, Aaronson PI, Ward JP. Hypoxic pulmonary vasoconstriction. Physiol Rev. 2012;92:367–520. doi: 10.1152/physrev.00041.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabima DM, Hacker TA, Chesler NC. Measuring right ventricular function in the normal and hypertensive mouse hearts using admittance-derived pressure-volume loops. Am J Physiol Heart Circ Physiol. 2010;299:H2069–2075. doi: 10.1152/ajpheart.00805.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabima DM, Roldan-Alzate A, Wang Z, Hacker TA, Molthen RC, Chesler NC. Persistent vascular collagen accumulation alters hemodynamic recovery from chronic hypoxia. J Biomech. 2012 doi: 10.1016/j.jbiomech.2011.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tozzi CA, Christiansen DL, Poiani GJ, Riley DJ. Excess collagen in hypertensive pulmonary arteries decreases vascular distensibility. Am J Respir Crit Care Med. 1994;149:1317–1326. doi: 10.1164/ajrccm.149.5.8173773. [DOI] [PubMed] [Google Scholar]
- Tozzi CA, Thakker-Varia S, Yu SY, Bannett RF, Peng BW, Poiani GJ, Wilson FJ, Riley DJ. Mast cell collagenase correlates with regression of pulmonary vascular remodeling in the rat. Am J Respir Cell Mol Biol. 1998;18:497–510. doi: 10.1165/ajrcmb.18.4.2536. [DOI] [PubMed] [Google Scholar]
- Vanderpool RR, Kim AR, Molthen R, Chesler NC. Effects of acute Rho kinase inhibition on chronic hypoxia-induced changes in proximal and distal pulmonary arterial structure and function. J Appl Physiol. 2010a;110:188–198. doi: 10.1152/japplphysiol.00533.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanderpool RR, Naeije R, Chesler NC. Impedance in isolated mouse lungs for the determination of site of action of vasoactive agents and disease. Ann Biomed Eng. 2010b;38:1854–1861. doi: 10.1007/s10439-010-9960-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker LA, Walker JS, Glazier A, Brown DR, Stenmark KR, Buttrick PM. Biochemical and myofilament responses of the right ventricle to severe pulmonary hypertension. Am J Physiol Heart Circ Physiol. 2011;301:H832–840. doi: 10.1152/ajpheart.00249.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Chesler NC. Pulmonary vascular wall stiffness: An important contributor to the increased right ventricular afterload with pulmonary hypertension. Pulm Circ. 2011a;1:212–223. doi: 10.4103/2045-8932.83453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Chesler NC. Role of collagen content and cross-linking in large pulmonary arterial stiffening after chronic hypoxia. Biomech Model Mechanobiol. 2011b;11:279–289. doi: 10.1007/s10237-011-0309-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Hacker TA, Chesler NC. Effects of collagen accumulation on proximal arterial stiffening and distal arterial narrowing during hypoxic pulmonary hypertension. American Thoracic Society International Conference; Philadelphia, PA. 2013a. [Google Scholar]
- Wang Z, Lakes RS, Eickhoff JC, Chesler NC. Effects of collagen deposition on passive and active mechanical properties of large pulmonary arteries in hypoxic pulmonary hypertension. Biomech Model Mechanobiol. 2013b doi: 10.1007/s10237-012-0467-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wauthy P, Pagnamenta A, Vassalli F, Naeije R, Brimioulle S. Right ventricular adaptation to pulmonary hypertension: an interspecies comparison. Am J Physiol Heart Circ Physiol. 2004;286:H1441–1447. doi: 10.1152/ajpheart.00640.2003. [DOI] [PubMed] [Google Scholar]
- Whittaker SRF, Winton FR. The apparent viscosity of blood flowing in the isolated hindlimb of the dog, and its variation with corpuscular concentration. J Physiol. 1933;78:339–369. doi: 10.1113/jphysiol.1933.sp003009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaiman A, Fijalkowska I, Hassoun PM, Tuder RM. One hundred years of research in the pathogenesis of pulmonary hypertension. Am J Respir Cell Mol Biol. 2005;33:425–431. doi: 10.1165/rcmb.F307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Luo Y, Liu ML, Wang J, Xu DQ, Dong MQ, Liu Y, Xu M, Dong HY, Zhao PT, Gao YQ, Li ZC. Macrophage migration inhibitory factor contributes to hypoxic pulmonary vasoconstriction in rats. Microvasc Res. 2012;83:205–212. doi: 10.1016/j.mvr.2011.09.014. [DOI] [PubMed] [Google Scholar]