

Abstract
Angiotensin II (AngII) acts on central angiotensin type 1 (AT1) receptors to increase water and saline intake. Prolonged exposure to AngII in cell culture models results in a desensitization of the AT1 receptor that is thought to involve receptor internalization, and a behavioral correlate of this desensitization has been shown in rats after repeated central injections of AngII. Specifically, rats given repeated injections of AngII drink less water than controls after a subsequent test injection of AngII. Under the same conditions, however, repeated injections of AngII have no effect on AngII-induced saline intake. Given earlier studies indicating that separate intracellular signaling pathways mediate AngII-induced water and saline intake, we hypothesized that the desensitization observed in rats may be incomplete, leaving the receptor able to activate mitogen-activated protein (MAP) kinases (ERK1/2), which play a role in AngII-induced saline intake without affecting water intake. In support of this hypothesis, we found no difference in MAP kinase phosphorylation after an AngII test injection in rats given prior treatment with repeated injections of vehicle, AngII, or Sar1,Ile4,Ile8-AngII (SII), an AngII analog that activates MAP kinase without G protein coupling. In addition, we found that pretreatment with the MAP kinase inhibitor U0126 completely blocked the desensitizing effect of repeated AngII injections on water intake. Furthermore, AngII-induced water intake was reduced similarly by repeated injections of AngII or SII. The results suggest that G protein-independent signaling is sufficient to produce behavioral desensitization of the angiotensin system and that the desensitization requires MAP kinase activation.
Introduction
Angiotensin II (AngII) plays an important role in mediating the coordinated behavioral and physiological responses to hypovolemia (Fitzsimons, 1998). Central injection of AngII causes robust increases in water and saline intake via stimulation of the angiotensin type 1 (AT1) receptor (Kirby et al., 1992; Sakai et al., 1994; Sakai et al., 1995; McKinley et al., 1996a; Weisinger et al., 1997). AT1 receptors are found in abundance in structures associated with the ventral portion of the lamina terminalis, such as the organum vasculosum of the lamina terminalis (OVLT) and the median preoptic nucleus (MnPO), and in the subfornical organ (SFO), which bulges into the third ventricle from the dorsal lamina terminalis (McKinley et al., 1986; McKinley et al., 1996b). These receptor populations are critical for mediating the increases in fluid intake generated by AngII (Simpson & Routtenberg, 1973, 1975; Buggy & Johnson, 1978; Fitts & Masson, 1990). In vitro, prolonged activation of the AT1 receptor reduces the efficacy of subsequent AngII (Sasamura et al., 1994; Thomas et al., 1996; Gebke et al., 1998; Thomas, 1999; Hunyady et al., 2000; Guo et al., 2001), and this process of desensitization appears to involve a rapid phosphorylation and internalization of the receptor (Thomas, 1999; Mehta & Griendling, 2007).
A small, but growing number of studies using rats shows that repeated introcerebroventricular (icv) injections of AngII over a short period of time reduce the dipsogenic (Quirk et al., 1988; Torsoni et al., 2004; Vento & Daniels, 2010; Zapparoli et al., 2011) and renal (Zapparoli et al., 2011) responses to subsequent AngII administration. Additional testing from our lab has shown that this reduction in the dipsogenic response to AngII, or behavioral desensitization, requires AT1 receptor activation (Vento & Daniels, 2010) and is not the result of some broader behavioral deficit (Vento et al., 2012). The effect of repeated injections of AngII likely involves an underlying change in AngII-responsiveness at the level of the receptor similar to that observed in vitro. The studies in rats, however, reveal an apparent contrast with the in vitro studies because repeated AngII administration does not affect AngII-induced saline intake (Vento & Daniels, 2010), indicating that the repeated injections of AngII do not cause the receptors to be completely unavailable as would be expected after the internalization demonstrated in vitro. Accordingly, the fact that water intake is reduced after repeated AngII, but saline intake remains normal, suggests that receptor internalization may not be the only mechanism by which AT1 receptor desensitization occurs.
The persistence of normal AngII-induced saline intake in the face of a desensitized water intake response may involve differences in the relative contributions of intracellular signaling pathways in the stimulation of these ingestive behaviors by AngII. More specifically, the dipsogenic and natriorexigenic actions of AngII appear to be mediated by separable intracellular signaling cascades (Daniels et al., 2005; Daniels et al., 2009). G protein coupling and the resultant protein kinase C (PKC) activation appear to be important for water intake stimulated by AngII (Fleegal & Sumners, 2003; Daniels et al., 2005; Daniels et al., 2009), whereas saline intake stimulated by AngII can occur independent of PKC signaling but requires activation of mitogen-activated protein (MAP) kinase (Daniels et al., 2005; Daniels et al., 2009). Given the divergence in signaling underlying the different ingestive behaviors stimulated by AngII and the finding that one ingestive behavior (water intake) is affected by repeated injections of AngII whereas the other (saline intake) is not, it seems plausible that repeated injections of AngII affect AT1-mediated signaling pathways differently. To test this hypothesis, using tissue that has previously been shown to exhibit AngII-induced changes in MAP kinase activation (Daniels et al., 2005), we tested for differences in phosphorylated MAP kinase in rats given repeated injections of AngII or Sar1,Ile4,Ile8-AngII (SII), an AngII analog that activates MAP kinase family members without affecting G protein-dependent pathways that are normally stimulated by the AT1 receptor (Miura & Karnik, 1999; Holloway et al., 2002; Daniels et al., 2005). We also tested the requirement for MAP kinase phosphorylation in AngII-induced desensitization by pretreating rats with the MAP kinase inhibitor U0126 before and during repeated injections of AngII. Additional experiments evaluated the possibility of AngII receptor-mediated desensitization in the absence of G protein coupling using repeated injections of SII. Taken together, the results of these experiments provide new information about the signaling requirements for AngII-induced behavioral desensitization and may provide important information about mechanisms underlying AT1 receptor function.
Methods
General methods
Ethical approval
The handling and care of laboratory animals conformed to the National Institutes of Health Guide for the Care and Use of Laboratory Animals and all animal use protocols were approved by the Institutional Animal Care and Use Committee of the State University of New York at Buffalo.
Experimental animals
Adult male Sprague Dawley rats (175-199 gm upon arrival from breeder) were obtained from Harlan Laboratories (Indianapolis, IN). Rats were individually housed in stainless steel, hanging wire-mesh cages, and maintained in a temperature- and humidity-controlled colony room on a 12:12 hr light:dark cycle. All experiments were performed early in the light phase of the light:dark cycle and rats were permitted ad libitum access to food and water, unless otherwise stated. Each rat was used in only one experiment.
Lateral ventricle cannula implantation
No fewer than 5 days after arrival from the breeder, rats were anesthetized by an intramuscular injection of a combination of ketamine (70 mg kg-1) and xylazine (5 mg kg-1). Their heads were fixed in a stereotaxic frame and a small incision was made in the scalp. A burr hole was made in the skull and a chronic indwelling cannula (33 ga, Plastics One, Roanoke, VA) aimed at the lateral ventricle (coordinates: 0.9 mm posterior to bregma, 1.4 mm lateral to midline, 1.8 mm ventral to dura) was implanted and affixed with bone screws and dental cement. Septocaine with epinephrine was applied topically to the skull before drilling and a single injection of carprofen (5 mg kg-1, sc) was given immediately after the procedure to minimize pain. Rats were given no fewer than 5 days to recover from surgery before proper cannula placement and AngII responsiveness were verified by injection of AngII [10 ng in 1 μl tris-buffered saline (TBS)]. Only rats that drank at least 6 ml in the 30 min after intracranial AngII were included in the experiments.
Drug injections
AngII (Bachem Bioscience Inc., King of Prussia, PA) and SII (Bachem Bioscience Inc., King of Prussia, PA) were diluted in TBS. U0126 (Promega US, Madison, WI) was diluted in dimethyl sulfoxide (DMSO). All injections were icv through an injector attached to water-filled PE 50 tubing using a Hamilton syringe (Hamilton Company, Reno, NV). The injector was left in place for approximately 30 s after each injection to allow for drug diffusion. In the experiments described below, rats were given repeated icv injections using our established injection protocol (Vento & Daniels, 2010) to explore the role of AT1-mediated intracellular signaling cascades in AngII-induced behavioral desensitization. Briefly, rats were administered 3 icv injections of either AngII (300 ng in 1 μl TBS) or vehicle (1 μl TBS), with 20 min between each injection; referred to as the “treatment regimen” for clarity. Twenty minutes after the final treatment regimen injection, all rats received a final “test injection” of AngII (100 ng in 1 μl TBS).
Data analysis
All data were analyzed using STATISTICA software (version 9.0; Statsoft, Tulsa, OK). The statistical tests used for each experiment are listed in the corresponding section of Experimental designs. For all experiments, statistically significant (p<0.05) main or interaction effects were further probed using Student Newman-Keuls post hoc tests.
Experimental designs
Experiment 1: Effect of repeated AngII receptor stimulation on AngII-induced MAP kinase activation
Rats (n=9 per group) received a treatment regimen of either AngII (3 × 300 ng in 1 μl TBS), SII (3 × 30 μg in 1 μl TBS), or vehicle (3 × 1 μl TBS) before all rats received a test injection of AngII (100 ng in 1 μl TBS). Food and water were removed immediately prior to the first treatment regimen injection and remained absent for the duration of the experiment. Five minutes after the test injection, rats were anesthetized by intramuscular injection of a combination of ketamine (70 mg kg-1) and xylazine (5 mg kg-1). Five minutes later (10 min after the test injection), rats were killed by decapitation. The brains then were rapidly removed and frozen in 2-methylbutane on dry ice. One hundred micron coronal sections were cut on a cryostat and a block of tissue (approximately 3 mm wide × 6 mm high) was isolated from each of eight brain sections to generate samples containing structures of the anterior or the posterior portions of the lamina terminalis. The lamina terminalis follows an anteroventral to posteriodorsal angle along the anterior wall of the third ventricle, therefore the tissue samples taken from the anterior brain sections contained populations of AngII receptors in structures associated with the ventral lamina terminalis, including the OVLT and the MnPO, whereas tissue samples from the more posterior brain sections contained structures associated with the dorsal portion of the lamina terminalis, such as the SFO (Figure 1).
Figure 1.
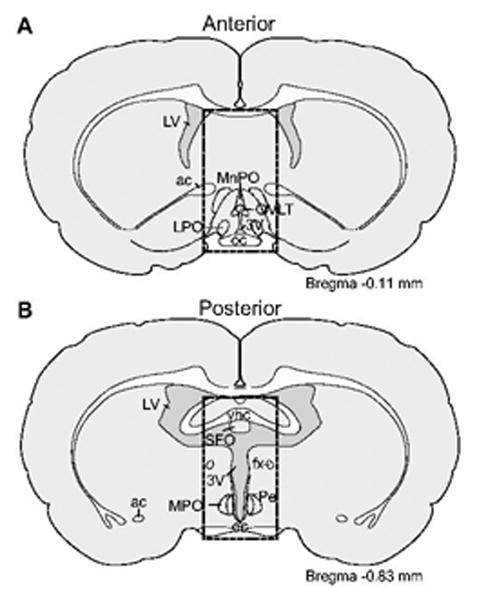
Illustration of the approximate boundaries of the microdissected tissue used to measure MAP kinase. One hundred micron tissue sections from the anterior (A) and posterior (D) lamina terminalis were dissected to include AngII-responsive structures in the ventral and the dorsal lamina terminalis, respectively. Atlas plates were modified from Swanson (2003). Abbreviations not used elsewhere: ac, anterior commissure; fx, fornix; LPO, lateral preoptic area; LV, lateral ventricle; MPO, medial preoptic nucleus; oc, optic chiasm; Pe, periventricular hypothalamus; 3V, third ventricle; ventral hippocampal commissure (vhc).
Tissue was homogenized in lysis buffer containing 20 mM Tris HCl, 1 M NaF, 0.1 M Na3VO4, 0.1 M Na3MoO4, 0.1 mM phenylarsin oxide, 20 μg aprotinin, 10 μg pepstatin, 10 μg leupeptin, and 45 mg sodium pyrophosphate. Homogenized samples were centrifuged at 13,200 rpm for 8 min. Supernatant was removed and protein concentration was assessed using the BCA assay method (Micro BCA Protein Assay Kit, Thermo Fisher Scientific, Inc., Rockford, IL). Samples then were standardized for protein content using lysis buffer, and Sample Reducing Agent and LDS Sample Buffer (Invitrogen, Life Technologies, Carlsbad, CA) were added. Samples were incubated at 70° C for 10 min before being loaded into NuPAGE 10% Bis-Tris Gels (Invitrogen, Life Technologies, Carlsbad, CA; 17.5-23.5 μg protein loaded per sample). One lane of each gel was loaded with Rainbow™ molecular weight markers (GE Healthcare, Piscataway, NJ, USA). Gels were transferred to a nitrocellulose membrane (Invitrogen, Life Technologies, Carlsbad, CA) and blocked overnight with non-fat dry milk supplemented with PBS and 10% thimerisol.
The membranes were processed for MAP kinase by Western blot analysis using a primary antibody directed against phosphorylated p44/42 MAP kinase (ERK1/2; 75 min at 1:1000; Cell Signaling Technologies, Inc., Danvers, MA) and peroxidase-conjugated donkey anti-mouse IgG (45 min at 1:1000; Jackson Immunoresearch Laboratories, Inc., West Grove, PA) as the secondary antibody. Blots were stripped by alternating washes of distilled water and 0.2 M NaOH, and re-probed with a primary antibody that recognizes both phosphorylated and unphosphorylated MAP kinase (total MAP kinase) (75 min at 1:2500; Cell Signaling Technologies, Inc., Danvers, MA) and peroxidase-conjugated donkey anti-rabbit IgG (45 min at 1:2500; Jackson Immunoresearch Laboratories, Inc., West Grove, PA) as the secondary antibody. Phosphorylated and total MAP kinase immunoreactivity was detected by chemiluminescence assay reagents (ECL Western Blotting Analysis System, GE Healthcare, Buckinghamshire, UK) in a ChemiDoc XRS imaging system (Bio-Rad Laboratories, Hercules, CA, USA). Quantity One 1-D analysis software (version 4.6.1.; Bio-Rad Laboratories, Hercules, CA, USA) was used to capture images and assess optical density and calculations of the ratio of phosphorylated:total MAP kinase were analyzed by one-way ANOVA.
Experiment 2: Effect of MAP kinase inhibition on AngII-induced behavioral desensitization
Rats (n=5-7 per group) were given 3 pretreatment injections of the MAP kinase inhibitor U0126 (10 mM in 0.5 μl DMSO) or vehicle (0.5 μl DMSO) with each injection separated by 20 min. Beginning immediately after the second pretreatment injection, rats received either an AngII (3 × 300 ng in 0.5 μl TBS) or a vehicle (3 × 0.5 μl TBS) treatment regimen. Twenty minutes after the final treatment regimen injection (40 min after the final pretreatment injection) all rats received a test injection of AngII (100 ng in 1 μl TBS) and subsequent 30 min water intake was measured. Food and water were removed immediately before the initial pretreatment injection and returned after the test injection. Water intake was assessed for 30 min immediately after the final AngII test injection using 100 ml graduated bottles with 1 ml gradations. Total intake was calculated as the volume of fluid remaining at a given time less the initial starting volume. Analysis was performed using a mixed design ANOVA (between-subjects effects of Pretreatment and Treatment Regimen and within-subjects effects of Time) on non-cumulative intake and two-way ANOVA (Pretreatment × Treatment Regimen) was used to assess differences in total water intake.
Experiment 3: Effect of repeated injections of the “biased” agonist, SII, on AngII-induced water intake
Using a counterbalanced, repeated measures design, rats (n=17) were given a treatment regimen of AngII (3 × 300 ng in 1 μl TBS), SII (3 × 30 μg in 1 μl TBS), or vehicle (3 × 1 μl TBS) prior to a test injection of AngII (100 ng in 1 μl TBS) and subsequent water intake was measured as described above. Each rat was exposed to each of the 3 experimental conditions (AngII, SII, and vehicle treatment regimens) with 4 days between each condition. Non-cumulative intake was analyzed by repeated measures two-way ANOVA (Time × Treatment Regimen) and total intake was analyzed by one-way repeated measures ANOVA.
Experiment 4: Time-course of the antagonistic effects of SII
Rats (n=7-8 per group) were given a single icv injection of SII (30 μg in 1 μl TBS) prior to an injection of AngII (100 ng in 1 μl TBS) given either 0, 10, 15, or 20 min later. Food and water were removed immediately before the initial injection of SII and returned after the AngII injection. Water intake was assessed for 30 min after injection of AngII using bottle weight (calculated as the initial bottle weight minus the final weight at the end of the experiment). Total intake was analyzed by one-way ANOVA
Results
No differences in activated MAP kinase were observed in either anterior or posterior portions of the lamina terminalis after a repeated AngII treatment regimen
Earlier studies show that the desensitization of fluid intake after repeated injections of AngII is incomplete (Vento & Daniels, 2010). Specifically, water intake is reduced after a repeated AngII treatment regimen, but saline intake is not. Given the suggestion by the divergent signaling hypothesis of AngII-induced fluid intake (Daniels, 2010) that MAP kinase is more relevant to AngII-induced saline intake than it is to AngII-induced water intake, we hypothesized that the persistent saline intake in rats given repeated injections of AngII is associated with persistent MAP kinase activation. To test this hypothesis, we performed Western blot analyses on brain tissue containing populations of AngII receptors along the lamina terminalis from rats that had received a treatment regimen of AngII (3 × 300 ng in 1 μl TBS), SII (3 × 30 μg in 1 μl TBS), or vehicle (3 × 1 μl TBS) before a test injection of AngII (100 ng in 1 μl TBS). As shown in Figure 2, we found no statistical difference in MAP kinase activity between groups in tissue dissected from anterior (F2,23=0.174, p=0.842; Figure 2C) or posterior (F2,24=0.218, p=0.806; Figure 2F) sections of the lamina terminalis microdissected from rat brains that were removed 10 min after the test injection of AngII.
Figure 2.
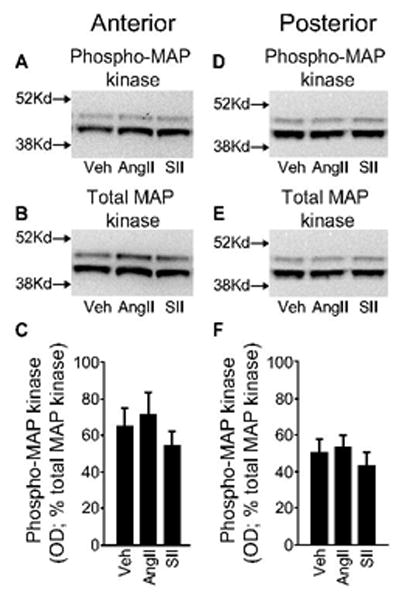
MAP kinase activity in tissue containing the ventral or the dorsal portion of the lamina terminalis after exposure to a treatment regimen of AngII, SII, or vehicle. Rats were given repeated injections of AngII (3 × 300 ng in 1 μl TBS), SII (3 × 30 μg in 1 μl TBS), or vehicle (3 × 1 μl TBS) before all rats received a test injection of AngII (100 ng in 1 μl TBS). Brains were removed and tissue containing either the ventral or the dorsal portion of the lamina terminalis was homogenized and analyzed by SDS-PAGE followed by Western blotting for phosphorylated and total p44/42 MAP kinase (ERK1/2). Representative Western blots of phosphorylated and total MAP kinase are shown in panels A and B and in D and E for anterior and posterior tissue, respectively. The approximate location of the two closest molecular weight markers is shown to the left of each image for phosphorylated and total MAP kinase. Optical density (OD) was measured and quantification of activated MAP kinase, expressed as the percent of total MAP kinase, is shown for the anterior (C) and posterior (F) samples. No group differences were detected in either region (p-values>0.05; n=9 per group).
Inhibition of MAP kinase blocks the desensitizing effect of a repeated AngII treatment regimen
Because previous studies using a neuronal culture model have implicated MAP kinase in AngII-induced phosphorylation of the AT1 receptor (Yang et al., 1997), and because receptor phosphorylation has been shown to be an important component of AT1 receptor desensitization in vitro (Thomas, 1999; Mehta & Griendling, 2007), we tested the requirement of MAP kinase activation for AngII-induced behavioral desensitization. To this end, rats were given 3 pretreatment injections of either U0126 (10 mM in 0.5 μl DMSO) or vehicle (0.5 μl DMSO), with each injection separated by 20 min. Beginning 20 min after the first pretreatment injection, rats received an AngII (3 × 300 ng in 0.5 μl TBS) or a vehicle (3 × 0.5 μl TBS) treatment regimen. Twenty minutes after the final treatment regimen injection (40 min after the final injection of U0126 or DMSO), all rats were given a test injection of AngII (100 ng in 1 μl TBS) and water intake was measured over the subsequent 30 min (for a timeline of the experimental design, see Figure 3A).
Figure 3.
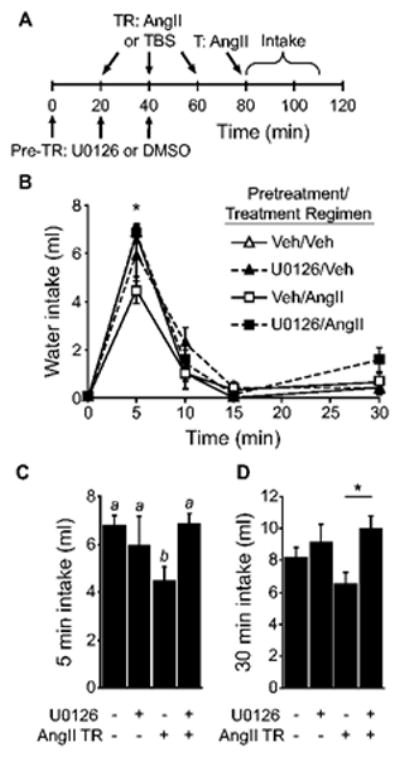
Pretreatment with the MAP kinase inhibitor, U0126, prevented the behavioral desensitization caused by repeated injections of AngII. A) Timeline of the experiment. B) There was a significant Time × Pretreatment × Treatment Regimen interaction and posthoc tests found that the effect was driven by differences in intake 5 min after the AngII test injection (*p<0.05; n=5-7 per group). As shown in panel C, pretreatment with U0126 prevented the effect of an AngII treatment regimen on 5-min water intake (p<0.05). Bars with different letters were significantly different. D) Thirty minute intake by rats given pretreatment with U0126 and repeated injections of AngII was greater than that by rats given repeated injections of AngII without U0126 (*p<0.05 U0126/AngII vs Veh/AngII).
As shown in Figure 3, U0126 blocked AngII-induced behavioral desensitization. Analysis of non-cumulative, binned water intake found a significant main effect of Pretreatment (F1,21=6.281, p<0.05) and a significant Time × Pretreatment × Treatment Regimen interaction (F3,63=2.914, p<0.05; Figure 3B). We did not, however, find a significant main effect of Treatment Regimen (F1,21=0.226, p=0.64) or a significant Pretreatment × Treatment Regimen interaction (F1,21=2.081, p=0.16). Differences in non-cumulative intake were significant in the 5 min bin, but not at any other time (Figure 3C). Post hoc probes of the 3-way interaction found that pretreatment with U0126 did not affect water intake by rats in the vehicle treatment regimen group (p=0.94). In the absence of U0126, administration of an AngII treatment regimen caused a reduction in AngII-induced water intake, but pretreatment with U0126 blocked this effect. Analysis of total 30 min water intake revealed a significant main effect of Pretreatment (F1,21=6.281, p<0.05, Figure 3D), but we did not find a statistically significant main effect of Treatment regimen (F1,21=0.226, p=0.64) or a significant Pretreatment × Treatment Regimen interaction (F1,21=2.081, p=0.16). Post hoc tests on the main effect of Pretreatment revealed that in rats given an AngII treatment regimen, water intake was greater after pretreatment with U0126 (p<0.05).
AT1 receptor-mediated G protein activity is not required for AngII-induced behavioral desensitization
Although the experiment above suggests that MAP kinase is required for the behavioral desensitization that results from repeated injections of AngII, it was important to determine if G protein-dependent processes are similarly required. To test this hypothesis, we used a repeated measure design to assess the desensitizing ability of SII, which stimulates AT1 receptors without engaging G protein-mediated signaling pathways. Controlling for order of treatment condition and providing 4 days between treatments, rats were given a treatment regimen of AngII (3 × 300 ng in 1 μl TBS), SII (3 × 30 μg in 1 μl TBS), or vehicle (3 × 1 μl TBS) before a test injection of AngII (100 ng in 1 μl TBS). Analysis of water intake after the test injection revealed a significant treatment effect in total 30 min water intake (F2,48=11.485, p<0.01; Figure 4A). Post hoc comparisons of 30 min intake showed rats given repeated injections of AngII or SII drank less water than was consumed by rats given repeated injections of vehicle (p<0.05). Analysis of non-cumulative, binned intake revealed a significant main effect of Treatment Regimen (F2,48=11.525, p<0.01) and a significant Time × Treatment Regimen interaction (F6,144=5.786, p<0.01; Figure 4B). Post hoc tests found these differences to be most prominent at 5 min (F2,48=7.583, p<0.01) and 10 min (F2,48=12.657, p<0.01) after the test injection (Figure 4C). Specifically, rats given a treatment regimen of AngII or SII drank less water than did rats in the vehicle treatment regimen group (p<0.05). Water intake by rats in the SII and AngII treatment regimen groups also was different at 5 min and 10 min after the test injection (p<0.05).
Figure 4.
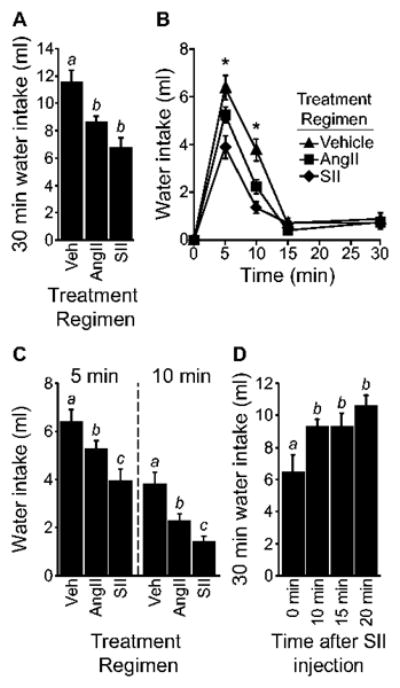
G protein activation is not required for AngII-induced behavioral desensitization. A) Rats given an AngII or an SII treatment regimen had reduced 30 min intake (p<0.05; n=17 per group). B) A significant difference in non-cumulative intake was observed at 5 min and 10 min after the test injection (*p<0.05). C) Rats given an AngII or an SII treatment regimen drank less than vehicle-treated rats at 5 and 10 min after the test injection (p<0.05). Rats treated with SII also drank less than AngII-treated rats in the 5 and 10 min time bins (p<0.05). D) SII caused a reduction in AngII-stimulated intake only when given immediately before AngII injection (p<0.05; n=7-8 per group). Bars with different letters are different from each other.
SII blocks the dipsogenic effect of AngII when both drugs are given simultaneously (Daniels et al., 2005). It was, therefore, important for us to determine if the difference in intake was the result of AngII-like behavioral desensitization instead of a lingering antagonism by SII that attenuated the effect of the test injection. A time-course analysis of the effect of SII on AngII-induced water intake, however, indicated that the intake-suppressive effects of SII did not persist long enough to support the hypothesis that SII was acting as a simple antagonist. Specifically, rats were given a single icv injection of SII (30 μg in 1 μl TBS) either 0, 10, 15, or 20 min before an icv injection of AngII (100 ng in 1 μl TBS). Analysis of 30 min water intake after the injection of AngII revealed a significant main effect of Time (F3,26=5.551, p<0.01, n=30; Figure 4D). Post hoc tests on the main effect showed that SII caused a reduction in water intake when administered immediately before AngII (p<0.05 vs. all other groups), but this effect was no longer evident when SII was given as soon as 10 min before AngII (all p-values>0.05), suggesting that the antagonist-like effects of SII do not last longer than 10 min and, therefore, most likely did not account for the differences in intake observed in the previous experiment.
Discussion
The present experiments tested the role of behaviorally relevant AT1-mediated intracellular signaling cascades in the phenomenon of AngII-induced behavioral desensitization. We found no evidence that repeated injections of AngII or SII changed the amount of phosphorylated MAP kinase after a test injection of AngII, suggesting that MAP kinase signaling persists in spite of the behavioral desensitization associated with repeated injections of AngII. Moreover, we found that MAP kinase activation was required for the development of behavioral desensitization after repeated injections of AngII. The experiments using SII indicated that G protein-mediated signaling pathways were not required for behavioral desensitization to AngII because the desensitizing effect of repeated injections of SII was similar to that caused by repeated injections of AngII, even though SII fails to stimulate G protein-mediated intracellular signaling (Miura & Karnik, 1999; Holloway et al., 2002; Daniels et al., 2005). Together, the results suggest that G protein-independent signaling pathways are sufficient to produce behavioral desensitization and that MAP kinase activation is a critical mediator of this effect.
A previous report showed that expression of suppressor of cytokine signaling (SOCS)-3 in rat hypothalamus plays an important role in AngII-induced behavioral desensitization and this may involve inhibitory feedback on JAK/STAT signaling cascades, thereby reducing the response to subsequent AngII (Torsoni et al., 2004). AngII has been shown to cause increases in phosphorylated JAK (Kodama et al., 1998) and STAT family members (Bhat & Baker, 1997) in rat cardiomyocytes and CHO-K1 cells, respectively. Moreover, the effect on STAT family members was shown to occur via activation of MAP kinases (Bhat & Baker, 1997). Therefore, the present studies may provide an important link in the signaling cascade involved in desensitization of the AT1 receptor in vivo. It is tempting to speculate that AngII stimulation of the AT1 receptor resulting in increases in activated MAP kinase may lead to phosphorylation of JAK/STAT family members, which then cause increases in gene expression and the formation of SOCS-3 that reduces further activation of the JAK/STAT pathway via negative feedback; however, additional research is needed to test this hypothesis.
The present experiments found that repeated injections of AngII or SII had no effect on MAP kinase activation after a test injection of AngII. It is important to note that we focused exclusively on tissue that was microdissected to include structures associated with the ventral lamina terminalis, including the OVLT and the MnPO, or the dorsal lamina terminalis, including the SFO. These sites were targeted because of their established role in mediating the dipsogenic actions of AngII (Epstein et al., 1970; Simpson & Routtenberg, 1973; Buggy et al., 1975; Simpson & Routtenberg, 1975; Buggy & Fisher, 1976; Hoffman & Phillips, 1976; Buggy & Johnson, 1978; Fitts & Masson, 1990; Cunningham et al., 1992) and because a previous study has shown increases in MAP kinase activity after icv AngII in tissue blocks that included these regions (Daniels et al., 2005). The present study, however, used a more refined tissue sampling method to test for differences in activated MAP kinase between regions that were examined collectively in the earlier study. The method used in the present studies allowed us to discriminate between structures of the ventral and dorsal lamina terminalis, but it should be noted that our samples likely included additional areas that bind AngII such as the suprachiasmatic nucleus (Saavedra et al., 1986). It also should be noted that several brain areas known to express AngII receptors and that likely play a role in the behavioral response to AngII were excluded from this analysis. Whether or not repeated injections of AngII differentially affect subsequent MAP kinase signaling in these structures remains an important question for future analyses.
Although the present studies suggest similar activation of MAP kinase under the experimental conditions, it is important to note that these studies were specifically designed to test for the effect of repeated injections of AngII, SII, or vehicle on levels of activated MAP kinase after all rats were given a test injection of AngII, and, therefore, the design does not directly test for activation of MAP kinase by AngII. Previous studies using cell culture models, however, have shown activation of MAP kinase after exposure to AngII (Sadoshima et al., 1995; Bhat et al., 1996), and icv injection of AngII results in increased MAP kinase activity in tissue blocks containing the entire lamina terminalis (Daniels et al., 2005). Nevertheless, the present finding that there were no group differences in activated MAP kinase after repeated AngII administration is consistent with the hypothesis that the MAP kinase response to AngII remains intact even though, under the same conditions, behavioral testing reveals a reduction in water intake. In light of this finding and previous research demonstrating the importance of MAP kinase activity for AngII-induced saline intake (Daniels et al., 2005; Daniels et al., 2009), it seems reasonable to hypothesize that our previous observation that normal saline intake persists in spite of a desensitized water intake response (Vento & Daniels, 2010) results from intact MAP kinase signaling, but additional studies are needed to address this question.
Although MAP kinase appears unimportant in the stimulation of water intake by AngII (Daniels et al., 2009), the present studies found that it is an important mediator of the behavioral desensitization that occurs with repeated injections of AngII. The dose of inhibitor used was relatively large, but it should be of little concern for two reasons: First, pretreatment with the MAP kinase inhibitor, U0126, increased water intake to levels similar to controls, whereas a decrease would be predicted if the dose were too high and generated a more general suppressive effect. Second, previous studies found that a single pre-treatment of U0126 has no effect on water intake stimulated by AngII (Daniels et al., 2009), so the behavioral effect here seems specific to the desensitization, not the primary effect of AngII. Accordingly, it seems reasonable to conclude that although MAP kinase is not involved in the stimulation of water intake after AngII, it is not completely uninvolved in the dipsogenic response.
In addition to supporting the hypothesis that MAP kinase is required for the behavioral effect of repeated injections of AngII, we found no evidence that G protein-dependent signaling pathways are required for desensitization of the drinking response to AngII. Specifically, administration of an SII treatment regimen caused a reduction in AngII-induced water intake similar to that caused by repeated injections of AngII. Previous reports demonstrate that SII binds AT1 receptors without engaging G protein-mediated events, including activation of PKC (Miura & Karnik, 1999; Holloway et al., 2002; Daniels et al., 2005). It, therefore, appears that G protein-independent pathways are sufficient to produce the behavioral desensitization observed after repeated injections of AngII, demonstrating another divergence in the signaling pathways that mediate the behavioral responses to AngII. We advise caution, however, before making a direct comparison between the effect of repeated injections of SII and repeated injections of AngII. This is mostly because the drugs were given at different doses and little work has been done to show the dose-response characteristics of SII in whole animal models. The dose of SII used here was based on a previous report that compared the behavioral effects of AngII and SII and was intended to compensate for the different binding affinities of the ligands (Miura & Karnik, 1999; Daniels et al., 2005). Nevertheless, the present findings allow the critical comparison between the effect of each of these compounds and the control condition. This comparison indicates that, although SII does not engage G protein-mediated signal transduction pathways, it does appear to produce behavioral desensitization similar to that observed after repeated injections of AngII. Accordingly, the present data suggest that G protein-dependent signaling is not required for the behavioral desensitization that occurs under these conditions.
Repeated injections of SII or AngII decreased water intake stimulated by a subsequent injection of AngII. There are a number of possible explanations for this effect other than receptor-mediated desensitization. Included among these alternative explanations are illness or malaise, a generalized motor impairment, and an exaggerated pressor response to the test injection of AngII. Although it is possible that SII and AngII act differently in this respect, we have shown that these explanations do not account for the effect of repeated AngII on water intake (Vento et al., 2012). Taken together with the finding that SII does not antagonize AngII-induced water intake when given as soon as 10 min before injection of AngII, the results suggest that G protein-independent mechanisms are sufficient to produce desensitization to repeated AngII.
The present findings add to a growing literature regarding the behavioral desensitization that occurs after repeated administration of AngII and provide further support for separable roles of AT1-mediated intracellular signaling pathways in the responses to AngII. Specifically, we demonstrated that although MAP kinase is not involved in the stimulation of water intake by central injection of AngII, it does appear to play an essential role in the desensitization of the dipsogenic response to AngII, and it appears to do so through G protein-independent signaling cascades.
Acknowledgments
Technical assistance and helpful comments regarding the manuscript were provided by Anikó Marshall, Naomi McKay, Elizabeth Mietlicki, Kimberly Plyler, and Jessica Santollo. Some data included here were presented in preliminary form at the annual meetings of the Society for the Study of Ingestive Behavior and the Society for Neuroscience. This research was supported by NIH award HL091911 awarded to DD.
References
- Bhat GJ, Abraham ST, Baker KM. Angiotensin II interferes with interleukin 6-induced Stat3 signaling by a pathway involving mitogen-activated protein kinase kinase 1. J Biol Chem. 1996;271:22447–22452. doi: 10.1074/jbc.271.37.22447. [DOI] [PubMed] [Google Scholar]
- Bhat GJ, Baker KM. Angiotensin II stimulates rapid serine phosphorylation of transcription factor Stat3. Mol Cell Biochem. 1997;170:171–176. doi: 10.1023/a:1006865721939. [DOI] [PubMed] [Google Scholar]
- Buggy J, Fisher AE. Anteroventral third ventricle site of action for angiotensin induced thirst. Pharmacol Biochem Behav. 1976;4:651–660. doi: 10.1016/0091-3057(76)90216-1. [DOI] [PubMed] [Google Scholar]
- Buggy J, Fisher AE, Hoffman WE, Johnson AL, Phillips MI. Ventricular obstruction: effect on drinking induced by intracranial injection of angiotensin. Science. 1975;190:72–74. doi: 10.1126/science.1166302. [DOI] [PubMed] [Google Scholar]
- Buggy J, Johnson AK. Angiotensin-induced thirst: effects of third ventricle obstruction and periventricular ablation. Brain Res. 1978;149:117–128. doi: 10.1016/0006-8993(78)90592-9. [DOI] [PubMed] [Google Scholar]
- Cunningham JT, Beltz T, Johnson RF, Johnson AK. The effects of ibotenate lesions of the median preoptic nucleus on experimentally-induced and circadian drinking behavior in rats. Brain Res. 1992;580:325–330. doi: 10.1016/0006-8993(92)90961-8. [DOI] [PubMed] [Google Scholar]
- Daniels D. Alan N. Epstein award: Intracellular signaling and ingestive behaviors. Physiol Behav. 2010;100:496–502. doi: 10.1016/j.physbeh.2010.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels D, Mietlicki EG, Nowak EL, Fluharty SJ. Angiotensin II stimulates water and NaCl intake through separate cell signalling pathways in rats. Exp Physiol. 2009;94:130–137. doi: 10.1113/expphysiol.2008.044446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels D, Yee DK, Faulconbridge LF, Fluharty SJ. Divergent behavioral roles of angiotensin receptor intracellular signaling cascades. Endocrinology. 2005;146:5552–5560. doi: 10.1210/en.2005-0774. [DOI] [PubMed] [Google Scholar]
- Epstein AN, Fitzsimons JT, Rolls BJ. Drinking induced by injection of angiotensin into the brain of the rat. J Physiol. 1970;210:457–474. doi: 10.1113/jphysiol.1970.sp009220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitts DA, Masson DB. Preoptic angiotensin and salt appetite. Behav Neurosci. 1990;104:643–650. doi: 10.1037//0735-7044.104.4.643. [DOI] [PubMed] [Google Scholar]
- Fitzsimons JT. Angiotensin, thirst, and sodium appetite. Physiol Rev. 1998;78:583–686. doi: 10.1152/physrev.1998.78.3.583. [DOI] [PubMed] [Google Scholar]
- Fleegal MA, Sumners C. Drinking behavior elicited by central injection of angiotensin II: roles for protein kinase C and Ca2+/calmodulin-dependent protein kinase II. Am J Physiol Regul Integr Comp Physiol. 2003;285:R632–640. doi: 10.1152/ajpregu.00151.2003. [DOI] [PubMed] [Google Scholar]
- Gebke E, Muller AR, Jurzak M, Gerstberger R. Angiotensin II-induced calcium signalling in neurons and astrocytes of rat circumventricular organs. Neuroscience. 1998;85:509–520. doi: 10.1016/s0306-4522(97)00601-5. [DOI] [PubMed] [Google Scholar]
- Guo DF, Sun YL, Hamet P, Inagami T. The angiotensin II type 1 receptor and receptor-associated proteins. Cell Res. 2001;11:165–180. doi: 10.1038/sj.cr.7290083. [DOI] [PubMed] [Google Scholar]
- Hoffman WE, Phillips MI. Regional study of cerebral ventricle sensitive sites to angiotensin II. Brain Res. 1976;110:313–330. doi: 10.1016/0006-8993(76)90405-4. [DOI] [PubMed] [Google Scholar]
- Holloway AC, Qian H, Pipolo L, Ziogas J, Miura S, Karnik S, Southwell BR, Lew MJ, Thomas WG. Side-chain substitutions within angiotensin II reveal different requirements for signaling, internalization, and phosphorylation of type 1A angiotensin receptors. Mol Pharmacol. 2002;61:768–777. doi: 10.1124/mol.61.4.768. [DOI] [PubMed] [Google Scholar]
- Hunyady L, Catt KJ, Clark AJ, Gaborik Z. Mechanisms and functions of AT(1) angiotensin receptor internalization. Regul Pept. 2000;91:29–44. doi: 10.1016/s0167-0115(00)00137-3. [DOI] [PubMed] [Google Scholar]
- Kirby RF, Thunhorst RL, Johnson AK. Effects of a non-peptide angiotensin receptor antagonist on drinking and blood pressure responses to centrally administered angiotensins in the rat. Brain Res. 1992;576:348–350. doi: 10.1016/0006-8993(92)90703-c. [DOI] [PubMed] [Google Scholar]
- Kodama H, Fukuda K, Pan J, Makino S, Sano M, Takahashi T, Hori S, Ogawa S. Biphasic activation of the JAK/STAT pathway by angiotensin II in rat cardiomyocytes. Circ Res. 1998;82:244–250. doi: 10.1161/01.res.82.2.244. [DOI] [PubMed] [Google Scholar]
- McKinley MJ, Allen A, Clevers J, Denton DA, Mendelsohn FA. Autoradiographic localization of angiotensin receptors in the sheep brain. Brain Res. 1986;375:373–376. doi: 10.1016/0006-8993(86)90761-4. [DOI] [PubMed] [Google Scholar]
- McKinley MJ, McAllen RM, Pennington GL, Smardencas A, Weisinger RS, Oldfield BJ. Physiological actions of angiotensin II mediated by AT1 and AT2 receptors in the brain. Clin Exp Pharmacol Physiol Suppl. 1996a;3:S99–104. [PubMed] [Google Scholar]
- McKinley MJ, Pennington GL, Oldfield BJ. Anteroventral wall of the third ventricle and dorsal lamina terminalis: headquarters for control of body fluid homeostasis? Clin Exp Pharmacol Physiol. 1996b;23:271–281. doi: 10.1111/j.1440-1681.1996.tb02823.x. [DOI] [PubMed] [Google Scholar]
- Mehta PK, Griendling KK. Angiotensin II cell signaling: physiological and pathological effects in the cardiovascular system. Am J Physiol Cell Physiol. 2007;292:C82–97. doi: 10.1152/ajpcell.00287.2006. [DOI] [PubMed] [Google Scholar]
- Miura S, Karnik SS. Angiotensin II type 1 and type 2 receptors bind angiotensin II through different types of epitope recognition. J Hypertens. 1999;17:397–404. doi: 10.1097/00004872-199917030-00013. [DOI] [PubMed] [Google Scholar]
- Quirk WS, Wright JW, Harding JW. Tachyphylaxis of dipsogenic activity to intracerebroventricular administration of angiotensins. Brain Res. 1988;452:73–78. doi: 10.1016/0006-8993(88)90010-8. [DOI] [PubMed] [Google Scholar]
- Saavedra JM, Israel A, Plunkett LM, Kurihara M, Shigematsu K, Correa FM. Quantitative distribution of angiotensin II binding sites in rat brain by autoradiography. Peptides. 1986;7:679–687. doi: 10.1016/0196-9781(86)90044-6. [DOI] [PubMed] [Google Scholar]
- Sadoshima J, Qiu Z, Morgan JP, Izumo S. Angiotensin II and other hypertrophic stimuli mediated by G protein-coupled receptors activate tyrosine kinase, mitogen-activated protein kinase, and 90-kD S6 kinase in cardiac myocytes. The critical role of Ca(2+)-dependent signaling. Circ Res. 1995;76:1–15. doi: 10.1161/01.res.76.1.1. [DOI] [PubMed] [Google Scholar]
- Sakai RR, He PF, Yang XD, Ma LY, Guo YF, Reilly JJ, Moga CN, Fluharty SJ. Intracerebroventricular administration of AT1 receptor antisense oligonucleotides inhibits the behavioral actions of angiotensin II. J Neurochem. 1994;62:2053–2056. doi: 10.1046/j.1471-4159.1994.62052053.x. [DOI] [PubMed] [Google Scholar]
- Sakai RR, Ma LY, He PF, Fluharty SJ. Intracerebroventricular administration of angiotensin type 1 (AT1) receptor antisense oligonucleotides attenuate thirst in the rat. Regul Pept. 1995;59:183–192. doi: 10.1016/0167-0115(95)00111-n. [DOI] [PubMed] [Google Scholar]
- Sasamura H, Dzau VJ, Pratt RE. Desensitization of angiotensin receptor function. Kidney Int. 1994;46:1499–1501. doi: 10.1038/ki.1994.429. [DOI] [PubMed] [Google Scholar]
- Simpson JB, Routtenberg A. Subfornical organ: site of drinking elicitation by angiotensin II. Science. 1973;181:1172–1175. doi: 10.1126/science.181.4105.1172. [DOI] [PubMed] [Google Scholar]
- Simpson JB, Routtenberg A. Subfornical organ lesions reduce intravenous angiotensin-induced drinking. Brain Res. 1975;88:154–161. doi: 10.1016/0006-8993(75)90965-8. [DOI] [PubMed] [Google Scholar]
- Swanson LW. Brain Maps: Structure of the Rat Brain. Elsevier, Amsterdam (Third revised edition) 2003 [Google Scholar]
- Thomas WG. Regulation of angiotensin II type 1 (AT1) receptor function. Regul Pept. 1999;79:9–23. doi: 10.1016/s0167-0115(98)00140-2. [DOI] [PubMed] [Google Scholar]
- Thomas WG, Thekkumkara TJ, Baker KM. Cardiac effects of AII. AT1A receptor signaling, desensitization, and internalization. Adv Exp Med Biol. 1996;396:59–69. [PubMed] [Google Scholar]
- Torsoni MA, Carvalheira JB, Calegari VC, Bezerra RM, Saad MJ, Gontijo JA, Velloso LA. Angiotensin II (AngII) induces the expression of suppressor of cytokine signaling (SOCS)-3 in rat hypothalamus - a mechanism for desensitization of AngII signaling. J Endocrinol. 2004;181:117–128. doi: 10.1677/joe.0.1810117. [DOI] [PubMed] [Google Scholar]
- Vento PJ, Daniels D. Repeated administration of angiotensin II reduces its dipsogenic effect without affecting saline intake. Exp Physiol. 2010;95:736–745. doi: 10.1113/expphysiol.2010.052191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vento PJ, Myers KP, Daniels D. Investigation into the specificity of angiotensin II-induced behavioral desensitization. Physiol Behav. 2012;105:1076–1081. doi: 10.1016/j.physbeh.2011.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisinger RS, Blair-West JR, Burns P, Denton DA, Tarjan E. Role of brain angiotensin in thirst and sodium appetite of rats. Peptides. 1997;18:977–984. doi: 10.1016/s0196-9781(97)00077-6. [DOI] [PubMed] [Google Scholar]
- Yang H, Lu D, Raizada MK. Angiotensin II-induced phosphorylation of the AT1 receptor from rat brain neurons. Hypertension. 1997;30:351–357. doi: 10.1161/01.hyp.30.3.351. [DOI] [PubMed] [Google Scholar]
- Zapparoli A, Figueiredo JF, Boer PA, Gontijo JA. Impaired dipsogenic and renal response to repetitive intracerebroventricular angiotensin II (AngII) injections in rats. J Renin Angiotensin Aldosterone Syst. 2011;12:161–168. doi: 10.1177/1470320310392617. [DOI] [PubMed] [Google Scholar]