

Abstract
Background
Many methods have been developed to identify disease genes and further module biomarkers of complex diseases based on gene expression data. It is generally difficult to distinguish whether the variations in gene expression are causative or merely the effect of a disease. The limitation of relying on gene expression data alone highlights the need to develop new approaches that can explore various data to reflect the casual relationship between network modules and disease traits.
Methods
In this work, we developed a novel network-based approach to identify putative causal module biomarkers of complex diseases by integrating heterogeneous information, for example, epigenomic data, gene expression data, and protein–protein interaction network. We first formulated the identification of modules as a mathematical programming problem, which can be solved efficiently and effectively in an accurate manner. Then, we applied our approach to colorectal cancer (CRC) and identified several network modules that can serve as potential module biomarkers for characterizing CRC. Further validations using three additional gene expression datasets verified their candidate biomarker properties and the effectiveness of the method. Functional enrichment analysis also revealed that the identified modules are strongly related to hallmarks of cancer, and the enriched functions, such as inflammatory response, receptor and signaling pathways, are specific to CRC.
Results
Through constructing a transcription factor (TF)-module network, we found that aberrant DNA methylation of genes encoding TF considerably contributes to the activity change of some genes, which may function as causal genes of CRC, and that can also be exploited to develop efficient therapies or effective drugs.
Conclusion
Our method can potentially be extended to the study of other complex diseases and the multiclassification problem.
Introduction
Complex diseases generally result from the intricate interactions among genetic, environmental and lifestyle factors at the macroscopic level.1 At the microscopic level complex diseases are typically caused by a combination of molecular perturbations and their interplay.2 During the past few decades, considerable efforts have been devoted to dissecting the individual gene biomarkers of complex diseases. Some biomarkers of human diseases have been successfully identified through genome-wide analysis of gene expression profiles.3 4 However, it is well accepted that genes or proteins within a cell do not function alone but interact with each other to form networks or pathways so as to carry out biological functions.5–7 Therefore, many methods have recently been developed systematically to identify network biomarkers or even dynamic network biomarkers based on gene expression data.8–16 These module-based methods have made much progress in identifying biomarkers for several cancers and other diseases.
Almost all existing methods rely on an underlying hypothesis that changes in gene expression may result in different phenotypes. However, it is often not possible to distinguish whether gene expression variations are causative or merely an effect of complex diseases.17 Moreover, recent studies have postulated that driver mutations coincide with a ‘genomic footprint’ in the form of a gene expression signature.18 19 The limitation of relying on gene expression data alone thus highlights the need to develop new approaches that can integrate various data to reflect the casual relationship between network modules and disease traits. In other words, as more and more information, such as known disease genes, copy number aberrations (CNA), and epigenomic data (eg, DNA methylation data), is available for a variety of diseases, it is increasingly necessary to combine them with gene expression data to identity causal modules of complex diseases. Some initial work has been performed to study this problem, for example, Kim et al20 introduced an approach simultaneously to identify causal genes and dysregulated pathways by combining CNA and gene expression data. Besides, the integration of multiple data sources has led to the discovery of biomarkers that are biologically validated.21 22 Much further effort is necessary to facilitate our understanding of complex diseases.
In this paper, by integrating multiple heterogeneous data, ie, known cancer genes, DNA methylation data, gene expression data and protein–protein interaction network, we developed a novel algorithm to identify causal network modules of a complex disease in an accurate manner. First, we define candidate causal genes as either known cancer genes or genes with differential methylation status based on DNA methylation data. Then, the activities of genes within a module are assumed to be altered by the candidate causal genes found in the same module, and the activity of a module is further defined based on the expression data of the candidate causal genes and their direct neighbors. Finally, we formulate the identification problem of causal modules underlying complex disease as a new mathematical programming model, which can be solved efficiently and effectively in an accurate manner. Using this method, we analyzed a colorectal cancer (CRC) dataset23 that includes paired gene expression data, as well as paired DNA methylation data. We identified several network modules and found that they can serve as effective module biomarkers for characterizing CRC patients. In addition, the validation results on three independent CRC gene expression datasets further confirmed the effectiveness of our method. Functional enrichment analysis also revealed that these identified modules are strongly related to hallmarks of cancer and inflammation, whose dysfunction may causatively result in CRC. Furthermore, by constructing the transcription factor (TF)-module network, we show that aberrant DNA methylation of genes encoding TF contributes to the activity variation of some genes, which may function as causal genes of CRC. Clearly, these findings not only provide clues to explain what causally results in the dysfunction of biological systems, but also help develop efficient therapies or effective drugs.
Materials and methods
Figure 1 illustrates the schematic flowchart of our method. The details of the procedure are described in the following subsections and the Results section.
Figure 1.

Schematic flowchart of our method. First, DNA methylation data, and known cancer genes (KCG) are exploited to define candidate causal genes (candidate causal genes), which are represented by nodes with black dashed border in the modules. Then, weighted protein–protein interaction (PPI) network, obtained by combining gene expression data and protein–protein interaction network, is used to cluster network modules. Furthermore, through ‘indexing’ (map the candidate causal genes to the proteins in the modules) and ‘assignment’ (map the normalized gene expression value of genes to the corresponding proteins), activity matrix of modules is obtained by defining the activity of modules Mij. Through introducing indicative variable xi and designing a classifier, we formulate the identification of causal modules as an integer linear programming problem. Finally, by solving it, we identify module biomarkers, which characterize complex diseases. This figure is only reproduced in colour in the online version.
Resources and datasets
We obtained DNA methylation data and gene expression data of CRC from NCBI GEO GSE25062 and GSE25070,23 respectively. We extracted 29 paired DNA methylation data of CRC and adjacent non-tumor tissue samples from GSE25062, and 26 paired gene expression data of CRC and adjacent non-tumor tissues from GSE25070. The preprocessing of probe level data was the same as that used in the original reference.23 If there are multiple probes corresponding to the same gene, we adopted the averaged intensity of these probes to represent the expression value of the gene.
In addition, we downloaded four independent datasets for validation, ie, gene expression data from three CRC cohorts GSE15960,24 GSE24514,25 and combined GSE867126 and GSE9348,27 and DNA methylation data from GSE17648. The strategy of preprocessing of these datasets is the same as described before.
Construction of a comprehensive human protein–protein interaction network
We applied a voting method to construct an ensemble protein–protein interaction network by integrating five curated human protein–protein interaction databases, ie, HPRD,28 BioGrid,29 IntAct,30 MINT31 and Reactome.32 Only interactions that are found in at least three of these databases were selected. The comprehensive protein–protein interaction network contains 7001 nodes with 19 188 edges. For a protein–protein interaction network G=(V,E), where V and E represent nodes and edges of the network, a weight w∈W is attached to an edge e∈E to construct the weighted protein–protein interaction network G=(V,E,W), and is calculated as follows:
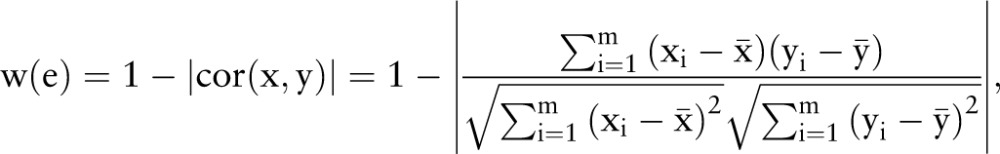 |
where x=(x1, …, xm) and y=(y1, …, ym) are two expression profiles of the two nodes of the edge e, 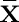 and
and 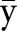 are the mean values of x and y, respectively.
are the mean values of x and y, respectively.
Determination of differential information
Because the tumor and non-tumor samples are paired in DNA methylation data or gene expression data, we employed the minimum multi-set cover strategy used in Kim et al20 to determine the differential information. In particular, first a gene is said to be differentially methylated between a tumor sample and its paired non-tumor sample if the difference of their β value (the measure of the level of DNA methylation) is more than 0.2.23 33 Then, if a gene is differentially methylated in more than half of the pair of tumor and non-tumor samples, this gene is said to be differentially methylated between the tumor and non-tumor samples.
Significance estimation of the related genes in the identified modules
To assess whether the genes in the identified modules are significantly enriched, we use the following formula to calculate the probability of a random overlap with a hypergeometric test:
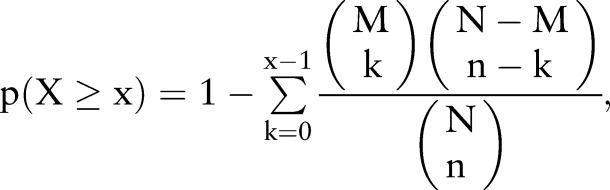 |
where N is the total number of genes in the reference set, M and n are the number of genes in two sets, and k is the number of the overlapped genes of these two sets.
Identification of causal modules by a mathematical programming model
First, we exploit DNA methylation and known cancer genes to define candidate causal genes. Because aberrant DNA methylation at CpG islands is considered to contribute to cancer initiation and progression,34 we define candidate causal genes as those genes that are differentially methylated between the tumor samples and non-tumor samples using DNA methylation data, or known cancer genes, which are collected from the genes annotated with ‘cancer’ in the DNA methylation chips, cancer gene census (CGC) database (2011-11-15 version),35 and tumor associated gene (TAG) database (2011–2011) (http://www.binfo.ncku.edu.tw/TAG/). Second, we decompose the protein–protein interaction network into modules by the Markov clustering algorithm36 and only consider those k modules that contain candidate causal genes. Third, normalized gene expression data are employed to define the activity of module Mi in the case of sample Sj as:
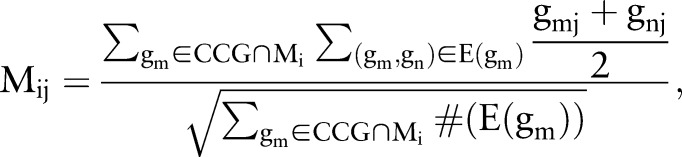 |
where 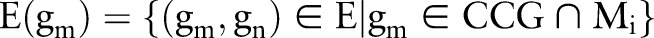 represents the set of those edges that connect to the candidate causal gene gm in the module Mi, #(E(gm)) means the number of edges in the set E(gm), and gmj corresponds to the normalized gene expression value of gene gm in sample Sj. In this way, we obtain the activity matrix with element Mij representing the activity of module Mi in the case of sample Sj. The indicative function is defined to indicate whether a module is selected or not as follows:
represents the set of those edges that connect to the candidate causal gene gm in the module Mi, #(E(gm)) means the number of edges in the set E(gm), and gmj corresponds to the normalized gene expression value of gene gm in sample Sj. In this way, we obtain the activity matrix with element Mij representing the activity of module Mi in the case of sample Sj. The indicative function is defined to indicate whether a module is selected or not as follows:
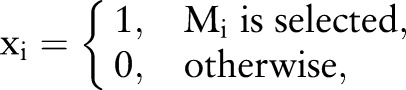 |
Then, we design a classifier to select causal modules as follows:
 |
where S, Scontrol, Scase,  , and
, and 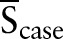 represent the sample, the non-tumor samples set, the tumor samples set, the center of the non-tumor samples set, and the center of the tumor samples set, respectively. Through simple calculation (see supplementary text S1, available online only), we can further express the conditions of the classifier as:
represent the sample, the non-tumor samples set, the tumor samples set, the center of the non-tumor samples set, and the center of the tumor samples set, respectively. Through simple calculation (see supplementary text S1, available online only), we can further express the conditions of the classifier as:
where C is a matrix function of Mij with each element Cij representing the jth module's contribution to the ith condition. Note that the above inequality is linear for (x1,x2, …, xk). The terms on the left side represent the classification ability of modules, ie, the more negative they are, the more clearly the modules are able to distinguish case and control samples.
We aim not only to classify the tumor and non-tumor samples based on the designed classifier, but also to identify the minimum number of modules in this classification process. Therefore, by combining the two objectives, we formulate the module identification problem as the following binary integer linear programming:
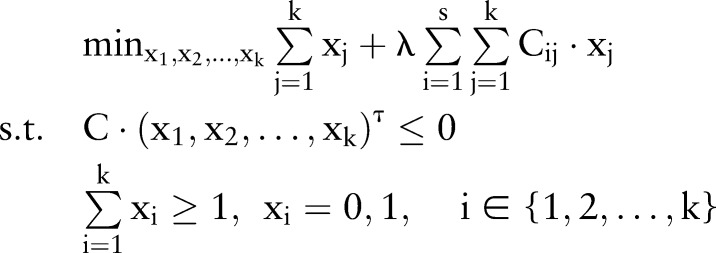 |
where s is the number of the samples. The first term in the objective function implies that we intend to minimize the number of modules, while the second term is used to characterize the classification ability of these modules, ie, we intend to maximize the classification ability of these modules (or minimize C (x1,x2, …, xk)τ). λ is a positive penalty parameter to control the trade-off between the number of modules and the classification ability of these modules.
Algorithm for solving binary integer linear programming problem
Clearly, the formulated problem is NP-hard. We turn to relax the constraints from binary variables xi∈{0,1} to continuous variables xi∈[0,1]. With such relaxations, we can adopt linear programming algorithm to solve the problem in an efficient manner. The experimental results show that such relaxation is both efficient and effective, ie, we almost always obtain integral solutions although it is not theoretically ensured. Here, we give a similar rule to determine how to choose λ as described in Zhao et al.37 Given λ, we define the classification ability of the identified modules from the constraints C (x1,x2, …, xk)τ≤0 as follows:
We have known that Cij indicates the jth module's contribution to the ith condition, so C (x1,x2, …, xk) represents all the conditions of the classifier resulting from all the modules. The smaller C (x1,x2, …, xk) are, the more clearly the modules are able to distinguish cancer and normal samples. Therefore, λ is chosen when CP attains the minimum value, and the resulting modules are regarded as the putative causal modules accordingly. In particular, we test λ by changing from 0 to 1 with the interval of 0.01 and choose λ corresponding to the minimum CP.
Results
Overview of causal module prediction
The flowchart of identifying causal modules is illustrated in figure 1. First, we collected a total of 936 known cancer genes from DNA methylation chips, CGC and TAG databases (see supplementary materials and methods, available online only). Among them, 830 known cancer genes were included in the gene expression data that we analyzed. By integrating these known cancer genes as well as 1392 differentially methylated genes identified in the DNA methylation data, we obtained 2145 candidate causal genes (77 genes belong to both known cancer genes and differentially methylated genes) (see figure 2A). Second, by clustering the protein–protein interaction network, we obtained 556 modules containing more than three genes, 343 of which contain at least one candidate causal gene. Then, following the flowchart in figure 1, we finally identified 17 causal modules by adjusting λ to be 0.13 (see supplementary figure S1, available online only), which minimizes the objective function in solving the integer linear programming problem (see online supplementary materials and methods, available online only). Details of the 17 causal modules are shown in supplementary figure S2 and table S1 (available online only).
Figure 2.
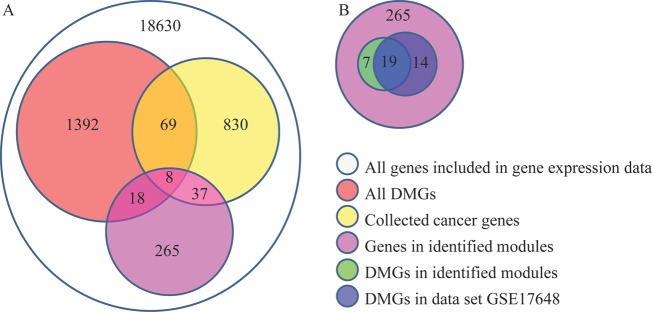
Overlaps of the predicted genes and other kinds of genes. (A) Overlaps between the predicted genes and differential methylated genes (DMG), collected cancer genes, and colorectal cancer genes, respectively. (B) overlap of differential methylated genes in this dataset and dataset GSE17648 in the identified modules. This figure is only reproduced in colour in the online version.
Causal modules act as putative biomarkers
To evaluate the quality of the identified causal modules in terms of distinguishing the tumor samples from the non-tumor samples, we exploited the module activity matrix of 17 modules (modules vs samples) to display the heat map in figure 3 by using hierarchical clustering. From figure 3, we found that these modules achieved high classification performance (100% accuracy), indicating that they may function as biomarkers or module biomarkers.
Figure 3.
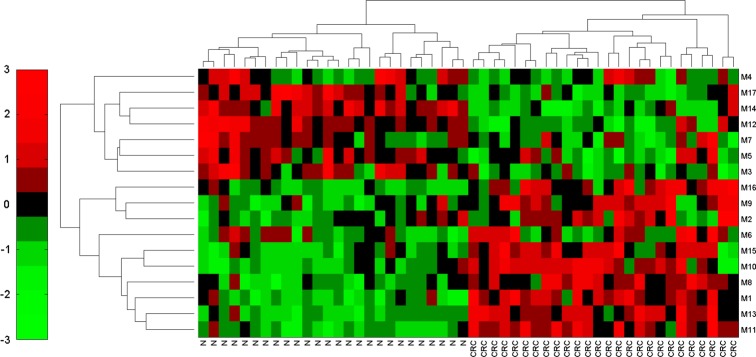
Dendrogram and heat map based on the identified causal modules. The row labels denote the module IDs, and column label ‘N’ represents normal samples, while ‘CRC’ stands for colorectal cancer samples. Colors represent the activity of the modules. Red indicates high activity, while green means low activity. This figure is only reproduced in colour in the online version.
We then examined the candidate causal genes (including both known cancer genes and differentially methylated genes) in the 17 modules and only 63 candidate causal genes (45 are known cancer genes and 26 are differentially methylated genes, figure 2A) were found (table 1). A hypergeometric test was used to evaluate the enrichment of these genes, in which we took all the genes included in gene expression data as the reference set. The statistical significance p values of known cancer genes, differentially methylated genes and candidate causal genes are 7.88×10−15 and 0.09, 1.29×10−8, respectively, from which we found that differentially methylated genes are not so significant. One reason may be that there are many false positives in differentially methylated genes.
Table 1.
List of known cancer genes, differentially methylated genes and CRC confirmed genes by literature search in the identified modules
| Known cancer genes | BIRC3 CBLC CCL2 CCNA2 CDK2 CLTCL1 CUL4A CYLD DDB2 DDX5 DHX16 EGFR ERBB2 ERCC2 ERCC3 ESR1 FANCA FANCC FANCE FANCF FANCG FGF4 FGF5 FGF6 FGF8 FGFR1 FGFR2 FGFR3 FZD7 GNA11 IRF4 LYN MALT1 MUC1 NCOA3 NRCAM PDGFRA PDGFRB PTEN SERPINI2 SETDB1 SKP2 SMG7 TFF1 THRB |
| Differentially methylated genes | CCL11 CCL2 CCL8 CELSR3 CNTN1 CNTN2 CNTNAP2 COL4A1 COL7A1 EPB41L3 ESR1 FCAR FGF4 FGF5 FGF8 GPX7 HNF4A IRF4 MEGF10 NR1H4 PAK7 PCDH17 PCDH8 PDGFRB POU4F2 TNFRSF1B |
| Confirmed CRC genes by literature search | EGFR38 ESR139 ERCC240 ERCC340 FGF441 CARD842 CCL243 CCNH44 CDK745 ERBB246 ESR247 FANCG48 GALNT1249 IKBKB50 LEPR51 PRDM252 PTEN53 TNF54 PIK3C2A55 |
CRC, colorectal cancer.
We validated the aberrant methylation status using an independent DNA methylation dataset GSE17648. In particular, we used the same strategy (see supplementary materials and methods, available online only) to detect differentially methylated genes in GSE17648, and identified 33 differentially methylated genes out of the genes in the identified modules. Furthermore, we found that a total of 19 differentially methylated genes were overlapped between the 26 differentially methylated genes in our study and 33 differentially methylated genes in the dataset GSE17648 (figure 2B), from which the p value is 7.77×10−15. This result showed that the overlap of differentially methylated genes in two datasets is significant and these tests further confirmed that the modules identified in this study are significant.
Functional enrichment of causal modules is related to hallmarks of cancer and is specific to CRC
We further analyzed the functional enrichment of the identified causal modules through a hypergeometric test by g:Profiler.56 The representative enriched GO terms in each module are presented in supplementary table S2 (available online only). These modules are mainly enriched in immune process, signaling pathways, cell communication, cell proliferation, apoptosis, cell cycle, cell division, DNA repair, etc. These processes highly correlate to the hallmarks of cancer and contribute to the major progression of cancer.57 58
We have showed that the enriched functions of the identified modules are highly connected to the hallmarks of cancer. In addition, we know that inflammatory response, receptors and signaling pathways are also very important in the progression of CRC.54 59 60 Therefore, it is necessary to check whether these functions are enriched in the identified modules. We found that not only individual genes are enriched in these functions, but also those modules (see supplementary data, available online only). Therefore, these enriched functions, ie, inflammatory response, receptor and signaling pathways of the identified modules are specific to CRC.
Prediction of novel causal genes for CRC
As described above, we have identified 17 modules to characterize CRC, and we have confirmed that several genes (see table 1) in these modules are specific to CRC through a literature search, which implies that the identified modules are highly relevant to CRC. Next, we predicted new causal genes for CRC by exploring the information on these modules from the perspective of a network. Through careful analysis of the modules and literature investigation (see supplementary data, available online only), we have predicted some novel causal genes for CRC, such as ESR2, PDGFRA, PDGFRB, FGF family, FGFR family, etc. Some newly confirmed CRC genes in the identified modules are also listed in table 1 (or see supplementary table S1, available online only).
Aberrant DNA methylation of genes encoding TF may also contribute to activity change of causal genes of CRC
Cooperative binding of TF has been shown to play a major role in maintaining the unmethylated status of CpG islands in health and disease.34 Therefore, if the genes encoding TF are aberrantly methylated, a series of processes related to disease may be initiated. From this perspective, we constructed a TF-module network in figure 4 by connecting a TF and a module if some genes of this module are targets of this TF.61 In the construction of the TF-module network, we used the transcriptional regulatory element database62 as a referenced TF-target database, and the widths of the edges in figure 4 are correlated to the number of connections between TF and the genes in the module. By checking the methylation information of these TF, we found that five TF are differentially methylated and are labeled as grey in figure 4. In particular, for example, four TF, ie, AR, FLI1, WT1, and PAX1, regulated EGFR in module M3, while AR also regulated ESR1 in module M4. More interestingly, ESR1 in module M4 is related to M3 as a TF in itself. In addition, AR and HOXA5 cooperatively regulated module M5. All of the five TF and/or their methylation status are highly associated with CRC.63–67 Therefore, we concluded that the aberrant methylation of these TF-coding genes may contribute to the activity change of CRC genes, such as ESR1, EGFR, FGF family, FGFR family, etc.
Figure 4.

The constructed transcription factor (TF)-module network. The nodes in the outside cycle represent TF, while the nodes in the inside cycle indicate modules. The grey TF mean that these TF are differentially methylated. This figure is only reproduced in colour in the online version.
Validation of the causal modules using other independent CRC datasets
We evaluated the effectiveness of the causal modules using gene expression data from another three independent CRC cohorts with 12 samples (six normal and six CRC samples from GSE15960),24 49 samples (15 normal and 34 CRC samples from GSE24514),25 and 146 samples (44 normal and 102 samples combined from GSE867126 and GSE9348).27 We exploited the identified causal modules to test whether they can classify the other three independent datasets. If these modules can successfully distinguish cancer samples from normal samples in these three independent datasets, it indicates that these identified modules can indeed serve as effective module biomarkers for characterizing CRC. The classification results are shown in supplementary figure S3 (available online only), figures 5 and 6, and the classification performances are evaluated by accuracy (Acc) and sensitivity (Se) (see supplementary data, available online only). From figure 5, it has a 89.8% accuracy for the level of sensitivity of 96.8% for the dataset GSE24514. Similarly, we determined that the accuracy is 98.6% for the level of sensitivity of 100% for the dataset combined from GSE8671 and GSE9348 in figure 6. These independent results provided additional evidence that the module-based biomarkers identified in this study could be used for CRC prediction, and further suggested the effectiveness of our method.
Figure 5.
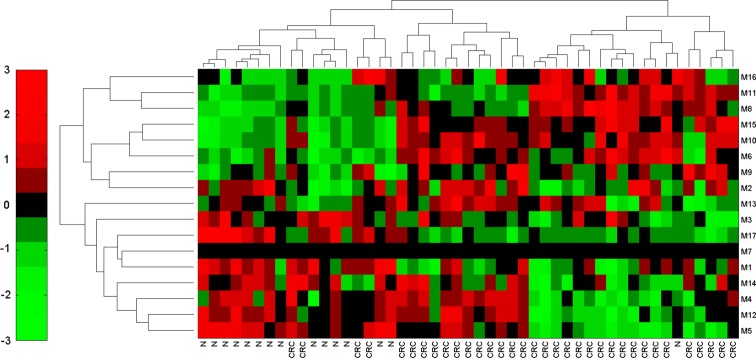
Dendrogram and heat map in the independent test dataset GSE24514 based on the identified causal modules. The row labels denote the module IDs, and column ‘N’ represents normal samples, while ‘CRC’ stands for colorectal cancer samples. The accuracy is 89.8% for the level of sensitivity of 96.8% (TP=30, FP=14, FN=1, TN=14). This figure is only reproduced in colour in the online version.
Figure 6.
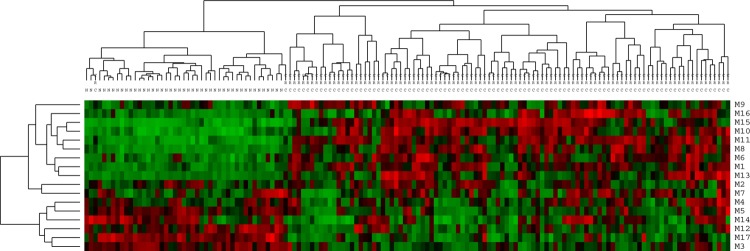
Dendrogram and heat map in the independent test dataset combined from GSE8671 and GSE9348 based on the identified causal modules. The row labels denote the module IDs, and column ‘N’ represents normal samples, while ‘CRC’ stands for colorectal cancer samples. The accuracy is 98.6% for the level of sensitivity of 100% (TP=100, FP=2, FN=0, TN=44). This figure is only reproduced in colour in the online version.
Discussion
Our method is novel in several aspects compared to previous methods. First, in contrast to the conventional methods relying on gene expression data alone, our method is able to reveal realistic causal relations between module biomarkers and phenotypes by integrating various heterogeneous data. One reason is that the gene expression observations alone are generally not sufficient to distinguish causative or responsive relations of a disease, while previous known disease genes, sequence or methylation data may underlie the cause of complex diseases. Second, technically, instead of those heuristic methods, our method formulated the identification of module biomarkers as a mathematical programming problem, which can be solved efficiently and effectively. Third, our method can include various previous information and other types of data easily, which may help improve the results in terms of reliability and accuracy.
In this work, we applied our method to study CRC and identified 17 causal modules. Through the heat map and by analyzing the genes inside the modules, we showed that these modules indeed can characterize CRC as putative biomarkers. In addition, we found that some newly identified cancer genes are not included in the 45 known cancer genes in these 17 causal modules. For example, phosphoinositide-3-kinase class 2α polypeptide (PIK3C2A), which plays an important role in signaling pathways involved in cell proliferation, oncogenic transformation, cell survival, cell migration, and intracellular protein trafficking, is considered as one of the drivers of CRC.55 Another gene, ESR2 (encoding estrogen receptor 2) in module M4, has been reported to be associated with the incidence of CRC among women.47 This result indicates that our method could identify new cancer genes besides known cancer genes. What is more, these modules were further exploited to classify the other three independent datasets, and the high accuracy of the prediction confirmed the effectiveness of our method.
Functional analysis reveals that these modules are mainly enriched in immune process, signaling pathways, inflammation, cell communication, cell proliferation, apoptosis, cell cycle, cell division, DNA repair, etc. These enriched processes, on the one hand, correspond highly to the hallmarks of cancer and contribute to the major progression of CRC. On the other hand, these modules are also enriched in inflammatory response, receptor and signaling pathways, which are specific to CRC. Furthermore, as another more specific character of CRC, colorectal tumors are known to present with a broad range of neoplasms and are predominantly epithelial-derived tumors.68 The identified modules are exactly correlated to this characteristic, which further provided additional evidence to confirm the causal modules of CRC. Therefore, we conclude that these modules are indeed causal modules of CRC.
Besides the known CRC genes in our identified modules, we also predicted some novel causal genes for CRC, such as ESR2, PDGFRA, PDGFRB, FGF family, FGFR family, etc. Some newly confirmed CRC genes in the identified modules are also listed in table 1 through literature search. However, some famous genes, such as TP53 (which is not differentially methylated or expressed),23 were not included in the identified modules. This limitation may be from the incomplete information we used, or the noisy data.
Finally, through constructing the TF-module network, we found that aberrant DNA methylation of genes encoding TF contributes to the activity variation of some genes, which may function as causal genes of CRC, and may be the targets of efficient therapies or effective drugs. In addition, we also confirmed the aberrant methylation status of the five TF in our study (figure 4) using an independent dataset. This validation further confirmed our prediction of the importance of these TF. Although we restricted our study to CRC in this paper, our method can potentially be extended to the study of other complex diseases. In addition, our framework can also be directly applied to multi-classification problems.
Conclusion
The identification of biomarkers can help the diagnosis and efficient treatment of complex diseases. In this paper, we presented a new network-based approach to identify putative causal module biomarkers of complex diseases by integrating various information, for example, epigenomic data, gene expression data and a protein–protein interaction network. The analysis based on such an integration can potentially lead to new insight into complex diseases at the systems level, through the identification of modules underlying complex diseases. In addition, the framework of the proposed method in this paper can also be modified to detect dynamic network biomarkers for the early diagnosis of complex diseases.16 The source code for the identification of modules is available on request from the authors.
Acknowledgments
The authors would like to thank Dr Tao Zeng (Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences) for his helpful discussions and suggestions.
Footnotes
Contributors: ZW wrote the paper, designed and performed the experiments, and analyzed the data. ZPL and ZL contributed materials and analysis tools. YZ conceived the experiments and wrote the paper. LC conceived and designed the experiments, and contributed materials and analysis tools.
Funding: This research was partly supported by the National Natural Science Foundation of China (NSFC) under grant nos 91029301, 61134013, 61072149, 31100949; by the Knowledge Innovation Program of the Chinese Academy of Sciences (CAS) with grant no. KSCX2-EW-R-01; by the Chief Scientist Program of Shanghai Institutes for Biological Sciences (SIBS) of CAS with grant no. 2009CSP002; by the Shanghai NSF under grant no. 11ZR1443100; by the Knowledge Innovation Program of SIBS of CAS with grant no. 2011KIP203; by the Shanghai Pujiang Program. This research was also partly supported by the National Center for Mathematics and Interdisciplinary Sciences of CAS; and by the FIRST program from JSPS initiated by CSTP.
Competing interests: None.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.Hunter DJ. Gene–environment interactions in human diseases. Nat Rev Genet 2005;6:287–98 [DOI] [PubMed] [Google Scholar]
- 2.Schadt EE. Molecular networks as sensors and drivers of common human diseases. Nature 2009;461:218–23 [DOI] [PubMed] [Google Scholar]
- 3.Alizadeh AA, Eisen MB, Davis RE, et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature 2000;403:503–11 [DOI] [PubMed] [Google Scholar]
- 4.Golub TR, Slonim DK, Tamayo P, et al. Molecular classification of cancer: class discovery and class prediction by gene expression monitoring. Science 1999;286:531–7 [DOI] [PubMed] [Google Scholar]
- 5.Barabasi AL, Oltvai ZN. Network biology: understanding the cell's functional organization. Nat Rev Genet 2004;5:101–13 [DOI] [PubMed] [Google Scholar]
- 6.Chen L, Wang RS, Zhang XS. Biomolecular networks: methods and applications in systems biology. Hoboken, New Jersey: John Wiley & Sons Inc, 2009 [Google Scholar]
- 7.Chen L, Wang R, Li C Aihara K. Modeling biomolecular networks in cells: structures and dynamics. London: Springer-Verlag, 2010 [Google Scholar]
- 8.de Lichtenberg U, Jensen LJ, Brunak S, et al. Dynamic complex formation during the yeast cell cycle. Science 2005;307:724–7 [DOI] [PubMed] [Google Scholar]
- 9.Segal E, Shapira M, Regev A, et al. Module networks: identifying regulatory modules and their condition-specific regulators from gene expression data. Nat Genet 2003;34:166–76 [DOI] [PubMed] [Google Scholar]
- 10.Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 2005;102:15545–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Van Leene J, Hollunder J, et al. Targeted interactomics reveals a complex core cell cycle machinery in Arabidopsis thaliana. Mol Syst Biol 2010;6:397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.He D, Liu ZP, Honda M, et al. Coexpression network analysis in chronic hepatitis B and C hepatic lesion reveals distinct patterns of disease progression to hepatocellular carcinoma. J Mol Cell Biol 2012;4:140–52 [DOI] [PubMed] [Google Scholar]
- 13.Liu X, Liu ZP, Zhao XM, et al. Identifying disease genes and module biomarkers by differential interactions. J Am Med Inform Assoc 2012;19:241–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Song W, Wang JG, Yang Y, et al. Rewiring drug-activated p53-regulatory network from suppressing to promoting tumorigenesis. J Mol Cell Biol 2012;4:197–206 [DOI] [PubMed] [Google Scholar]
- 15.Wen Z, Liu ZP, Yan Y, et al. Identifying responsive modules by mathematical programming an application to budding yeast cell cycle. PLoS One 2012;7:e41854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen L, Liu R, Liu ZP, et al. Detecting early-warning signals for sudden deterioration of complex diseases by dynamical network biomarkers. Sci Rep 2012;2:342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Iorns E, Lord CJ, Turner N, et al. Utilizing RNA interference to enhance cancer drug discovery. Nat Rev Drug Discov 2007;6:556–68 [DOI] [PubMed] [Google Scholar]
- 18.Akavia UD, Litvin O, Kim J, et al. An integrated approach to uncover drivers of cancer. Cell 2010;143:1005–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schadt EE, Lamb J, Yang X, et al. An integrative genomics approach to infer causal associations between gene expression and disease. Nat Genet 2005;37:710–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim YA, Wuchty S, Przytycka TM. Identifying causal genes and dysregulated pathways in complex diseases. PLoS Comput Biol 2011;7:e1001095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Aerts S, Lambrechts D, Maity S, et al. Gene prioritization through genomic data fusion. Nat Biotechnol 2006;24:537–44 [DOI] [PubMed] [Google Scholar]
- 22.Lee Y, Yang X, Huang Y, et al. Network modeling identifies molecular functions targeted by miR-204 to suppress head and neck tumor metastasis. PLoS Comput Biol 2010;6:e1000730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hinoue T, Weisenberger DJ, Lange CP, et al. Genome-scale analysis of aberrant DNA methylation in colorectal cancer. Genome Res 2012;22:271–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Galamb O, Spisak S, Sipos F, et al. Reversal of gene expression changes in the colorectal normal-adenoma pathway by NS398 selective COX2 inhibitor. Br J Cancer 2010;102:765–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Alhopuro P, Sammalkorpi H, Niittymaki I, et al. Candidate driver genes in microsatellite-unstable colorectal cancer. Int J Cancer 2012;130:1558–66 [DOI] [PubMed] [Google Scholar]
- 26.Sabates-Bellver J, Van der Flier LG, de Palo M, et al. Transcriptome profile of human colorectal adenomas. Mol Cancer Res 2007;5:1263–75 [DOI] [PubMed] [Google Scholar]
- 27.Hong Y, Downey T, Eu KW, et al. A ‘metastasis-prone’ signature for early-stage mismatch-repair proficient sporadic colorectal cancer patients and its implications for possible therapeutics. Clin Exp Metastasis 2010;27:83–90 [DOI] [PubMed] [Google Scholar]
- 28.Peri S, Navarro JD, Kristiansen TZ, et al. Human protein reference database as a discovery resource for proteomics. Nucleic Acids Res 2004;32(Database issue):D497–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stark C, Breitkreutz BJ, Reguly T, et al. BioGRID: a general repository for interaction datasets. Nucleic Acids Res 2006;34(Database issue):D535–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hermjakob H, Montecchi-Palazzi L, Lewington C, et al. IntAct: an open source molecular interaction database. Nucleic Acids Res 2004;32(Database issue):D452–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ceol A, Chatr Aryamontri A, Licata L, et al. MINT, the molecular interaction database: 2009 update. Nucleic Acids Res 2010;38(Database issue):D532–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Matthews L, Gopinath G, Gillespie M, et al. Reactome knowledgebase of human biological pathways and processes. Nucleic Acids Res 2009;37(Database issue):D619–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bibikova M, Le J, Barnes B, et al. Genome-wide DNA methylation profiling using Infinium((R)) assay. Epigenomics 2009;1:177–200 [DOI] [PubMed] [Google Scholar]
- 34.Gebhard C, Benner C, Ehrich M, et al. General transcription factor binding at CpG islands in normal cells correlates with resistance to de novo DNA methylation in cancer cells. Cancer Res 2010;70:1398–407 [DOI] [PubMed] [Google Scholar]
- 35.Futreal PA, Coin L, Marshall M, et al. A census of human cancer genes. Nat Rev Cancer 2004;4:177–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Enright AJ, Van Dongen S, Ouzounis CA. An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res 2002;30:1575–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhao XM, Wang RS, Chen L, et al. Uncovering signal transduction networks from high-throughput data by integer linear programming. Nucleic Acids Res 2008;36:e48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Poole EM, Curtin K, Hsu L, et al. Genetic variability in EGFR, Src and HER2 and risk of colorectal adenoma and cancer. Int J Mol Epidemiol Genet 2011;2:300–15 [PMC free article] [PubMed] [Google Scholar]
- 39.Saito S, Kato J, Hiraoka S, et al. DNA methylation of colon mucosa in ulcerative colitis patients: correlation with inflammatory status. Inflamm Bowel Dis 2011;17:1955–65 [DOI] [PubMed] [Google Scholar]
- 40.Mort R, Mo L, McEwan C, et al. Lack of involvement of nucleotide excision repair gene polymorphisms in colorectal cancer. Br J Cancer 2003;89:333–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ikeda S, Sasazuki S, Natsukawa S, et al. Screening of 214 single nucleotide polymorphisms in 44 candidate cancer susceptibility genes: a case-control study on gastric and colorectal cancers in the Japanese population. Am J Gastroenterol 2008;103:1476–87 [DOI] [PubMed] [Google Scholar]
- 42.Mockelmann N, von Schonfels W, Buch S, et al. Investigation of innate immunity genes CARD4, CARD8 and CARD15 as germline susceptibility factors for colorectal cancer. BMC Gastroenterol 2009;9:79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Erreni M, Mantovani A, Allavena P. Tumor-associated macrophages (TAM) and inflammation in colorectal cancer. Cancer Microenviron 2011;4:141–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Huang WY, Berndt SI, Kang D, et al. Nucleotide excision repair gene polymorphisms and risk of advanced colorectal adenoma: XPC polymorphisms modify smoking-related risk. Cancer Epidemiol Biomarkers Prev 2006;15:306–11 [DOI] [PubMed] [Google Scholar]
- 45.Kweekel DM, Antonini NF, Nortier JW, et al. Explorative study to identify novel candidate genes related to oxaliplatin efficacy and toxicity using a DNA repair array. Br J Cancer 2009;101:357–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li N, Bu X, Wu P, et al. The “HER2-PI3K/Akt-FASN Axis” regulated malignant phenotype of colorectal cancer cells. Lipids 2012;47:403–11 [DOI] [PubMed] [Google Scholar]
- 47.Honma N, Arai T, Takubo K, et al. Oestrogen receptor-beta CA repeat polymorphism is associated with incidence of colorectal cancer among females. Histopathology 2011;59:216–24 [DOI] [PubMed] [Google Scholar]
- 48.de Angelis PM, Fjell B, Kravik KL, et al. Molecular characterizations of derivatives of HCT116 colorectal cancer cells that are resistant to the chemotherapeutic agent 5-fluorouracil. Int J Oncol 2004;24:1279–88 [PubMed] [Google Scholar]
- 49.Guda K, Moinova H, He J, et al. Inactivating germ-line and somatic mutations in polypeptide N-acetylgalactosaminyltransferase 12 in human colon cancers. Proc Natl Acad Sci U S A 2009;106:12921–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Curtin K, Wolff RK, Herrick JS, et al. Exploring multilocus associations of inflammation genes and colorectal cancer risk using hapConstructor. BMC Med Genet 2010;11:170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pechlivanis S, Bermejo JL, Pardini B, et al. Genetic variation in adipokine genes and risk of colorectal cancer. Eur J Endocrinol 2009;160:933–40 [DOI] [PubMed] [Google Scholar]
- 52.de Vogel S, Wouters KA, Gottschalk RW, et al. Genetic variants of methyl metabolizing enzymes and epigenetic regulators: associations with promoter CpG island hypermethylation in colorectal cancer. Cancer Epidemiol Biomarkers Prev 2009;18:3086–96 [DOI] [PubMed] [Google Scholar]
- 53.Slattery ML, Herrick JS, Lundgreen A, et al. Genetic variation in a metabolic signaling pathway and colon and rectal cancer risk: mTOR, PTEN, STK11, RPKAA1, PRKAG2, TSC1, TSC2, PI3K and Akt1. Carcinogenesis 2010;31:1604–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kraus S, Arber N. Inflammation and colorectal cancer. Curr Opin Pharmacol 2009;9:405–10 [DOI] [PubMed] [Google Scholar]
- 55.Wood LD, Parsons DW, Jones S, et al. The genomic landscapes of human breast and colorectal cancers. Science 2007;318:1108–13 [DOI] [PubMed] [Google Scholar]
- 56.Reimand J, Kull M, Peterson H, et al. g:Profiler–a web-based toolset for functional profiling of gene lists from large-scale experiments. Nucleic Acids Res 2007;35(Web Server issue):W193–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011;144:646–74 [DOI] [PubMed] [Google Scholar]
- 58.Cavallo F, De Giovanni C, Nanni P, et al. 2011: the immune hallmarks of cancer. Cancer Immunol Immunother 2011;60:319–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Grossmann AH, Samowitz WS. Epidermal growth factor receptor pathway mutations and colorectal cancer therapy. Arch Pathol Lab Med 2011;135:1278–82 [DOI] [PubMed] [Google Scholar]
- 60.Rizzo A, Pallone F, Monteleone G, et al. Intestinal inflammation and colorectal cancer: a double-edged sword? World J Gastroenterol 2011;17:3092–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li J, Lenferink AE, Deng Y, et al. Identification of high-quality cancer prognostic markers and metastasis network modules. Nat Commun 2010;1:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jiang C, Xuan Z, Zhao F, et al. TRED: a transcriptional regulatory element database, new entries and other development. Nucleic Acids Res 2007;35(Database issue):D137–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gu S, Papadopoulou N, Nasir O, et al. Activation of membrane androgen receptors in colon cancer inhibits the prosurvival signals Akt/bad in vitro and in vivo and blocks migration via vinculin/actin signaling. Mol Med 2011;17:48–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Xu XL, Yu J, Zhang HY, et al. Methylation profile of the promoter CpG islands of 31 genes that may contribute to colorectal carcinogenesis. World J Gastroenterol 2004;10:3441–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Oster B, Thorsen K, Lamy P, et al. Identification and validation of highly frequent CpG island hypermethylation in colorectal adenomas and carcinomas. Int J Cancer 2011;129:2855–66 [DOI] [PubMed] [Google Scholar]
- 66.Kanai M, Hamada J, Takada M, et al. Aberrant expressions of HOX genes in colorectal and hepatocellular carcinomas. Oncol Rep 2010;23:843–51 [PubMed] [Google Scholar]
- 67.Cassinotti E, Melson J, Liggett T, et al. DNA methylation patterns in blood of patients with colorectal cancer and adenomatous colorectal polyps. Int J Cancer 2012;131:1153–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hu B, Elinav E, Flavell RA. Inflammasome-mediated suppression of inflammation-induced colorectal cancer progression is mediated by direct regulation of epithelial cell proliferation. Cell Cycle 2011;10:1936–9 [DOI] [PubMed] [Google Scholar]