

Abstract
OX40 is a member of the TNFR superfamily that has potent costimulatory properties. Although the impact of blockade of the OX40-OX40L pathway has been well documented in models of autoimmune disease, its effect on the rejection of allografts is less well defined.
Here we show that the alloantigen-mediated activation of naïve and memory CD4+ T cells results in the induction of OX40 expression and that blockade of OX40-OX40L interactions prevents skin allograft rejection mediated by either subset of T cells. Moreover, a blocking anti-OX40 was found to have no effect on the activation and proliferation of T cells, but rather effector T cells failed to accumulate in peripheral lymph nodes and subsequently migrate to skin allografts. This was found to be the result of an enhanced degree of cell death amongst proliferating effector cells. In clear contrast, blockade of OX40-OX40L interactions at the time of exposure to alloantigen enhanced the ability of regulatory T cells to suppress T cell responses to alloantigen by supporting rather than diminishing regulatory T cell survival.
These data show that OX40-OX40L signalling contributes to the evolution of the adaptive immune response to an allograft via the differential control of alloreactive effector and regulatory T cell survival. Moreover, these data serve to further highlight OX40 and OX40L as therapeutic targets to assist the induction of tolerance to allografts and self-antigens.
Keywords: mice, costimulation, CD4 T cells, Treg, OX40, transplantation
Introduction
The provision of costimulatory signals is critical for optimal T cell activation (1). OX40 (CD134) is a 50kDa transmembrane protein belonging to the tumour necrosis factor receptor (TNFR) superfamily which possesses such costimulatory properties (2, 3). OX40 is upregulated on T cells following activation (4), whilst Treg constitutively express OX40 (5, 6). This is in agreement with the proposed role of OX40-OX40L interactions i.e. in the propagation of the response, rather than in initial T cell priming. The ligand for OX40 (OX40L) is predominantly restricted to APC such as B cells, macrophages, microglia, dendritic cells (DC) and endothelial cells (7-13). Signalling via OX40-OX40L interactions during effector T cell responses has been shown to lead to enhanced T cell survival (14, 15), cytokine production (16) and increased numbers of memory CD4+ T cells (17). These data have been corroborated using human CD4+ T cells cultured in vitro (18). Experimental models of autoimmunity and inflammation have shown a clear role for OX40-OX40L as blocking the interaction attenuates disease progression or severity. For example, administration of anti-OX40L in a mouse model of collagen induced arthritis ameliorated disease severity however failed to prevent the expansion of collagen-reactive T cells. There was a significant inhibition of IFN-γ production from T cells isolated from the dLN and collagen-specific IgG2a antibody production in the serum (19). In contrast, anti-OX40L mAb has been demonstrated to have no impact on the rejection of full MHC mismatched islet allografts in mice (20). Similarly, blocking the OX40-OX40L pathway (using an OX40-Ig fusion protein) in a mouse model of cardiac transplantation was shown to be ineffective at prolonging allograft survival across a full MHC mismatch. However, prolonged cardiac allograft survival was observed (MST 14 days vs >100 days) when donor and recipient were mismatched at only minor histocompatability antigen loci (21). These data provide a clear precedent for the utilisation of the OX40-OX40L costimulatory pathway in rejection however this appears to be contingent on suboptimal or low frequency T cell responses. This is borne out by the finding that when OX40 blockade is used in combination with interruption of other costimulatory pathways, such as CD40/CD154, CD28/CD80/CD86, a more pronounced impact on skin allograft survival has been observed due to perturbation of the expansion or persistence of alloreactive effector T cells (17, 22-24).
It has also been suggested that OX40-OX40L has a diametric role on effector T cells and Foxp3+ regulatory T cells (Treg) i.e. OX40 signalling enhances effector T cell responses whilst inhibits the generation of inducible Treg (iTreg) from naïve CD4+ T cells. So et al demonstrated that OX40 signalling and a low dose of antigen (moth cytochrome C) suppressed the differentiation of naïve TCR-transgenic CD4+ T cells into FoxP3+ Treg (25). More recently, Xiao et al have elegantly shown that engagement of OX40 in naïve recipients results in expansion of Treg, although these in vitro expanded cells function as poor suppressor cells due to a deficiency in IL-2 (26).
In addition to OX40 signalling impacting naive and regulatory T cell responses OX40 signalling has also been shown to be required to sustain memory T cell responses. For example, Gaspal et al elegantly demonstrated that OX40 signalling in concert with CD30 signals were required for productive secondary antibody responses (27). CD30−/− OX40−/− T cells had similar proliferation compared to wild-type controls but these double deficient T cells failed to survive in vivo (27).
Memory T cells (Tm) participate in the response to allografts and are not restricted to patients that have received prior sensitisation with alloantigen in the form of a previous transplant, blood transfusion or pregnancy. Indeed, it has been shown that a subset of pre-existent Tm, generated as a result of previous encounter with either infectious or environmental antigens, cross-react to alloantigen (a process termed heterologous immunity) (28). Moreover, the presence of high numbers of donor-reactive Tm prior to transplantation has been found to be detrimental to transplant survival regardless of whether this is induced by costimulatory molecule blockade or conventional immunosuppressive agents (29, 30). In addition, Hong et al have shown a combination of immune modulating agents (i.e. anti-OX40, anti-CD154, anti-IL-2 complex and rapamycin) failed to significantly prolong islet allograft survival mediated by memory T cells, whilst significant allograft survival was observed when mediated by naïve T cells (31). Therefore, the development of drugs which could inhibit Tm as well as other cells in the immune repertoire would clearly be beneficial in preventing allograft rejection as well as autoimmunity.
Here we demonstrate that blockade of the OX40-OX40L costimulation pathway prevents the activation and accumulation of naïve and memory CD4+ T cells in response to alloantigen, using a model of skin allograft rejection. In contrast, OX40 blockade promotes the survival of Treg following re-activation and enhances suppression of T cell responses to alloantigen in vitro and in vivo. Therefore, these data demonstrate that OX40-OX40L interactions differentially control the survival of effector/memory CD4+ T cells and Treg in response to alloantigen.
Materials and Methods
Mice
TEa TCR transgenic mice (C57BL/6 background; H2b) produce CD4+ T cells that are reactive to a 17-mer peptide of the BALB/c H2IEα chain presented in the context of H2IAb i.e. presented by B6 MHC Class II via the indirect pathway (32). The TEa mice were a kind gift from Dr W.Gao and Prof. T. Strom (Boston, USA). CBA.Ca RAG-1 knockout (CBA RAG−/−; H2k) mice were a kind gift from Prof. D. Kioussis (Mill Hill, London, UK). C57BL/6 RAG-1 knockout (B6 RAG−/−; H2b), CBA/Ca (CBA; H2k), C57BL/6 (H2b), BALB/cB6 F1 mice were bred and housed in the Biomedical Services Unit at the John Radcliffe Hospital (Oxford, UK). All mice were used at 6-12 weeks old at time of first procedure and all studies were performed in accordance with the Animals (Scientific Procedures) Act 1986.
Skin Transplantation
Full thickness tail skin (1cm2) was transplanted onto the left flank of recipient mice one day after adoptive transfer of cells as previously described (33). Grafts were covered with sterile dressings for 7 days and graft viability was assessed daily. Rejection was defined as complete loss of viable donor skin.
Antibody treatment in vivo
Mice were treated with PEGylated Fab Control (A33 Fab PEG, UCB Pharma, UK) or anti-OX40 Fab PEG (710 Fab PEG, UCB Pharma, UK) at 2.5mg/dose subcutaneously twice weekly for 4 weeks post-transplantation. The anti-OX40 Fab PEG has been shown to be able to completely block OX40L binding to OX40 on activated mouse T cells (data not shown) and has no agonistic properties. PEGylated Fab fragments have been chosen for use in these studies as the addition of a polyethylene glycol (PEG) molecule extends the in vivo half life of these reagents and lowers the immunogenicity allowing them to be used effectively in experimental and clinical settings.
To generate alloantigen-reactive Treg, wild-type CBA mice were treated with 200μg of non-depleting anti-CD4 mAb (YTS177) i.v. on day −28 and −27. In addition on day −27, mice also received 250μl of C57BL/6 whole blood (i.v.). On day 0, mice possessed a population of CD4+CD25+ T cells capable of suppressing donor-alloantigen specific T cell responses (34, 35).
Generation of Memory T cells
TEa memory T cells (Tm) were generated by injecting 1×106 sorted naïve TEa CD4+ T cells on day −1 into B6 Rag−/− mice. On days 0, 7, 14 mice received 1×107 BALB/cB6F1 splenocytes. CBA polyclonal Tm were generated by allogeneic splenocyte challenge (B6) on days 0 and 7. Following the final alloantigen challenge all mice were left for 50-100 days before being used in subsequent experiments. Upon harvest, spleen and mLN were removed from mice containing Tm and the percentage of live Tm was determined by flow cytometry. Typically >95% of T cells were Tm (expressed CD44; data not shown).
Lymphocyte isolation, cell sorting and adoptive transfer
Naïve (CD44−) or memory (CD44+) CD4+ T cells were purified from the spleen and mesenteric lymph nodes (mLN) of naïve or alloantigen primed wild-type (CBA) or B6 RAG−/− (reconstituted with TEa CD4+ T cells) mice. Single cell leukocyte suspensions were generated and erythrocytes removed by osmotic shock. Leukocytes were stained with CD4-APC, CD44-PE, CD45RB-Pacific Blue (or Vα2 for isolation of TEa T cells) (eBiosciences) before the naïve CD4+ T cell population was sorted using a FACSAria cell sorter (BD Biosciences). Typically CD4+ T cells were isolated to >98% purity. Cells for adoptive transfer were resuspended in PBS and injected intravenously into syngeneic RAG−/− mice one day prior to skin transplantation.
Labelling of cells for analysis of proliferation
Leukocytes were resuspended to either 5×107 cells/ml or 1×106 cells/ml in serum free media before incubation for 10 minutes with 5μM CFSE (Molecular Probes Inc, Netherlands) or Cell Trace violet (Invitrogen), respectively. Cells were washed in ice-cold RPMI 1640 (Invitrogen Life Technologies) then resuspended in PBS (Oxoid, Oxford) prior to adoptive transfer.
Flow cytometry analysis
A single cell suspension was prepared from spleen and mLN and cell surface staining was conducted as previously described (15). All samples were acquired immediately on a FACSCanto (BD Biosciences) and analysed using FACSDiva software (BD Biosciences). For intracellular staining, cells were surface stained as above and then washed in FACS buffer. Cells were then incubated in 500μl of Fix/Perm Buffer (Transcription Factor buffer Kit; BD Biosciences) for 1 hour at 4°C. This incubation was followed by two wash steps in wash buffer (Transcription Factor buffer Kit; BD, UK) before incubation with FoxP3-APC and Bcl-2-FITC for 30 minutes incubation at 4°C. After incubation, cells were washed once in permeabilisation buffer. The absolute number of TEa CD4+ T cells was determined using synthetic fluorescent beads (CaliBRITE beads, BD Biosciences) as described previously (36).
Isolation of CD25+ and CD25− CD4+ T cells
A single cell suspension enriched for T cells was prepared and enriched for CD4+ T cells. After CD4+ purification by Dynal beads, cells were stained with anti-CD25-PE (ebioscience) for 30 minutes at 4°C. Cells were washed once in PBS/2% FCS before incubation with anti-PE beads (Miltenyi Biotech, Germany). Cells were washed and loaded onto a MACS column on a MACS magnet. CD25−CD4+ T cells were isolated by negative selection (collected from flow through), and CD25+CD4+ T cells were isolated by positive selection (those attached to the column), according to the manufacturer’s instructions.
CD11c+ Isolation
A single cell suspension (~2×108/ml in PBS/2% FCS) from spleens of naïve B6 mice were incubated with anti-B220 (RA36β2) at a final concentration of 10μg/ml and incubated for 45-60 minutes at 4°C. Cells were washed once and re-suspended in 10% FCS/PBS. Sheep anti-rat coated Dynabeads (Dynal A.S, Norway) were added at a ratio of one bead per cell and incubated on a rotating wheel at 4°C for 30 minutes. Residual cells incubated with anti-CD11c (Miltenyi Biotech) for 30 minutes at 4°C, before being isolated by positive selection using a magnet. The resulting enriched cell population was then counted, purity checked and re-suspended at the required concentration for further use.
In vitro suppression assay
5×105 CD4+CD25− responder T cells (isolated from naïve CBA mice) were labelled with violet proliferation dye (Invitrogen) and incubated with purified CD11c+ cells and varying numbers of CFSE-labelled Treg. Treg were isolated from mice which had received 177/DST and either anti-OX40 or PEG control on days −28 and −25. Proliferation of the CD4+CD25− responder T cells was assessed by flow cytometry on day 5.
Histology
Skin grafts were embedded into paraffin wax and cut into 4μm sections before being loaded onto a Lab Vision Autostainer (Lab Vision Corporation, USA) for staining with rabbit anti-CD3 antibody at room temperature (Abcam). After washing, sections were incubated with a biotinylated donkey anti-rabbit secondary antibody (Jackson Immunoresearch) and then labelled streptavidin biotin (Dako) before being incubated with the DAB for 5 minutes. The cell nuclei were counterstained with haematoxylin. The sections were then dehydrated through graded ethanol, cleared in Histoclear (Fischer Scientific) and cover slipped before being analysed. Automatic image analysis was performed using Definiens Tissue Studio (Definiens, Germany).
Statistical Analysis
Data were analysed using the Student’s t-test using Prism (version 5; Graph Pad software) and are expressed as mean ± standard deviation (SD). Kaplan-Meier survival graphs were constructed and log rank comparisons of the groups were used to calculate P values. Values for p < 0.05 were considered statistically significant.
Results
CD4+ T cells upregulate OX40 after stimulation with alloantigen in vitro, whilst Treg constitutively express OX40
Before the role of OX40-OX40L interactions in CD4+ T cell responses to alloantigen was investigated we sought to determine the expression pattern of OX40 and OX40L on CD4+ T cells (naïve and memory) by stimulating naïve CBA splenocytes with allogeneic irradiated B6 splenocytes in vitro. Polyclonal naïve CD4+ T cells (gated on total CD4+ T cells) were found to upregulate expression of OX40 from day 2 of culture, with maximal expression on day 5 (Figure 1A), whereas no expression of OX40L was found at any time-point following activation (Figure 1A). Similarly, polyclonal Tm (CD3+CD4+CD44+) stimulated under the same conditions also expressed OX40 from day 3 (Figure 1B), but did not express OX40L at any time (Figure 1B). TEa Tm also showed a similar profile of expression for OX40 (data not shown).
Figure 1. Effector and memory T cells express OX40 after activation, while Treg constitutively express OX40.
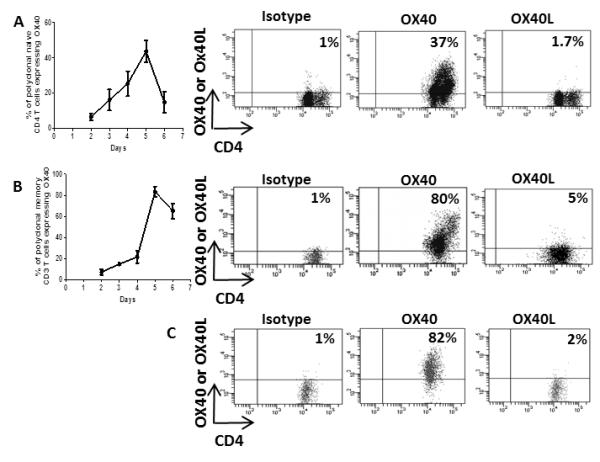
2×105 naïve or memory polyclonal T cells were cultured in vitro with 4×105 allogeneic irradiated stimulator splenocytes. Expression of OX40 and OX40L on naïve (total CD4+ T cells) (A) and memory (CD3+CD4+CD44+) B) polyclonal CD4+ T cells and naïve CD25+CD4+Foxp3+ Treg (C) was determined by flow cytometry. Quadrant gates were set using an isotype control antibody. Results are expressed as mean % ± SD, performed in triplicate and are representative of 3 independent experiments.
The expression of OX40 and OX40L was also analysed on CD4+CD25+FoxP3+ T cells (Treg). Treg sorted from CBA mice were found to express OX40 without activation (Figure 1C) but did not express OX40L (Figure 1C). After activation with alloantigen, Treg expressed OX40 to similar levels, but remained OX40L− (data not shown).
Blockade of OX40-OX40L interactions attenuates skin allograft rejection mediated by naïve and memory CD4+ T cells
It has been previously shown that both naïve and memory T cells express OX40 and that OX40 signalling is required for allograft rejection in certain settings (15, 17). In light of these data, we next wanted to test whether anti-OX40 could prevent the rejection of skin allografts in a model where rejection was elicited by traceable, alloantigen-reactive CD4+ T cells. To this end, 1×105 naïve or memory TCR-Transgenic (TEa) CD4+ T cells were adoptively transferred into syngeneic Rag−/− mice and one day later the mice received a skin allograft (BALB/cB6 F1). Mice were treated twice weekly with PEG control or anti-OX40 from the time of transplant until day 28 post-transplant. Mice reconstituted with naïve TEa T cells and given PEG control acutely rejected their skin allografts, whilst allograft survival was significantly prolonged in mice treated with anti-OX40 (MST 49 vs. 18.5 days; p< 0.0004) (Figure 2A). Similarly, mice reconstituted with TEa Tm and treated with PEG control rejected their skin allografts acutely, while treatment with anti-OX40 was found to markedly prolong skin allograft survival (MST 89.5 vs. 18 days; p<0.0007; Figure 2C).
Figure 2. Blocking the OX40-OX40L pathway prevents skin allograft rejection mediated by naïve and memory CD4+ T cells.

1×105 naïve TEa (A), naïve polyclonal (B) memory TEa (C) memory polyclonal (D) CD4+ T cells were adoptively transferred into syngeneic Rag−/− mice. One day later, mice received an allogeneic skin graft (BALB/cB6 F1 or B6 respectively) and either anti-OX40 or PEG control was administered s.c. at 100mg/kg twice weekly for 4 weeks. Kaplan-Meier survival curves show skin allograft rejection kinetics. (n= 6-8 mice per group).
Although OX40 blockade resulted in significant attenuation of skin allograft rejection mediated by naïve or memory TEa T cells to rule out any effects that could be attributable to the use of TCR-transgenic T cells we sought to confirm these results using polyclonal naïve and memory T cells. To this end, naïve or memory polyclonal CD4+ T cells were transferred into syngeneic Rag−/− mice. The following day mice received an H2Kb+ (B6) skin allograft together with either anti-OX40 or PEG control (twice weekly for 4 weeks).
Administration of anti-OX40 given from the time of skin transplant significantly prolonged allograft survival in mice that had received naïve polyclonal CD4+ T cells compared to the PEG control group (MST 97 vs.14 days; p< 0.0006; Figure 2B). This dramatic impact of OX40 blockade on graft survival was also seen in a second strain combination when polyclonal B6 CD4+ T cells were adoptively transferred into syngeneic Rag−/− recipients and mice received an allogeneic skin transplant (CBA; MST 67 vs. 13 days; p< 0.0067; Supplementary Figure 1). Mice reconstituted with polyclonal CD4+ Tm and treated with PEG control rejected skin allografts acutely. In contrast, skin allograft survival was significantly prolonged when mice received allograft and anti-OX40 (MST 51.5 vs. 15.5 days; p<0.0005; Figure 2D).
Taken together, these data demonstrate that the OX40-OX40L pathway plays a key role in facilitating the rejection of allografts by both naïve and memory CD4+ T cells, which is consistent with other studies.
Anti-OX40 prevents accumulation of naïve and memory CD4+ T cells in the draining lymph nodes following allogeneic skin transplantation
It has previously been suggested that OX40-OX40L costimulation promotes effector T cell survival (14, 37) therefore we next examined whether this was also the case in the context of T cell responses to allografts. Our previous studies have shown that large numbers of primed effector/memory CD4+ T cells could be detected specifically in the draining lymph nodes (dLN) by 15 days after allogeneic skin transplantation (data not shown). Therefore, next we assessed the impact of OX40-OX40L interactions on the clonal expansion of donor-reactive TEa T cells following allogeneic skin transplantation in vivo. 1×105 TEa T cells were adoptively transferred into syngeneic Rag−/− mice, and one day later such mice received either a syngeneic or allogeneic skin graft. Mice receiving an allograft were either given PEG control or anti-OX40 twice weekly until harvested at day 15.
In the presence of PEG control a significant expansion of primed effector/memory TEa T cells was found in the dLN after an allogeneic skin graft (TEa cell number 198562 ± 91555), whilst little expansion occurred in the non-draining contralateral lymph node (cLN) (TEa number 9628 ± 7212) (Figure 3A). The expansion of TEa T cells in the dLN was shown to be allo-specific as there was no expansion of TEa T cells in the dLN after syngeneic skin graft transplantation (Figure 3A). In contrast, anti-OX40 treatment was found to prevent the accumulation of TEa T cells in the dLN (TEa cell number 23146 ± 18585; 91±10% inhibition; p<0.0095; Figure 3A) 15 days after skin allografting. Similar results were obtained after adoptive transfer of TEa Tm (Supplementary Figure 2A).
Figure 3. Blocking OX40 inhibits clonal expansion of CD4+ T cells after allogeneic skin transplantation.
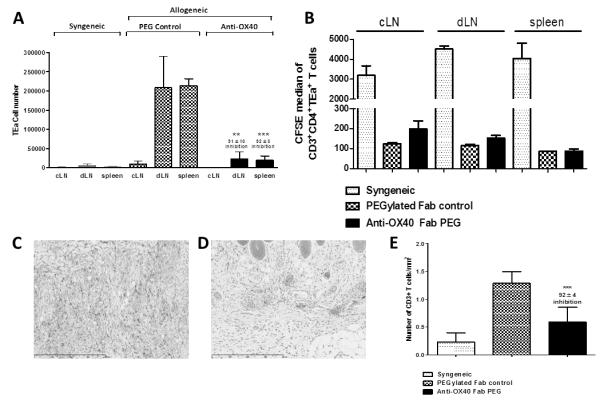
1×105 naïve TEa T cells were adoptively transferred into syngeneic Rag−/− mice. 1 day later, mice received an allogeneic skin graft and anti-OX40 or PEG control, administered s.c at 100mg/kg twice weekly for 15 days. (A) Number of TEa T cells ± SD in the cLN, dLN and spleen after syngeneic or allogeneic skin transplantation (B) Proliferation, measured by CFSE dilution, of TEa T cells in the cLN, dLN and spleen. (C-E) Skin grafts were analysed by immunohistochemistry for the presence of CD3+ T cell infiltration. Photomicrographs are representative of 3 non-serial sections per graft. (C) TEa T cells + PEG control (D) TEa T cells + anti-OX40 (E) Enumeration of CD3 infiltration after reconstitution with TEa T cells. Results are expressed as mean number ± SD, performed in triplicate and are representative of 2 independent experiments. The unpaired student t test was used to compare differences between anti-OX40 and PEG treated mice where ** represents p<0.01, *** represents p<0.001. (n = 4 mice per group).
Following allogeneic skin transplantation, alloreactive T cells become activated, proliferate and accumulate in the dLN before migrating to the allograft and to the spleen where they reside as long-lived effector/memory T cells. Therefore, as expected, an expanded population of TEa T cells was found in the spleen after allogeneic skin transplantation however this re-distribution of effector/memory TEa T cells was also inhibited by anti-OX40 treatment at day 15 (anti-OX40: 19278 ± 11296 vs PEG control: 213722 ± 18008 TEa T cells; 92±5% inhibition; p<0.0001; Figure 3A).
Alloreactive effector T cells do not require OX40 blockade for optimal proliferation
The attenuated T cell accumulation seen on administration of anti-OX40 could be due to a requirement for OX40 for optimal proliferation or that OX40 is required to maintain the survival of activated effector cells. To distinguish between these possibilities we looked at the proliferation of alloreactive T cells (as judged by the loss of CFSE) following transplantation in the presence or absence of anti-OX40 blockade. TEa T cells were found to have proliferated specifically in the dLN by 15 days after transplantation (Figure 3B). Interestingly, despite a marked diminution in the number of TEa T cells (Figure 3A) anti-OX40 did not affect the initial proliferation of alloreactive T cells (Figure 3B). TEa T cells in mice that had received a syngeneic skin graft or in the cLN of allograft recipients maintained high levels of CFSE, consistent with the fact these cells had not proliferated or expanded by homeostatic proliferation (Figure 3B). Experiments where TEa Tm rather than naïve T cells were transferred yielded similar results (Supplementary Figure 2B).
Anti-OX40 inhibits T cell infiltration of skin allografts
Anti-OX40 inhibited the accumulation but not the initial proliferation of CD4+ T cells in the dLN following allogeneic skin transplantation. To rule out the possibility that anti-OX40 had enhanced the migration to other lymphoid tissues and in particular to the allograft, we harvested skin allografts from mice treated with anti-OX40 or PEG control at day 15. Skin allografts were analysed for the presence of CD3+ T cells by immunohistochemistry as the only T cells present in these mice were the transferred naïve or memory TEa T cells (Figure 3C-D and Supplementary Figure 2C-D). We found a dramatic reduction in the number of TEa cells that had infiltrated skin allografts after treatment with anti-OX40 (Figure 3C-E) which is consistent with anti-OX40 preventing the survival of effector CD4+ T cells rather than inducing enhanced migration of effector T cell to the skin allografts.
OX40 is required for the survival of activated, alloreactive CD4+ T cells
To confirm this finding we next looked ex vivo at dLN taken from mice that had received skin allografts. To this end, 1×105 TEa T cells were adoptively transferred into syngeneic Rag−/− recipients and the following day mice received a skin allograft. Mice were either treated with PEG control or anti OX40 twice weekly, before being sacrificed on day 13 for analysis by flow cytometry. This time-point provided an opportunity to observe cell death in the dLN before the cells were deleted.
Cells from the dLN were analysed by annexin V and 7AAD viability stains to assess the proportion of live/dead TEa T cells. The number and percentage of live cells after anti-OX40 treatment was found to be reduced after anti-OX40 treatment compared to in control mice (Figures 4A and B). OX40 blockade increased the proportion of pre-apoptotic (Annexin V+7AAD−) (Figure 4C) but had no impact on the proportion of apoptotic (Annexin V+7AAD+) TEa T cells in the dLN (Figure 4D), which correlated with our in vitro findings in CD8+ T cells (15), and with others in CD4+ T cells (14, 37) suggesting that OX40 signals were required for the survival of activated, alloreactive CD4+ T cells.
Figure 4. Anti-OX40 increases cell death in the dLN after skin allograft transplantation.
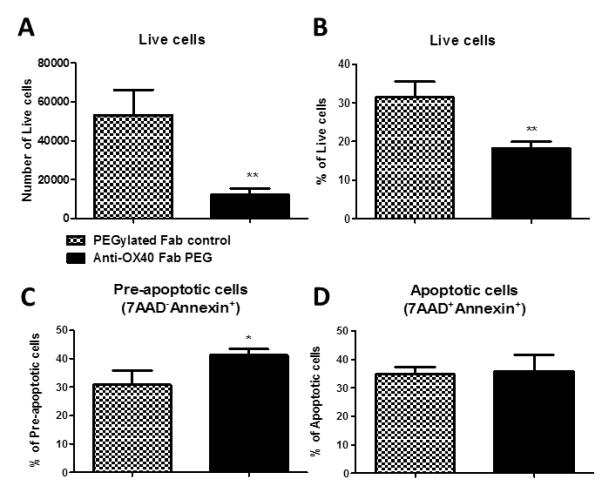
1×105 naïve TEa CD4+ T cells were adoptively transferred into syngeneic Rag−/− mice. One day later, mice received an allogeneic skin graft and either anti-OX40 or PEG control, administered s.c. at 100mg/kg and sacrficed on day 13. dLN were analysed for live (A and B), pre apoptotic (annexin V+7AAD−) (C) and apoptotic (AnnexinV+7AAD+) (D) CD4+ T cells. Results are expressed as mean ± SD, performed in triplicate ** represents p<0.01.
OX40 blockade enhances the potency of Treg
It has previously been suggested that there may be a differential impact of OX40-OX40L signalling on effector/memory T cells and Treg. Therefore we next wanted to investigate the impact of OX40 blockade on alloreactive Treg responses. Our laboratory has previously developed a protocol to generate alloreactive Treg in vivo by the administration of a non-depleting anti-CD4 mAb (YTS177; 177) and donor-specific transfusion of whole blood (DST) to mice (35, 38).
Using this protocol we determined whether OX40 blockade enhanced or attenuated the number or activity of Treg generated by 177/DST in vivo. To this end, alloreactive Treg were generated in vivo following administration of 177/DST in the presence or absence of anti-OX40 (days −28 and −25). Treg were isolated on day 0 and tested for their relative ability to suppress an alloreactive T cell response in vitro or skin allograft rejection in vivo (Figure 5).
Figure 5. Treg generated in the absence of OX40 signalling have a greater suppressive capacity in vivo.
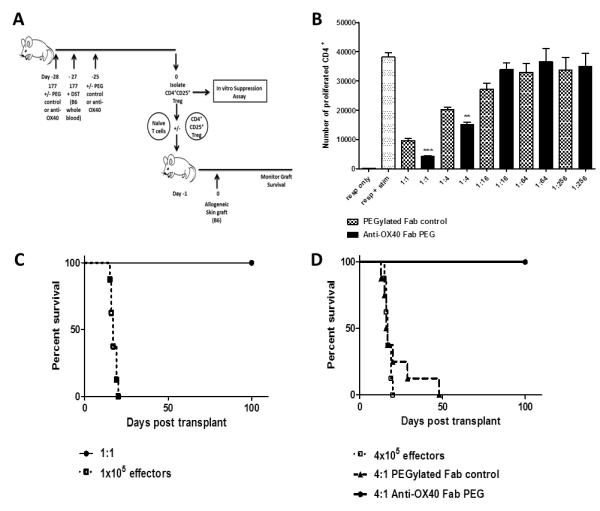
A) Naïve mice received 200μg of non-depleting anti-CD4 mAb (YTS177) and DST on days −28 and −27. Anti-OX40 or PEG control were administered at 100mg/kg s.c. on days −28 and −25. Treg (CD4+CD25+) were purified on day 0 from mice treated with 177/DST and either labelled with CFSE for use in an in vitro suppression assay or co-transferred in vivo with CD4+CD25− T cells (naïve T cells) purified from un-manipulated CBA mice. Purified Treg and naïve T cells were adoptively transferred into syngeneic Rag−/− recipient mice at varying ratios. The following day mice received a skin allograft (H2b). B) Naïve responder T cells (CD4+CD25−) were cultured with stimulator B6 CD11c+ cells with and without varying ratios of Treg (purified from mice that had received 177/DST with and without anti-OX40). Cultures were analysed after 6 days of culture by flow cytometry. Results are expressed as mean cell number ± SD, performed in triplicate (n = 6 mice per group (pooled)) and representative of 2 independent experiments). ** represents p<0.01. *** represents p<0.0001. C) Kaplan-Meier survival curves show skin allograft rejection kinetics. A) Allograft survival after reconstitution with 1×105 naïve T cells (effectors) and 1×105 Treg and (1:1 ratio). B) Allograft survival after transfer with 4×105 naïve T cells (effectors) and 1×105 Treg (4:1 ratio). In both A) and B) a group of mice was included that received naïve T cells alone. (n = 4-8 mice per group and data pooled between 2 independent experiments).
The addition of alloreactive PEG control Treg markedly inhibited the proliferation of CD4+CD25− responder T cells to CD11c+ allogeneic stimulator cells at a 1:1 ratio (Figure 5B). With decreasing numbers of PEG control Treg (i.e. increased ratio of naïve:Treg) suppression was lost in a dose dependent manner (Figure 5B). Importantly, Treg generated in the absence of OX40 signalling showed enhanced suppression compared to PEG control Treg at ratios of both 1:1 and 1:4 (Figure 5B). Treg generated in the presence of absence of anti-OX40 had comparable levels of FoxP3 (data not shown).
Similarly, 177/DST+PEG control generated Treg were able to control skin allograft rejection at a 1:1 but not at a 4:1 (effector:Treg) ratio with naïve T cells when transferred to Rag−/− mice (Figure 5C). However, blockade of OX40 at the time of 177/DST resulted in the generation of Treg that remained able to prevent skin allograft rejection when transferred in a 4:1 ratio with naïve T cells (MST >100 days vs. 16.5 days; p<0.0003; Figure 5C). These data clearly demonstrate that blockade of OX40 interactions increased rather than decreased the potency of alloreactive CD4+CD25+ Treg.
Alloreactive Treg generated in the absence of OX40 signalling demonstrate enhanced survival compared to Treg generated in an OX40 sufficient environment
OX40 blockade was found to significantly enhance cell death in CD4+ (Figure 4) and CD8+ alloreactive (15), effector T cells which led us to investigate whether OX40 blockade had a distinct impact on the survival of Treg which may explain the resultant increase in potency. To address this, the number of live Treg (that had been generated by 177/DST in the presence or absence of OX40-OX40L interactions) present after 5 days of culture with syngeneic responder (CD4+CD25−) T cells and allogeneic CD11c+ splenocytes was determined. Importantly, Treg from anti-OX40 treated or control treated animals, were used at the same number per well. 177/DST Treg generated in the absence of OX40 signalling had a significant survival advantage both in terms of number (Treg number: anti-OX40, 34527 ± 3368 vs PEG control, 5934 ± 384; Figure 6A) and percentage of live cells (% Treg: anti-OX40, 46.4% ± 0.8 vs PEG control, 12.3 ± 1.1; Figure 6B) compared to Treg generated in PEG control treated mice. This survival advantage was evident at all the ratios of responders to Treg following reactivation with allogeneic splenocytes in vitro.
Figure 6. Alloreactive Treg generated in the presence of anti-OX40 exhibit enhanced survival upon re-activation.

Mice received 200μg YTS177 and a DST on days −28 and −27. Anti-OX40 or PEG control were administered at 100mg/kg s.c. on days −28 and −25. Treg (CD4+CD25+) were purified on day 0 from mice treated with 177/DST and labelled with CFSE. 1×105 naïve CBA responder T cells (CD4+CD25−) were cultured with B6 stimulator CD11c+ cells with and without Treg, added at varying ratios to the responder cells. Cultures were analysed for number (A) or % (B) live Treg at day 6 or Bcl-2 expression (days 3-5) by flow cytometry. Results are expressed as mean number or % ± SD, n = 6 mice per group (pooled) and representative of 2 independent experiments. * represents p<0.05, ** represents p<0.001.
Finally, given that Treg generated in the absence of OX40-OX40L interactions demonstrated enhanced survival following reactivation (Figure 6A and B) we examined the expression of the anti-apoptotic molecule Bcl-2 in such cultures. Treg generated in the absence of OX40 signalling were found to have a significantly higher number of Bcl-2+ Treg at day 4 (Bcl-2+ Treg cell number 2410 ± 255 vs. 844 ± 559; p< 0.0116) and day 5 (Bcl-2+ Treg cell number 7943 ± 594 vs. 3466 ± 627; p< 0.0009) compared to 177/DST+PEG control generated Treg (Figure 6C).
Discussion
Alloimmune responses are complex and require orchestration of a number of different cell types in order to elicit rejection. Given the significant role played by costimulatory molecules in T cell activation, costimulation blockade provides a promising adjunctive or alternative therapy to the currently licenced immunosuppressive drugs used to prevent graft rejection and autoimmunity (1). A number of costimulatory molecules which are required for optimal naïve T cell responses are also expressed by Treg, for example, CD28 signalling has been reported as being required for the optimal suppressive function of Treg (39). However, not all costimulatory molecules promote Treg-mediated suppression, for example, signalling through GITR (a TNFR superfamily member) which is expressed on Treg (like OX40) has been shown to result in a loss of Treg function (40, 41). Importantly blockade of OX40 and GITR has a similar impact on effector T cells and results in a delay in disease onset or attenuated severity in models of diabetes (42, 43). Therefore, data such as these have led to the possibility that certain costimulatory molecules may be utilised in different ways by conventional and regulatory T cells.
In agreement with the literature, OX40 was found to be expressed on activated, but not naïve, CD4+ T cells (Figure 1). Furthermore, the delayed kinetics of expression (OX40 was not expressed until 3 days after activation) was consistent with a role for OX40-OX40L in effector and memory T cell generation i.e. in the propagation of the response, rather than in initial T cell priming. Blocking OX40 markedly impacted the ability of alloreactive naïve and memory CD4+ T cells to elicit rejection of skin allografts in vivo (Figure 2). Furthermore, blockade of the OX40-OX40L pathway prevented the accumulation of alloreactive CD4+ T cells in the dLN (Figure 3 and Supplementary Figure 2). These in vivo studies demonstrate that CD4+ T cells undergo enhanced activation induced cell death in the absence of OX40 signalling (Figure 4). Therefore, OX40-OX40L interactions appeared to provide critical survival signals for effector/memory CD4+ T cells rather than being essential for initial T cell activation and differentiation, which is in agreement with other studies to date (14, 15, 37). These data are also in agreement with studies blocking OX40-OX40L in autoimmune diseases EAE, where Nohara et al used a proteolipid protein induced model of EAE and administration of anti-OX40L ameliorated disease. These studies showed a dramatic reduction in the infiltration of mononuclear and CD4+ T cells into the CNS (44).
Our data are in agreement with previous reports where OX40−/− T cells were shown to become normally activated and expanded (45, 46) but failed to maintain high levels of anti-apoptotic Bcl-2 family members such as Bcl-2 and Bcl-XL (14, 47) thus inhibiting the ability to maintain clonal expansion over time, thereby preventing the generation or sustenance of memory T cell responses (46, 48). It has been shown recently that maintenance of high PKB activity is an essential downstream signal of OX40, and is involved in the control of survival of CD4+ T cells through the up-regulation of anti-apoptotic Bcl-2 family members (37). CD4+ T cells that lack OX40 are unable to maintain high levels of anti-apoptotic molecules demonstrating a clear link between NFκB signalling and cell survival (49).
Our experiments with alloreactive naïve and memory CD4+ T cells also concur with published data whereby administration of agonistic OX40 enhanced primary T cell expansion and survival which in turn increased the frequency of memory T cells (46, 50, 51). These data suggested that the absence of OX40 gave rise to a defect in survival or in their ability for long term division. The impaired survival of OX40−/− T cells could be reversed by the addition of peptide inhibitors of caspases (pan-caspase inhibitor or specific inhibitors to caspase-3 or -9) (14) suggesting that signalling via OX40-OX40L prevents caspase-mediated apoptosis. However in our studies we did not discriminate between caspase dependent and independent apoptosis.
The upregulation of anti-apoptotic molecules (Bcl-XL and Bcl-2) to promote survival after T cell antigen recognition and OX40 costimulation is not unique to OX40 and had also been demonstrated when costimulation was provided by other Ig and TNFR family members such as CD28 (52) and 4-1BB (53, 54). Our data clearly showed OX40 signalling provided a critical survival signal for naïve effector CD4+ T cells but importantly was also key for memory CD4+ T cells. Tm are now accepted as being a barrier to tolerance induction, as a number of strategies (e.g. CD28 and/or CD154 blockade) that are successful at preventing allograft rejection by naïve T cells (using mouse models in pathogen free conditions) (55-57) fail to induce tolerance in rodents which have been pre-sensitised with donor alloantigen (28, 58). In addition, Heeger et al showed that alloreactive Tm were present in patients prior to kidney transplantation and that increased number of Tm cells were associated with an increased incidence of acute rejection (29). These and other reports provide evidence that Tm are critical to the success of allograft survival, therefore developing strategies to inhibit Tm would be beneficial in preventing allograft rejection and tolerance induction.
It is particularly noteworthy that the absence of OX40-OX40L signalling resulted in significantly prolonged allograft survival despite the availability of numerous other costimulatory ligands such as CD80/CD86 (59) and CD40 (60, 61) on donor APC that could provide survival signals to the T cells (59). It therefore appears that OX40 plays a dominant and indispensable role in the continued survival of alloantigen-activated T cells and the ability to mediate graft rejection. However it must be noted that the prolongation of skin allograft survival in the absence of OX40-OX40L signalling was limited to only situations where a small number of T cells was transferred, as anti-OX40 failed to impact allograft survival in immunocompetent mice (data not shown).
Importantly, while anti-OX40 continued to be administered allografts remained free from rejection, however, grafts were rejected after the cessation of therapy (Figure 2). It therefore appears that blockade of OX40-OX40L interactions failed to induce tolerance. Other evidence for the lack of tolerance induction is borne out of the observation that skin allografts were rejected after the anti-OX40 therapy was discontinued and the reagent cleared from the systemic circulation (data not shown).
The failure of OX40 blockade to induce tolerance could be due to a number of factors. Firstly, not all activated CD4+ T cells expressed OX40 (Figure 1), suggesting some T cells are not dependent on the OX40-OX40L pathway. Numerous other costimulatory molecules within the Ig and TNFR superfamilies can make up for the lack of OX40 signalling due to the high degree of redundancy within the system.
Secondly, in this adoptive transfer model not all T cells simultaneously encounter alloantigen following transplantation for example naïve alloreactive T cells can be detected up to 50 days post transplantation despite allograft rejection (N.D Jones; personal communication). As naïve T cells do not express OX40 these cells may be able to elicit rejection upon alloantigen recognition and activation following the clearance of anti-OX40.
Lastly, an important caveat of this adoptive transfer model is that purified naïve CD4+ T cells were transferred to Rag−/− knockout mice therefore these animals have no alloreactive Treg. Indeed the TCR-transgenic T cells used in these studies were devoid of T cells with a regulatory phenotype (data not shown) and the polyclonal T cells were isolated as CD4+CD44−CD45RBhi cells to ensure Tm and Treg were excluded. Although a strict sorting strategy was used, it is possible that T cells could be converted into Treg (so called inducible Treg) by OX40 blockade, although we would suggest that this is unlikely as all the allografts were rejected following clearance of anti-OX40 (Figure 2). Treg have been shown to be critical for the maintenance of long-term graft survival following costimulatory molecule blockade in immunocompetent mice raising the possibility that tolerance would never be attainable in this model because of their absence (62).
The data presented here raises the question whether anti-OX40 could impact Treg function as Treg constitutively express OX40, unlike conventional T cells. We demonstrate that the exposure of Treg to OX40 blockade and alloantigen generates Treg that are more potent at suppressing T cell responses to alloantigen in vitro and in vivo compared to those generated in the presence of OX40 signalling.
In our studies, OX40 blockade promoted, rather than suppressed, the ability of Treg generated in vivo under the cover of anti-CD4 and DST to regulate allo-specific immune responses by increasing the survival of Treg (Figures 5 and 6). Other groups have also shown that OX40-OX40L can impact Treg function (26, 63) while signalling via OX40-OX40L in effector T cells results in enhanced survival and generation of productive memory T cell responses (4, 46, 64). These data suggest that OX40-OX40L interactions have a dichotomous role on effector/memory T cells and Treg. Burocchi et al has also suggested a diametric role for OX40-OX40L whereby signalling via OX40 inhibited Treg mediated suppression and enhanced effector T cell activation in a tumour model (65). Other in vitro data has suggested that signalling through OX40-OX40L results in a reduction in FoxP3 expression (26, 63). Our data whilst consistent with these reports additionally reveals that OX40 signalling diminishes the survival of activated Treg which clearly impacts on their potency to suppress alloimmune responses in vitro and in vivo (Figures 5 and 6).
There is now strong supportive evidence that FoxP3− T cells can be converted into FoxP3+ T cells (iTreg) which have suppressive function in the periphery (66, 67). Indeed, it has been previously shown that YTS177/DST generated alloreactive Treg are generated in part through conversion of naïve T cells to iTreg and in part through the expansion/priming of naturally occurring Treg that are cross-reactive with donor alloantigen (68, 69). Therefore, further experiments will be required to ascertain whether anti-OX40 impacts nTreg or iTreg or both, however, So et al provided evidence that OX40 signalling could antagonise FoxP3 induction (25) suggesting that OX40 signalling may impact the induction of FoxP3 expression and therefore iTreg generation.
Enhanced Treg survival may be only part of the story and OX40 blockade may have other effects on Treg function which were not examined in our studies which could also increase Treg potency. Therefore further studies would be required to dissect the role of OX40-OX40L on molecules that have been associated with Treg function such as IL-10 and TGF-β. Analysis of Treg isolated from tumour sites demonstrated that tumour resident Treg exhibited a reduction in IL-10 production in the presence of OX40 signalling (65) which correlated with a decrease in the transcription factor interferon regulatory factor 1 (Irf1). This has also been found to be associated with IL-10 production from Treg isolated from the lamina propria (70). These data suggest further analyses of molecules such as TGF-β, IL-10 and CTLA-4 could provide further dissection on the mechanisms by which OX40-OX40L impact Treg and their suppressive capacity.
OX40 signalling has been described to alter Treg positioning and homing. Following administration of agonistic OX40, Treg exhibited an increase in the expression of CCR8 and CD103 and a decrease in CCR4 (65). However these findings have not been replicated in disease models for example as OX40−/− Treg express comparable levels of CD103 to wild-type Treg and in a T cell transfer induced colitis model OX40−/− Treg were able to accumulate in the mLN and lamina propria (71), thus making the interpretation of the transcriptome data more complex. These analyses interrogate patterns of expression at a specific time of OX40 signalling and not on the long term impact of OX40 signalling.
In summary, we have shown that OX40-OX40L interactions are important for the sustained expansion/survival of alloreactive naïve and memory CD4+ T cells during skin allograft rejection. However, in clear contrast, mice exposed to alloantigen in the absence of OX40 signalling results in the generation of alloreactive Treg that demonstrate increased potency in their ability to suppress alloreactive T cell responses as a result of enhanced survival. This differs from other reports which show that signalling via OX40-OX40L results in a downregulation of FoxP3 or induction of iTreg (63, 72). These data have confirmed a clear diametric role for OX40 in the response of effector/memory T cells and Treg to allografts that could be exploited as part of tolerance induction therapies to combat transplant rejection and re-establish tolerance in autoimmunity. In addition, blockade of OX40-OX40L has proven successful in mouse models of autoimmune diseases (44, 73) and allergic inflammation (48, 74) thereby providing other disease areas which the blockade of OX40 may have a therapeutic effect. In particular there is a growing body of evidence that OX40 signalling affects Th2 (75) and more recently Th9 differentiation (76) so targeting OX40 in allergic inflammation may also be beneficial.
Supplementary Material
Acknowledgements
The authors thank the staff of the Biomedical Services Unit for excellent husbandry and animal care.
This work was supported by grants from the MRC (Case Studentship to GK) and The Wellcome Trust.
Abbreviations
- cLN
Contralateral lymph node
- DC
Dendritic cell
- dLN
Draining lymph node
- Irf1
Interferon regulatory factor 1
- mLN
Mesenteric lymph node
- PEG
Polyethylene glycol
- PKB
Protein kinase B
- SD
Standard deviation
- Tm
Memory T cell
- TNFR
Tumour Necrosis Factor Receptor
- TRAF
Tumour Necrosis Factor Receptor Associated Factor
- Treg
Regulatory T cell
Footnotes
GK – designed experiments, performed experiments, analyzed data and wrote the paper. GK is supported by a CASE Studentship (MRC and UCB Pharma collaboration).
KJW – provided reagents and was involved in writing of the paper.
FFA – provided reagents and edited paper.
NDJ – provided reagents, designed experiments, analyzed data and was involved in writing the paper.
Conflict of Interest: None
References
- 1.Kinnear G, Jones ND, Wood KJ. Costimulation Blockade: Current Perspectives and Implications for Therapy. Transplantation. 2012 doi: 10.1097/TP.0b013e31826d4672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Godfrey WR, Fagnoni FF, Harara MA, Buck D, Engleman EG. Identification of a human OX-40 ligand, a costimulator of CD4+ T cells with homology to tumor necrosis factor. J Exp Med. 1994;180:757–762. doi: 10.1084/jem.180.2.757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Akiba H, Atsuta M, Yagita H, Okumura K. Identification of rat OX40 ligand by molecular cloning. Biochemical and biophysical research communications. 1998;251:131–136. doi: 10.1006/bbrc.1998.9376. [DOI] [PubMed] [Google Scholar]
- 4.Gramaglia I, Weinberg AD, Lemon M, Croft M. OX40 ligand: a potent costimulatory molecule for sustaining primary CD4 T cell responses. Journal of Immunology. 1998;161:6510–6517. [PubMed] [Google Scholar]
- 5.Takeda I, Ine S, Killeen N, Ndhlovu LC, Murata K, Satomi S, Sugamura K, Ishii N. Distinct roles for the OX40-OX40 ligand interaction in regulatory and nonregulatory T cells. Journal of Immunology. 2004;172:3580–3589. doi: 10.4049/jimmunol.172.6.3580. [DOI] [PubMed] [Google Scholar]
- 6.Valzasina B, Guiducci C, Dislich H, Killeen N, Weinberg AD, Colombo MP. Triggering of OX40 (CD134) on CD4(+)CD25+ T cells blocks their inhibitory activity: a novel regulatory role for OX40 and its comparison with GITR. Blood. 2005;105:2845–2851. doi: 10.1182/blood-2004-07-2959. [DOI] [PubMed] [Google Scholar]
- 7.Miura S, Ohtani K, Numata N, Niki M, Ohbo K, Ina Y, Gojobori T, Tanaka Y, Tozawa H, Nakamura M, et al. Molecular cloning and characterization of a novel glycoprotein, gp34, that is specifically induced by the human T-cell leukemia virus type I transactivator p40tax. Mol Cell Biol. 1991;11:1313–1325. doi: 10.1128/mcb.11.3.1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Baum PR, Gayle RB, 3rd, Ramsdell F, Srinivasan S, Sorensen RA, Watson ML, Seldin MF, Baker E, Sutherland GR, Clifford KN, et al. Molecular characterization of murine and human OX40/OX40 ligand systems: identification of a human OX40 ligand as the HTLV-1-regulated protein gp34. EMBO J. 1994;13:3992–4001. doi: 10.1002/j.1460-2075.1994.tb06715.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stuber E, Neurath M, Calderhead D, Fell HP, Strober W. Cross-linking of OX40 ligand, a member of the TNF/NGF cytokine family, induces proliferation and differentiation in murine splenic B cells. Immunity. 1995;2:507–521. doi: 10.1016/1074-7613(95)90031-4. [DOI] [PubMed] [Google Scholar]
- 10.Stuber E, Strober W. The T cell-B cell interaction via OX40-OX40L is necessary for the T cell-dependent humoral immune response. J Exp Med. 1996;183:979–989. doi: 10.1084/jem.183.3.979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ohshima Y, Tanaka Y, Tozawa H, Takahashi Y, Maliszewski C, Delespesse G. Expression and function of OX40 ligand on human dendritic cells. J Immunol. 1997;159:3838–3848. [PubMed] [Google Scholar]
- 12.Brocker T, Gulbranson-Judge A, Flynn S, Riedinger M, Raykundalia C, Lane P. CD4 T cell traffic control: in vivo evidence that ligation of OX40 on CD4 T cells by OX40-ligand expressed on dendritic cells leads to the accumulation of CD4 T cells in B follicles. European Journal of Immunology. 1999;29:1610–1616. doi: 10.1002/(SICI)1521-4141(199905)29:05<1610::AID-IMMU1610>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 13.Imura A, Hori T, Imada K, Ishikawa T, Tanaka Y, Maeda M, Imamura S, Uchiyama T. The human OX40/gp34 system directly mediates adhesion of activated T cells to vascular endothelial cells. J Exp Med. 1996;183:2185–2195. doi: 10.1084/jem.183.5.2185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rogers PR, Song J, Gramaglia I, Killeen N, Croft M. OX40 promotes Bcl-xL and Bcl-2 expression and is essential for long-term survival of CD4 T cells. Immunity. 2001;15:445–455. doi: 10.1016/s1074-7613(01)00191-1. [DOI] [PubMed] [Google Scholar]
- 15.Kinnear G, Wood KJ, Marshall D, Jones ND. Anti-OX40 prevents effector T-cell accumulation and CD8+ T-cell mediated skin allograft rejection. Transplantation. 2010;90:1265–1271. doi: 10.1097/TP.0b013e3181fe5396. [DOI] [PubMed] [Google Scholar]
- 16.Flynn S, Toellner KM, Raykundalia C, Goodall M, Lane P. CD4 T cell cytokine differentiation: the B cell activation molecule, OX40 ligand, instructs CD4 T cells to express interleukin 4 and upregulates expression of the chemokine receptor, Blr-1. Journal of Experimental Medicine. 1998;188:297–304. doi: 10.1084/jem.188.2.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vu MD, Clarkson MR, Yagita H, Turka LA, Sayegh MH, Li XC. Critical, but conditional, role of OX40 in memory T cell-mediated rejection. J Immunol. 2006;176:1394–1401. doi: 10.4049/jimmunol.176.3.1394. [DOI] [PubMed] [Google Scholar]
- 18.Ukyo N, Hori T, Yanagita S, Ishikawa T, Uchiyama T. Costimulation through OX40 is crucial for induction of an alloreactive human T-cell response. Immunology. 2003;109:226–231. doi: 10.1046/j.1365-2567.2003.01648.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yoshioka T, Nakajima A, Akiba H, Ishiwata T, Asano G, Yoshino S, Yagita H, Okumura K. Contribution of OX40/OX40 ligand interaction to the pathogenesis of rheumatoid arthritis. European Journal of Immunology. 2000;30:2815–2823. doi: 10.1002/1521-4141(200010)30:10<2815::AID-IMMU2815>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 20.Wu T, Hering B, Kirchof N, Sutherland D, Yagita H, Guo Z. The effect of OX40/OX40L and CD27/CD70 pathways on allogeneic islet graft rejection. Transplant Proc. 2001;33:217–218. doi: 10.1016/s0041-1345(00)01980-1. [DOI] [PubMed] [Google Scholar]
- 21.Curry AJ, Chikwe J, Smith XG, Cai M, Schwarz H, Bradley JA, Bolton EM. OX40 (CD134) blockade inhibits the co-stimulatory cascade and promotes heart allograft survival. Transplantation. 2004;78:807–814. doi: 10.1097/01.tp.0000131670.99000.54. [DOI] [PubMed] [Google Scholar]
- 22.Yuan X, Salama AD, Dong V, Schmitt I, Najafian N, Chandraker A, Akiba H, Yagita H, Sayegh MH. The role of the CD134-CD134 ligand costimulatory pathway in alloimmune responses in vivo. J Immunol. 2003;170:2949–2955. doi: 10.4049/jimmunol.170.6.2949. [DOI] [PubMed] [Google Scholar]
- 23.Demirci G, Amanullah F, Kewalaramani R, Yagita H, Strom TB, Sayegh MH, Li XC. Critical role of OX40 in CD28 and CD154-independent rejection. J Immunol. 2004;172:1691–1698. doi: 10.4049/jimmunol.172.3.1691. [DOI] [PubMed] [Google Scholar]
- 24.Habicht A, Najafian N, Yagita H, Sayegh MH, Clarkson MR. New insights in CD28-independent allograft rejection. Am J Transplant. 2007;7:1917–1926. doi: 10.1111/j.1600-6143.2007.01886.x. [DOI] [PubMed] [Google Scholar]
- 25.So T, Croft M. Cutting edge: OX40 inhibits TGF-beta- and antigen-driven conversion of naive CD4 T cells into CD25+Foxp3+ T cells. Journal of immunology. 2007;179:1427–1430. doi: 10.4049/jimmunol.179.3.1427. [DOI] [PubMed] [Google Scholar]
- 26.Xiao X, Gong W, Demirci G, Liu W, Spoerl S, Chu X, Bishop DK, Turka LA, Li XC. New insights on OX40 in the control of T cell immunity and immune tolerance in vivo. Journal of immunology. 2012;188:892–901. doi: 10.4049/jimmunol.1101373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gaspal FM, Kim MY, McConnell FM, Raykundalia C, Bekiaris V, Lane PJ. Mice deficient in OX40 and CD30 signals lack memory antibody responses because of deficient CD4 T cell memory. Journal of immunology. 2005;174:3891–3896. doi: 10.4049/jimmunol.174.7.3891. [DOI] [PubMed] [Google Scholar]
- 28.Adams AB, Williams MA, Jones TR, Shirasugi N, Durham MM, Kaech SM, Wherry EJ, Onami T, Lanier JG, Kokko KE, Pearson TC, Ahmed R, Larsen CP. Heterologous immunity provides a potent barrier to transplantation tolerance. Journal of Clinical Investigation. 2003;111:1887–1895. doi: 10.1172/JCI17477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Heeger PS, Greenspan NS, Kuhlenschmidt S, Dejelo C, Hricik DE, Schulak JA, Tary-Lehmann M. Pretransplant frequency of donor-specific, IFN-gamma-producing lymphocytes is a manifestation of immunologic memory and correlates with the risk of posttransplant rejection episodes. Journal of Immunology. 1999;163:2267–2275. [PubMed] [Google Scholar]
- 30.Nadazdin O, Boskovic S, Murakami T, Tocco G, Smith RN, Colvin RB, Sachs DH, Allan J, Madsen JC, Kawai T, Cosimi AB, Benichou G. Host alloreactive memory T cells influence tolerance to kidney allografts in nonhuman primates. Sci Transl Med. 2011;3:86ra51. doi: 10.1126/scitranslmed.3002093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hong J, Yeom HJ, Lee E, Han KH, Koo TY, Cho B, Ro H, Oh KH, Ahn C, Yang J. Islet allograft rejection in sensitized mice is refractory to control by combination therapy of immune-modulating agents. Transpl Immunol. 2013 doi: 10.1016/j.trim.2013.01.005. [DOI] [PubMed] [Google Scholar]
- 32.Firpo EJ, Kong RK, Zhou Q, Rudensky AY, Roberts JM, Franza BR. Antigen-specific dose-dependent system for the study of an inheritable and reversible phenotype in mouse CD4+ T cells. Immunology. 2002;107:480–488. doi: 10.1046/j.1365-2567.2002.01540.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Billingham RE, P. M. The technique of free skin grafting in mammals. Journal of Experimental Biology. 1951;28:385–402. [Google Scholar]
- 34.Karim M, Feng G, Wood KJ, Bushell AR. CD25+CD4+ regulatory T cells generated by exposure to a model protein antigen prevent allograft rejection: antigen-specific reactivation in vivo is critical for bystander regulation. Blood. 2005;105:4871–4877. doi: 10.1182/blood-2004-10-3888. [DOI] [PubMed] [Google Scholar]
- 35.Kingsley CI, Karim M, Bushell AR, Wood KJ. CD25+CD4+ regulatory T cells prevent graft rejection: CTLA-4- and IL-10-dependent immunoregulation of alloresponses. Journal of immunology. 2002;168:1080–1086. doi: 10.4049/jimmunol.168.3.1080. [DOI] [PubMed] [Google Scholar]
- 36.Jones ND, Carvalho-Gaspar M, Luo S, Brook MO, Martin L, Wood KJ. Effector and memory CD8+ T cells can be generated in response to alloantigen independently of CD4+ T cell help. J Immunol. 2006;176:2316–2323. doi: 10.4049/jimmunol.176.4.2316. [DOI] [PubMed] [Google Scholar]
- 37.Song J, Salek-Ardakani S, Rogers PR, Cheng M, Van Parijs L, Croft M. The costimulation-regulated duration of PKB activation controls T cell longevity. Nature Immunology. 2004;5:150–158. doi: 10.1038/ni1030. [DOI] [PubMed] [Google Scholar]
- 38.Karim M, Kingsley CI, Bushell AR, Sawitzki BS, Wood KJ. Alloantigen-induced CD25+CD4+ regulatory T cells can develop in vivo from CD25-CD4+ precursors in a thymus-independent process. Journal of immunology. 2004;172:923–928. doi: 10.4049/jimmunol.172.2.923. [DOI] [PubMed] [Google Scholar]
- 39.Salomon B, Lenschow DJ, Rhee L, Ashourian N, Singh B, Sharpe A, Bluestone JA. B7/CD28 costimulation is essential for the homeostasis of the CD4+CD25+ immunoregulatory T cells that control autoimmune diabetes. Immunity. 2000;12:431–440. doi: 10.1016/s1074-7613(00)80195-8. [DOI] [PubMed] [Google Scholar]
- 40.Shimizu J, Yamazaki S, Takahashi T, Ishida Y, Sakaguchi S. Stimulation of CD25(+)CD4(+) regulatory T cells through GITR breaks immunological self-tolerance. Nature immunology. 2002;3:135–142. doi: 10.1038/ni759. [DOI] [PubMed] [Google Scholar]
- 41.McHugh RS, Whitters MJ, Piccirillo CA, Young DA, Shevach EM, Collins M, Byrne MC. CD4(+)CD25(+) immunoregulatory T cells: gene expression analysis reveals a functional role for the glucocorticoid-induced TNF receptor. Immunity. 2002;16:311–323. doi: 10.1016/s1074-7613(02)00280-7. [DOI] [PubMed] [Google Scholar]
- 42.You S, Poulton L, Cobbold S, Liu CP, Rosenzweig M, Ringler D, Lee WH, Segovia B, Bach JF, Waldmann H, Chatenoud L. Key role of the GITR/GITRLigand pathway in the development of murine autoimmune diabetes: a potential therapeutic target. PLoS One. 2009;4:e7848. doi: 10.1371/journal.pone.0007848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Martin-Orozco N, Chen Z, Poirot L, Hyatt E, Chen A, Kanagawa O, Sharpe A, Mathis D, Benoist C. Paradoxical dampening of anti-islet self-reactivity but promotion of diabetes by OX40 ligand. J Immunol. 2003;171:6954–6960. doi: 10.4049/jimmunol.171.12.6954. [DOI] [PubMed] [Google Scholar]
- 44.Nohara C, Akiba H, Nakajima A, Inoue A, Koh CS, Ohshima H, Yagita H, Mizuno Y, Okumura K. Amelioration of experimental autoimmune encephalomyelitis with anti-OX40 ligand monoclonal antibody: a critical role for OX40 ligand in migration, but not development, of pathogenic T cells. J Immunol. 2001;166:2108–2115. doi: 10.4049/jimmunol.166.3.2108. [DOI] [PubMed] [Google Scholar]
- 45.Lee SW, Park Y, Song A, Cheroutre H, Kwon BS, Croft M. Functional dichotomy between OX40 and 4-1BB in modulating effector CD8 T cell responses. Journal of immunology. 2006;177:4464–4472. doi: 10.4049/jimmunol.177.7.4464. [DOI] [PubMed] [Google Scholar]
- 46.Gramaglia I, Jember A, Pippig SD, Weinberg AD, Killeen N, Croft M. The OX40 costimulatory receptor determines the development of CD4 memory by regulating primary clonal expansion. Journal of immunology. 2000;165:3043–3050. doi: 10.4049/jimmunol.165.6.3043. [DOI] [PubMed] [Google Scholar]
- 47.Song A, Tang X, Harms KM, Croft M. OX40 and Bcl-xL promote the persistence of CD8 T cells to recall tumor-associated antigen. Journal of Immunology. 2005;175:3534–3541. doi: 10.4049/jimmunol.175.6.3534. [DOI] [PubMed] [Google Scholar]
- 48.Salek-Ardakani S, Song J, Halteman BS, Jember AG, Akiba H, Yagita H, Croft M. OX40 (CD134) controls memory T helper 2 cells that drive lung inflammation. The Journal of experimental medicine. 2003;198:315–324. doi: 10.1084/jem.20021937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Song J, So T, Croft M. Activation of NF-kappaB1 by OX40 contributes to antigen-driven T cell expansion and survival. J Immunol. 2008;180:7240–7248. doi: 10.4049/jimmunol.180.11.7240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Maxwell JR, Weinberg A, Prell RA, Vella AT. Danger and OX40 receptor signaling synergize to enhance memory T cell survival by inhibiting peripheral deletion. J Immunol. 2000;164:107–112. doi: 10.4049/jimmunol.164.1.107. [DOI] [PubMed] [Google Scholar]
- 51.Ruby CE, Redmond WL, Haley D, Weinberg AD. Anti-OX40 stimulation in vivo enhances CD8+ memory T cell survival and significantly increases recall responses. European journal of immunology. 2007;37:157–166. doi: 10.1002/eji.200636428. [DOI] [PubMed] [Google Scholar]
- 52.Boise LH, Minn AJ, Noel PJ, June CH, Accavitti MA, Lindsten T, Thompson CB. CD28 costimulation can promote T cell survival by enhancing the expression of Bcl-XL. Immunity. 1995;3:87–98. doi: 10.1016/1074-7613(95)90161-2. [DOI] [PubMed] [Google Scholar]
- 53.Lee HW, Park SJ, Choi BK, Kim HH, Nam KO, Kwon BS. 4-1BB promotes the survival of CD8+ T lymphocytes by increasing expression of Bcl-xL and Bfl-1. Journal of immunology. 2002;169:4882–4888. doi: 10.4049/jimmunol.169.9.4882. [DOI] [PubMed] [Google Scholar]
- 54.Cannons JL, Lau P, Ghumman B, DeBenedette MA, Yagita H, Okumura K, Watts TH. 4-1BB ligand induces cell division, sustains survival, and enhances effector function of CD4 and CD8 T cells with similar efficacy. Journal of immunology. 2001;167:1313–1324. doi: 10.4049/jimmunol.167.3.1313. [DOI] [PubMed] [Google Scholar]
- 55.van Maurik A, Wood KJ, Jones ND. Impact of both donor and recipient strains on cardiac allograft survival after blockade of the CD40-CD154 costimulatory pathway. Transplantation. 2002;74:740–743. doi: 10.1097/00007890-200209150-00026. [DOI] [PubMed] [Google Scholar]
- 56.Larsen CP, Elwood ET, Alexander DZ, Ritchie SC, Hendrix R, Tucker-Burden C, Cho HR, Aruffo A, Hollenbaugh D, Linsley PS, Winn KJ, Pearson TC. Long-term acceptance of skin and cardiac allografts after blocking CD40 and CD28 pathways. Nature. 1996;381:434–438. doi: 10.1038/381434a0. [DOI] [PubMed] [Google Scholar]
- 57.Turka LA, Linsley PS, Lin H, Brady W, Leiden JM, Wei RQ, Gibson ML, Zheng XG, Myrdal S, Gordon D, et al. T-cell activation by the CD28 ligand B7 is required for cardiac allograft rejection in vivo. Proceedings of the National Academy of Sciences of the United States of America. 1992;89:11102–11105. doi: 10.1073/pnas.89.22.11102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Valujskikh A, Pantenburg B, Heeger PS. Primed allospecific T cells prevent the effects of costimulatory blockade on prolonged cardiac allograft survival in mice. American Journal of Transplantation. 2002;2:501–509. doi: 10.1034/j.1600-6143.2002.20603.x. [DOI] [PubMed] [Google Scholar]
- 59.Mukherjee S, Maiti PK, Nandi D. Role of CD80, CD86, and CTLA4 on mouse CD4(+) T lymphocytes in enhancing cell-cycle progression and survival after activation with PMA and ionomycin. Journal of leukocyte biology. 2002;72:921–931. [PubMed] [Google Scholar]
- 60.Tsubata T, Wu J, Honjo T. B-cell apoptosis induced by antigen receptor crosslinking is blocked by a T-cell signal through CD40. Nature. 1993;364:645–648. doi: 10.1038/364645a0. [DOI] [PubMed] [Google Scholar]
- 61.Akifusa S, Ohguchi M, Koseki T, Nara K, Semba I, Yamato K, Okahashi N, Merino R, Nunez G, Hanada N, Takehara T, Nishihara T. Increase in Bcl-2 level promoted by CD40 ligation correlates with inhibition of B cell apoptosis induced by vacuolar type H(+)-ATPase inhibitor. Exp Cell Res. 1998;238:82–89. doi: 10.1006/excr.1997.3848. [DOI] [PubMed] [Google Scholar]
- 62.Banuelos SJ, Markees TG, Phillips NE, Appel MC, Cuthbert A, Leif J, Mordes JP, Shultz LD, Rossini AA, Greiner DL. Regulation of skin and islet allograft survival in mice treated with costimulation blockade is mediated by different CD4+ cell subsets and different mechanisms. Transplantation. 2004;78:660–667. doi: 10.1097/01.tp.0000130449.05412.96. [DOI] [PubMed] [Google Scholar]
- 63.Vu MD, Xiao X, Gao W, Degauque N, Chen M, Kroemer A, Killeen N, Ishii N, Chang Li X. OX40 costimulation turns off Foxp3+ Tregs. Blood. 2007;110:2501–2510. doi: 10.1182/blood-2007-01-070748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bansal-Pakala P, Halteman BS, Cheng MH, Croft M. Costimulation of CD8 T cell responses by OX40. J Immunol. 2004;172:4821–4825. doi: 10.4049/jimmunol.172.8.4821. [DOI] [PubMed] [Google Scholar]
- 65.Burocchi A, Pittoni P, Gorzanelli A, Colombo MP, Piconese S. Intratumor OX40 stimulation inhibits IRF1 expression and IL-10 production by Treg cells while enhancing CD40L expression by effector memory T cells. European journal of immunology. 2011;41:3615–3626. doi: 10.1002/eji.201141700. [DOI] [PubMed] [Google Scholar]
- 66.Apostolou I, von Boehmer H. In vivo instruction of suppressor commitment in naive T cells. The Journal of experimental medicine. 2004;199:1401–1408. doi: 10.1084/jem.20040249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cobbold SP, Castejon R, Adams E, Zelenika D, Graca L, Humm S, Waldmann H. Induction of foxP3+ regulatory T cells in the periphery of T cell receptor transgenic mice tolerized to transplants. Journal of immunology. 2004;172:6003–6010. doi: 10.4049/jimmunol.172.10.6003. [DOI] [PubMed] [Google Scholar]
- 68.Francis RS, Feng G, Tha-In T, Lyons IS, Wood KJ, Bushell A. Induction of transplantation tolerance converts potential effector T cells into graft-protective regulatory T cells. European journal of immunology. 2011;41:726–738. doi: 10.1002/eji.201040509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Oliveira VG, Caridade M, Paiva RS, Demengeot J, Graca L. Sub-optimal CD4+ T-cell activation triggers autonomous TGF-beta-dependent conversion to Foxp3+ regulatory T cells. European journal of immunology. 2011;41:1249–1255. doi: 10.1002/eji.201040896. [DOI] [PubMed] [Google Scholar]
- 70.Feuerer M, Hill JA, Kretschmer K, von Boehmer H, Mathis D, Benoist C. Genomic definition of multiple ex vivo regulatory T cell subphenotypes. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:5919–5924. doi: 10.1073/pnas.1002006107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Piconese S, Pittoni P, Burocchi A, Gorzanelli A, Care A, Tripodo C, Colombo MP. A non-redundant role for OX40 in the competitive fitness of Treg in response to IL-2. European journal of immunology. 2010;40:2902–2913. doi: 10.1002/eji.201040505. [DOI] [PubMed] [Google Scholar]
- 72.Xiao X, Kroemer A, Gao W, Ishii N, Demirci G, Li XC. OX40/OX40L costimulation affects induction of Foxp3+ regulatory T cells in part by expanding memory T cells in vivo. Journal of immunology. 2008;181:3193–3201. doi: 10.4049/jimmunol.181.5.3193. [DOI] [PubMed] [Google Scholar]
- 73.Yoshioka T, Nakajima A, Akiba H, Ishiwata T, Asano G, Yoshino S, Yagita H, Okumura K. Contribution of OX40/OX40 ligand interaction to the pathogenesis of rheumatoid arthritis. Eur J Immunol. 2000;30:2815–2823. doi: 10.1002/1521-4141(200010)30:10<2815::AID-IMMU2815>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 74.Humphreys IR, Walzl G, Edwards L, Rae A, Hill S, Hussell T. A critical role for OX40 in T cell-mediated immunopathology during lung viral infection. J Exp Med. 2003;198:1237–1242. doi: 10.1084/jem.20030351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Akiba H, Miyahira Y, Atsuta M, Takeda K, Nohara C, Futagawa T, Matsuda H, Aoki T, Yagita H, Okumura K. Critical contribution of OX40 ligand to T helper cell type 2 differentiation in experimental leishmaniasis. Journal of Experimental Medicine. 2000;191:375–380. doi: 10.1084/jem.191.2.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Xiao X, Balasubramanian S, Liu W, Chu X, Wang H, Taparowsky EJ, Fu YX, Choi Y, Walsh MC, Li XC. OX40 signaling favors the induction of T(H)9 cells and airway inflammation. Nature immunology. 2012 doi: 10.1038/ni.2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.