

Abstract
Background and significance
Intellectual disability is a condition characterized by significant limitations in cognitive abilities and social/behavioral adaptive skills and is an important reason for pediatric, neurologic, and genetic referrals. Approximately 10% of protein-encoding genes on the X chromosome are implicated in intellectual disability, and the corresponding intellectual disability is termed X-linked ID (XLID). Although few mutations and a small number of families have been identified and XLID is rare, collectively the impact of XLID is significant because patients usually are unable to fully participate in society.
Objective
To reveal the molecular mechanisms of various intellectual disabilities and to suggest small molecules which by binding to the malfunctioning protein can reduce unwanted effects.
Methods
Using various in silico methods we reveal the molecular mechanism of XLID in cases involving proteins with known 3D structure. The 3D structures were used to predict the effect of disease-causing missense mutations on the folding free energy, conformational dynamics, hydrogen bond network and, if appropriate, protein-protein binding free energy.
Results
It is shown that the vast majority of XLID mutation sites are outside the active pocket and are accessible from the water phase, thus providing the opportunity to alter their effect by binding appropriate small molecules in the vicinity of the mutation site.
Conclusions
This observation is used to demonstrate, computationally and experimentally, that a particular condition, Snyder-Robinson syndrome caused by the G56S spermine synthase mutation, might be ameliorated by small molecule binding.
Keywords: In Silico Modeling, Experimenatal Measurements, Mental Disorders, Missense Mutation, Small Molecule Screening
Background and significance
It is known that missense mutations cause many human diseases and underlie the natural differences between individuals by affecting the structure, function, interactions, and other properties of expressed proteins. In some cases, one altered allele can cause a major health problem. These diseases are usually rare but serious. Other conditions, such as heart disease and cancer, are genetically complex, with alleles from several genes contributing to the disorder. The ability to predict whether a given missense mutation causes disease or is harmless would be very important for early detection in patients with a high risk of developing a particular illness. More useful would be the ability to offer a specific treatment to reduce or completely eliminate the effects of disease-causing missense mutations. This work focuses on further understanding the molecular mechanisms of various intellectual disabilities (rare X-linked conditions) and on developing the methodology and in silico tools for altering their phenotype by binding small molecules to the corresponding proteins.
Over the past few years, significant progress has been made in building databases of disease-causing missense mutations1–4 and sequentially using these classified mutations for the early identification of patients at risk.5–8 However, very little work has been done to develop possible treatments, perhaps due to a lack of understanding of the pathogenesis of the effects caused by the mutations.9 10 Another possible reason is that the common approach in drug discovery is to design competitive inhibitors that alter the effect of the disease.11–15 However, such an approach may not be successful in designing drugs capable of reversing the effects of disease-causing mutations because most of these effects cannot be fixed by simply inhibiting a particular reaction. Instead, drug design should be preceded by a detailed investigation of the effects caused by the mutation, followed by analysis to determine which effects result in disease, and lastly, by a search for small molecules that can restore the wild type (WT) function of the corresponding protein by binding to it.
Rare diseases are particularly promising targets for exploring the possibility of reversing the effects of disease-causing missense mutations. First, the fact that a particular disease is typically caused by a single mutation makes analysis of the effects much easier as compared to a complex disease, and thus reduces the ambiguity of in silico modeling. Second, the observation that different mutations within the same gene result in the same phenotype suggests that these mutations have similar effects on the target protein's function and its interactions, thus provides the opportunity for in silico modeling to identify the dominant molecular effects causing the disease. Third, the fact that a disease is caused by a malfunction of a particular protein makes it easier to find a treatment that will reverse the effect. Because the target protein is known, the effects caused by missense mutations will be pinpointed by in silico modeling and small molecules bound to the target protein might be able to reverse the effect. Fourth, the fact that the disease is caused by a single mutation in a particular protein allows experiments be designed and carried out to directly test in silico generated hypotheses and to follow the effects induced by small molecule binding.
Identification of genetic disorders is a crucial step for both providing an early diagnosis of disease predisposed patients (personalized diagnostics)16–20 and prescribing the optimal drug (personalized medicine).21 22 However, identifying disease-causing genetic differences and distinguishing them from harmless variations is difficult.23–26 Typically, a disease-causing variant is identified by searching databases of known disease-causing genetic defects or by machine learning algorithms. However, the success of both of these approaches depends on the database of known disease-causing mutations and neither method can classify new mutations. Thus, in terms of early diagnosis, a new disease-causing DNA variant or a rare mutation may not be identified by these methods and consequently the patient will not be treated. At the other end of the spectrum are methods utilizing first principle approaches. While these methods should be able to detect new disease-causing mutations, typically they are not used for screening as they are too time consuming. To bridge this gap, approaches based on first principle calculations, but much faster and adjusted to match experimental data, have been developed.27 Such combined approaches have several advantages over existing methods and offer several additional outcomes, which can be extremely useful in personalizing treatment for patients as: (a) the proposed approaches are not based on a comparison, but on first principle approaches, so they can detect defects potentially causing disease even in cases that are not previously identified in other patients; (b) they avoid ambiguities associated with predicting the effects of multiple mutations in terms of their additivity or compensation (neutralization); and (c) they also provide details of the molecular mechanisms of the effects, resulting in more effective personalized treatment.
Currently, most medical research efforts are directed towards the study and treatment of common diseases, for example cancer research. Rare diseases are frequently referred to as ‘neglected’ and receive much less attention from the scientific and pharmaceutical communities. Although these rare disorders affect few individuals, they can have devastating effects. They are sometimes severe and affected patients generally cannot fully participate in society. For patients and their families, progress in understanding the origin of disease brings hope for the development of treatment to reduce the burden of the condition. This work reports the preliminary results of research to mitigate the effects of X-linked disease-causing mutations by small molecule binding, an approach used by other groups to alter the stability of proteins28 29 or protein signaling.30
Methods
Calculating the changes in folding free energy
One of the main characteristics of each biological macromolecule is its stability, typically referred to as the free energy of folding/unfolding. There are several distinct approaches for computing changes in the folding free energy following a single mutation, ranging from detailed free energy perturbation methods,31 32 to statistical potentials,33–35 and to methods based on machine learning,36 37 reviewed in Potapov et al,38 Gromiha et al,39 and Khan and Vihinen.40 Free energy perturbation methods are physically sound but are quite time consuming and thus are unsuitable for evaluating the effects of hundreds of mutations. Methods based on statistical potentials are fast, but depend on the training set used to deliver the statistical potentials. On the other hand, machine learning methods are fast and suitable for large scale predictions, but do not reveal the reason for the effects, leaving very little information for analysis of the molecular causes of the disease. Therefore, we developed our own method suitable for application to a large number of mutations while at the same time revealing the atomic details of changes caused by the mutation. The method is termed the scaled Molecular Mechanics Generalized Born (sMMGB) approach and is based on the following formula27:
 |
1 |
where G(folded) is the total potential energy of the folded state and G(unfolded) is the total potential energy of the unfolded state. The energy, G(unfolded), of the unfolded state, is split into two terms, G0(unfolded) and Gx(unfolded), as discussed in our previous work.41–43 Gx(unfolded) is the energy of the unfolded state of ‘x’ residue segments at the center of the mutation site, while G0(unfolded) is the energy of the unfolded state of the rest of protein (figure 1). Under such an assumption, G0(unfolded) is identical for WT protein and mutants, cancels out in equation (2), and therefore does not need to be calculated.
Figure 1.

Approach for modeling folded and unfolded states.
The folding energy change due to a mutation is calculated with the following equation:
 |
2 |
where ΔΔG(folding_mutation) represents the folding energy change due to a mutation, and ΔG(folding_WT) and ΔG(folding_mutant) are the folding energy of the WT protein and the mutant, respectively.
It is recognized that the above MMGB method tends to overestimate the magnitude of the predicted free energy changes (compared with the experimental data), mostly because the entropy is not included, and therefore the predicted ΔΔG may have to be scaled to match the experimentally determined changes in the folding free energy. In our previous work27 we devised an optimization procedure to minimize the root mean square deviation (RMSD) between the scaled calculated results and experimental data by scaling the energies via a linear function with two adjustable parameters. The optimal values of the adjustable parameters were obtained by minimizing the RMSD of the predictions against experimental data derived from the ProTherm website (Thermodynamic Database for Proteins and Mutants: http://gibk26.bio.kyutech.ac.jp/jouhou/Protherm/protherm_search.html).44 45 Benchmarking resulted in RMSD=1.78 kcal/mol and the slope of the linear regression fit between the experimental data and the calculations was 1.04 on a set of 1109 mutations.27
Modeling conformational dynamics with molecular dynamics simulations
Using snapshots of the WT and the corresponding mutant, the structural dynamics are evaluated via the RMSD. The snapshots are superimposed on the starting structures as described in our previous work43 in order to calculate the time averaged RMSD of the entire structure or selected set of amino acids as:
 |
3 |
where ‘n’ is the snapshot number, which has an important property: it tends asymptotically to a particular value and thus can be used to assess if the quantity of interest was properly estimated in the particular simulation time window. Once RMSD reaches a steady value, for both the WT and the mutant, the simulations are considered to have converged.
Modeling ionization states and the corresponding hydrogen bond network
The Multi-Conformation Continuum Electrostatics program (http://134.74.90.158)46–48 is utilized to calculate the pKa's of ionizable groups for both the WT and mutant structures and thus allows the pKa shifts induced by mutation to be calculated per titratable amino acid:
| 4 |
where ΔpKai(mut) is the pKa shift of amino acid i due to the mutation, and pKai(WT) and pKai(mutant) are the pKa values of amino acid i in the WT protein and the mutant protein, respectively. As result, one can calculate the following quantity:
 |
5 |
where Sum(ΔpKa) is the global pKa shift of the whole protein due to the mutation, which was used to predict the effect of mutation on the protonation states of the entire protein.
Calculating changes in binding free energy
Binding affinity is another important characteristic of native proteins, as most proteins interact with another partner(s) or form a dimer of higher level assemblage. In our previous work,41 42 49 the binding free energy (ΔΔG(binding)) was calculated as the difference between the total potential energy of the complex minus the total potential energy of the separated monomers (figure 2):
Figure 2.
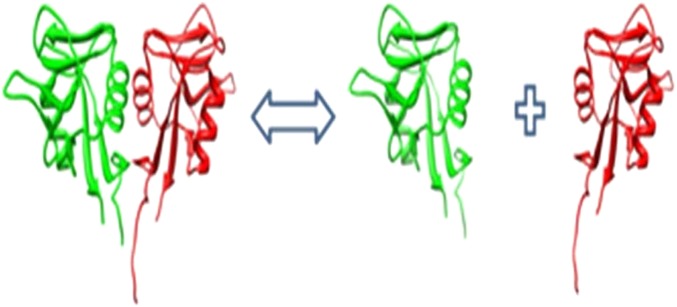
Approach for calculating binding.
 |
6 |
where ΔG(complex) is the potential energy of the complex, and ΔG(monomer_A) and ΔG(monomer_B) are the potential energies of monomers ‘A’ and ‘B’, respectively. The energies were calculated using TINKER software50 utilizing the MMGB method. Entropy was considered to be the same for the monomer and the complex and thus cancels out in equation (6), and so the change in the potential energy from the unbound to bound states was considered to be the free energy of binding. These calculations are done for WT structures and for each mutant and the difference, which evaluates the effect of the mutation on the affinity (binding free energy), is calculated as49:
 |
7 |
where ΔΔG(binding_WT) is the binding energy of the WT monomers and ΔΔG(binding_mutant) is the binding energy of the corresponding mutant.
Virtual screening for small molecules
The structures of the mutants were analyzed to identify surface cavities close the mutation site. These areas and their associated structures were used for in silico screening of small molecules to determine the best candidates and select those where binding alters the disease-causing effects. We screened a focused protein–protein interaction inhibitor compounds collection (taken from ChemBridge: http://www.chembridge.com/index.php) that was filtered for drug-likeness using the software FAF-Drugs2.51 The 3D structures of small molecules were generated using the web server Frog2.52 In silico screening was conducted using the software AutoDock Vina53 54 employing a stochastic docking algorithm and an empirical scoring function.
The candidate small molecules were ranked according to binding affinity and the top ranked 500 molecules were considered. Finally, 10 potential molecules were manually selected based on expert evaluation of molecule burial and hydrogen bond formation.
Experimental measurements of the activity of selected mutants in the presence of small molecules
The enzyme spermine synthase (SMS) mutant, G56S, which causes Snyder-Robinson syndrome (SRS), was studied to determine the effect of small molecule binding. The 10 best small molecule candidates from the virtual screening, being hydrophobic, were dissolved in 5% BSA solution so the experiments could be conducted in water. The G56S mutant protein itself had little activity compared with WT (1.3% of WT activity) (figure 3, left panel). However, the addition of 5% BSA increased G56S mutant activity by 122.2% (a 2.22-fold increase in activity) (figure 3, right panel).
Figure 3.
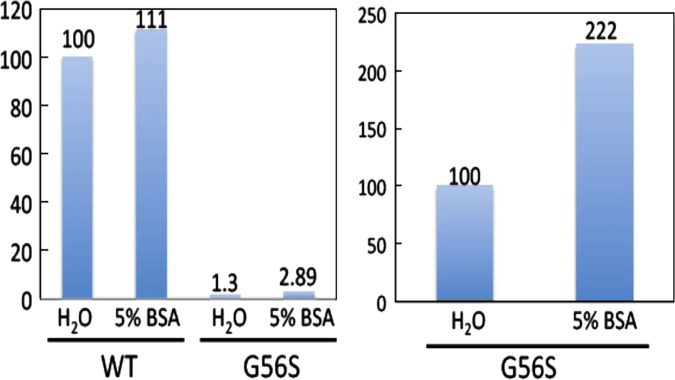
Activity of the wild type (WT) protein and G56S mutant with and without 5% BSA.
Activity was measured by following the production of spermine from spermidine in 100 mM sodium phosphate buffer (pH 7.5) in the presence of 0.1 mM dcAdoMet as the propylamine donor.55 Reactions were run for 60 min and polyamines were extracted in 10% trichloroacetic acid. The extracts were directly injected onto the o-phthalaldehyde postcolumn ion-exchange HPLC system.56 As no differences were found when the experiments were repeated, no further independent measurements were carried out.57 58
Results
In this section, we describe the results of our in silico investigations of the molecular effects associated with three types of intellectual disability (SRS, CLIC2-related intellectual disability, and creatine deficiency syndrome), and then select one particular missense mutation causing SRS in order to identify small molecule candidates for binding via virtual screening and test the effect experimentally.
Snyder-Robinson syndrome
It was shown by Schwartz and co-workers41 59–61 that SRS is caused by missense mutations in the spermine synthase gene that encodes a protein (SMS) of 529 amino acids.58 We investigated, in silico, the molecular effect of three known missense mutations in SMS (G56S, V132G, and I150T) which have been clinically identified as causing the condition (figure 4). We utilized the methods and approaches described above to determine the effects of these mutations on SMS stability, flexibility, and interactions. Among the findings was the suggestion that the catalytic residue, Asp276, should be protonated before binding the substrates.41 The pKa calculations revealed an important consequence of the I150T mutation, predicting that it causes pKa changes in WT SMS involving titratable residues interacting with the decarboxylated S-adenosylmethionine (MTA) substrate. The I150T missense mutation was also found to decrease the stability of the C-terminal domain and to induce structural changes in the vicinity of the MTA binding site. The other two missense mutations, G56S and V132G, are distant from the active site and do not perturb its WT properties, but do affect the stability of both the monomers and the dimer (figure 4). In particular, the G56S mutation is predicted to greatly reduce dimer affinity and thus prevent dimer formation. Since dimerization is essential for SMS function, altering WT dimer stability would have a dramatic effect on SMS function.58
Figure 4.
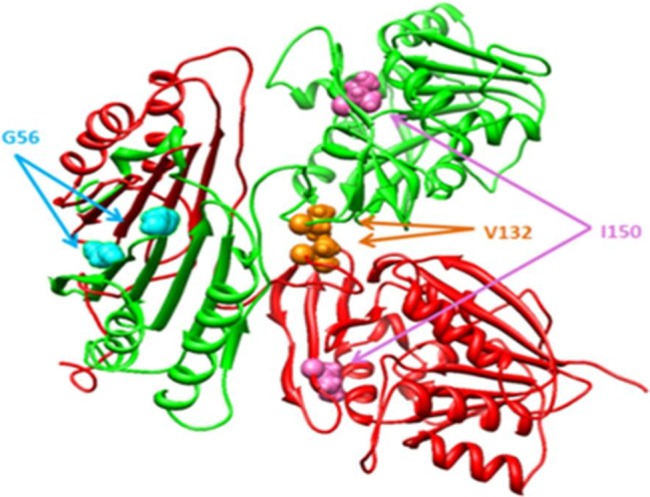
The 3D structure of SMS. The mutations sites are indicated with colored balls.
These findings reveal that none of the mutations, including I150T, is directly involved in catalysis and their effect on SMS function is indirect. Particularly interesting is the mutation G56S, which is distant from the binding pocket and was found to primarily affect the dimerization of SMS, and to have almost no effect on other SMS properties. The N-terminal domain, where the mutation site is located, is not active and its role is simply to facilitate dimer formation. However, if binding of a small molecule can restore the WT affinity of the dimer, the G56S mutant will then function similarly to the WT. This will be explored in the next sections below.
X-linked intellectual disability caused by a CLIC2 gene defect
The CLIC2 gene codes for a protein, CLIC2, which is believed to be both a chloride channel and a calcium channel regulator.62–65 Our investigation was based on the recent discovery of a missense mutation (H101Q) in the CLIC2 gene on Xq28 in a male with X-linked intellectual disability (XLID) and not found in healthy individuals.66 At the same time, numerous nsSNPs (non-synonymous SNPs) have been reported in the CLIC2 gene in healthy individuals, indicating that the CLIC2 protein can tolerate amino acid substitutions and still be fully functional (figure 5). Our modeling predicted that H101Q, in contrast with other nsSNPs, (a) rigidifies the joint loop which is important for the normal functioning of CLIC2, (b) stabilises the overall 3D structure of CLIC2 and thus reduces the possibility of large conformational change expected to occur when CLIC2 moves from a soluble to a membrane form, and (c) removes the positively charged residue, H101, which may be important for the membrane association of CLIC2 and interaction with ryanodine receptors. The results of in silico modeling, in conjunction with the polymorphism analysis, suggest that p.H101Q indeed is a disease-causing mutation, the first suggested in the CLIC family,66 as recently confirmed experimentally.67
Figure 5.

The 3D structure of CLIC2 protein. The mutation sites are indicated with colored balls.
Very little is known about the function(s) of CLIC2, but two critical requirements are known: it has to interact with the ryanodine receptor in order to regulate calcium transport and it must be inserted into the membrane bilayer to function as a chloride channel. However, the molecular details of these reactions are currently not available. Our modeling of the CLIC2–ryanodine receptor indicates that H101 is very close to the interface of the complex and is situated in a positive potential. Replacement of positively charged His with polar but uncharged Gln is expected to dramatically change CLIC–ryanodine receptor interactions. However, WT binding affinity, in principle, can be rescued by small molecule binding at the interface of the CLIC2–ryanodine receptor. The other event, insertion into the lipid membrane and functioning as a chloride channel, is much more difficult to model. Initial investigations suggest that this would require significant conformational change associated with the N-terminal domain of CLIC2 and formation of a dimer/trimer within the membrane. Such a large conformational change would require rearrangement of the N-terminal domain of CLIC2, which in turn would require that the ‘joint loop’ (figure 5) be able to adopt different conformations as well. The H101Q mutation is predicted to rigidify the loop, which would therefore need to be destabilized in order to rescue WT flexibility.
Creatine deficiency syndrome
The intellectual disability associated with creatine deficiency syndrome is caused by malfunctioning of a protein coded by the SLC6A8 gene. The structure of the protein is not available experimentally and so for the purposes of this work, it was built in silico by homology modeling. The mutations were mapped onto the model but none were found to be or be close to the active site residue. Many of the mutation sites are exposed to the water phase. The results of in silico modeling of the changes in the folding free energy indicate that some mutations make the protein more, while others make it less stable by a significant amount of energy (figure 6). Previous investigations have shown that over-stabilization and destabilization are equally disadvantageous for WT function. Two types of missense mutations are known for this protein: disease-causing missense mutations, c.1361C>T (p.P544L), c.259G>A (p.G87R), c.1169C>T (p.P390L), c.1011C>G (p.C337W), c.1161C>T (p.P554L), c.1141G>C (p.G381R), c.1271G>A (p.G424D), c.1067G>T (p.G356V), c.1171C>T (p.R391W), c.1145C>T (p.P382L), G132V, C491W, and c.757G>C (p.G253R), which are located in the highly conserved sequence regions, except for c.1210G>C (p.A404P); and five harmless missense mutations, c.11A>G (p.K4R), c.76G>A (p.G26R), c.544G>A (p.V182M), c.1678A>G (p.M560V), and c.1185G>A (p.V629I), which are located in sequence regions which are not highly conserved. The results of the energy calculations were able to predict all of the harmless mutations (region 2 in figure 6) and most of the disease-causing mutations (regions 1 and 3 in figure 6). These energy regions in figure 6 were assigned using a threshold of 5 kcal/mol, which causes both stabilization and destabilization of the protein. This threshold was selected to include in region 2 all harmless mutations, but it is obvious that such a value is protein specific and cannot be predicted without experimental data. It can be seen that disease-causing mutations either strongly destabilize the protein (region 1) or in some cases (region 3) make it more stable. In both cases, this results in a significant deviation of the WT characteristics of the protein. Some of the known disease-causing mutations are predicted to cause very small changes in the folding free energy (region 2), of the same order of magnitude as the harmless mutations. Therefore, the disease-causing effect associated with these mutations does not impact the stability, but perhaps something related to their position in the 3D structure of the protein inserted into the membrane (note that in this work the membrane was not included in the modeling). Further investigations are needed to elucidate the pathogenic effect of the disease-causing mutations listed in region 2.
Figure 6.

Predicted folding free energy changes for the SLC6A8 protein for each of 17 mutations.
A significant fraction of disease-causing mutations are located the loops connecting the SLC6A8 helices and are accessible from the water phase. Therefore, restoring the WT stability of SLC6A8 is plausible by targeting the mutation sites with small molecules, the binding of which would reduce the effects shown in figure 6.
Virtual screening for small molecules for the G56S SMS mutant
To illustrate the possibility of neutralizing disease-causing effects, we choose a particular mutant protein, the G56S SMS mutant causing SRS, and conducted virtual screening to identify small drug-like molecules capable of stabilizing the dimer. Our hypothesis is that such stabilization will increase the activity of the mutant G56S SMS. To this end we targeted in silico a large druggable zone that was identified on the mutant dimer (shown in figure 7). The pocket is situated between the interfaces of N-terminal domains of SMS and is accessible from the water phase. Binding a small molecule into the pocket would fill the gap between the interfaces and remove the water, and thus could increase dimer affinity. For this purpose we analyzed 10 docking poses per molecule for the top ranked 500 molecules, looking for shape complementarity, hydrogen bonds, and hydrophobic interactions between the mutant dimer and the docked ligands. We subjected the 10 top compounds to in vitro essays (see next paragraph) and identified three molecules which experimentally increased the activity of the mutant G56S SMS. Figure 7 shows the best poses of the three molecules docked on the dimer. As can be seen, the three molecules anchor in the same cavity close to the mutation site. Several ‘hot spot’ residues are involved in the predicted protein–ligand interactions. The binding mode of molecule 5790328 suggests: (a) hydrogen bond interactions with H81 from the opposite chain (indicated as H462 in figure 7) (the protonated ND1) and P3 (P16 in the SMS sequence) (the backbone CO); (b) aromatic interactions with Y79 from the opposite chain (indicated as Y460 in figure 7) and Y76 (Y91 in the SMS sequence); and (c) hydrophobic contacts with the V2 (V15 in the SMS sequence) side chain. The same polar and hydrophobic interactions are suggested for molecule 7754012 (aromatic contacts with Y76 (Y91 in the SMS sequence)). The third molecule 9129729 is found to have two hydrogen bonds with D59 from the opposite chain (indicated as D440 in figure 7) (the backbone CO and the side chain). Again V2 (V15 in the SMS sequence) and P3 (P16 in the SMS sequence) are suggested to be involved in hydrophobic interactions with 9129729. The three predicted binding modes strongly support the identified potential druggable pocket, which is further validated experimentally in the next paragraph.
Figure 7.

Best poses of the three active molecules docked on the mutant G65S dimer. Residues are numbered according to the SMS sequence, not according to the 3C6K PDB structure (see text).
Experimental investigation of the effect of small molecules on the activity of the G56S SMS mutant
The 10 top small molecule candidates identified by virtual screening above were purchased and experiments carried out to determine their effect on G56S SMS mutant activity (see the Methods section for details). The results of experimental investigations are very promising, as can be seen in figure 7. The vertical axis of the graph shows activity normalized to 100% for the G56S SMS mutant without the binding of small molecules (left-most bar in figure 8). The horizontal axis indicates the small molecule ID number. It can be seen that some of the small molecules increased the activity of the G56S SMS mutant by more than 30% (9129729, 7754012, and 5790327). Although none of the small molecules were able to completely restore the WT activity of SMS, the trend is clear: activity is increased by small molecules present in the solution. Further work is underway on larger set of small molecules to investigate their physical binding to SMS.
Figure 8.

Activity of the G56S mutant with small molecules bound. The left-most bar is the reference activity of the G56S mutant without small molecules present.
Conclusions
Three mutations implicated in XLID were studied in this work based on the 3D structure of the corresponding protein. It was shown that none of the mutations in this particular set affect the active site or directly participate in the corresponding biochemical reaction. Some of the mutations were predicted to decrease WT stability and the binding affinity of the corresponding protein, while others were calculated to increase stability. This indicates that any substantial deviation away from WT characteristics can cause disease, but there is no clear metric to determine how large the deviation needs to be. In some cases, a change in stability or binding affinity caused by a kcal/mol can have a significant effect; in other cases, it may have no impact on the function of the corresponding protein. Instead of addressing this difficult question, one can ask ‘can we restore the WT characteristics by binding small molecules to the mutant?’ Indeed it should be possible, since almost all mutation sites studied in this work are accessible from the water phase. We demonstrate for a particular case that the disease-causing effect can be reduced by small molecule binding to the mutant. The results of in silico modeling were experimentally tested and it was shown that some small molecules indeed increase the activity of the malfunctioning protein by more than 30%.
The presented approach is dependent on the existence of a 3D structure of the corresponding protein, either experimentally determined or capable of being modeled accurately. In some cases, particularly those involving large scaffolding or membrane proteins, the 3D structure may not be available, and thus the proposed methodology will not be applicable. Without a 3D structure, the free energy calculations and structural analysis of the effects caused by the mutations cannot be carried out, nor can structure-based virtual screening for small molecule candidates. Alternative bioinformatics approaches should be developed and tested.
The proposed approach assumes that the disease-causing effects can be reversed through small molecule binding. Indeed, for the G56S SMS mutant it was experimentally shown that some small molecules increase G56S activity. The validity of this methodology has also been confirmed by other researchers.28–30 However, in many other cases the disease-causing mutation may truncate the mutation resulting in a truncated protein, which includes instances of proteins lacking the active site residues. Even where the mutant is a full length protein, the mutation can have a devastating effect on the WT properties of the protein, for example, a hydrophilic mutation in the hydrophobic core of the protein. Obviously such cases cannot be repaired by small molecule binding.
In terms of small molecule binding, binding requires the existence of an appropriate binding cavity or a patch of residues. The binding epitope in this work was sought close to the mutation site, but, in principle, this should not be a limitation. Due to the cooperation of intra- and inter-molecular interactions, it can be speculated that even binding away from the mutation site could introduce the compensating effect. However, finding a small molecule which binds with high affinity to the target mutant and at the same time affects the mutant's properties in the desired fashion is a difficult goal and may not be achievable in all cases.
Acknowledgments
ZZ thanks the Chateaubriand Fellowship, which is supported by the Embassy of France in the USA.
Footnotes
Funding : The work of ZZ and EA was supported in part by NIH, NLM, grant number 1R03LM009748.
Competing interests: None.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.Stenberg KA, Riikonen PT, Vihinen M. KinMutBase, a database of human disease-causing protein kinase mutations. Nucleic Acids Res 2000;28:369–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.van Triest HJ, Chen D, Ji X, et al. PhenOMIM: an OMIM-based secondary database purported for phenotypic comparison. Conf Proc IEEE Eng Med Biol Soc 2011;2011:3589–92 [DOI] [PubMed] [Google Scholar]
- 3.Amberger J, Bocchini CA, Scott AF, et al. McKusick's Online Mendelian Inheritance in Man (OMIM). Nucleic Acids Res 2009;37():D793–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Amladi S. Online Mendelian Inheritance in Man ‘OMIM’. Indian J Dermatol Venereol Leprol 2003;69:423–4 [PubMed] [Google Scholar]
- 5.Gaspar H, Michel-Calemard L, Morel Y, et al. Prenatal diagnosis of autosomal recessive polycystic kidney disease (ARPKD) without DNA from an index patient in a current pregnancy. Prenat Diagn 2006;26:392–3 [DOI] [PubMed] [Google Scholar]
- 6.Vergopoulos A, Knoblauch H, Schuster H. DNA testing for familial hypercholesterolemia: improving disease recognition and patient care. Am J Pharmacogenomics 2002;2:253–62 [DOI] [PubMed] [Google Scholar]
- 7.Aguilera I, Garcia-Lozano JR, Munoz A, et al. Mitochondrial DNA point mutation in the COI gene in a patient with McArdle's disease. J Neurol Sci 2001;192:81–4 [DOI] [PubMed] [Google Scholar]
- 8.Forslund O, Nordin P, Andersson K, et al. DNA analysis indicates patient-specific human papillomavirus type 16 strains in Bowen's disease on fingers and in archival samples from genital dysplasia. Br J Dermatol 1997;136:678–82 [PubMed] [Google Scholar]
- 9.Xiao X, Shao S, Ding Y, et al. An application of gene comparative image for predicting the effect on replication ratio by HBV virus gene missense mutation. J Theor Biol 2005;235:555–65 [DOI] [PubMed] [Google Scholar]
- 10.Takamiya O, Kimura S. Molecular mechanism of dysfunctional factor VII associated with the homozygous missense mutation 331Gly to Ser. Thromb Haemost 2005;93:414–19 [DOI] [PubMed] [Google Scholar]
- 11.Moroy G, Martiny VY, Vayer P, et al. Toward in silico structure-based ADMET prediction in drug discovery. Drug Discov Today 2012;17:44–55 [DOI] [PubMed] [Google Scholar]
- 12.Kalyaanamoorthy S, Chen YP. Structure-based drug design to augment hit discovery. Drug Discov Today 2011;16:831–9 [DOI] [PubMed] [Google Scholar]
- 13.Guido RV, Oliva G. Structure-based drug discovery for tropical diseases. Curr Top Med Chem 2009;9:824–43 [DOI] [PubMed] [Google Scholar]
- 14.Lundstrom K. An overview on GPCRs and drug discovery: structure-based drug design and structural biology on GPCRs. Methods Mol Biol 2009;552:51–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nicola G, Abagyan R. Structure-based approaches to antibiotic drug discovery. Curr Protoc Microbiol 2009;Chapter 17:Unit17 2 [DOI] [PubMed] [Google Scholar]
- 16.Curran ME, Platero S. Diagnostics and personalized medicine: observations from the World Companion Diagnostics Summit. Pharmacogenomics 2011;12:465–70 [DOI] [PubMed] [Google Scholar]
- 17.Schmidt C. Challenges ahead for companion diagnostics. J Natl Cancer Inst 2012;104:14–15 [DOI] [PubMed] [Google Scholar]
- 18.Nikolcheva T, Jager S, Bush TA, et al. Challenges in the development of companion diagnostics for neuropsychiatric disorders. Expert Rev Mol Diagn 2011;11:829–37 [DOI] [PubMed] [Google Scholar]
- 19.Ross JS. Cancer biomarkers, companion diagnostics and personalized oncology. Biomark Med 2011;5:277–9 [DOI] [PubMed] [Google Scholar]
- 20.Becker R, Jr, Mansfield E. Companion diagnostics. Clin Adv Hematol Oncol 2010;8:478–9 [PubMed] [Google Scholar]
- 21.Hoggatt J. Personalized medicine—trends in molecular diagnostics: exponential growth expected in the next ten years. Mol Diagn Ther 2011;15:53–5 [DOI] [PubMed] [Google Scholar]
- 22.Parkinson DR, Johnson BE, Sledge GW. Making personalized cancer medicine a reality: challenges and opportunities in the development of biomarkers and companion diagnostics. Clin Cancer Res 2012;18:619–24 [DOI] [PubMed] [Google Scholar]
- 23.Spencer DH, Bubb KL, Olson MV. Detecting disease-causing mutations in the human genome by haplotype matching. Am J Hum Genet 2006;79:958–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ware JS, Walsh R, Cunningham F, et al. Paralogous annotation of disease-causing variants in Long QT syndrome genes. Hum Mutat 2012;33:1188–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wei X, Ju X, Yi X, et al. Identification of sequence variants in genetic disease-causing genes using targeted next-generation sequencing. PLoS One 2011;6:e29500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sanders SS. Large-scale sequencing to identify disease causing variants in X-linked mental retardation. Clin Genet 2010;77:35–6 [DOI] [PubMed] [Google Scholar]
- 27.Zhang Z, Wang L, Gao Y, et al. Predicting folding free energy changes upon single point mutations. Bioinformatics 2012;28:664–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Boeckler FM, Joerger AC, Jaggi G, et al. Targeted rescue of a destabilized mutant of p53 by an in silico screened drug. Proc Natl Acad Sci USA 2008;105:10360–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ray SS, Nowak RJ, Brown RH, et al. Jr Jr. Small-molecule-mediated stabilization of familial amyotrophic lateral sclerosis-linked superoxide dismutase mutants against unfolding and aggregation. Proc Natl Acad Sci USA 2005;102:3639–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Grant BJ, Lukman S, Hocker HJ, et al. Novel allosteric sites on Ras for lead generation. PLoS One 2011;6:e25711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sun YC, Veenstra DL, Kollman PA. Free energy calculations of the mutation of Ile96–>Ala in barnase: contributions to the difference in stability. Protein Eng 1996;9:273–81 [DOI] [PubMed] [Google Scholar]
- 32.Wickstrom L, Gallicchio E, Levy RM. The linear interaction energy method for the prediction of protein stability changes upon mutation. Proteins 2012;80:111–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dehouck Y, Kwasigroch JM, Gilis D, et al. PoPMuSiC 2.1: a web server for the estimation of protein stability changes upon mutation and sequence optimality. BMC Bioinformatics 2011;12:151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Van Durme J, Delgado J, Stricher F, et al. A graphical interface for the FoldX forcefield. Bioinformatics 2011;27:1711–2 [DOI] [PubMed] [Google Scholar]
- 35.Schymkowitz J, Borg J, Stricher F, et al. The FoldX Web Server: an online force field. Nucleic Acids Res 2005;33(Web Server issue):W382–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kang S, Chen G, Xiao G. Robust prediction of mutation-induced protein stability change by property encoding of amino acids. Protein Eng Des Sel 2009;22:75–83 [DOI] [PubMed] [Google Scholar]
- 37.Casadio R, Compiani M, Fariselli P, et al. Predicting free energy contributions to the conformational stability of folded proteins from the residue sequence with radial basis function networks. Proc Int Conf Intell Syst Mol Biol 1995;3:81–8 [PubMed] [Google Scholar]
- 38.Potapov V, Cohen M, Schreiber G. Assessing computational methods for predicting protein stability upon mutation: good on average but not in the details. Protein Eng Des Sel 2009;22:553–60 [DOI] [PubMed] [Google Scholar]
- 39.Gromiha MM, Huang LT. Machine learning algorithms for predicting protein folding rates and stability of mutant proteins: comparison with statistical methods. Curr Protein Pept Sci 2011;12:490–502 [DOI] [PubMed] [Google Scholar]
- 40.Khan S, Vihinen M. Performance of protein stability predictors. Hum Mutat 2010;31:675–84 [DOI] [PubMed] [Google Scholar]
- 41.Zhang Z, Teng S, Wang L, et al. Computational analysis of missense mutations causing Snyder-Robinson syndrome. Hum Mutat 2010;31:1043–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang Z, Norris J, Schwartz C, et al. In silico and in vitro investigations of the mutability of disease-causing missense mutation sites in spermine synthase. PloS one 2011;6:e20373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Witham S, Takano K, Schwartz C, et al. A missense mutation in CLIC2 associated with intellectual disability is predicted by in silico modeling to affect protein stability and dynamics. Proteins 2011;79:2444–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bava KA, Gromiha MM, Uedaira H, et al. ProTherm, version 4.0: thermodynamic database for proteins and mutants. Nucleic Acids Res 2004;32(Database issue):D120–1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen ZY, Shie JL, Tseng CC. Gut-enriched Kruppel-like factor represses ornithine decarboxylase gene expression and functions as checkpoint regulator in colonic cancer cells. J Biol Chem 2002;277:46831–9 [DOI] [PubMed] [Google Scholar]
- 46.Alexov E, Gunner MR. Modeling the first electron transfer from QA to QB in reaction centers from the bacteria Rb. Biophysical Journal 1999;76:A240–A40 [Google Scholar]
- 47.Alexov EG, Gunner MR. Incorporating protein conformational flexibility into the calculation of pH-dependent protein properties. Biophysical Journal 1997;72:2075–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Georgescu RE, Alexov EG, Gunner MR. Combining conformational flexibility and continuum electrostatics for calculating pK(a)s in proteins. Biophys J 2002;83:1731–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Teng S, Madej T, Panchenko A, et al. Modeling effects of human single nucleotide polymorphisms on protein-protein interactions. Biophys J 2009;96:2178–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ren P, Ponder JW. Tinker polarizable atomic multipole force field for proteins. Abstr Pap Am Chem S 2002;224:U473–U73 [Google Scholar]
- 51.Lagorce D, Maupetit J, Baell J, et al. The FAF-Drugs2 server: a multi-step engine to prepare electronic chemical compound collections. Bioinformatics 2011;27:2018–20 [DOI] [PubMed] [Google Scholar]
- 52.Miteva MA, Guyon F, Tuffery P. Frog2: Efficient 3D conformation ensemble generator for small compounds. Nucleic Acids Res 2010;38(Web Server issue):W622–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sandeep G, Nagasree KP, Hanisha M, et al. AUDocker LE: A GUI for virtual screening with AUTODOCK Vina. BMC Res Notes 2011;4:445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem 2010;31:455–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wiest L, Pegg AE. Assay of spermidine and spermine synthases. Methods Mol Biol 1998;79:51–7 [DOI] [PubMed] [Google Scholar]
- 56.Shirahata A, Takahashi N, Beppu T, et al. Effects of inhibitors of spermidine synthase and spermine synthase on polyamine synthesis in rat tissues. Biochem Pharmacol 1993;45:1897–903 [DOI] [PubMed] [Google Scholar]
- 57.Wu H, Min J, Zeng H, et al. Crystal structure of human spermine synthase: implications of substrate binding and catalytic mechanism. J Biol Chem 2008;283:16135–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Korolev S, Ikeguchi Y, Skarina T, et al. The crystal structure of spermidine synthase with a multisubstrate adduct inhibitor. Nat Struct Biol 2002;9:27–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schwartz CE, Wang X, Stevenson RE, et al. Spermine synthase deficiency resulting in X-linked intellectual disability (Snyder-Robinson syndrome). Methods Mol Biol 2011;720:437–45 [DOI] [PubMed] [Google Scholar]
- 60.Becerra-Solano LE, Butler J, Castaneda-Cisneros G, et al. A missense mutation, p.V132G, in the X-linked spermine synthase gene (SMS) causes Snyder-Robinson syndrome. Am J Med Genet A 2009;149A:328–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.de Alencastro G, McCloskey DE, Kliemann SE, et al. New SMS mutation leads to a striking reduction in spermine synthase protein function and a severe form of Snyder-Robinson X-linked recessive mental retardation syndrome. J Med Genet 2008;45:539–43 [DOI] [PubMed] [Google Scholar]
- 62.Meng X, Wang G, Viero C, et al. CLIC2-RyR1 interaction and structural characterization by cryo-electron microscopy. J Mol Biol 2009;387:320–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cromer BA, Gorman MA, Hansen G, et al. Expression, purification, crystallization and preliminary X-ray diffraction analysis of chloride intracellular channel 2 (CLIC2). Acta Crystallogr Sect F Struct Biol Cryst Commun 2007;63(Pt 11):961–3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cromer BA, Gorman MA, Hansen G, et al. Structure of the Janus protein human CLIC2. J Mol Biol 2007;374:719–31 [DOI] [PubMed] [Google Scholar]
- 65.Heiss NS, Poustka A. Genomic structure of a novel chloride channel gene, CLIC2, in Xq28. Genomics 1997;45:224–8 [DOI] [PubMed] [Google Scholar]
- 66.Witham S, Takano K, Schwartz C, et al. A missense mutation in CLIC2 associated with intellectual disability is predicted by in silico modeling to affect protein stability and dynamics. Proteins 2011;79:2444–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Takano K, Liu D, Tarpey P, et al. An X-linked channelopathy with cardiomegaly due to a CLIC2 mutation enhancing ryanodine receptor channel activity. Hum Mol Genet 2012;21:4497–507 [DOI] [PMC free article] [PubMed] [Google Scholar]