

Abstract
Background
An enduring challenge in personalized medicine lies in selecting the right drug for each individual patient. While testing of drugs on patients in large trials is the only way to assess their clinical efficacy and toxicity, we dramatically lack resources to test the hundreds of drugs currently under development. Therefore the use of preclinical model systems has been intensively investigated as this approach enables response to hundreds of drugs to be tested in multiple cell lines in parallel.
Methods
Two large-scale pharmacogenomic studies recently screened multiple anticancer drugs on over 1000 cell lines. We propose to combine these datasets to build and robustly validate genomic predictors of drug response. We compared five different approaches for building predictors of increasing complexity. We assessed their performance in cross-validation and in two large validation sets, one containing the same cell lines present in the training set and another dataset composed of cell lines that have never been used during the training phase.
Results
Sixteen drugs were found in common between the datasets. We were able to validate multivariate predictors for three out of the 16 tested drugs, namely irinotecan, PD-0325901, and PLX4720. Moreover, we observed that response to 17-AAG, an inhibitor of Hsp90, could be efficiently predicted by the expression level of a single gene, NQO1.
Conclusion
These results suggest that genomic predictors could be robustly validated for specific drugs. If successfully validated in patients’ tumor cells, and subsequently in clinical trials, they could act as companion tests for the corresponding drugs and play an important role in personalized medicine.
Keywords: Cancer Genomics, Pharmacogenomics, Drug Discovery, Biomarkers, Machine Learning, Peronalized Medicine
Introduction
The advent of personalized medicine raises many practical challenges. In order to develop targeted therapies for individuals, one must resort to the lengthy and expensive process of drug development and validation in clinical trials. While clinical trials are the only way truly to assess drug efficacy and toxicity, the scarcity of resources is not conducive to the testing of the hundreds of drugs that are currently under development.1 Substantial efforts have therefore been made to streamline drug development and to optimize selection of the best drug regimen for each individual patient. One possible approach consists of directly measuring the sensitivity of a patient's tumor cells to a drug of interest in two/three-dimensional in-vitro cultures2 and in-vivo models such as mouse xenograft and genetically engineered mouse models.3 This approach has the potential of capturing most of the relevant biological features of a patient's tumor, and therefore providing better models to test drug sensitivity. However, such an approach is costly, time consuming and hardly scalable to screen dozens or hundreds of drugs in parallel.
Another approach proposed by several research groups during the past decade is to build genomic predictors of drug response from large panels of cancer cell lines instead.4–10 Most studies relied on high-throughput screening technologies to investigate the sensitivity of these cancer cell lines to numerous drugs. Once the genomic profiles of these cell lines are measured (single nucleotide polymorphisms (SNP) or gene expression profiles, for instance), one can build statistical models predictive of drug response based on genomic data. Such models could then be used to predict the sensitivity of a patient's tumor based on its genomic profile.11 Assuming that these predictive models yield clinical relevance, they could be used to screen the sensitivity of a given patient's tumor to numerous drugs in parallel quickly at virtually no cost. This explains in main part the enthusiasm of the bioinformatics and biomedical communities in building preclinical models of drug response.
The NCI60 cell line panel and associated drug screen programs (CellMiner12 and developmental therapeutics program)11 13 pioneered the use of cancer cell lines to link drug sensitivity to genomic data.6 12 14 The analysis of NCI60 led to the discovery of mutations in BRAF and EGFR, which are now known to be clinically relevant in predicting response to vemurafenib and other kinase inhibitors.15 16 Despite these advances, a large number of cancer drugs has not yet been linked to specific genomic features that could otherwise have been used as biomarkers for assessing the selective therapeutic effectiveness of such drugs.17 In March 2012, two large-scale studies published in Nature extended this initial dataset by generating pharmacogenomic data for several hundreds of cancer cell lines, allowing a broader representation of the genomic diversity of human cancers.4 5 Analyses of these data are promising in improving our understanding of the mechanisms of action of well-established and new drugs. This may in turn lead to robust companion test development for these drugs. This, however, is not a trivial task, as we are increasingly coming to understand that it is not individual genes but rather biological pathways that drive the development of a particular phenotype (response to an anticancer therapy for instance). Given the complexity of the task and the risk of artifactual discovery, there is an urgent need for combining large pharmacogenomic datasets to build robust multivariate genomic predictors and for validating them on fully independent data.
In this study, we use two large pharmacogenomic datasets and compare different approaches in constructing models predictive of sensitivity to multiple anticancer drugs. To the best of our knowledge, this is the first time that both these datasets are analyzed in a single study, which should provide us with sufficient sample size for both building robust predictors and validating them in large, fully independent datasets. The generation of genomic predictors of drug response in the preclinical setting like the models we validated in our study and their incorporation into cancer clinical trial design could accelerate the emergence of ‘personalized’ therapeutic regimens18 and therefore dramatically improve cancer therapy.
Materials and methods
Cell lines and drug sensitivity
In order to develop robust genomic predictors of response to anticancer drugs, we collected, curated, and annotated published datasets of two recent large-scale preclinical studies, namely the cancer genome project (CGP),5 and cancer cell line encyclopedia (CCLE).4 This large compendium of datasets, which includes 1718 gene expression profiles of 1299 distinct cell lines, was used to build and validate genomic predictors of sensitivity to 16 drugs present both in CGP and CCLE datasets (table 1). In each of these projects, cell line drug sensitivity was measured as the concentration at which the drug inhibited 50% of the cellular growth (IC50);4 we represent drug sensitivity S as S=−log10 (x/1 000 000) where x is the IC50 measured in micromolar (μM) units, as is common practice in the field of pharmacology. It is important to note that in the CCLE study, the maximum concentration for inactive compounds was used instead of the IC50. As IC50 values are formally impossible to estimate for these inactive compounds, these values (56% of the total number of IC50 measurements) were replaced with NA (missing values) to avoid biases in our analyses.
Table 1.
Anticancer drugs analyzed in the CGP
| Compound | Target(s) | Class | Organization |
|---|---|---|---|
| Erlotinib | EGFR | Kinase inhibitor | Genentech |
| Lapatinib | EGFR, HER2 | Kinase inhibitor | GlaxoSmithKline |
| PHA-665752 | c-MET | Kinase inhibitor | Pfizer |
| Crizotinib | c-MET, ALK | Kinase inhibitor | Pfizer |
| TAE684 | ALK | Kinase inhibitor | Novartis |
| Nilotinib | Abl/Bcr-Abl | Kinase inhibitor | Novartis |
| AZD0530 | Src, Abl/Bcr-Abl, EGFR | Kinase inhibitor | AstraZeneca |
| Sorafenib | Flt3, C-KIT, PDGFRbeta, RET, Raf kinase B, Raf kinase C, VEGFR-1, KDR, FLT4 | Kinase inhibitor | Bayer |
| PD-0332991 | CDK4/6 | Kinase inhibitor | Pfizer |
| PLX4720 | RAF | Kinase inhibitor | Plexxikon |
| PD-0325901 | MEK | Kinase inhibitor | Pfizer |
| AZD6244 | MEK | Kinase inhibitor | AstraZeneca |
| Nutlin-3 | MDM2 | Other | Roche |
| 17-AAG | HSP90 | Other | Bristol-Myers Squibb |
| Paclitaxel | β-Tubulin | Cytotoxic | Bristol-Myers Squibb |
| Irinotecan | Topoisomerase I | Cytotoxic | Pfizer |
Among the 131 drugs analyzed in the cancer genome project (CGP), 16 were also analyzed in the cancer cell lines encyclopedia.
Gene expression data
Raw gene expression profiles (Affymetrix CEL format) for 727 (CGP) and 991 (CCLE) cell lines were retrieved respectively from ArrayExpress (E-MTAB-783), and the CCLE website (http://www.broadinstitute.org/ccle/). These gene expression data were normalized with frozen RMA19 using the bioconductor chip description file packages (hgu133plus2 for CCLE and hthgu133a for CGP). Affymetrix probesets were further annotated with biomaRt.20 For each unique Entrez gene ID, we used the R package jetset to select the best probeset21 such that a gene is represented by only one probeset. Subsequent analyses were restricted to the probesets common to the CCLE and CGP datasets, for a total of 12 172 probesets/genes.
Analysis pipeline
As illustrated in figure 1, in order to estimate the feasibility of predicting outcomes on an independent dataset, we first performed a prevalidation analysis,22 which consisted of 10 repetitions of 10-fold cross-validation for each of the models and for each of the drugs in the CGP dataset. We then trained each of the models with the full CGP dataset for each of the drugs and tested them on each of the two following subsets of the CCLE dataset: subset containing only cell lines that are common to both the CGP and CCLE datasets (COMMON), and subset containing only cell lines that are different between them (NEW). This furthers the notion of building robust models that are portable and generalizable across multiple datasets.
Figure 1.

Experimental design of the study. First we performed a prevalidation analysis of the genomic predictors for 16 anticancer drugs using cross-validation in the training set cancer genome project (CGP). We then built genomic predictors using the full training set and evaluate their performance in the cancer cell line encyclopedia (CCLE), a fully independent validation dataset. We split the validation dataset into two parts containing the cell lines that are present in the training set (COMMON) or cell lines that are unique to the CCLE dataset (NEW).
Predictive models
To build genomic predictors of response to anticancer drugs we implemented five linear methods of increasing complexity:
SINGLEGENE: The gene most correlated with the outcome (IC50), as estimated using Spearman correlation, is used to fit a univariate regression model.
RANKENSEMBLE: This method uses ranking based on correlation to select the most relevant genes and then uses an ensemble approach to combine the corresponding univariate regression models. We used the simplest scheme of ensemble combination as it consists of averaging the predictions computed by each univariate model (all models therefore have the same weight in the combination).23
RANKMULTIV: Based on the same ranking as RANKENSEMBLE, most relevant genes are selected and subsequently used to fit a multivariate regression model.
MRMR: uses the minimum-redundancy maximum-relevance (mRMR) technique24 25 to select the most relevant and less redundant genes to include in a multivariate regression model.
ELASTICNET: Elastic net is an efficient, widely used regularized regression technique,26 27 which was used in both CCLE and CGP original publications.4 5
For RANKENSEMBLE, RANKMULTIV, and MRMR, the number of selected genes was fixed to 30 to decrease the computational time, to facilitate the comparison between methods and because this ‘signature’ size has been reported as a good trade-off between relevance and model complexity in published comparative studies of predictive modeling from gene expression data.28 29 For ELASTICNET, we used the same approach as Barretina et al4 by selecting the optimal regularization parameters α ε (0.2, 1.0) (10 values were tested) and λ=eγ, where γ ε (−6, 5) (250 values tested), by optimizing the mean squared error of the model in inner 10-fold cross-validation.
Results
We used two large pharmacogenomic datasets, referred to as CGP5 and CCLE,4 which include 1718 gene expression profiles of 1299 distinct cell lines, to build and validate genomic predictors of sensitivity to 16 drugs (table 1). Drug sensitivity (also referred to as drug response), was measured based on the drug concentration that induced an absolute growth inhibition of 50% (IC50).
There exists a plethora of machine learning methods to construct predictive models from high dimensional data such as gene expression profiles. In this study, we decided to focus on a set of five linear methods of increasing complexity. The first method (SINGLEGENE) is the simplest as it consists of a univariate regression model using as input the gene correlating strongest with the outcome (IC50). Because multiple genes are expected to be required to predict sensitivity to most of the drugs accurately, SINGLEGENE will mostly serve as a benchmark, allowing us to identify the drugs for which multivariate models do not perform significantly better than a simple univariate model. The RANKENSEMBLE and RANKMULTIV methods use a ranking procedure to select the most relevant genes based on their correlation with outcome. For RANKENSEMBLE each selected gene is used to fit regression models, which are further combined using a simple ensemble approach in which models’ predictions are averaged. For RANKMULTIV, all the selected genes are used to fit a multivariate regression model. These models, although multivariate by nature, do not take into account redundancy during feature selection, as ranking is a filtering technique.30 31 Therefore, we implemented the MRMR feature selection, as it selects the set of genes most correlated with the outcome while minimizing redundancy across selected genes;24 25 these genes are subsequently used in a multivariate regression model. Finally, we used ELASTICNET,26 27 an efficient, well-established regularized regression technique, which was used in the original publications of both the CCLE and CGP studies.4 5
We compared the five genomic predictive models in the training set (CGP) using a cross-validation framework consisting of 10 repetitions of 10-fold cross-validations (figure 1). We first selected the 1000 genes exhibiting the highest variance in the training set in order to reduce data dimensionality. We then prevalidated predictors’ performance by computing the concordance index, which is a generalization of the area under the receiving characteristics operating curve.32 The concordance index estimates the probability that, for a random pair of cell lines, the model predicts correctly which is the most and less sensitive cell line; a random predictive would yield an index of 0.5 while a perfect predictor yields an index of 1. As can be seen in figure 2, we observed a significant and relatively good performance for nine out of 16 drugs (erlotinib, lapatinib, AZD0530, PD-0332991, PD-0325901, AZD6244, nutlin 3, 17-AAG, and irinotecan), the MRMR and ELASTICNET yielding a concordance index greater than 0.60 (p<0.05). Among these, genomic predictors for lapatinib and 17-AAG exhibited the highest performance, the best ones yielding R>0.69 (figure 2). However, we were unable to build efficient predictors for seven of the drugs (concordance index ≤ 0.6), namely PHA_665752, crizotinib, TAE684, nilotinib, sorafenib, PLX4720 and paclitaxel.
Figure 2.
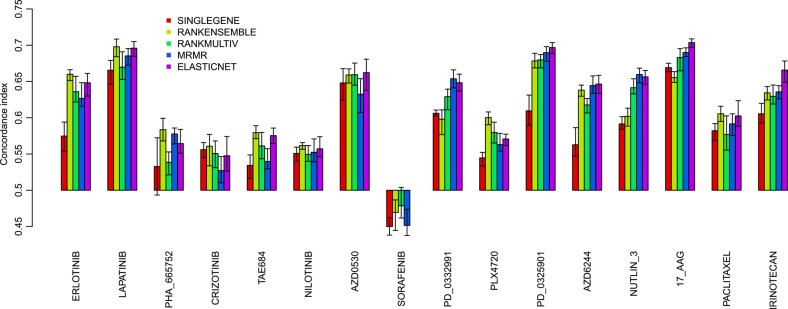
Mean prediction performance of the five genomic predictors evaluated in 10 repetitions of 10-fold cross-validation in the training set cancer genome project (CGP), as quantified by the concordance index between the predicted and observed IC50 values. The error bars represent the 95% confidence interval of the performance computed during the 10 repetitions of cross-validation.
We further validated the performance of the genomic predictors in CCLE, a fully independent dataset.4 We divided the test set (CCLE) into two subsets: the first subset (COMMON) contains the cell lines that were also present in the training set (CGP), comprising 419 cell lines, which can therefore be considered as biological replicates; the second subset (NEW) contains the cell lines that were not analyzed in CGP, comprising 572 cell lines (figure 1). The latter validation set is the most challenging as it allows us to address whether the genomic predictors are generalizable to new biological samples. As we can see in figure 3, for biological replicates (CCLE COMMON), the performance of the models predictive for irinotecan, 17-AAG, PD-0325901, and PLX4720 is close to what was estimated in our prevalidation study (concordance index>0.6 for MRMR and ELASTICNET predictors). More importantly, this is also the case for the CCLE NEW subset (figure 4), thereby demonstrating the generalization of these genomic predictors. Although predictors for AZD0530 and lapatinib performed well in prevalidation (figure 2) this was not the case when validating on new biological samples (CCLE COMMON and NEW; figures 3 and 4).
Figure 3.
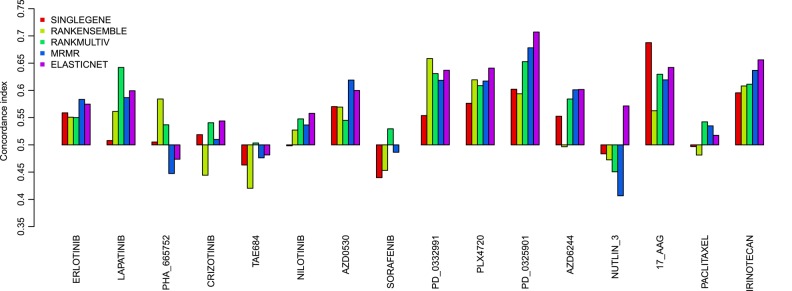
Prediction performance of the five genomic predictors in the validation set (cancer cell line encyclopedia COMMON) composed of the 419 cell lines that are also present in the training set cancer genome project. Prediction performance is quantified by the concordance index between the predicted and observed IC50 values.
Figure 4.

Prediction performance of the five genomic predictors in the validation set (cancer cell line encyclopedia NEW) composed of the 572 cell lines that are not present in the training set cancer genome project. Prediction performance is quantified by the concordance index between the predicted and observed IC50 values.
Discussion
During the past decade much effort has been made to build efficient genomic predictors of drug response from preclinical data.11 Until recently the number of cell lines and their lineage diversity used in published studies were insufficient to develop robust predictors. To address this issue, Garnet et al5 and Barretina et al4 published two large pharmacogenomic datasets of unprecedented size, including almost 1000 cell lines, each of which are screened on clinically relevant drug compounds (CGP and CCLE). However, these datasets have only been analyzed in isolation; we therefore seized this opportunity and combined the datasets generated in these two important studies in order to build and validate robustly the genomic predictors of drug response.
Following five modeling approaches, we built and validated genomic predictors for 16 drugs. In a prevalidation setting, we were able to build performant predictors for nine out of 16 drugs (erlotinib, lapatinib, AZD0530, PD-0332991, PD-0325901, AZD6244, nutlin 3, 17-AAG, and irinotecan) while the response of the remaining seven drugs, namely PHA_665752, crizotinib, TAE684, nilotinib, sorafenib, PLX4720 and paclitaxel, could not be reliably predicted in the training set. This can be explained by the fact that our study relies solely on gene expression to predict drug response, whereas genotyping data might be highly predictive for specific drugs. For example, Barretina et al4 successfully predicted response to paclitaxel in their own prevalidation analysis while our predictors were unable to achieve comparable performances. Similarly, Eng et al33 analyzed both SNP and gene expression data measured on the NCI60 cell line panel and showed a significant gain of predictive power by including SNP data compared to gene expressions alone.
We further validated the performance of predictors for response to irinotecan, 17-AAG, PD-0325901, and PLX4720 in two large independent datasets. On the contrary, predictors for AZD0530 and lapatinib did not perform well on independent datasets calling into question their reliability. Interestingly, the genomic predictors for PD-0332991 performed well in prevalidation and in CCLE COMMON but not in CCLE NEW, suggesting the mechanism of action of this drug strongly depends on cell lineage.
The purpose of our study was also to compare different modeling approaches; however, it is beyond the scope of our work to identify which would be the best model for each drug we analyzed. The main differences in the five predictors we implemented can be described as follows: whether they are univariate (SINGLEGENE) or multivariate (the rest); and whether there is redundancy among the selected genes (RANKENSEMBLE and RANKMULTIV) or not (MRMR and ELASTICNET). As expected, given the putative complexity of the biological processes underlying drug response, we observed that multivariate models consistently outperform the univariate model. Improvement over univariate model is substantial for multivariate predictors of response to irinotecan, PD-0325901, PD-0332991 and AZD6244. In the cases of PD-0325901 and AZD6244, both drugs target mitogen-activated protein kinase (MAPK/ERK kinase or MEK; table 1). The threonine/tyrosine specificity of MEK is key to its implication in the MAPK signaling pathway, which is frequently activated in human tumors. This pathway involves a relatively large number of genes, supporting the use of multivariate models in predictive analysis. Among the genes with the highest predictive contributions (ie, a high coefficient absolute value) in all models for both drugs, we observe an enrichment of FXYD5, SPRY2 and LGALS3BF (see supplementary table S1, available online only). This overlap between the independent analyses of all models in both drugs suggests that a combination of those genes’ expression levels is likely to yield a robust predictor for sensitivity to MEK-targeting drugs. SPRY2 also emerged as one of the top predictors of cell response to the drug AZD6244 across all models but the univariate model SINGLEGENE. It is known that SPRY2 inhibits cell growth and differentiation by specifically inhibiting the Ras/Raf/MAPK pathway.34 Moreover, the identification of SPRY2 concurs with the findings of Barretina et al.4
Our results suggest that response to 17-AAG could be robustly predicted using a single gene, namely NQO1 (see supplementary file 1) available online only). 17-AAG works as an Hsp90 inhibitor, leading to the depletion of oncogenic proteins such as Raf-1 and p53.35 Given its ansamycin benzoquinone structure, 17-AAG is reduced into a potent antitumor metabolite by the NAD(P)H:quinone oxidoreductase (NQO1).36 Indeed, the positive coefficient attributed to NQO1 in all models (see supplementary file 1, available online only) indicates that cell lines with higher levels of NQO1 expression have an increased sensitivity to 17-AAG. This concurs with multiple association studies in which a polymorphism in NQO1 (rs1800566), causing a decrease of enzymatic activity, has been shown to be linked to a higher risk of gastrointestinal tract cancer.37–39 In addition, Barretina et al4, using CCLE as the training set, independently identified NQO1 as a potential biomarker for sensitivity to Hsp90 inhibitors.
Conclusion
In our study we were able to build and validate robust predictors of response to irinotecan, 17-AAG, PD-0325901, and PLX4720. While multivariate models usually outperformed univariate models, it was not the case for the drug 17-AAG, whose response can be reliably predicted by a single gene. Although these genomic predictors exhibited promising performance in large, independent preclinical datasets, we now need to assess their clinical relevance by testing them first ex vivo on patients’ tumor cells, then in clinical trials. If successful these predictors could be used as companion tests to improve therapeutic benefits by identifying the subpopulation of patients likely to respond to a panel of drugs of interest.
Our study could be extended in several ways. First, we relied solely on gene expression data whereas mutation data are available for both the CGP and CCLE datasets; we expect a joint analysis of gene expression and mutation data to improve prediction performance for some drugs, such as paclitaxel.4 Second, more modeling approaches could be integrated; non-linear models such as k-nearest neighbors or support vector machine with non-linear kernels might yield better prediction for some of the difficult drugs (sorafenib or crizotinib, for instance). Finally, NCI60,6 a small but high quality dataset, could be added as a validation set even though the NCI60 cell lines are all part of CGP and CCLE datasets, and drug mapping between NCI60 and CGP or CCLE is complicated by the fact that only drugs’ national service center numbers are provided in CellMiner.12
Acknowledgments
The authors would like to thank the investigators of the cancer genome project, the cancer cell line encyclopedia and the CellMiner database who have made their invaluable data available to the scientific community.
Footnotes
Funding: SPC, NDJ and NH were supported by the start-up fund of Benjamin Haibe-Kains. JQ was supported by a grant from the National Library of Medicine of the US National Institutes of Health (R01 LM010129-01) and by a grant from the Claudia Adams Barr Program in Innovative Basic Cancer Research. GB and CO were supported by the Belgian French Community ARC (Action de Recherche Concertée) funding.
Competing interests: None.
Provenance and peer review: Commissioned; externally peer reviewed.
Data sharing statement: All data are publicly available from from ArrayExpress (E-MTAB-783), and the Broad Institute website (http://www.broadinstitute.org/ccle/).
References
- 1.Tauzin B. More than 800 medicines and vaccines in testing offer hope in the fight against cancer. Washington, DC: Medicines in Development for Cancer, 2009 [Google Scholar]
- 2.Griffith LG, Swartz MA. Capturing complex 3D tissue physiology in vitro. Nature 2006;7:211–24 [DOI] [PubMed] [Google Scholar]
- 3.Richmond A, Su Y. Mouse xenograft models vs GEM models for human cancer therapeutics. Dis Model Mech 2008;1:78–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barretina J, Caponigro G, Stransky N, et al. The cancer cell line encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012; 483:603–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Garnett MJ, Edelman EJ, Heidorn SJ, et al. Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature 2012;483:570–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shoemaker RH. The NCI60 human tumour cell line anticancer drug screen. Nat Rev Cancer 2006;6:813–23 [DOI] [PubMed] [Google Scholar]
- 7.Heiser LM, Wang NJ, Talcott CL, et al. Integrated analysis of breast cancer cell lines reveals unique signaling pathways. Genome Biol 2009;10:R31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kutalik Z, Beckmann JS, Bergmann S. A modular approach for integrative analysis of large-scale gene-expression and drug-response data. Nat Biotech 2008;26:531–9 [DOI] [PubMed] [Google Scholar]
- 9.Sirota M, Dudley JT, Kim J, et al. Discovery and preclinical validation of drug indications using compendia of public gene expression data. Sci Transl Med 2011;3:96ra77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cohen AL, Soldi R, Zhang H, et al. A pharmacogenomic method for individualized prediction of drug sensitivity. Mol Syst Biol 2011;7:1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Caponigro G, Sellers WR. Advances in the preclinical testing of cancer therapeutic hypotheses. Nat Rev Drug Discov 2011;10:179–87 [DOI] [PubMed] [Google Scholar]
- 12.Shankavaram UT, Varma S, Kane D, et al. CellMiner: a relational database and query tool for the NCI-60 cancer cell lines. BMC Genomics 2009;10:277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Collins JM. Developmental Therapeutics Program NCI/NIHhttp://dtp.nci.nih.gov/ (accessed 1 Oct 2012).
- 14.Hann MM, Oprea TI. Pursuing the leadlikeness concept in pharmaceutical research. Curr Opin Chem Biol 2004;8:255–63 [DOI] [PubMed] [Google Scholar]
- 15.McDermott U, Sharma SV, Dowell L, et al. Identification of genotype-correlated sensitivity to selective kinase inhibitors by using high-throughput tumor cell line profiling. Proc Natl Acad Sci U S A 2007;104:19936–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chapman PB, Hauschild A, Robert C, et al. BRIM-3 Study Group Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med 2011;364:2507–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McDermott U, Settleman J. Personalized cancer therapy with selective kinase inhibitors: an emerging paradigm in medical oncology. J Clin Oncol 2009;27:5650–9 [DOI] [PubMed] [Google Scholar]
- 18.Macconaill LE, Garraway LA. Clinical implications of the cancer genome. J Clin Oncol 2010;28:5219–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McCall MN, Bolstad BM, Irizarry RA. Frozen robust multiarray analysis (fRMA). Biostatistics 2010;11:242–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Durinck S, Moreau Y, Kasprzyk A, et al. BioMart and Bioconductor: a powerful link between biological databases and microarray data analysis. Bioinformatics 2005;21:3439–40 [DOI] [PubMed] [Google Scholar]
- 21.Li Q, Birkbak NJ, Győrffy B, et al. Jetset: selecting the optimal microarray probe set to represent a gene. BMC Bioinformatics 2011;12:474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Höfling H, Tibshirani R. A study of pre-validation. Ann Appl Stat 2008;2:643–64 [Google Scholar]
- 23.Kittler J, Hatef M, Duin R, et al. On combining classifiers. IEEE Trans Pattern Anal Mach Intell 1998;20:226–38 [Google Scholar]
- 24.Ding C, Peng H. Minimum redundancy feature selection from microarray gene expression data. J Bioinform Comput Biol 2005;3:185–205 [DOI] [PubMed] [Google Scholar]
- 25.Meyer PE, Kontos K, Lafitte F, et al. Information-theoretic inference of large transcriptional regulatory networks. EURASIP J Bioinform Syst Biol 2007:79879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Friedman J, Hastie T, Tibshirani R. Regularization paths for seneralized linear models via coordinate descent. J Stat Softw 2010;33:1–22 [PMC free article] [PubMed] [Google Scholar]
- 27.Zou H, Hastie T. Regularization and variable selection via the elastic net. J Royal Stat Soc Series B (Stat Methodol) 2005;67:301–20 [Google Scholar]
- 28.Dudoit S, Fridlyand J, Speed TP. Comparison of discrimination methods for the classification of tumors using gene expression data. J Am Stat Assoc 2002;97:77–87 [Google Scholar]
- 29.Haibe-Kains B, Desmedt C, Sotiriou C, et al. A comparative study of survival models for breast cancer prognostication based on microarray data: does a single gene beat them all? Bioinformatics 2008;24:2200–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Guyon I, Elisseeff A. An introduction to variable and feature selection. J Mach Learn Res 2003;3:1157–82 [Google Scholar]
- 31.Kohavi R, John GH. Wrappers for feature subset selection. Artif Intell 1997;97:273–324 [Google Scholar]
- 32.Harrell FJ, Lee K, Mark D. Multivariable prognostic models: issues in developing models, evaluating assumptions and adequacy, and measuring and reducing errors. Stat Med 1996;15:361–87 [DOI] [PubMed] [Google Scholar]
- 33.Eng L, Ibrahim-zada I, Jarjanazi H, et al. Bioinformatic analyses identifies novel protein-coding pharmacogenomic markers associated with paclitaxel sensitivity in NCI60 cancer cell lines. BMC Med Genomics 2011;4:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wong K-K. Recent developments in anti-cancer agents targeting the Ras/Raf/ MEK/ERK pathway. Recent Pat Anticancer Drug Discov 2009;4:28–35 [DOI] [PubMed] [Google Scholar]
- 35.Sankhala KK, Mita MM, Mita AC, et al. Heat shock proteins: a potential anticancer target. Curr Drug Targets 2011;12:2001–8 [DOI] [PubMed] [Google Scholar]
- 36.Guo W, Reigan P, Siegel D, et al. Formation of 17-allylamino-demethoxygeldanamycin (17-AAG) hydroquinone by NAD(P)H:quinone oxidoreductase 1: role of 17-AAG hydroquinone in heat shock protein 90 inhibition. Cancer Res 2005;65:10006–15 [DOI] [PubMed] [Google Scholar]
- 37.Ding R, Lin S, Chen D. Association of NQO1 rs1800566 polymorphism and the risk of colorectal cancer: a meta-analysis. Int J Colorectal Dis 2012;27:885–92 [DOI] [PubMed] [Google Scholar]
- 38.Malik MA, Zargar SA, Mittal B. Role of NQO1 609C>T and NQO2 -3423G>A gene polymorphisms in esophageal cancer risk in Kashmir valley and meta analysis. Mol Biol Rep 2012;39:9095–104 [DOI] [PubMed] [Google Scholar]
- 39.Yang F-Y, Guan Q-K, Cui Y-H, et al. NAD(P)H quinone oxidoreductase 1 (NQO1) genetic C609T polymorphism is associated with the risk of digestive tract cancer: a meta-analysis based on 21 case-control studies. Eur J Cancer Prev 2012;21:432–41 [DOI] [PubMed] [Google Scholar]