

Abstract
Background
While genome-wide association studies (GWAS) of complex traits have revealed thousands of reproducible genetic associations to date, these loci collectively confer very little of the heritability of their respective diseases and, in general, have contributed little to our understanding the underlying disease biology. Physical protein interactions have been utilized to increase our understanding of human Mendelian disease loci but have yet to be fully exploited for complex traits.
Methods
We hypothesized that protein interaction modeling of GWAS findings could highlight important disease-associated loci and unveil the role of their network topology in the genetic architecture of diseases with complex inheritance.
Results
Network modeling of proteins associated with the intragenic single nucleotide polymorphisms of the National Human Genome Research Institute catalog of complex trait GWAS revealed that complex trait associated loci are more likely to be hub and bottleneck genes in available, albeit incomplete, networks (OR=1.59, Fisher's exact test p<2.24×10−12). Network modeling also prioritized novel type 2 diabetes (T2D) genetic variations from the Finland–USA Investigation of Non-Insulin-Dependent Diabetes Mellitus Genetics and the Wellcome Trust GWAS data, and demonstrated the enrichment of hubs and bottlenecks in prioritized T2D GWAS genes. The potential biological relevance of the T2D hub and bottleneck genes was revealed by their increased number of first degree protein interactions with known T2D genes according to several independent sources (p<0.01, probability of being first interactors of known T2D genes).
Conclusion
Virtually all common diseases are complex human traits, and thus the topological centrality in protein networks of complex trait genes has implications in genetics, personal genomics, and therapy.
Keywords: Protein Interactions, SNPs, Complex Disease Inheritance, Adult Onset Diabetes, Crohn's Disease
Introduction
In spite of the vast number of reproducibly associated polymorphisms that genome-wide association studies (GWAS) have found for complex traits, their underlying biological mechanisms remain elusive, and they have in many cases not been able to achieve their intended goal: to discover and understand the functional underpinnings of complex traits. Moreover, while those studies have contributed to a comprehensive catalog of disease-associated loci (http://www.genome.gov/gwastudies), the polymorphisms each contribute only marginally to the heritability of the disease1 and have afforded us little new knowledge of the trait's key functional mechanisms.
To address this issue, two approaches have been used to increase statistical power and unveil additional single nucleotide polymorphisms (SNP) buried in the large lists of polymorphisms published in GWAS. The predominantly data-driven approach consists of the integration or meta-analysis of multiple GWAS datasets comprising tens of thousands of samples.2 The second approach, knowledge-driven, consists of incorporating external mechanistic facts into enhanced statistical genetic models before the GWAS case–control analysis. SNP have thus been prioritized using biological functions,3 canonical pathways4–8 or expression quantitative trait loci (eQTL) associations.9 10 Surprisingly, protein interactions have not yet been fully exploited for characterizing SNP mechanistically for complex diseases. So far, they have been implicated in the inter-trait modularity of Mendelian disorders,11 cancer,12 and have successfully predicted novel disease genes from the interaction partners of known causal disease genes cataloged in the online Mendelian inheritance in man (OMIM).13 Conclusively, protein interactions and network modeling are thought to be key techniques for shedding new light on the poorly understood complex genetic basis of many common disorders.14
The protein interaction networks help reveal the importance of disease-associated variations, their host genes, protein products and interacting partners. Topologically central genes or proteins characterized as ‘hubs’, or ‘bottlenecks’ are those within a network defined as the top most connected proteins or those with the topmost ‘betweenness scores’,15 respectively (cut-off of 20%). Furthermore, proteins that lie close to each other in a network are more likely to have similar functions16 and further support the protein interaction networks (PIN) analysis of variations associated with complex traits with poorly understood genetic etiology. First interactors of known disease or phenotype genes have been shown to be more likely to be involved in the same disease or biological process,13 17 and thus mutations in genes interacting with disease genes tend to lead to similar disease phenotypes.18 Therefore, direct protein interactions or first interactors can be used to estimate functional linkage between novel candidate disease genes and those with established pathophysiological biology.
Studies have shown that shared genes associated with multiple diseases (pleiotropic effects) are more topologically central than specific genes associated with only one disease.19 Cancer disease genes tend to encode in central hubs of highly interconnected modules within protein–protein interaction networks.20 Those findings suggest that at least some types of disease genes may possess some topology characteristics in protein–protein interactions. Here, we hypothesized that protein interaction modeling of GWAS findings, especially the topological features of proteins in the network, could help unveil the genetic architecture of complex diseases. We first confirm the topological centrality of trait-associated SNP found in the National Human Genome Research Institute (NHGRI) online catalog, which contains significant variations published in GWAS.21 Second, we propose a novel predictive network modeling of protein interactions that reprioritizes intragenic SNP associated with a complex disorder according to GWAS, and using conservative empirical controls, we computationally identify unexpected pairs of interactors in type 2 diabetes (T2D). We evaluate the predicted SNP against an independent gold standard, and confirm a significantly increased OR of correct findings among network-reprioritized SNP compared to the original GWAS prioritization. Furthermore, we evaluate the topology of the predicted network constructed from available, albeit incomplete sources, and show increased presence of hub and bottleneck proteins of the host genes consistent with properties described in the NHGRI catalog of SNP–trait associations. Therefore, the molecular mechanisms supporting the contribution of protein interactions to complex traits are systematically identified. The results, reported here, strongly imply that the statistical signals inferred from GWAS have not yet been exhausted and can be revealed without requiring massive increased sample sizes.
Methods
Supplementary table S10 (available online only) recapitulates the abbreviations and key concepts (eg, host genes, etc) also defined in this paper at their first occurence.
Datasets used in this study
GWAS and protein interaction datasets and their preparations are summarized in tables 1 and 2, respectively. The preparation of gold standard GWAS SNP utilized to select the optimal network model of protein interactions is described below in the ‘Gold standard’ section of the Methods. Finally, the independent non-GWAS sources of biologically validated T2D genes are described in table 3.
Table 1.
GWAS datasets, intragenic SNP and their corresponding host genes
| Sources | Samples | Genetic | |||
|---|---|---|---|---|---|
| Cases | Controls | Intragenic SNP (out of total SNP) | Host genes | ||
| T2D GWAS | FUSION | 1161 | 1174 | 137 248 (315 635) | 16 711 |
| WTCCC | 2000 | 3000 | 187 842 (447 306) | 15 367 | |
| IBDGC GWAS | IBDGCi | 561 | 563 | 134 257 (306 835) | 16 539 |
| IBDGCii | 407 | 432 | |||
Two type 2 diabetes (T2D) genome-wide association study (GWAS) datasets are used, Finland–USA Investigation of NIDDM Genetics (FUSION),22 and Wellcome Trust Case Control Consortium (WTCCC).23 A third GWAS dataset is used, the Inflammatory Bowel Disease Genetics Consortium (IBDGC) with non-Jewish, European ancestry and with Jewish ancestry. Results for IBDGC GWAS were obtained using database of genotypes and phenotypes (dbGAP; executable file dbGaP.archive.4825.part1.exe).24 All single nucleotide polymorphism (SNP) flat files were downloaded on October 19, 2009 from the single nucleotide polymorphism database (dbSNP), (ftp://ftp.ncbi.nih.gov/snp/organisms/human_9606/ASN1_flat/) and the SNP were extracted along with their corresponding HUGO gene symbols and functions in order to map each SNP to a host gene. The host genes of intragenic SNP were defined by genomic boundaries extending from 200 kb upstream (5′ side) to 0.5 kb downstream (3′ side) of the gene. All files contained 306 835 SNP in total and their corresponding p values, ranging from 2.6×10−16 to 1. All SNP were ranked according to each GWAS prioritization—no individual data were required for this study. Each host gene can be paired with one or more intragenic SNP, which were assigned to a host gene by selecting the best GWAS-ranked SNP p value among all SNP annotated to the gene. GWAS-ranked host genes harboring the GWAS-ranked SNP were systematically organized into overlapping sets by selecting the top 25, 50, 100, 150, 250, 500, 600, 700, 800, 1000, and 1100 ranked genes (host gene cut-off) to compose each set, respectively.
Table 2.
Protein interaction datasets
| Protein interaction datasets details | |
|---|---|
| Retrieved from | http://string.embl.de |
| STRING versions used | V.6.3 (Jan 2007) |
| V.8.2 (May 2010) | |
| Resulting dataset | 14 025 Distinct human proteins |
| 492 087 Protein–protein interactions |
The protein interaction networks is downloaded using the search tool for the retrieval of interacting genes, STRING.26 To ensure the independence of the network from the published genome-wide association study results, protein interactions derived only from text mining were removed (see supplementary methods, available online only).
Table 3.
Independent sources (non-GWAS) used for validation of SPAN-reprioritized SNP (enrichment statistics of figure 5C)
| Independent (non–GWAS) sources of T2D genes | Retrieved from | Dataset details |
|---|---|---|
| OMIM | 97 T2D-annotated genes, by querying NIDDM (MIM ID 125853; December 14, 2010) | See supplementary tables S5 and S6 (available online only) |
| IPA | http://www.ingenuity.com; V.9.0 | See supplementary table S6 (available online only) |
| Literature evidence | Pubmed | See supplementary table S6 (available online only) |
| Literature evidence for biological processes | KEGG; GO; Reactome http://www.genome.jp/kegg/ http://www.geneontology.org/ http://www.reactome.org/ |
See supplementary table S6 (available online only) |
The host genes within the optimal SPAN model of type 2 diabetes (T2D) were analyzed for enrichment in 97 T2D reported genes within online Mendelian inheritance in man (OMIM).27 Eighty-three of these genes were also found within the protein interaction database (see supplementary table S5, available online only), and used as a basis for enrichment evaluation using Fisher's exact test (figure 5C). Such a Fisher's exact test enrichment evaluation was also conducted against the T2D genes curated in the ingenuity pathway analysis application (IPA, figure 5C), and the hubs and bottleneck genes of the overall protein interaction network (figure 6). We qualitatively annotated the potential T2D-related functions of host genes within the optimal SPAN model of T2D from a review of the literature and canonical pathways in the Kyoto encyclopedia of genes and genomes (KEGG),28 gene ontology (GO)29 and Reactome30 (figure 5C and see supplementary table S6, available online only).
GWAS, genome-wide association studies; NIDDM, non-insulin-dependent diabetes mellitus; SNP, single nucleotide polymorphism.
Experimental design
First, we demonstrate that topological protein network properties from available, albeit incomplete, networks are significantly enriched in complex disease traits. This finding serves to establish the merit of developing and evaluating a predictive model of complex trait polymorphisms anchored on protein interactions. Next, we describe in detail how we extend the SPAN model12 to reprioritize statistically unexpected host gene interactions among modestly ranked intragenic SNP of GWAS. This method is evaluated with three independent GWAS involving two different complex traits in order to show its robustness and reproducibility. Third, we report the GWAS SNP reprioritized by a genetically constrained protein interaction model of T2D derived from independent SPAN analyses of two GWAS (outlined in figure 1). We further evaluate these results according to different network centrality metrics of these reprioritized SNP host genes: hubness, bottleneckness, their overlap with validated T2D genes discovered by biologists, and their number of direct interactors to the host genes of validated T2D SNP that were not used to select the optimal model.
Figure 1.

Outline for prioritizing single nucleotide polymorphisms (SNP) using protein network modeling. This figure provides details on the empirical control of the SPAN network modeling, and the reprioritized SNP in the network models. Step 1: The entire set of SNP measured on a genome-wide association studies (GWAS) array is annotated with respect to host genes as intragenic or intergenic using dbSNP129. Step 2: The p values of the observed ranked host genes are determined based on the lowest p value of the GWAS of origin being assigned to each host gene that may have more than one SNP (Wellcome Trust Case Control Consortium (WTCCC), Finland–USA Investigation of Non-Insulin-Dependent Diabetes Mellitus Genetics (FUSION) or Inflammatory Bowel Disease Genetics Consortium). Step 3: For host genes of the observed ranked list that can be mapped to protein interactions, the observed interactions are carried at different cut-offs (n=i) each yielding a different network model (see Methods section). Step 4: In parallel, 1000 control bootstraps are constructed at each cut-off of the ranked gene list in the following way: SNP are sampled from the total number of SNP in the array and assigned the corresponding p value of the observed distribution for that rank. The intragenic SNP among them are assembled at different cut-offs to generate an equal number of distinct host genes as the observed set at that cut-off. The lowest GWAS p value is assigned to each host gene. Step 5: In each bootstrap, the number of first interactors of host genes from the control set is calculated in the same way as previously described in step 3 for the observed set (at each cut-off). Finally, the ‘SPAN analysis’ produces a reprioritized p value for each SNP associated with a host gene of the observed set using the observed node degree of a host gene at a certain cut-off and its corresponding empirical control. PIN, protein interaction networks.
Empirical control for protein network model
To conduct an empirical control for T2D network analysis, intragenic SNP were resampled (1000 bootstraps) to create empirical SNP lists and derived their corresponding list of host genes (figure 1 and see supplementary methods, available online only).
Protein interaction network modeling: using SPAN to reprioritize GWAS-ranked host genes
Protein network models are constructed by analyzing the protein interactions among GWAS-ranked host genes at different cut-offs (figure 1 and see supplementary table S1, available online only). We used SPAN,12 25 to prioritize host genes by estimating the expected frequency (p value) of single (individual) protein interactions occurring between these top GWAS-ranked host genes using empirical controls. In particular, GWAS-ranked host genes from each GWAS were individually analyzed using SPAN and controlled by bootstrapping SNP. Briefly, SPAN is a network model in which the observed number of interactions between the host genes of GWAS-prioritized intragenic SNP is compared with an expected distribution through permutation resampling (see supplementary methods, available online only). First, we calculated the likelihood of obtaining the same number of interactions in the empirical distribution that were found for each host gene in the observed network (empirical SPAN frequency). Then, GWAS-ranked host genes contained in each set created at each cut-off were reprioritized using this calculated empirical SPAN frequency. By design, this model is controlled for topological properties such as bottleneckness and hubness. The resulting statistically significant genes at the best cut-off were then aggregated to form an ‘optimal network model of the GWAS’.
Reference trait-associated SNP and gold standards
SNP–trait associations data, identified for T2D and Crohn's disease (independently) from GWAS, were collected from the NHGRI online catalog (http://www.genome.gov/gwastudies/; excel format on 5 November 2010),21 and then intragenic SNP are determined (figure 2). The set of selected intragenic T2D SNP were used to assess the accuracy of network models constructed for the Wellcome Trust Case Control Consortium (WTCCC) and the Finland–USA Investigation of Non-Insulin-Dependent Diabetes Mellitus Genetics (FUSION), respectively (figure 2, see supplementary tables S2 and S3, available online only). The set of selected intragenic Crohn's disease in the Inflammatory Bowel Disease Genetics Corsortium (IBDGC) GWAS platform as well as in the PIN database was used to assess the accuracy of our Crohn's network models (see supplementary table S4, available online only).
Figure 2.

Construction of the gold standard of single nucleotide polymorphisms (SNP) from type 2 diabetes (T2D)-associated SNP obtained from independent genome-wide association studies (GWAS) for validation of our prioritized SNP. Step 1: All SNP present in the National Human Genome Research Institute (NHGRI) GWAS catalog were taken as an initial step. Step 2: SNP were filtered based on their associated trait to select only T2D-associated SNP. Among 76 SNP, 42 are intragenic SNP and correspond to 22 host genes according to dbSNP annotations. Step 3: dbSNP annotations were used to determine genomic locations of the SNP and select only the intragenic ones. Step 4: From the list of intragenic T2D SNP, only the SNP that met the following criteria were selected: they existed in both the protein interaction networks (PIN) and the respective GWAS of interests’ platform (Wellcome Trust Case Control Consortium (WTCCC) Affymetrix GeneChip 500 k and Finland–USA Investigation of Non-Insulin-Dependent Diabetes Mellitus Genetics (FUSION) Illumina Infinium II Human Hap 300 Beadchip V.1.0) and were selected as T2D-associated SNP by another GWAS.
Optimal SPAN model of T2D and its evaluation
For each GWAS, the best OR was used to determine the optimal network model and its associated parameters (host gene cut-off, empirical SPAN frequency; figure 5B,C). In order to improve statistical power the optimal network models of FUSION and WTCCC were combined and we quantitatively evaluated the relevant reprioritized host genes within the optimal SPAN model of T2D in three ways: their probability of being first interactors of known T2D genes (empirical edgetic p value; see supplementary methods and table S10, available online only), the enrichment of T2D reported genes from authoritative databases, and their enrichment in hubs and bottleneck genes.
Figure 5.

Validated type 2 diabetes (T2D) genes and their first interactors are enriched in the optimal SPAN model of T2D. Optimal SPAN modeling independently prioritized 12 genes in Finland–USA Investigation of Non-Insulin-Dependent Diabetes Mellitus Genetics (FUSION) (A) and 10 in Wellcome Trust Case Control Consortium (WTCCC) (B). (C) The combined optimal SPAN Model of T2D contains 21 genes, 12 from FUSION (green), 10 from WTCCC (blue), where one gene (KCNJ11) is shared and is a known T2D gene from the National Human Genome Research Institute (NHGRI) catalog (see supplementary table S1, available online only). Aside from the obvious relatedness between these studies via KCNJ11, we identify three additional interactions between the independently prioritized networks: PIN4 and MAP3K1 from FUSION interact with CDC42 of WTCCC, while DLL1 from FUSION interacts with LFNG of WTCCC with statistical significance (p=0.0041, FDR=0.0001). Interestingly, these biomolecular networks were already the most connected within their respective studies (eg, within FUSION: DLL1–KCNJ10–KCNJ11 and MAP3K1–AXIN1; within WTCCC: ZNF284–CDC42–ARHGEF12). (D) The prioritized model comprises more first interactors of known T2D genes reported in the NHGRI than predicted using conservative control (empirical distribution p =0.015, see Methods section). These interactions to known NHGRI T2D genes as a proxy evaluation are reported to illustrate the biological functional relatedness to T2D, as NOTCH2, KCNQ1 and PPARG were not among host genes of NHGRI known T2D single nucleotide polymorphisms that prioritized this network (see Methods section, figure 2). Panel (E) illustrates that known T2D genes are enriched in the optimal SPAN model of T2D. The functions of the 21 genes were also evaluated against different types of T2D datasets independent from the NHGRI: including online Mendelian inheritance in man (OMIM), ingenuity pathway analysis, and in manually curated literature sources (see supplementary table S6, available online only). Among 83 T2D genes reported in OMIM, two were significantly enriched in the prioritized network (OR 29.5, p=0.003). Among the five genes that directly interact with NHGRI T2D gold standard genes, three have been reported in the literature as associated to T2D. Two genes, KCNJ11 and MAP3K1, were confirmed to be T2D genes by OMIM (OR 29.5, p=0.003, Fisher's exact test) and canonical pathway analysis (IPA ‘type ii diabetes mellitus signaling’, p=0.0004). In addition, an intensive review of the literature provided evidence for T2D associations for eight genes (ARHGEF12, AXIN1, KCNJ10, MAP3K1, CREM, TACR3, SNAP29 and ZBTB17) and T2D biological process associations for two genes (PROK2, obesity; TRIM7, glycogen metabolism). One gene, ART1, was reported in a type 1 diabetes (T1D)-related study, highlighting the possibility of T1D contamination by incorrect diagnosis in the original GWAS or the potential for overlapping etiology in some subset of patients.
Hub and bottleneck enrichment analysis of SNP sets
The hubness or bottleneckness of an intragenic SNP was defined based on the connectivity of its host gene where hubs are defined as those genes within the top 20% of degree distribution (node degree) and bottlenecks as genes in the top 20% of betweenness scored proteins using an established algorithm (http://www.gersteinlab.org/proj/bottleneck/).15 To investigate whether hub and bottleneckness properties are more likely to be enriched in complex disease traits, we utilized the entire set of SNP in the NHGRI catalog of SNP–trait associations. The enrichment is tested using Fisher's exact test and for each intragenic SNP in dbSNP two criteria were used: whether the SNP is cataloged in NHGRI (or GWAS for T2D and Crohn's); and whether the host gene of the SNP is a hub and/or bottleneck gene. The background SNP utilized in this study are from dbSNP.
Results
Host genes of complex trait-associated SNP are enriched in hub and bottleneck centrality properties in currently available networks
We conducted a thorough analysis of hubness and bottleneckness for complex trait-associated intragenic SNP and report these two topological properties for the 1390 trait SNP from the NHGRI GWAS catalog21 (see Methods section and figure 3). Conclusively, complex trait SNP are significantly more likely to lie in hub genes (OR 1.59, p=2.24×10−12; Fisher's exact test) and bottleneck genes (OR 1.56, p=1.90×10−12; Fisher's exact test) when using the human intragenic SNP of dbSNP whose host genes occur in the PIN as a background, albeit these networks comprise some ascertainment-biased interactions that may favor this hypothesis (although we systematically removed publication-based interactions). Considering that hub and bottleneck proteins have been associated with several key biological functions and gene expression dynamics,15 it is not surprising that complex trait-associated SNP are enriched in these network topology properties.
Figure 3.

National Human Genome Research Institute (NHGRI) intragenic single nucleotide polymorphisms (SNP) are enriched in hub and bottleneck host genes. This figure compares the proportion of intragenic complex trait SNP for which the corresponding imputed host gene protein determined to be a hub or bottleneck using the NHGRI catalog as a control. In addition, intragenic SNP from the Illumina HumanMap 300 Beadchips and from HapMap population of Utah residents with northern and eastern European ancestry from the CEPH collection (CEU) were also used as controls and were binned according to their minor allelic frequency. (A) The proportion of complex trait SNP that reside in hub genes is significantly higher than that of the two controls. At least 23.02% of NHGRI SNP are in hub genes compared with 16% of SNP, at most, in the Illumina HumanMap 300 Beadchips and in HapMap (p=8.8×10−8 Mann–Whitney U test), shown by a dashed line. (B) The proportion of complex trait associated SNP in bottleneck genes is also significantly higher than that of the two controls. 26% of NHGRI SNP reside in bottleneck genes while 19%, at most, do so in the background platforms (dashed line, p=5.2×10−8, Mann–Whitney U test). Similar enrichment results seen for that of the 20% cut-off have been obtained for three other cut-offs, specifically at 5%, 10% and 30% (data not shown), demonstrating the robustness of these findings. Importantly, the proportion of hub and bottleneck host genes is consistent regardless of minor allele frequency (data not shown). To examine the difference in the enrichment of centrality genes found for single gene disorders and complex traits, we compared the genes found in representative datasets for each type of disorder: Online Mendelian inheritance in man and the NHGRI catalog, respectively. Complex disease genes were less likely to be central genes in a network than single gene inheritance disease genes when using the standard 20% cut-off for hub and bottleneck genes, as well as at smaller and larger cut-offs as mentioned above (data not shown).
GWAS SNP reprioritized by protein interaction models are more likely to be validated in ulterior studies
Trait-associated intragenic SNP prioritized by GWAS are stratified according to their original GWAS rank and are translated into their host genes that serve to constrain the protein interaction modeling genetically (figures 4 and 5 and see supplementary methods, available online only). The statistical likelihood, or empirical SPAN frequency, of obtaining the number of observed interactors for each host gene in the genetically constrained network was introduced as a second parameter. The empirical SPAN frequency is equal to the number of times the exact (or greater) node degree (count of direct protein interactors among the prioritized host genes of SNP) for any given protein is found in an empirical distribution made of a 1000 random samplings of GWAS intragenic SNP (converted to host genes) that is the same node degree found in the actual given gene set.
Figure 4.

Genome-wide association studies (GWAS) single nucleotide polymorphisms (SNP) reprioritized by protein network models are more predictive of trait-sssociated SNP. SPAN analysis repeatedly identifies secondary ranked GWAS host genes at a reproducible cut-off. In particular, at a cut-off of GWAS-ranked host genes equal to 600 and with an empirical SPAN frequency of 0.1% or less, SPAN obtained the maximum OR in both type 2 diabetes (T2D) studies. The OR (y-axis) correspond to the probability of identifying a gold standard SNP among SNP prioritized either in the original unmodified GWAS-ranked SNP (dotted black line) or those reprioritized by SPAN network models (colored lines) under different cut-offs of GWAS-ranked host genes that serve as inputs to the models (x-axis, see Methods section) in datasets Finland–USA Investigation of Non-Insulin-Dependent Diabetes Mellitus Genetics (FUSION) (A), Wellcome Trust Case Control Consortium (WTCCC), and Inflammatory Bowel Disease Genetics Consortium (IBDGC) (C). The gold standard is derived from trait-associated SNP of the National Human Genome Research Institute collection that were discovered and validated in an ulterior and independent GWAS. The number of SNP in the GWAS corresponding to each host gene cut-off is shown in parenthesis of the x-axis (eg, a host gene may have more than one associated SNP within the cut-off range, see supplementary methods, available online only). We ranked the host genes according to the lowest trait-associated probability among their corresponding ranked SNP at a certain cut-off. Furthermore, the likelihood of finding the number of observed interactions for each host gene in the list is calculated in each network as a ‘frequency’ by empirical models and the threshold is used to produce various models is illustrated by different colored lines. SPAN analysis robustly and reproducibly reprioritized gold standard genes when varying the protein interaction confidence criteria (see supplementary figure S1, available online only), and when defining more or less SNP as intragenic upstream and downstream of the host gene (see supplementary figure S2, available online only). Furthermore, SPAN outperformed straightforward pathway enrichment at different cut-offs (see supplementary figure S3, available online only).
We analyzed two independent T2D GWAS, FUSION22 and WTCCC.23 In FUSION, the OR of the control was highest at 150 GWAS-ranked host genes, and from 250 on showed a linear decrease with respect to the size of the GWAS-ranked host gene set (figure 4A). From 500 on, every subsequent SPAN model improved the GWAS-ranked SNP OR. The SPAN model at 600 GWAS-ranked host genes with a frequency of 0.1% or less yielded the best OR of 2512 (10-fold increase; p=0.00059; Fisher's exact test). In WTCCC, the OR of the baseline control showed linear decreases with respect to the size of the GWAS-ranked host gene set in between each peak at 50, 500 and 800 (figure 4B). From 600 host genes on, the SPAN model with a frequency of 0.1% or less yielded the best OR of 1585 (p=0.00073; Fisher's exact test), with 46% improvement compared to the baseline. We also examined the Crohn's disease GWAS data to determine if the T2D GWAS result is trait specific or could also be extend to this disorder. A SPAN model of Crohn's disease at 700 showed the best accuracy for re-capturing gold standards with the OR of 3981 for IBDGC (figure 4C). We selected the model at 600 GWAS-ranked host genes with a frequency of 0.1% or less as the optimal network as it consistently yielded the best OR. This model contains 12, 10, and one host gene(s) from FUSION (figure 5A), WTCCC (figure 5B), and KCNJ11 (rs5215), which is common between both studies (see supplementary tables S1 and S7, available online only). Taken together, these results demonstrate the scalability of the network modeling and we also conducted additional studies to demonstrate its robustness (see supplementary figures S1 and S2B, available online only).
SPAN reprioritized genes interact directly with known T2D genes
Interestingly, first interactors of known disease genes have been shown to be more likely to be true disease genes,13 17 18 and can serve as candidate genes for further analysis (figure 5C,D). Furthermore, closer distances between two proteins in a network have been shown to signify increased functional similarity.16 Therefore, we can infer that first interactors of known T2D genes are more likely to be related to the pathophysiology of the disease. We analyzed then our combined optimal network model for biological and functional relatedness. We also evaluated how many of our prioritized host genes were first interactors of NHGRI known T2D genes. Importantly, these genes were not used to prioritize our network; rather, they were considered as established T2D genes by the NHGRI, and served as markers for GWAS result reprioritization. In our combined optimal networks of WTCCC and FUSION (figure 5C and see supplementary table S8, available online only), five genes (KCNJ11, KCNJ10, LFNG, MAP3K1, and ZBTB17) were shown to interact directly with three gold standard genes (NOTCH2, KCNQ1, and PPARG, figure 5D). Using an empirical distribution of random interactions (edge) between 21 reprioritized host genes and 12 gold standards (13 known T2D genes reported in the NHGRI catalog excluding the overlapping gene KCNJ11), we calculated their probability of being first interactors of known T2D genes. In the optimal network model, five direct interactions with T2D gold standard genes were identified to be significant (0.0036<edgetic p<0.0538; see supplementary methods and table S9, available online only). This proxy estimation further supports our network model, since the candidate T2D associated genes connect directly with gold standards more than expected by chance. In addition, biologically or clinically validated T2D genes are enriched in the optimal SPAN model of T2D (figure 5E and see supplementary table S6, available online only).
Hub and bottleneck genes are enriched in the optimal T2D GWAS network
By definition, the overall protein interaction network includes 20% hubs and 20% bottleneck genes. In the optimal SPAN protein interaction network model of T2D, we observe 29% hubs and 33% bottleneck genes (figure 6). Therefore, the topological centrality of reprioritized host genes of the optimal T2D GWAS network concurs with the broader observation that hubs and bottlenecks are enriched in GWAS-associated complex diseases (figure 3).
Figure 6.
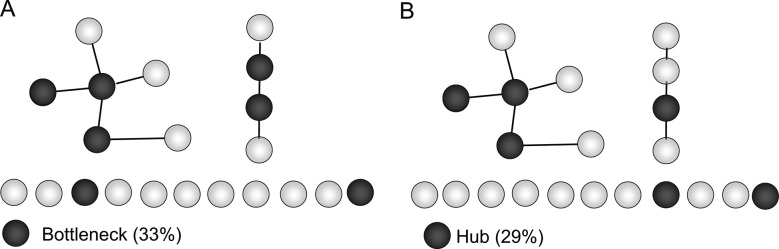
Hub and bottleneck genes are enriched in the optimal type 2 diabetes (T2D) genome-wide association studies (GWAS) network. The optimal SPAN model comprising reprioritized single nucleotide polymorphisms (SNP) and their corresponding host genes was determined using the Finland–USA Investigation of Non-Insulin-Dependent Diabetes Mellitus Genetics and Wellcome Trust Case Control Consortium GWAS (figure 4A,B, best OR). Reprioritized T2D-associated SNP are presented in supplementary table S1 (available online only) and their corresponding 21 host genes are illustrated in panels A and B as black when annotated as a hub or bottleneck, respectively. By definition, the entire set of 14 025 proteins (492 087 distinct interactions) in the protein interaction network contains 20% hubs and 20% bottleneck genes. The optimal T2D network model is significantly enriched in both hubs and bottlenecks and an empirical statistic was used to conservatively control for network topology (see Methods section). Therefore, the observed enrichment is supportive of an increased topological centrality among host genes of highly ranked GWAS SNP. OR and p values were calculated using Fisher's exact test.
Discussion
As GWAS reveal minimal biological mechanisms underlying discovered SNP, known molecular functions or pathways have been implemented as straightforward, high-throughput post-GWAS analyzes.31 Wang et al7 demonstrated that pathway approaches, which take into account multiple SNP, can provide additional value to GWAS data. Furthermore, Baranzini et al5 demonstrated that modestly prioritized SNP from two multiple sclerosis GWAS could reveal statistical associations at the canonical pathway level. In addition, pathway approaches have elucidated the potential utility of protein interaction analyses of GWAS data,7 which have been confirmed in yeast using eQTL data,10 32 and used to identify subnetworks that may contribute to complex human traits using text mining knowledge.33 Convincingly, the discovery of new biological modules is possible by utilizing protein interaction network approaches along with GWAS knowledge; however, no previous studies have provided insight into the network topology associated with complex traits.
Edgetic perturbation: a model for complex trait analysis
Zhong and colleagues11 established that an ample proportion of disease gene alterations, reported in OMIM, in fact cause changes in protein interactions, deemed edgetic perturbations, rather than complete protein (or node) removal. In our analysis, we extend this principle to examine the host genes of intragenic SNP that confer various effects on their final protein products. For example, synonymous or intronic SNP may cause aberrant alternative splicing,34 35 which has been shown to be an important mechanism in disease,36 37 and that may affect both expression levels and protein sequence. Furthermore, SNP may alter expression by being located in the promoter region or the 3′ untranslated region targeted by microRNA, which can decrease the stability or reduce the level of interaction enough to produce an edgetic change.11 Goldstein's group also demonstrated that prioritized SNP in GWAS may be markers of a common variant in linkage disequilibrium (LD) with the SNP, as well as a marker of rare variants creating synthetic associations that are attributed to common variants.38 We thus propose that SPAN prioritized intragenic SNP are possible markers of either mechanism: a common variant or rare genetic variants. Therefore, genetic alterations that underlie edgetic perturbations are not limited to non-synonymous mutations, but may also include those that cause alternative types of changes in protein functionality and interrelationships. In this vein, our method not only focuses on edgetic changes by examining protein interactions, but also incorporates this idea by analyzing intragenic SNP that cause various or unknown alterations including synonymous and non-synonymous mutations.
SPAN analysis repeatedly identifies secondary ranked GWAS host genes at a reproducible cut-off
Apart from the topmost prioritized SNP (generally top 10), it is now known that GWAS lack reproducibility of prioritized SNP that are ranked lower than 10 according to their GWAS p value. This indicates that the SNP ranking, taken in isolation, is not sufficiently precise (positive prediction value) to identify novel or important disease genes. Interestingly, the SPAN modeling method reproducibly identified two biomolecular modules comprising sets of interacting genes in two independent T2D GWAS (figure 5A,B). Based on edgetic and topological evaluations, the application of protein interaction information appears to benefit our understanding of T2D GWAS host genes that impact or are potentially true disease-related genes. We have shown that meaningful polymorphisms are likely to be present in secondary ranked signals (200–800 according to their GWAS p values among half a million SNP) identified by GWAS, and were consistently found in the same range for both complex traits. Using this approach and protein knowledge can bring us closer to developing a more comprehensive understanding of GWAS results by highlighting potentially essential disease-related signals.
SPAN results provide the rationale for investigating edgetic models of complex diseases
Our network model of WTCCC and FUSION T2D GWAS results has revealed 21 potential biological candidate T2D genes. As an example, three SPAN-prioritized genes were both supported by ulterior sources and were found to be first interactors of T2D gold standard genes (figure 5D), and thus may serve as quintessential T2D candidate genes: KCNJ10, MAP3K1 and ZBTB17. A comprehensive literature review for the 21 gene set revealed possible biological explanations for 13 genes (see supplementary table S6, available online only). In combination, our results suggest that examination of GWAS results using protein interactions may provide a theoretical stepping stone for gaining biological insight into complex diseases and may be useful for GWAS follow-up studies in general.
Enrichment of hubs and bottlenecks in complex traits and multiple controls
Focusing on edgetic functionality is supported by the theory that the importance of a hub is in fact related to the loss of a hub's essential protein interactions (edges), rather than the loss of a hub gene itself.39 Genes that have increased connectivity within the network confer greater biological effects, and are in fact three times more likely to be essential than those with a limited number of connections.40 However, depending on the biological scale of the measurement, the positive or negative association between diseases and hubness remains complex and controversial due to the heterogeneity of the studies. For example, the enrichment of hub genes is shown Barabasi et al18 and others for diseases of Mendelian inheritance in OMIM,41 42 which is attributable to the subset of disease genes that are also essential.43 Similar results are reproduced in the current study for traits and diseases of complex inheritance (figure 3).
To ensure that the enrichment of disease-associated SNP in hub or bottleneck genes was not due to any of the following straightforward genetic properties, we confirm that hub or bottleneck genes were not more likely to be prioritized in SPAN because of gene length, higher numbers of SNP per gene, and automatically containing SNP with lower GWAS-ranked p values (data not shown). Furthermore, we conducted several studies to address other biological explanations or biases of the association of PIN topological centrality to complex disease that may transpire at a higher level, including: LD/haplotype analysis: hub and bottleneck genes are not found to enrich in long LD/haplotype blocks; the correlation between node degree and the recombination rate of a host gene was inconsequential (spearman correlation r=0.0257, p=0.00345; see supplementary methods, available online only); gene expression: difference in gene expression hub/non-hub genes are minimal and not significant in two T2D gene expression datasets; and selection bias (experimental or sociological).
Further analysis of GWAS findings with their function and protein interactions has shown an excess modularity on a genome scale. Indeed, by applying this SPAN network analysis to the entire NHGRI catalog of SNP–trait associations, and showing a significant enrichment in network centrality (figure 4), this model is designed to highlight the possible importance of network centrality genes and their interactions in complex diseases in general. Subsequently, we showed that our T2D model shows the same enrichment of topological properties seen for the NHGRI data, further suggesting that this approach may identify regulatory networks perturbed in complex disease. Taken together, these results show that protein interaction modeling may prioritize genes that are more biologically essential, and imply that our method can be extended to examine other complex diseases that have been investigated by GWAS.
Study limitations
The protein interaction-centric approach of our study targets intragenic and near-intragenic SNP and, by design, does not take into account signals that exist in intergenic regions. Furthermore, the interactions contained within STRING inherently contain their own biases that vary for each type of interaction evidence. For example, interactions based on experimental-derived data may have a sociological selection bias because the proteins studied are generally those for which there is great interest (ie, disease associated). In addition, many different types of experimental methods utilized for text mining are based on interaction results, and although they were only included in STRING V.6.3 in our study, these studies are not restricted specifically to large-scale studies that were unbiased in their selection of baits and preys. We also recognize that the association of centrality genes to disease could possibly be due to their higher than physiologically normal level of gene expression; however, we verified that there is no statistical association between gene expression and T2D hub genes (data not shown). In addition, a lower range of assayed proteins through high-throughput methods could confound our analysis through selection bias.
Conclusions
Network modeling has the potential to tease out the elusive aspects of complex disease biology and highlight critical signals that would otherwise have remained buried in the deluge of GWAS results. As new methods are required to identify true disease-critical genes accurately, additional investment in SPAN analysis is warranted to generalize further the usage of network modeling and protein interactions. By exploring the importance of topological centrality and edgetic perturbation in complex traits, we have shown the value of network modeling for increasing our understanding of how proteins and genes interact and differ under conditions of health and disease. Network modeling can provide a platform for evaluating the molecular interactions and the multitude of variations that in combination lead to complex traits and disease development, and for facilitating the integration of multiple scales of biological complexity. With a predictive model, we demonstrate the utility of protein interactions to enhance genetic inquiry of complex diseases. For example, these approaches could contribute to GWAS in prioritizing SNP ab initio in a way analogous to that of eQTL.9 In personal genomics, predictive models of the implications of rare variants in health are required and cannot by design be validated in cohorts at the single rare-variant level. Topological properties of protein networks may, however, provide useful insights for inferring their functions at the individual patient level. For instance, first interactors with known T2D genes were demonstrated to be enriched in conservatively controlled models and could be utilized to implicate ‘families of nucleotide polymorphisms’ across multiple patients to obtain statistical power. This approach establishes their contribution to disease and thus also infers the function of rare variants. Finally, conventional therapy for complex diseases focuses on single targets or well-established molecular pathways. The knowledge that interactions are not only important in cancer and single-gene inheritance disease, but also in other complex diseases, provides an opportunity for future paradigm-shifting, unbiased network-targeted therapy, in which proteins and their interactions are modeled more extensively for improved drug development and the earlier identification of toxicities that can be overlooked under the current single-gene paradigm.
In conclusion, the topological centrality of protein interactions has been well established in two types of genetic inheritance: in Mendelian inheritance by edgetic perturbation models11 as well as in somatic mutations of cancer.44 Here, we observe similar results for 310 complex traits documented in the NHGRI catalog of SNP–trait associations and show that SNP are significantly enriched in two key protein interaction properties: hubs and bottlenecks within currently available networks. We also observe these enrichments in topological findings specifically in the prioritized T2D protein interaction networks constrained by GWAS-identified genetic signals. Taken together, these results support the hypothesis that hub and bottleneck proteins are also central in complex trait inheritance that merits further investigation as larger, more statistically powered and less sociologically biased sets of genome-wide interactions become available. Because common diseases comprise a large subclass of complex traits, the topological architecture of the protein networks associated with their inheritable polymorphisms has implications in genetics, personal genomics, and in therapy.
Supplementary Material
Acknowledgments
The authors would like to thank Dr M Boehnke for graciously providing the extensive prioritized list of SNP from FUSION. WTCCC was downloaded during the period when it was publicly available.
Footnotes
Contributors The work of HL, JL, ER, JLC and YAL was completed partly at The University of Chicago.
Funding: This research was supported in part by NIH grants R01 MH090937, U01 GM61393, KL2TR000431, K22LM008308, UL1TR000050, and UL1RR029879. The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Competing interests: None.
Ethics approval: Ethics approval was granted by the institutional review board of The University of Chicago.
Provenance and peer review: Not commissioned; externally peer reviewed.
Data sharing statement: The SPAN algorithm written in R language and bioconductor is made available at https://bitbucket.org/lussierlab/faime-opensource.
References
- 1.Manolio TA, Collins FS, Cox NJ, et al. Finding the missing heritability of complex diseases. Nature 2009;461:747–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Naj AC, Jun G, Beecham GW, et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer's disease. Nat Genet 2011;43:436–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Province MA, Borecki IB. Gathering the gold dust: methods for assessing the aggregate impact of small effect genes in genomic scans. Pac Symp Biocomput 2008;13:190–200 [PubMed] [Google Scholar]
- 4.Lesnick TG, Papapetropoulos S, Mash DC, et al. A genomic pathway approach to a complex disease: axon guidance and Parkinson disease. PLoS Genet 2007;3:e98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baranzini SE, Galwey NW, Wang J, et al. Pathway and network-based analysis of genome-wide association studies in multiple sclerosis. Hum Mol Genet 2009;18:2078–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Torkamani A, Topol EJ, Schork NJ. Pathway analysis of seven common diseases assessed by genome-wide association. Genomics 2008;92:265–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang K, Li M, Bucan M. Pathway-based approaches for analysis of genomewide association studies. Am J Hum Genet 2007;81:1278–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Braun R, Buetow K. Pathways of distinction analysis: a new technique for multi-SNP analysis of GWAS data. PLoS Genet 2011;7:e1002101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nicolae DL, Gamazon E, Zhang W, et al. Trait-associated SNPs are more likely to be eQTLs: annotation to enhance discovery from GWAS. PLoS Genet 2010;6:e1000888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Litvin O, Causton HC, Chen BJ, et al. Modularity and interactions in the genetics of gene expression. Proc Natl Acad Sci U S A 2009;106:6441–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhong Q, Simonis N, Li QR, et al. Edgetic perturbation models of human inherited disorders. Mol Syst Biol 2009;5:321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee Y, Yang X, Huang Y, et al. Network modeling identifies molecular functions targeted by miR-204 to suppress head and neck tumor metastasis. PLoS Comput Biol 2010;6:e1000730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Oti M, Snel B, Huynen MA, et al. Predicting disease genes using protein-protein interactions. J Med Genet 2006;43:691–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Han JD. Understanding biological functions through molecular networks. Cell Res 2008;18:224–37 [DOI] [PubMed] [Google Scholar]
- 15.Yu H, Kim PM, Sprecher E, et al. The importance of bottlenecks in protein networks: correlation with gene essentiality and expression dynamics. PLoS Comput Biol 2007;3:e59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sharan R, Ulitsky I, Shamir R. Network-based prediction of protein function. Mol Syst Biol 2007;3:88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hartwell LH, Hopfield JJ, Leibler S, et al. From molecular to modular cell biology. Nature 1999;402(6761 Suppl.):C47–52 [DOI] [PubMed] [Google Scholar]
- 18.Barabasi AL, Gulbahce N, Loscalzo J. Network medicine: a network-based approach to human disease. Nat Rev Genet 2011;12:56–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chavali S, Barrenas F, Kanduri K, et al. Network properties of human disease genes with pleiotropic effects. BMC Syst Biol 2010;4:78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rambaldi D, Giorgi FM, Capuani F, et al. Low duplicability and network fragility of cancer genes. Trends Genet 2008;24:427–30 [DOI] [PubMed] [Google Scholar]
- 21.Hindorff LA, Sethupathy P, Junkins HA, et al. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc Natl Acad Sci U S A 2009;106:9362–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Scott LJ, Mohlke KL, Bonnycastle LL, et al. A genome-wide association study of type 2 diabetes in Finns detects multiple susceptibility variants. Science 2007;316:1341–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.The Wellcome Trust Case Control Consortium Genome-wide association study of 14 000 cases of seven common diseases and 3000 shared controls. Nature 2007;447:661–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Duerr RH, Taylor KD, Brant SR, et al. A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science 2006;314:1461–3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen J, Sam L, Huang Y, et al. Protein interaction network underpins concordant prognosis among heterogeneous breast cancer signatures. J Biomed Inform 2010;43:385–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jensen LJ, Kuhn M, Stark M, et al. STRING 8—a global view on proteins and their functional interactions in 630 organisms. Nucleic Acids Res 2009;37(Database issue):D412–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McKusick–Nathans Institute of Genetic Medicine JHUB MaNCfBI, National Library of Medicine (Bethesda, MD) (downloaded Dec. 14, 2010) Online Mendelian Inheritance in Man, OMIM (TM). http://omim.org/entry/125853
- 28.Ogata H, Goto S, Sato K, et al. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res 1999;27:29–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ashburner M, Ball CA, Blake JA, et al. Gene ontology: tool for the unification of biology. Nat Genet 2000;25:25–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Croft D, O'Kelly G, Wu G, et al. Reactome: a database of reactions, pathways and biological processes. Nucleic Acids Res 2011;39(Database issue):D691–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shen TH, Carlson CS, Tarczy-Hornoch P. SNPit: a federated data integration system for the purpose of functional SNP annotation. Comput Methods Programs Biomed 2009;95:181–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Suthram S, Beyer A, Karp RM, et al. eQED: an efficient method for interpreting eQTL associations using protein networks. Mol Syst Biol 2008;4:162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Krauthammer M, Kaufmann CA, Gilliam TC, et al. Molecular triangulation: bridging linkage and molecular-network information for identifying candidate genes in Alzheimer's disease. Proc Natl Acad Sci U S A 2004;101:15148–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pagani F, Baralle FE. Genomic variants in exons and introns: identifying the splicing spoilers. Nat Rev Genet 2004;5:389–96 [DOI] [PubMed] [Google Scholar]
- 35.Cartegni L, Chew SL, Krainer AR. Listening to silence and understanding nonsense: exonic mutations that affect splicing. Nat Rev Genet 2002;3:285–98 [DOI] [PubMed] [Google Scholar]
- 36.Heinzen EL, Ge D, Cronin KD, et al. Tissue-specific genetic control of splicing: implications for the study of complex traits. PLoS Biol 2008;6:e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kalnina Z, Zayakin P, Silina K, et al. Alterations of pre-mRNA splicing in cancer. Genes Chromosomes Cancer 2005;42:342–57 [DOI] [PubMed] [Google Scholar]
- 38.Dickson SP, Wang K, Krantz I, et al. Rare variants create synthetic genome-wide associations. PLoS Biol 2010;8:e1000294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.He X, Zhang J. Why do hubs tend to be essential in protein networks? PLoS Genet 2006;2:e88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jeong H, Mason SP, Barabasi AL, et al. Lethality and centrality in protein networks. Nature 2001;411:41–2 [DOI] [PubMed] [Google Scholar]
- 41.Xu J, Li Y. Discovering disease-genes by topological features in human protein-protein interaction network. Bioinformatics 2006;22:2800–5 [DOI] [PubMed] [Google Scholar]
- 42.Goh KI, Cusick ME, Valle D, et al. The human disease network. Proc Natl Acad Sci U S A 2007;104:8685–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tu Z, Wang L, Arbeitman MN, et al. An integrative approach for causal gene identification and gene regulatory pathway inference. Bioinformatics 2006;22:e489–96 [DOI] [PubMed] [Google Scholar]
- 44.Pujana MA, Han JD, Starita LM, et al. Network modeling links breast cancer susceptibility and centrosome dysfunction. Nat Genet 2007;39:1338–49 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.