

Abstract
Invariant natural killer T (iNKT) cells are innate T lymphocytes that specifically recognize α-linked glycosphingolipids (α-GSLs) as antigens presented by CD1d molecules. Activating iNKT cells by administering α-GSLs improves disease outcomes in murine cancer models and, thus, there is great interest in the clinical potential of these lipids for treating human cancers. However, humans possess several other CD1 isoforms that are not present in mice and it is not clear whether these CD1 molecules, which also bind lipids, affect human iNKT cell responses. We demonstrate here that CD1c, which is co-expressed with CD1d on blood dendritic cells and on a fraction of B cells, is able to present α-galactosylceramide (α-GalCer) as a weak agonist to human iNKT cells, and that the presence of CD1c synergistically enhances α-GalCerdependent activation of iNKT cells by CD1d. Primary human B cells expressing CD1c induced stronger iNKT cell responses to α-GalCer than the CD1c- subset, and an antibody against CD1c inhibited iNKT cell cytokine secretion. These results suggest that therapeutic activation of human iNKT cells by α-GSLs will be driven preferentially by CD1c+ cell types. Thus, B cell neoplasias that co-express CD1c and CD1d may be particularly susceptible to α-GSL therapy, and cancer vaccines using α-GSLs as adjuvants may be most effective when presented by CD1c+ antigen-presenting cells.
Keywords: human, iNKT cells, CD1 molecules, α-GalCer
Introduction
Invariant natural killer T (iNKT) cells are a population of innate T lymphocytes that are conserved in most mammalian species, including mice and humans. They recognize lipids and glycolipids as antigens presented by CD1d molecules and are able to promote anti-tumor immune responses (1). The anti-tumor effects of iNKT cells are thought to be mainly due to their early production of Th1 cytokines such as IFN-γ, and to their ability to activate antigen-presenting cells (APCs) to secrete IL-12p70 via co-stimulation by CD40L (2-5). These iNKT cell functions enhance the tumoricidal effects of natural killer (NK) cells and cytotoxic T lymphocytes (CTLs), and also lead to improved cell-mediated responses to tumor vaccines (6-9). Thus, iNKT cells carry out adjuvant-like functions that could be exploited in clinical contexts to enhance human anti-tumor immunity.
An attractive approach to engaging the functions of iNKT cells clinically is to specifically activate them in vivo by administering a cognate antigen. The prototypical antigens recognized by iNKT cells comprise an unusual class of glycolipids called α-glycosphingolipids (α-GSLs), in which a sugar is linked in an α-anomeric conformation to the polar head group of the lipid (10). Administration of antigens of this type, which include a potent activator of iNKT cells called α-galactosylceramide (α-GalCer), has been shown in murine models to produce marked immunological activation in vivo, and studies with knockout mice have established that the effects of α-GalCer treatment are completely dependent on the presence of iNKT cells and CD1d molecules (11, 12). Studies of human subjects have confirmed that administration of α-GalCer-pulsed dendritic cells can result in the activation of human iNKT cells in vivo (13-15). Moreover, based on the ability of iNKT cells that have been activated by α-GalCer to promote tumor rejection in murine model systems, a number of clinical trials have been performed to investigate using α-GalCer for treatment of human cancers (16-19). Finally, there is also growing interest in the possibility of using α-GSLs to co-activate iNKT cells when tumor vaccines are administered as a means of generating more potent effector and memory T cell responses against tumor antigens (20). Thus, a substantial amount of accumulated data suggests that administration of α-GSLs, such as α-GalCer or related compounds, may be a viable strategy to elicit or enhance anti-tumor immune responses in human patients.
However, most investigations of the anti-tumor effects of α-GSLs have been performed in murine model systems. Whereas mice only possess the CD1d isoform, humans express five different CD1 genes (21). These additional CD1 isoforms also bind lipidic ligands (i.e., lipids, glycolipids, lipopeptides) and three of the additional isoforms (CD1a, CD1b, and CD1c) have been shown to present lipidic antigens to T cells (22). There is some specialization in the antigen-presenting functions of human CD1 molecules in that they tend to sample lipids from distinct compartments within the cell and may preferentially bind different types of lipids (23, 24). Yet, it is also clear that different CD1 isoforms are capable of binding and presenting some of the same lipids. For example, a sphingolipid called sulfatide is able to bind and be presented by either CD1a, CD1b, CD1c, or CD1d molecules (25). Thus, it seems likely that, in contrast to murine model systems, human CD1d competes to some extent with other CD1 isoforms for lipid binding, and it is possible that pharmacological activation of human iNKT cells using α-GSLs may be affected by the expression of other CD1 molecules. To provide a better understanding of how best to utilize α-GSLs as pharmacological agents for humans, here we have investigated the impact of the CD1c molecule on α-GalCer-dependent activation of human iNKT cells.
Results
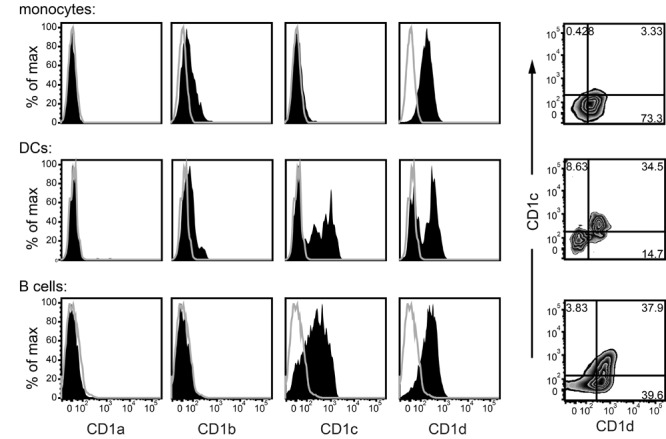
The main human cell types known to constitutively express CD1d are monocytes, myeloid dendritic cells (DCs), and B cells (26). Primary monocytes are typically nearly 100% positive for CD1d, with little or no expression of other CD1 isoforms (Figure 1, top panels). DCs within human peripheral blood are divided into CD1d+ and CD1d- subsets, with almost all of the CD1d+ DCs also expressing CD1c (Figure 1, middle panels). Most peripheral blood B cells are positive for CD1d, and a fraction of these also express CD1c (Figure 1, bottom panels). Hence, as human CD1d molecules are most often co-expressed with CD1c, we focused our analysis on the effects of this CD1 isoform.
Figure 1. Co-expression of CD1c and CD1d on human peripheral blood cell types. Flow cytometric analysis of CD1 expression on primary cells from human peripheral blood. Light scatter and specific antibody staining were used to identify monocytes, DCs, and B cells. Filled histograms show CD1 staining, grey lines show staining by an isotype-matched negative control antibody. Contour plots on the right show the distribution of CD1c vs. CD1d within the monocyte, DC, and B cell populations, respectively. Results are representative of analyses of six different donors.
Binding of α-GalCer by CD1c
To investigate whether CD1c can bind an α-GSL that is antigenic for iNKT cells, recombinant CD1c-Fc or CD1d-Fc fusion proteins or a negative control IgG were coated onto a microtiter plate. The wells were then washed, blocked with BSA, and incubated with titrated concentrations of biotinylated α-GalCer dissolved in PBS containing 0.1% BSA. Binding of the biotinylated α-GalCer was assessed using a streptavidin-enzyme conjugate. The CD1c-Fc and CD1d-Fc fusion proteins showed similar concentration-dependent signals for biotinylated α-GalCer that were clearly above the binding observed for the negative control IgG that was incubated with the same concentrations of biotinylated α-GalCer (Figure 2A). These results suggested that recombinant human CD1c and CD1d molecules have a similar ability to passively bind the α-GalCer glycolipid.
Figure 2. Ability of CD1c to access and bind α-GalCer molecules. (A) Binding of α-GalCer to CD1c and CD1d. Microtiter plates coated with CD1c-Fc or CD1d-Fc fusion proteins or an isotype-matched negative control mAb (neg IgG) were incubated with PBS/BSA solutions containing the indicated concentrations of biotinylated α-GalCer (shaded symbols). Open symbols on the left show the assay backgound from wells that were incubated with PBS/BSA alone. The amount of biotinylated α-GalCer captured on the plate in each condition was detected using a streptavidin-enzyme conjugate; for any given biotin-α-GalCer concentration, the difference between the streptavidin signal obtained from wells coated with CD1-Fc molecules and those coated with negative control IgG is considered to indicate specific binding of the biotinylated α-GalCer to CD1 molecules. Error bars (not always visible on the scale shown) show the standard deviations of the means. Data are representative of 6 independent experiments. (B) Intracellular co-localization of CD1c and CD1d. HeLa cells stably transduced with both CD1c and CD1d were analyzed by immunofluorescence microscopy. The left panel shows the CD1c staining (red), the middle panel shows CD1d staining (green), and the right panel shows the two superimposed. (C) Impact of saposins on α-GalCer loading into CD1c molecules. Recombinant CD1d or CD1c molecules were preloaded with the ganglioside GT1b, then purified CD1-lipid complexes were incubated with a 4.5-molar excess of α-GalCer alone (passive loading) or in the presence of the indicated saposin proteins. Native isoelectric focusing was performed to visualize displacement of GT1b from CD1 molecules by α-GalCer, which is seen as a “downwards” shift (from acidic to basic) of the migration of the CD1 protein band due to the loss of the negatively charged GT1b species. Percentages under the lanes represent the amount of the total CD1 protein signal that has shifted to the displaced band, as estimated from the band intensities. Lanes shown for CD1d on the right are reproduced from reference (30).
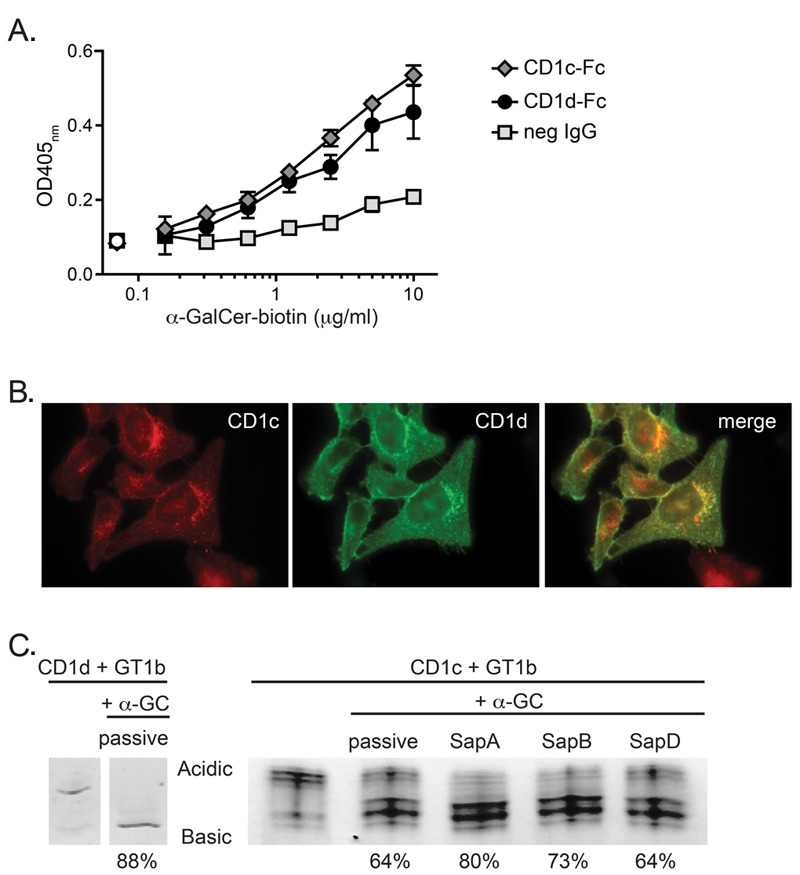
Although it is possible for lipids to load into CD1 molecules at the cell surface, much of the physiological uptake of lipids by CD1 molecules is thought to occur in endocytic compartments. Because the cytoplasmic tails of human CD1d and CD1c molecules both express a binding site for the AP-2 adaptor protein (27), they are expected to traffic similarly within cells and to show an overlapping distribution in endosomal vesicles and, thus, intracellular CD1c molecules might compete with CD1d for α-GalCer binding. We performed fluorescence microscopy to confirm the subcellular co-localization of CD1d and CD1c. HeLa cells stably expressing both isoforms revealed cell surface fluorescence and a punctate pattern of intracellular staining for both CD1c and CD1d (Figure 2B). There appeared to be substantial co-localization of the intracellular CD1c staining with that of CD1d in co-expressing cells (Figure 2B), suggesting that the two CD1 isoforms are indeed often present in the same vesicles.
Since lipid transfer proteins called saposins are thought to play a major role in mediating lipid binding to CD1 molecules within intracellular vesicles (28, 29), we investigated the impact of these accessory proteins on the binding of α-GalCer to CD1c molecules. To do this, we used isoelectric focusing to analyze the ability of α-GalCer to displace a charged lipid (i.e., the ganglioside GT1b) from the CD1 protein. We had previously observed that α-GalCer loading into recombinant CD1d molecules did not require saposins (30) (relevant bands are reproduced in Figure 2C). Analysis of recombinant CD1c molecules revealed that there was also substantial passive loading of α-GalCer into CD1c, and the addition of saposin A or B enhanced the loading (Figure 2C). Thus, although saposins may not be absolutely required for α-GalCer binding to CD1c molecules, the intracellular loading of this lipid may be facilitated by saposins A and B. Together, these results provide strong evidence that both CD1d and CD1c molecules are able to bind α-GalCer.
Presentation of α-GalCer by CD1c
Competition for α-GalCer by CD1c molecules co-expressed on CD1d+ APCs might be expected to result in diminished iNKT cell activation because the CD1c molecules might act as a sink for α-GalCer molecules and, thus, reduce the amount of lipid that is presented by CD1d. However, comparison of the crystal structures of human CD1c and CD1d molecules indicates that the overall conformations of the α1 and α2 helices of the two proteins are similar, with the notable exception that the C-terminal end of the α1 helix appears disordered in the CD1c structure and, therefore, this region may be quite structurally divergent from that of CD1d (31, 32) (Figure 3A). With the exception of two residues in the disordered loop, the chemical characteristics of most of the amino acid side chains that have been identified as important iNKT T cell receptor (TCR) contacts in CD1d are conserved in CD1c (33) (Figure 3A). There is also structural homology between CD1c and CD1d residues in the α-helices that help to coordinate the head group of the α-GalCer molecule (Figure 3A). Thus, based on these structural data, an alternative possibility is that the TCRs of iNKT cells might be able to cross-reactively recognize lipids presented by CD1c molecules.
Figure 3. iNKT cell recognition of α-GSL presented by CD1c. (A) Comparison of the molecular structures of human CD1d and CD1c. Overlayed ribbons represent the main chain atoms of human CD1d in orange (31) and CD1c in yellow (32). Amino acid side chains of the indicated CD1 residues are shown as ball and stick images with carbon atoms shown in orange or yellow, nitrogen atoms in blue, and oxygen atoms in red. α-GalCer bound to CD1d is shown with carbon atoms colored green. The bottom part of the figure shows an alignment of amino acid sequences of the α-helices shown in the figure for human and murine CD1d compared to CD1c. Boxed residues make contacts with α-GalCer in human and murine CD1d crystal structures. Shaded residues have been implicated as CD1d TCR contacts, with those that had a strong impact on TCR binding shown in red, moderate impact in orange, lesser impact in yellow, and slight impact in grey (33). (B) iNKT cell recognition of α-GalCer presented by CD1c-transfected APCs. Untransfected or CD1c-transfected K562 cells were pulsed with the indicated concentrations of α-GalCer (left plot) and untransfected, CD1c-transfected, or CD1d-transfected K562 cells were pulsed with 200 ng/ml α-GalCer or treated with vehicle alone (right plot). The plots show the amount of cytokine secretion as quantitated by ELISA by human iNKT cell clones exposed to the indicated K562 cells. Similar results were obtained in seven independent experiments. p values were calculated using a two-tailed unpaired t test. (C) Histograms showing flow cytometric staining of untransfected (dashed line), CD1c-transfected (yellow), or CD1d-transfected (blue) K562 cells that were stained with an antibody against CD1d (left plot) or against CD1c (right plot). (D) iNKT cell recognition of α-GalCer presented by recombinant CD1c molecules. iNKT cells were exposed to immobilized CD1c-Fc or CD1d-Fc fusion proteins that had been pulsed with α-GalCer or vehicle, and GM-CSF secretion was quantitated by standardized ELISA. p values were calculated using a two-tailed unpaired t test. Similar results were obtained in multiple independent assays using 10 different iNKT cell clones. (E) Comparison of the cytokine secretion responses of individual iNKT cell clones to α-GSL presented by CD1c-Fc vs. CD1d-Fc. The plot on the right shows the α-GSL-induced iNKT cell cytokine secretion normalized by the vehicle response (stimulation index) for CD1d vs. CD1c. Each symbol represents the mean from 2-4 independent experiments using the indicated iNKT cell clone.

To test this, we investigated the responses of iNKT cell clones to CD1c or CD1d transfectants. CD1c-transfected K562 cells that were pulsed with α-GalCer induced clearly detectable iNKT cell cytokine secretion in a dose-dependent manner, whereas the untransfected K562 cells that were similarly treated with α-GalCer elicited little or no detectable cytokine secretion (Figure 3B, left plot). CD1d-transfected K562 cells typically stimulate a modest cytokine secretion response by iNKT cell clones even when they have not been pulsed with α-GalCer, which is thought to be due to iNKT cell recognition of cellular antigens (Figure 3B, right plot). In contrast, CD1c-transfected K562 cells that were not pulsed with α-GalCer induced no detectable response (Figure 3B, right plot), suggesting that CD1c-mediated presentation of self-antigens is not sufficient to activate human iNKT cell clones. This is consistent with results from previous experiments, in which we also did not see any evidence that human iNKT cells were able to recognize the self lipid lysophosphatidycholine (LPC) presented by CD1c molecules (30). We chose to use K562 cells transfected with individual CD1 isoforms for these experiments because we have found that, in contrast to many other human cell lines, untransfected K562 cells do not appear to express endogenous CD1d molecules. However, when we performed flow cytometric analysis of the transfected K562 cells using antibodies against CD1d, we typically saw very weak signal for the CD1c-transfected cells (Figure 3C, left plot). Notably, we did not typically see any signal from the CD1c antibody staining the CD1d-transfected K562 cells (Figure 3C, right plot). This weak CD1d signal on the CD1c transfectant is most likely due to a small amount of cross-reactivity of anti-CD1d mAbs for the CD1c molecules expressed by this transfectant, however, we could not rule out that it is due to upregulation of endogenous CD1d molecules as a result of ectopic CD1c expression.
Therefore, to confirm the ability of CD1c molecules to present α-GalCer to human iNKT cells, we used purified recombinant CD1c glycoproteins as the antigen-presenting molecules. CD1c-Fc or CD1d-Fc fusion proteins were immobilized on microtiter plates and pulsed with α-GalCer or vehicle, then washed and tested for their ability to stimulate cytokine secretion by human iNKT cell clones. Human iNKT cell clones consistently showed responses to α-GalCer-treated CD1c-Fc that were significantly above those elicited by vehicle-treated CD1c (Figure 3D, left plot). The iNKT cell cytokine secretion responses to CD1c-Fc molecules were dependent on the concentration of α-GSL lipid added, and were not simply due to auto-presentation of the lipid antigen, since iNKT cells incubated in wells coated with a negative control IgG that were similarly treated with α-GSL produced little or no detectable GM-CSF (Figure 3D, right plot). These results formally established that α-GalCer presentation by CD1c molecules is able to stimulate functional responses by human iNKT cells.
However, we noted that CD1c-mediated presentation of saturating doses of α-GSLs stimulated lower levels of cytokine secretion from the iNKT cells than CD1d-mediated presentation, and that higher lipid doses were required in order to see detectable iNKT cell responses to CD1c (Figure 3E, left panel). Presentation of α-GalCer by CD1c-Fc molecules typically stimulated about 10- to 100-fold less cytokine secretion from iNKT cell clones than presentation by CD1d-Fc molecules, and the strength of CD1cmediated and CD1d-mediated responses showed an approximate correlation (Figure 3E, right panel). These results suggested that α-GalCer presented by CD1c serves as a weaker agonist for iNKT cells than the complex of α-GalCer with CD1d.
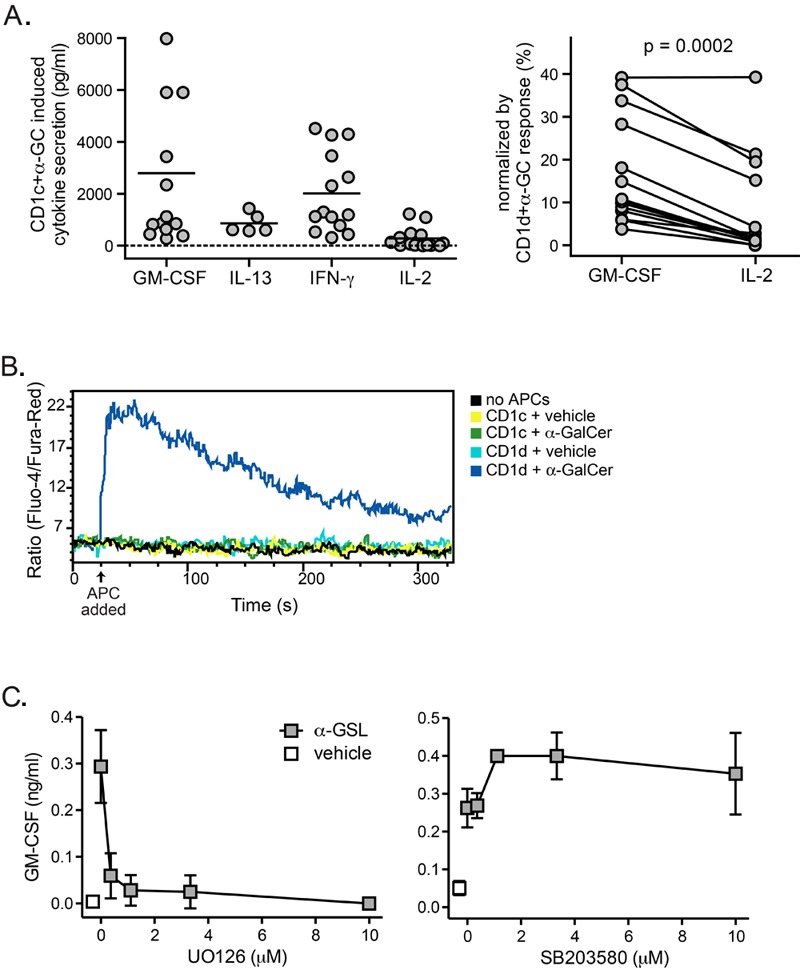
We have previously observed that human iNKT cell recognition of autoantigens presented by CD1d molecules elicits a biased cytokine secretion response compared to recognition of CD1d-α-GalCer complexes, probably because autoantigens serve as weaker TCR agonists than α-GalCer (34). We investigated the nature of the iNKT cell cytokine response to CD1c-mediated presentation of α-GalCer and observed that the iNKT cells were stimulated to produce multiple different cytokines (Figure 4A, left panel), although this route of activation appeared relatively more efficient for inducing GM-CSF secretion than IL-2 secretion (Figure 4A, right panel). Our previous studies indicated that iNKT cell secretion of GM-CSF does not require the induction of a strong cytoplasmic calcium flux and instead depends mainly on MAPK signaling, while IL-2 production is much more dependent on calcium signaling (34). Consistent with this, CD1c-mediated α-GalCer presentation stimulated little or no detectable cytoplasmic calcium flux by iNKT cell clones (Figure 4B). However, iNKT cell cytokine secretion in response to lipid-loaded CD1c-Fc molecules was almost completely abrogated by the MEK inhibitor U0126 (Figure 4C, left panel), suggesting that recognition of CD1c-α-GalCer complexes induces a MAPK signaling response that depends on phosphorylation of ERK. Notably, addition of inhibitors of p38 MAPK kinases did not inhibit iNKT cell responses to α-GalCer presented by CD1c (Figure 4C, right panel), further indicating that the cytokine response to α-GalCer-loaded CD1c molecules depends on AP-1 transcriptional activity, which is selectively targeted by U0126-mediated inhibition of MEK kinases. Together, these results suggest that stimulation by CD1c molecules presenting α-GalCer delivers a comparatively weak TCR stimulus to iNKT cells that activates MAPK signaling but induces little cytoplasmic calcium flux.
Figure 4. α-GalCer presentated by CD1c acts as a weak agonist. (A) Cytokines produced by human iNKT cell clones in response to α-GalCer presentation by CD1cmolecules. The symbols each represent independent analyses of human iNKT cell responses, and include data from clones J3N.5, JC2.8, J24L.17, GG1.2, and the DaDu polyclonal line. The plot on the left shows the amount of each cytokine produced in response to α-GalCer-pulsed CD1c-transfected K562 cells. The plot on the right shows paired GM-CSF and IL-2 responses from individual iNKT cells normalized by their responses in the same experiments to α-GalCer pulsed CD1d-transfected K562 cells. (B) Analysis of iNKT cell calcium flux. iNKT cells were labeled with the calcium indicator dyes Fluo-4 and Fura-Red and stimulated by contact with CD1c- or CD1d-transfected K562 cells that were pulsed with α-GalCer (200 ng/ml) or vehicle. Ensuing intracellular calcium levels were assessed by flow cytometry. The data shown are from one representative experiment out of three. (C) Effect of MAPK inhibitors on iNKT cell GM-CSF secretion in response to α-GSL presented by plate-bound CD1c-Fc molecules. The left plot shows the effect of the MEK inhibitor U0126, and the right plot shows that of the p38 MAPK inhibitor SB203580. Similar results were observed in 6 independent experiments.
Functional impact of CD1c-mediated presentation of α-GalCer
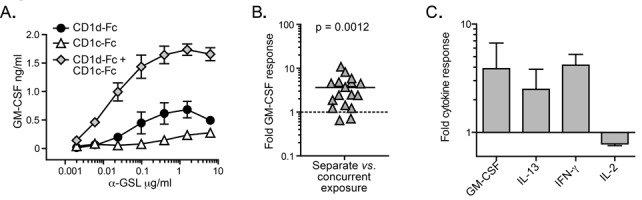
As CD1c is mainly expressed on cells that also express CD1d, we next investigated how co-expression of CD1c and CD1d might impact iNKT cell activation by α-GalCer compared to expression of CD1d alone. Microtiter plate wells were coated with a suboptimal concentration of CD1d-Fc (0.05 μg/well), either alone or in the presence of a 10-fold excess of CD1c-Fc (i.e., 0.5 μg/well), or with 0.5 μg/well CD1c-Fc alone. The plates were then incubated with titrated concentrations of α-GalCer and used to stimulate human iNKT cell clones. The presence of CD1c-Fc along with the suboptimal CD1d-Fc produced substantially more cytokine secretion by iNKT cells than exposure to the same concentration of CD1d-Fc alone (Figure 5A). Moreover, the combination of CD1d and CD1c appeared to have a synergistic effect, with the amount of cytokine released when both fusion proteins were present being significantly greater than the sum of the amounts produced in response to either fusion protein alone (Figure 5B). This synergistic enhancement of iNKT cell cytokine secretion by concurrent exposure to CD1c was observed for GM-CSF, IL-13, and IFN-γ, but not for IL-2 (Figure 5C).
Figure 5. Synergistic effect of concurrent stimulation by α-GSL-loaded CD1c and CD1d molecules. (A) Stimulation by the combination of both CD1c and CD1d. Microtiter plate wells were coated with 0.5 μg/well CD1c-Fc fusion protein, or 0.05 μg/well CD1d-Fc fusion protein, or a combination of 0.05 μg/well CD1d-Fc and 0.5 μg/ well CD1c-Fc fusion protein, incubated with α-GSL, and used to stimulate iNKT cell clones. GM-CSF production was quantitated by ELISA. Similar results were obtained in 13 independent experiments. (B) Compiled results from all 13 experiments showing the amount of GM-CSF produced in response to the combination of CD1c-Fc and CD1d-Fc divided by the sum of the responses to each CD1-Fc molecule alone. The p value comparing the results to a hypothetical median of 1 was calculated using a two-tailed Wilcoxon signed-rank test. (C) Comparison of the effect of concurrent stimulation by α-GSL-loaded CD1c and CD1d on iNKT cell production of four different cytokines. The plot shows the amount of each cytokine produced in a given experiment in response to the combination of CD1c-Fc and CD1d-Fc divided by the sum of the responses to each CD1-Fc molecule alone. Results are from independent analyses of two different iNKT cell clones.
These results suggested that CD1c+ APCs might more efficiently activate α-GalCer-dependent responses by iNKT cells than CD1c- APCs. To test this, we flow cytometrically sorted CD1c-high or CD1c- B cells from fresh peripheral blood and then tested them for their ability to stimulate α-GalCer-dependent cytokine secretion by human iNKT cell clones. CD1c-high B cells reproducibly induced greater iNKT cell cytokine secretion than their CD1ccounterparts (Figure 6A). Moreover, addition of an anti-CD1c mAb to total peripheral blood B cells resulted in reduced but not abrogated iNKT cell cytokine secretion (Figure 6B), which is consistent with a role for CD1c on B cells in augmenting iNKT cell responses to α-GalCer. Notably, inclusion of an anti-CD1d antibody typically resulted in almost complete abrogation of iNKT cell cytokine secretion (Figure 6B), suggesting that CD1c-mediated presentation of α-GalCer on primary B cells is not strong enough to induce an independent response.
Figure 6. Enhanced α-GalCer-dependent activation by CD1c+ B cells. (A) Comparison of iNKT cell activation by CD1c+ vs. CD1c- B cells. Peripheral blood B cells were flow cytometrically sorted according to CD1c expression, then pulsed with 10 ng/ml α-GalCer or vehicle, and used to stimulate iNKT cell clones. GM-CSF secretion was detected by ELISA. The p value was calculated using a two-tailed unpaired t test. Results are representative of 4 independent experiments using two different iNKT cell clones. (B) Blocking of iNKT cell activation by an anti-CD1c antibody. Purified peripheral blood B cells were pulsed with the indicated concentrations of α-GalCer and used to activate iNKT cell clones in the presence of the indicated antibodies. Results are representative of 3 independent experiments using two different iNKT cell clones. (C) α-GalCer dependent activation of primary CD1c+ vs. CD1c- B cells. Freshly isolated PBMCs were incubated with α-GalCer or vehicle for 48 hours, and B cell expression of CD69 was assessed by flow cytometry. Black histograms show CD69 staining for the α-GalCer condition, grey histograms show the vehicle condition, and the dotted line indicates staining by a negative control antibody. The scatter plot shows the fold α-GalCer-dependent CD69 upregulation on CD1c+ vs. CD1c- B cells from 11 independent analyses (vehicle-treated B cell CD69 expression was subtracted from α-GalCer-treated, and the percent CD69+ of the CD1c+ B cells was divided by that of the CD1c- B cells). The p value comparing the results to a hypothetical median of 1 was calculated using a two-tailed Wilcoxon signed-rank test.
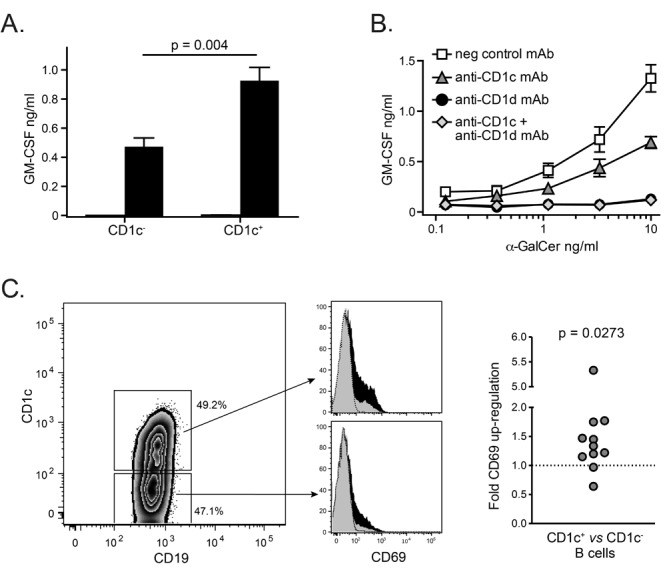
Finally, since iNKT cells are known to co-stimulate APCs via expression of ligands such as CD40L, we investigated the activation of CD1c+ and CD1c- B cells by primary iNKT cells. Freshly isolated peripheral blood mononuclear cells (PBMCs) were incubated with α-GalCer or vehicle for 48 hours, and CD69 expression on B cells was evaluated by flow cytometry. Incubation with α-GalCer led to increased frequencies of B cells expressing CD69 compared to vehicle treatment in all of the samples tested, and the CD1c+ B cells consistently showed more α-GalCer-dependent CD69 upregulation than the CD1c- B cells in the same sample (Figure 6C). These results support the premise that CD1c+ B cells are more efficient partners for the α-GalCer-mediated responses of primary human iNKT cells.
Discussion
The results presented here uncover a surprising complexity to the human CD1 antigen-presenting system: that the co-expression of CD1c isoforms on APCs may positively impact the functional responses of CD1d-restricted iNKT cells. The ability of CD1c molecules to bind α-GalCer and the overlap in the intracellular localization of CD1d and CD1c suggest that in APCs that co-express these CD1 molecules, there is likely to be competition between CD1c and CD1d for binding to α-GSLs. Such competition might be expected to reduce the efficiency of iNKT cell activation by diminishing the number of cognate α-GalCer-CD1d complexes available for TCR recognition. However, our results reveal for the first time that the canonical Vα24+/Vβ11+ T cell receptors expressed by human iNKT cells appear to have an unexpected degree of cross-reactivity for α-GalCer presented by CD1c molecules. Because of this, CD1c-mediated presentation of α-GalCer functions as a weak TCR stimulus for iNKT cells.
Although recognition of CD1c-α-GalCer complexes may have little functional impact in isolation, it is clear that CD1c molecules are mainly present on APCs that also express CD1d, and our results suggest that in this context, the weak agonism provided by CD1c-mediated presentation of α-GalCer may sub-stantially enhance iNKT cell responses. We speculate that the synergistic impact of concurrent CD1c- and CD1d-mediated presentation of α-GalCer that we observed may be explained as follows: the strong TCR agonism produced by CD1d-mediated presentation of α-GalCer is probably required to induce efficient calcium signaling in iNKT cells and, thus, CD1c-mediated presentation in the absence of CD1d elicits only modest iNKT cell responses; however, in the presence of limiting amounts of CD1d, CD1c-mediated presentation of α-GalCer may serve to increase the strength of the iNKT cell MAPK signaling and, thus, increase the iNKT cell response beyond that delivered by limiting the CD1d alone. According to this model, CD1c-mediated presentation of α-GalCer does not activate a different iNKT cell signaling pathway than CD1d-mediated presentation, but instead has its effect by increasing the amount of MAPK signaling that occurs in the presence of the small number of high affinity TCR interactions delivered by the CD1d-α-GalCer complexes. This model resembles findings that MHC molecules loaded with self-peptides that are weak TCR agonists appear to play an important role in enhancing T cell activation by small numbers of high affinity ligands (35-37). Hence, rather than simply competing for α-GSL binding and, thereby, diminishing iNKT cell activation, co-expression of CD1c in the presence of low levels of CD1d may enhance the sensitivity of human iNKT cells to α-GalCer-mediated activation.
Consistent with this possibility, we observed greater cytokine secretion by human iNKT cells in response to CD1c+ primary B cells that were pulsed with α-GalCer than to their CD1c- counterparts. The further observation that inclusion of an anti-CD1c antibody diminished the amount of iNKT cell cytokine secretion supports the possibility that α-GalCer presentation by CD1c molecules contributes to the iNKT cell response to primary B cells. However, it is important to note that CD1c+ B cells may also differ in other ways that affect iNKT cell activation. For example, we have observed that CD1c+ B cells typically appear to have slighly higher levels of CD1d expression (approximately 1.5-fold) than CD1c- B cells (data not shown). Additionally, we and others (38) have observed that whereas the CD1c- B cell population in peripheral blood comprises of about 80% naїve B cells with the remainder made up mostly of class-switched memory B cells, the CD1c+ population typically includes a sizeable fraction (about 40%) of B cells with a marginal zone phenotype (JG and JM, unpublished results). Thus, other APC factors beyond CD1c-mediated presentation of α-GalCer may contribute to enhanced iNKT cell responsiveness to CD1c+ B cells.
Nevertheless, our results suggest that CD1c expression may serve as a biomarker for cell types (i.e., a fraction of the blood DCs and the CD1c+ B cell subset) that are likely to be particularly potent in presenting α-GSLs to iNKT cells. This has important clinical implications since CD1c and CD1d co-expression (which is a feature of some human B cell neoplasias) might thus be associated with better responses to α-GSL therapy, whereas downregulation of either CD1c or CD1d might impede responsiveness to α-GSLs. Moreover, these findings suggest that CD1c+ DCs are likely to be better APCs for use in vaccination protocols that aim to engage the adjuvant functions of iNKT cells by co-presentation of α-GSLs along with peptide antigens.
Since the immunological effects of iNKT cells are thought to result not only from their own cytokine production but also from their ability to activate APCs, an important area for future investigation will be to determine how CD1d+ APCs that coexpress CD1c differ functionally from their counterparts that are CD1c-. For example, we have found that CD1c+ B cells from human peripheral blood show higher expression levels of CD24, which is a marker that has been found to be highly expressed on human B cells that have regulatory functions (39) (data not shown). Thus, CD1c expression by human B cells may be characteristic of particular subsets that have specialized functions, and administration of α-GSLs may therefore selectively enhance the activity of these B cells.
It is also an important point that CD1c and CD1d expression on myeloid DCs and B cells is differentially regulated by compounds that affect PPAR and RAR transcription factors (38). Activators of these transcription factors include endogenous ligands that may be constitutively present at low levels such as lysophosphatidic acid and cardiolipin, as well as oxidized forms of prostaglandins and hydroxyoctadecadienoic acids (i.e., PGJ2, 15d-PGJ2, 9-HODE, and 13-HODE) that are characteristic of inflammatory states (40). Additionally, compounds such as thiazolidinedione drugs that have been used clinically to treat patients with type 2 diabetes (e.g., rosiglitazone) are activators of PPAR transcription factors and can markedly affect the expression of CD1 molecules on human cell types (41). Thus, both physiological and pharmacological compounds present during inflammatory conditions can differentially modulate CD1d and CD1c expression on APCs and might affect human immune responses to α-GSLs. Therefore, in order to develop α-GSLs as therapeutic agents for clinical use in humans, it will be important to understand not only the interactions of iNKT cells with APCs that typically co-express CD1d and CD1c, but also how the expression of these antigen-presenting molecules becomes altered during inflammation.
Acknowledgments
Major funding for this work was provided by National Institutes of Health grant R01AI07494 to JEG. GSB acknowledges support in the form of a Royal Society Wolfson Research Merit Award from the Medical Research Council and The Wellcome Trust (084923/B/08/7).
Materials and methods
Flow cytometric analysis of CD1 expression
Protocols involving the collection and use of human tissues were approved by the University of Wisconsin Minimal Risk Institutional Review Board, and written informed consent was obtained from all blood donors. Human PBMCs were purified by density gradient centrifugation, blocked with 25% human serum, and stained for flow cytometric analysis using commercially available fluorescently labeled antibodies. B cells were identified by expression of CD19, and monocytes and DCs were identified by expression of CD14 or CD11b, compared to CD11c.
Fluorescence microscopy of CD1-transfected APCs
HeLa cells stably expressing CD1c and CD1d were generated using lentivirus-mediated gene transfer using the vector pHAGEpuro (42), followed by puromycin drug selection. Cells on coverslips were washed with saline buffer (PBS) and fixed with 4% paraformaldehyde and permeablized with 0.5% saponin in PBS and 3% BSA. The cells were then incubated with an unlabeled antibody against CD1d (CD1d51) followed by Alexa 594-conjugated secondary antibody (Molecular Probes), then with an antibody against CD1c (F10/21A3) that had been preincubated with Zenon Alexa 488-conjugated FAb fragments (Molecular Probes).
Analysis of α-GalCer binding to CD1 molecules
Recombinant human CD1-Fc fusion proteins were prepared as previously described (43). CD1 fusion proteins or the negative control IgG (clone UPC-10) were dissolved in PBS and coated onto high protein binding microtiter plates at a concentration of 10 μg/ml in a volume of 50 μl. After coating, the plates were washed once with PBS, then blocked with PBS containing 0.1% BSA. Synthetic α-GalCer (KRN7000) that was modified near the polar head group by the covalent addition of a biotin with an ether linkage was sonicated in a heated water bath for 20 minutes, then diluted at the indicated concentrations into PBS containing 0.1% BSA. The fusion protein and negative control IgG-coated wells were incubated at 37°C with 50 μl PBS/BSA containing the biotinylated α-GalCer, or with PBS/BSA alone. The wells were then thoroughly washed with PBS and bound biotinylated α-GalCer was detected using a streptavidin-alkaline phosphatase conjugate (Zymed).
Analysis of the impact of saposins on α-GalCer loading was carried out using an assay that has been described previously, which measures displacement of bound ganglioside GT1b from recombinant CD1c (30, 44). Briefly, a 6-His-tagged construct of the human CD1c ectodomain was co-expressed with human β2-microglobulin using a baculovirus insect expression system. CD1c protein was purified using Ni-NTA resin, followed by size-exclusion chromatography over a Superdex 200 column (GE Healthcare). The CD1c was preloaded with purified trisialoganglioside GT1b (Matreya), then incubated with a 4.5-fold molar excess of α-GalCer alone or in the presence of the indicated recombinant human saposin proteins. The species were then separated according to charge on a native isoelectric focusing gel (PhastGel IEF, GE Healthcare), and protein bands were visualized by Coomassie stain. To quantitate the amount of GT1b displacement by α-GalCer observed in the passive loading condition compared to the saposin conditions, the CD1c band intensities at the GT1b-undisplaced and -displaced positions were estimated using ImageJ software. The intensity of the displaced band was divided by the sum of the displaced and undisplaced band intensities and multiplied by 100 to generate the percent of the CD1c that had undergone displacement of GT1b.
iNKT cell activation by α-GalCer presented by CD1c or CD1d
Human iNKT cell clones were established and maintained as described previously (45). Synthetic glycosphingolipids (KRN7000, C20:2) were prepared as described (46, 47) and sonicated for at least 20 minutes at 37°C prior to use. Recombinant CD1d-Fc and CD1c-Fc fusion proteins were prepared as described previously (30). CD1-Fc fusion proteins or negative control IgG were coated onto high protein binding 96-well microtiter plates in the presence of 0.05 μg anti-LFA-1 mAb (clone HI111, BioLegend) and used to stimulate iNKT cell clones as previously described (30, 45). For APC-mediated activation of iNKT cells, K562 cells were transfected with cDNA-encoding CD1d or CD1c as described (48). CD1c/K562 transfectants were a kind gift of Dr. Annemieke de Jong, Brigham and Women’s Hospital and Harvard Medical School. Alternatively, total B cells were purified from freshly isolated PBMCs by magnetic sorting using CD19 microbeads (Miltenyi Biotec) or CD1c+ and CD1c- B cells were flow cytometrically sorted based on CD19 and CD1c staining. APCs were pulsed with α-GalCer or vehicle for 2-4 hours, then used to stimulate iNKT cells as previously described (49).
Calcium flux
iNKT cells were incubated in culture medium lacking IL-2 for 18-24 hours before performing the calcium flux assay. iNKT cells were labeled with the calcium indicator dyes Fluo-4 and Fura-Red (Invitrogen). Flow cytometric data on iNKT cells alone were acquired for 20 seconds to establish a baseline, then the indicated APCs were added, and the cells were centrifuged for 10 seconds to initiate contact. Cells were resuspended by brief vortexing, and data were acquired for the following 5 minutes. Data analysis was performed using FlowJo (TreeStar) software.
B cell upregulation of CD69
PBMCs were purified from fresh blood by density gradient centrifugation and incubated at 37°C and 5% CO2 for 48 hours with 200 ng/ml α-GalCer or vehicle (0.05% Tween 20 in PBS). Samples were then blocked with 25% human serum and stained for flow cytometric analysis using commercially available fluorescently-labeled antibodies.
References
- 1.Terabe M, Berzofsky JA. The role of NKT cells in tumor immunity. Adv Cancer Res. 2008;101:277–348. doi: 10.1016/S0065-230X(08)00408-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cui J, Shin T, Kawano T, Sato H, Kondo E, Toura I, Kaneko Y, Koseki H, Kanno M, Taniguchi M. Requirement for Valpha14 NKT cells in IL-12-mediated rejection of tumors. Science. 1997;278:1623–1626. doi: 10.1126/science.278.5343.1623. [DOI] [PubMed] [Google Scholar]
- 3.Kitamura H, Iwakabe K, Yahata T, Nishimura S, Ohta A, Ohmi Y, Sato M, Takeda K, Okumura K, Van Kaer L, Kawano T, Taniguchi M, Nishimura T. The natural killer T (NKT) cell ligand alphagalactosylceramide demonstrates its immunopotentiating effect by inducing interleukin (IL)-12 production by dendritic cells and IL-12 receptor expression on NKT cells. J Exp Med. 1999;189:1121–1128. doi: 10.1084/jem.189.7.1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Smyth MJ, Thia KY, Street SE, Cretney E, Trapani JA, Taniguchi M, Kawano T, Pelikan SB, Crowe NY, Godfrey DI. Differential tumor surveillance by natural killer (NK) and NKT cells. J Exp Med. 2000;191:661–668. doi: 10.1084/jem.191.4.661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Smyth MJ, Crowe NY, Pellicci DG, Kyparissoudis K, Kelly JM, Takeda K, Yagita H, Godfrey DI. Sequential production of interferon-gamma by NK1.1(+) T cells and natural killer cells is essential for the antimetastatic effect of alpha-galactosylceramide. Blood. 2002;99:1259–1266. doi: 10.1182/blood.v99.4.1259. [DOI] [PubMed] [Google Scholar]
- 6.Ishihara S, Nieda M, Kitayama J, Osada T, Yabe T, Kikuchi A, Koezuka Y, Porcelli SA, Tadokoro K, Nagawa H, Juji T. Alpha-glycosylceramides enhance the antitumor cytotoxicity of hepatic lymphocytes obtained from cancer patients by activating CD3-CD56+ NK cells in vitro. J Immunol. 2000;165:1659–1664. doi: 10.4049/jimmunol.165.3.1659. [DOI] [PubMed] [Google Scholar]
- 7.Fujii S, Shimizu K, Smith C, Bonifaz L, Steinman RM. Activation of natural killer T cells by alpha-galactosylceramide rapidly induces the full maturation of dendritic cells in vivo and thereby acts as an adjuvant for combined CD4 and CD8 T cell immunity to a coadministered protein. J Exp Med. 2003;198:267–279. doi: 10.1084/jem.20030324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Silk JD, Hermans IF, Gileadi U, Chong TW, Shepherd D, Salio M, Mathew B, Schmidt RR, Lunt SJ, Williams KJ, Stratford IJ, Harris AL, Cerundolo V. Utilizing the adjuvant properties of CD1d-dependent NK T cells in T cell-mediated immunotherapy. J Clin Invest. 2004;114:1800–1811. doi: 10.1172/JCI22046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chung Y, Qin H, Kang CY, Kim S, Kwak LW, Dong C. An NKT-mediated autologous vaccine generates CD4 T-cell dependent potent antilymphoma immunity. Blood. 2007;110:2013–2019. doi: 10.1182/blood-2006-12-061309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kawano T, Cui J, Koezuka Y, Toura I, Kaneko Y, Motoki K, Ueno H, Nakagawa R, Sato H, Kondo E, Koseki H, Taniguchi M. CD1drestricted and TCR-mediated activation of valpha14 NKT cells by glycosylceramides. Science. 1997;278:1626–1629. doi: 10.1126/science.278.5343.1626. [DOI] [PubMed] [Google Scholar]
- 11.Burdin N, Brossay L, Kronenberg M. Immunization with alphagalactosylceramide polarizes CD1-reactive NK T cells towards Th2 cytokine synthesis. Eur J Immunol. 1999;29:2014–2025. doi: 10.1002/(SICI)1521-4141(199906)29:06<2014::AID-IMMU2014>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 12.Carnaud C, Lee D, Donnars O, Park SH, Beavis A, Koezuka Y, Bendelac A. Cutting edge: Cross-talk between cells of the innate immune system: NKT cells rapidly activate NK cells. J Immunol. 1999;163:4647–4650. [PubMed] [Google Scholar]
- 13.Nieda M, Okai M, Tazbirkova A, Lin H, Yamaura A, Ide K, Abraham R, Juji T, Macfarlane DJ, Nicol AJ. Therapeutic activation of Valpha24+Vbeta11+ NKT cells in human subjects results in highly coordinated secondary activation of acquired and innate immunity. Blood. 2004;103:383–389. doi: 10.1182/blood-2003-04-1155. [DOI] [PubMed] [Google Scholar]
- 14.Chang DH, Osman K, Connolly J, Kukreja A, Krasovsky J, Pack M, Hutchinson A, Geller M, Liu N, Annable R, Shay J, Kirchhoff K, Nishi N, Ando Y, Hayashi K, Hassoun H, Steinman RM, Dhodapkar MV. Sustained expansion of NKT cells and antigen-specific T cells after injection of alpha-galactosyl-ceramide loaded mature dendritic cells in cancer patients. J Exp Med. 2005;201:1503–1517. doi: 10.1084/jem.20042592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chang DH, Liu N, Klimek V, Hassoun H, Mazumder A, Nimer SD, Jagannath S, Dhodapkar MV. Enhancement of ligand-dependent activation of human natural killer T cells by lenalidomide: therapeutic implications. Blood. 2006;108:618–621. doi: 10.1182/blood-2005-10-4184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Motohashi S, Nakayama T. Clinical applications of natural killer T cell-based immunotherapy for cancer. Cancer Sci. 2008;99:638–645. doi: 10.1111/j.1349-7006.2008.00730.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schneiders FL, Scheper RJ, von Blomberg BM, Woltman AM, Janssen HL, van den Eertwegh AJ, Verheul HM, de Gruijl TD, van der Vliet HJ. Clinical experience with alpha-galactosylceramide (KRN7000) in patients with advanced cancer and chronic hepatitis B/C infection. Clin Immunol. 2011;140:130–141. doi: 10.1016/j.clim.2010.11.010. [DOI] [PubMed] [Google Scholar]
- 18.Taniguchi M, Tashiro T, Dashtsoodol N, Hongo N, Watarai H. The specialized iNKT cell system recognizes glycolipid antigens and bridges the innate and acquired immune systems with potential applications for cancer therapy. Int Immunol. 2010;22:1–6. doi: 10.1093/intimm/dxp104. [DOI] [PubMed] [Google Scholar]
- 19.Dhodapkar MV. Harnessing human CD1d restricted T cells for tumor immunity: progress and challenges. Front Biosci. 2009;14:796–807. doi: 10.2741/3278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cerundolo V, Silk JD, Masri SH, Salio M. Harnessing invariant NKT cells in vaccination strategies. Nat Rev Immunol. 2009;9:28–38. doi: 10.1038/nri2451. [DOI] [PubMed] [Google Scholar]
- 21.Martin LH, Calabi F, Milstein C. Isolation of CD1 genes: a family of major histocompatibility complex-related differentiation antigens. Proc Natl Acad Sci U S A. 1986;83:9154–9158. doi: 10.1073/pnas.83.23.9154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kasmar A, Van Rhijn I, Moody DB. The evolved functions of CD1 during infection. Curr Opin Immunol. 2009;21:397–403. doi: 10.1016/j.coi.2009.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cohen NR, Garg S, Brenner MB. Antigen presentation by CD1 lipids, T cells, and NKT cells in microbial immunity. Adv Immunol. 2009;102:1–94. doi: 10.1016/S0065-2776(09)01201-2. [DOI] [PubMed] [Google Scholar]
- 24.Salio M, Silk JD, Cerundolo V. Recent advances in processing and presentation of CD1 bound lipid antigens. Curr Opin Immunol. 2010;22:81–88. doi: 10.1016/j.coi.2009.12.008. [DOI] [PubMed] [Google Scholar]
- 25.Shamshiev A, Gober HJ, Donda A, Mazorra Z, Mori L, De Libero G. Presentation of the same glycolipid by different CD1 molecules. J Exp Med. 2002;195:1013–1021. doi: 10.1084/jem.20011963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Exley M, Garcia J, Wilson SB, Spada F, Gerdes D, Tahir SM, Patton KT, Blumberg RS, Porcelli S, Chott A, Balk SP. CD1d structure and regulation on human thymocytes, peripheral blood T cells, B cells and monocytes. Immunology. 2000;100:37–47. doi: 10.1046/j.1365-2567.2000.00001.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gumperz JE. The ins and outs of CD1 molecules: bringing lipids under immunological surveillance. Traffic. 2006;7:2–13. doi: 10.1111/j.1600-0854.2005.00364.x. [DOI] [PubMed] [Google Scholar]
- 28.Zhou D, Cantu C, 3rd, Sagiv Y, Schrantz N, Kulkarni AB, Qi X, Mahuran DJ, Morales CR, Grabowski GA, Benlagha K, Savage P, Bendelac A, Teyton L. Editing of CD1d-bound lipid antigens by endosomal lipid transfer proteins. Science. 2004;303:523–527. doi: 10.1126/science.1092009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kang SJ, Cresswell P. Saposins facilitate CD1d-restricted presentation of an exogenous lipid antigen to T cells. Nat Immunol. 2004;5:175–181. doi: 10.1038/ni1034. [DOI] [PubMed] [Google Scholar]
- 30.Fox LM, Cox DG, Lockridge JL, Wang X, Chen X, Scharf L, Trott DL, Ndonye RM, Veerapen N, Besra GS, Howell AR, Cook ME, Adams EJ, Hildebrand WH, Gumperz JE. Recognition of lyso-phospholipids by human natural killer T lymphocytes. PLoS Biol. 2009;7:e1000228. doi: 10.1371/journal.pbio.1000228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pellicci DG, Patel O, Kjer-Nielsen L, Pang SS, Sullivan LC, Kyparissoudis K, Brooks AG, Reid HH, Gras S, Lucet IS, Koh R, Smyth MJ, Mallevaey T, Matsuda JL, Gapin L, McCluskey J, Godfrey DI, Rossjohn J. Differential recognition of CD1d-alphagalactosyl ceramide by the V beta 8.2 and V beta 7 semi-invariant NKT T cell receptors. Immunity. 2009;31:47–59. doi: 10.1016/j.immuni.2009.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Scharf L, Li NS, Hawk AJ, Garzon D, Zhang T, Fox LM, Kazen AR, Shah S, Haddadian EJ, Gumperz JE, Saghatelian A, Faraldo-Gómez JD, Meredith SC, Piccirilli JA, Adams EJ. The 2.5 a structure of CD1c in complex with a mycobacterial lipid reveals an open groove ideally suited for diverse antigen presentation. Immunity. 2010;33:853–862. doi: 10.1016/j.immuni.2010.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wun KS, Borg NA, Kjer-Nielsen L, Beddoe T, Koh R, Richardson SK, Thakur M, Howell AR, Scott-Browne JP, Gapin L, Godfrey DI, McCluskey J, Rossjohn J. A minimal binding footprint on CD1dglycolipid is a basis for selection of the unique human NKT TCR. J Exp Med. 2008;205:939–949. doi: 10.1084/jem.20072141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang X, Chen X, Rodenkirch L, Simonson W, Wernimont S, Ndonye RM, Veerapen N, Gibson D, Howell AR, Besra GS, Painter GF, Huttenlocher A, Gumperz JE. Natural killer T-cell autoreactivity leads to a specialized activation state. Blood. 2008;112:4128–4138. doi: 10.1182/blood-2008-05-157529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wulfing C, Sumen C, Sjaastad MD, Wu LC, Dustin ML, Davis MM. Costimulation and endogenous MHC ligands contribute to T cell recognition. Nat Immunol. 2002;3:42–47. doi: 10.1038/ni741. [DOI] [PubMed] [Google Scholar]
- 36.Krogsgaard M, Li QJ, Sumen C, Huppa JB, Huse M, Davis MM. Agonist/endogenous peptide-MHC heterodimers drive T cell activation and sensitivity. Nature. 2005;434:238–243. doi: 10.1038/nature03391. [DOI] [PubMed] [Google Scholar]
- 37.Krogsgaard M, Juang J, Davis MM. A role for “self ” in T-cell activation. Sem Immunol. 2007;19:236–244. doi: 10.1016/j.smim.2007.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Allan LL, Stax AM, Zheng DJ, Chung BK, Kozak FK, Tan R, van den Elzen P. CD1d and CD1c expression in human B cells is regulated by activation and retinoic acid receptor signaling. J Immunol. 2011;186:5261–5272. doi: 10.4049/jimmunol.1003615. [DOI] [PubMed] [Google Scholar]
- 39.Blair PA, Norena LY, Flores-Borja F, Rawlings DJ, Isenberg DA, Ehrenstein MR, Mauri C. CD19(+)CD24(hi)CD38(hi) B cells exhibit regulatory capacity in healthy individuals but are functionally impaired in systemic Lupus Erythematosus patients. Immunity. 2010;32:129–140. doi: 10.1016/j.immuni.2009.11.009. [DOI] [PubMed] [Google Scholar]
- 40.Leslie DS, Dascher CC, Cembrola K, Townes MA, Hava DL, Hugendubler LC, Mueller E, Fox L, Roura-Mir C, Moody DB, Vincent MS, Gumperz JE, Illarionov PA, Besra GS, Reynolds CG, Brenner MB. Serum lipids regulate dendritic cell CD1 expression and function. Immunology. 2008;125:289–301. doi: 10.1111/j.1365-2567.2008.02842.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Szatmari I, Pap A, Ruhl R, Ma JX, Illarionov PA, Besra GS, Rajnavolgyi E, Dezso B, Nagy L. PPARgamma controls CD1d expression by turning on retinoic acid synthesis in developing human dendritic cells. J Exp Med. 2006;203:2351–2362. doi: 10.1084/jem.20060141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mostoslavsky G, Fabian AJ, Rooney S, Alt FW, Mulligan RC. Complete correction of murine Artemis immunodeficiency by lentiviral vector-mediated gene transfer. Proc Natl Acad Sci U S A. 2006;103:16406–11641. doi: 10.1073/pnas.0608130103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gumperz JE, Miyake S, Yamamura T, Brenner MB. Functionally distinct subsets of CD1d-restricted natural killer T cells revealed by CD1d tetramer staining. J Exp Med. 2002;195:625–636. doi: 10.1084/jem.20011786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cantu C, 3rd, Benlagha K, Savage PB, Bendelac A, Teyton L. The paradox of immune molecular recognition of alpha-galactosylceramide: low affinity, low specificity for CD1d, high affinity for alpha beta TCRs. J Immunol. 2003;170:4673–4682. doi: 10.4049/jimmunol.170.9.4673. [DOI] [PubMed] [Google Scholar]
- 45.Brigl M, van den Elzen P, Chen X, Meyers JH, Wu D, Wong CH, Reddington F, Illarianov PA, Besra GS, Brenner MB, Gumperz JE. Conserved and heterogeneous lipid antigen specificities of CD1drestricted NKT cell receptors. J Immunol. 2006;176:3625–3634. doi: 10.4049/jimmunol.176.6.3625. [DOI] [PubMed] [Google Scholar]
- 46.Yu KO, Im JS, Molano A, Dutronc Y, Illarionov PA, Forestier C, Fujiwara N, Arias I, Miyake S, Yamamura T, Chang YT, Besra GS, Porcelli SA. Modulation of CD1d-restricted NKT cell responses by using N-acyl variants of alpha-galactosylceramides. Proc Natl Acad Sci U S A. 2005;102:3383–3388. doi: 10.1073/pnas.0407488102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Veerapen N, Leadbetter EA, Brenner MB, Cox LR, Besra GS. Synthesis of a novel alpha-galactosyl ceramide haptenated-lipid antigen, a useful tool in demonstrating the involvement of iNKT cells in the production of antilipid antibodies. Bioconjug Chem. 2010;21:741–747. doi: 10.1021/bc9005255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gumperz JE. Generation of HLA class I transfected target cell lines. Methods Mol Biol. 2000;121:49–60. doi: 10.1385/1-59259-044-6:49. [DOI] [PubMed] [Google Scholar]
- 49.Chen X, Wang X, Keaton JM, Reddington F, Illarionov PA, Besra GS, Gumperz JE. Distinct endosomal trafficking requirements for presentation of autoantigens and exogenous lipids by human CD1d molecules. J Immunol. 2007;178:6181–6190. doi: 10.4049/jimmunol.178.10.6181. [DOI] [PubMed] [Google Scholar]