

Abstract
IRX-2 is a cytokine-based biologic agent that has the potential to enhance antitumor immune responses. We investigated whether IRX-2 can protect T cells from tumor-induced apoptosis. Tumor-derived microvesicles (MV) expressing FasL were purified from supernatants of tumor cells and incubated with activated CD8+ T cells. MV induced significant CD8+ T-cell apoptosis, as evidenced by Annexin binding (64.4±6.4%), caspase activation (58.1±7.6%), a loss of mitochondrial membrane potential (82.9±3.9%) and DNA fragmentation. T-cell pretreatment with IRX-2 prevented apoptosis. IRX-2-mediated cytoprotection was dose and time dependent and was comparable to effects of IL-2, IL-7 or IL-15. IRX-2 prevented MV-induced downregulation of JAK3 and TCRζ chain and induced STAT5 activation in T cells. IRX-2 prevented MV-induced Bax and Bim upregulation (P<0.005–0.05), prevented cytochrome c release and Bid cleavage, and concurrently restored the expression of Bcl-2, Bcl-xL, FLIP and Mcl-1 (P<0.005–0.01) in T cells. In addition, IRX-2 reversed MV-induced inhibition of the PI3K/Akt pathway. An Akt inhibitor (Akti-1/2) abrogated protective effects of IRX-2, suggesting that Akt is a downstream target of IRX-2 signaling. Thus, ex vivo pretreatment of CD8+ T cells with IRX-2 provided potent protection from tumor-induced apoptosis. IRX-2 application to future cancer biotherapies could improve their effectiveness by bolstering T-cell resistance to tumor-induced immunosuppression.
Keywords: cytokines, apoptosis, lymphocyte survival, PI3K/Akt, IRX-2
Tumor-induced local and systemic immune suppression is commonly seen in patients with advanced malignancies. It is mediated by a variety of tumor-driven mechanisms, among which is the targeted apoptosis of effector T cells.1 In previous studies we observed a high level of apoptosis in tumor-infiltrating lymphocytes and T lymphocytes in the peripheral circulation of head and neck squamous-cell carcinoma (HNSCC) and melanoma patients.2,3 We demonstrated that CD8+ T cells are more sensitive to apoptosis than CD4+ cells, and that the effector and tumor-specific subpopulations of CD8+ T cells are preferentially targeted for apoptosis.4
We and others have associated apoptosis of these antitumor effector cells with expression of FasL on the surface of tumor cells.5 Many human tumors express FasL and can eliminate activated Fas+ effector lymphocytes through the Fas/FasL pathway.6 FasL expression on tumor cells of different origin has been shown to correlate with patient prognosis negatively.7,8 Tumors can release membrane-associated FasL through secretion of microvesicles (MV), thereby providing an explanation for spontaneous apoptosis of T lymphocytes observed in the peripheral circulation of patients with cancer.9–11 We showed previously that MV detected in sera of patients with oral carcinoma induced caspase-3 cleavage, DNA fragmentation, cytochrome c release, loss of mitochondrial membrane potential (MMP) and TCRζ-chain downregulation in activated T lymphocytes.11 Thus, the activity of tumor-derived MV might significantly contribute to the dysfunction and death of effector T cells in cancer patients. The loss of these cells could be responsible for inadequate antitumor function, and by extension, for inadequate immune responses to cancer vaccines.
The immunotherapeutic agent IRX-2 is a multitarget biologic agent containing physiological quantities of IL-1β, IL-2, IL-6, IL-8, GM-CSF, interferon-γ (IFNγ) and tumor necrosis factor-α (TNFα). Initial clinical trials have shown that IRX-2-based immunotherapy in HNSCC patients increased both apparent disease-free survival and overall survival12 (JW Hadden, unpublished data). The underlying molecular mechanisms of IRX-2-mediated effects include increased T-cell proliferation and enhanced dendritic cell (DC) activation and maturation.13 These data suggest that the promising clinical effects of IRX-2 are the result of more effective antitumor T-cell responses. The objective of this work was to test whether IRX-2 could additionally act as a survival factor able to protect T cells from tumor-induced apoptosis mediated by MV or other tumor-derived products.
Results
IRX-2 protects both Jurkat T cells and primary T lymphocytes from cell death mediated by a variety of apoptosis-inducing agents
To determine whether IRX-2 protects T cells from apoptosis mediated by tumor-derived MV, we preincubated CD8+ FasL-sensitive Jurkat cells with IRX-2 for 24 h and subsequently treated them with 10 μg of tumor-derived MV. Co-incubation of Jurkat cells with MV caused marked apoptosis, as demonstrated by enhanced FITC-VAD-FMK staining indicative of caspase activation (Figure 1a and c) and Annexin (ANX) binding (Figure 1b and d). On preincubation of Jurkat cells with IRX-2, the MV-induced apoptosis was significantly reduced (Figure 1a–d).
Figure 1.

Preincubation with IRX-2 decreases CD8+ Jurkat cell apoptosis induced by various agents. CD8+ Jurkat cells were pretreated with IRX-2 (1 : 3 dilution ~4 ng/ml or 90 IU/ml IL-2) for 24 h and then treated with tumor-derived MV (10 μg), CH-11 Ab (400 ng/ml) or staurosporine (1 μg/ml) for 3 h. (a) Caspase activation was detected by FITC-VAD-FMK staining and flow cytometry. The gate was set on CD8+ Jurkat cells. Dot plots shown are representative of five independent experiments. (b). CD8+ Jurkat cells were analyzed for Annexin V binding by flow cytometry. Dead cells (7-AAD+) were excluded, and the gate was set on 7-AAD− CD8+ Jurkat cells. (c) Mean percentage ± S.D. of FITC-VAD-FMK+ Jurkat T cells following incubation with various apoptosis-inducing agents. (d) Mean percentage ± S.D. of Annexin-V-positive/7-AAD− Jurkat cells following incubation with various apoptosis-inducing agents. Results shown in c and d are representative of three independent experiments (*P<0.05; **P<0.002). In each case, apoptosis levels±IRX-2 pretreatment are compared. (e, f) Antiapoptotic effects of IRX-2 are time and concentration dependent. CD8+ Jurkat cells were pretreated with IRX-2 (1 : 3 dilution) before MV incubation for increasing periods of time (0–24 h) or with increasing IRX-2 concentrations for a 24 h period, and subsequently treated with MV (10 μg) for 3 h. Activation of caspases was analyzed by measuring FITC-VAD-FMK binding by flow cytometry. (e) Time-course analysis. (f) Concentration-course analysis. Results are mean percentages±S.D. of four independent experiments
Interestingly, we found that IRX-2 was effective not only against MV-induced apoptosis, but also provided protection to Jurkat cells against FasL-induced (CH-11 Ab) and cytotoxic drug-induced (staurosporine) apoptosis (Figure 1c and d).
IRX-2-mediated protection from MV- or CH-11 Ab-induced apoptosis was also observed when using primary blood-derived CD8+ and CD4+ T lymphocytes, as indicated by decreases in caspase activation (Table 1) and ANX binding (data not shown) in these T cells. CD8+ T cells showed a significantly greater sensitivity to MV-induced apoptosis than CD4+ T cells, but in both subsets, IRX-2 pretreatment provided protection against MV-induced apoptosis (Table 1). Taken together, these findings indicate that IRX-2 can effectively protect primary T cells and T-cell lines not only from MV- or anti-Fas Ab-induced apoptosis but also from intrinsic apoptosis associated with staurosporine-induced mitochondrial changes.
Table 1.
IRX-2 protects activated peripheral blood CD8+ and CD4+ T lymphocytes from MV- and Fas-induced apoptosisa
| CD8+ cells |
CD4+ cells |
|||
|---|---|---|---|---|
| FITC-VAD-FMK+ cells | P-valueb | FITC-VAD-FMK+ cells | P-valueb | |
| Untreated cells | 14.8±4.8 | — | 12.6±2.3 | — |
| No IRX-2+CH-11 Ab | 52.8±4.9 | — | 41.2±9.8 | — |
| +IRX-+CH-11 Ab | 15.0±3.5 | 0.0010 | 15.4±6.1 | 0.0510 |
| No IRX-2+MV | 71.0±8.0 | — | 45.7±1.6 | — |
| +IRX-2+MV | 26.5±10.3 | 0.0006 | 13.9±6.4 | 0.0211 |
Activated primary CD8+ or CD4+ cells were preincubated with IRX-2 for 24 h (at 1 : 3 final dilution = 90 IU/ml IL-2) and were then treated with 10 μg MV or CH-11 Ab at 400 ng/ml for an additional 24 h. Cells were analyzed for caspase activation by FITC-VAD-FMK binding by flow cytometry. Results are mean percentages±S.D. of positive cells obtained in three independent experiments.
The P-values are for differences between IRX-2 untreated and +IRX-2 treated cells
IRX-2-mediated protection from apoptosis is time and concentration dependent
CD8+ Jurkat cells were next preincubated with IRX-2 for various time periods or with increasing concentrations of IRX-2 and subsequently treated with MV for 3 h (Figure 1e and f). IRX-2-mediated blocking of MV-induced apoptosis was time dependent, as extending the time of the preincubation with IRX-2 intensified its protective effects (Figure 1e). Preincubation of T cells with different IRX-2 concentrations showed a dose-dependent inhibition of apoptosis caused by MV (Figure 1f). At the highest possible concentration (i.e., undiluted IRX-2), IRX-2 completely inhibited the induction of apoptosis by MV (Figure 1f). IRX-2 alone had no effect on Jurkat cell apoptosis.
IRX-2 protects T cells from MV-induced apoptosis at various steps of the apoptotic cascade
Next, the ability of IRX-2 to inhibit downstream steps in the apoptotic process was evaluated by co-incubating Jurkat cells with MV. We previously determined that the protection observed in this system was similar to that seen with primary T cells in response to several apoptosis-inducing agents tested. MV treatment of Jurkat cells led to a very strong increase in the mean fluorescent intensity (MFI) of caspase-3/-7-FAM, a dye that binds most efficiently to activated caspase-3 and caspase-7, the main effector caspases of both receptor- and mitochondria-mediated apoptosis (Figure 2a and b). Pretreatment of Jurkat cells with IRX-2 completely prevented the induction of caspase activity, similar to the irreversible caspase inhibitor, zVAD, and the anti-Fas neutralizing monoclonal antibody (mAb), ZB4. Caspase-3 activation in these cells by MV was also detected by western immunoblots, where a significant processing of the prodomain and a simultaneous increase of the active cleaved form (p18) was observed in MV-treated Jurkat T cells over 24 h (Figure 2c, lanes 3 and 4). IRX-2 pretreatment effectively blocked the generation of the active subunit (Figure 2c, lanes 5 and 6).
Figure 2.

IRX-2 blocks MV-induced activation of caspases-3 and -7 and a loss of mitochondrial membrane potential (MMP) in CD8+ Jurkat cells. (a) Activation of caspases-3 and -7 in CD8+ Jurkat cells was assessed by flow cytometry for caspase-3/-7-FAM binding. Results of a representative experiment out of five experiments performed are shown. (b) Mean fluorescence intensity (MFI) of caspase-3/-7-FAM expression was determined in CD8+ Jurkat cells that were either untreated, incubated with 10 μg MV for 3 h or pretreated for 24 h with IRX-2 (1 : 3 dilution) and then incubated with MV for 3 h. CD8+ Jurkat cells were also co-incubated with MV and 20–100 μM of the pan-caspase inhibitor Z-VAD-FMK (zVAD) or co-incubated with MV and the anti-Fas neutralizing mAb ZB4 (10 μg/ml) (controls). Results are the mean MFI± S.D. of three independent experiments. (c) Western immunoblots showing caspase-3 activation in CD8+ Jurkat cells. The cells were either untreated, treated with IRX-2 (1 : 3 dilution) for 24 h (+ IRX), treated with MV (10 μg) for 3 h (+ MV 3 h) or 24 h (+ MV 24 h) or preincubated with IRX-2 for 24 h and then treated with MV (10 μg) for 3 or 24 h (+ IRX→MV). Whole-cell lysates of the cells were separated on SDS-PAGE and transferred to PVDF membranes for subsequent western blotting. Activation of caspase-3 is shown as a decrease in the inactive proform and the appearance of the active subunits p17 and p10. Results are representative of three independent western blots. (d) CD8+ Jurkat cells were analyzed by flow cytometry for a decrease in red fluorescence of the cationic dye, JC-1, indicating a loss of MMP. Percentages of JC-1-red-negative cells were determined in untreated CD8+ Jurkat cells, after 3 h treatment with MV (10 μg) alone or after 24 h pretreatment with IRX-2 (1 : 3 dilution) +MV. Cells treated with 50 μM carbonyl cyanide 3-chlorophenylhydrazone (CCCP), a protonophore that dissipates the H+ gradient across the inner mitochondrial membrane, were used as a positive control. The data are from a representative flow cytometry experiment out of five experiments performed. (e) Effects of MV alone, IRX + MV or MV in combination with Z-VAD-FMK or ZB4 on a loss of MMP in Jurkat cells. The data are means±S.D. of five independent experiments (*P<0.005; **P<0.002 compared to samples without IRX-2)
In addition, IRX-2 blocked the MV-induced loss of MMP in Jurkat T cells (Figure 2d and e). This effect was similar to that mediated by the two inhibitors z-VAD and ZB4 (Figure 2e), although zVAD-mediated inhibition was partial, even when high concentrations (up to 100 μM) were used. This result suggests that MV may also induce mitochondrial depolarization in a caspase-independent manner. Finally, IRX-2 pretreatment significantly reduced the MV-induced nuclear DNA fragmentation as detected by TUNEL assays (Figure 3A and B), representing an advanced step in the apoptotic process. We have, therefore, confirmed that the protective effect of IRX-2 impacts many of the relevant steps that culminate in MV-mediated T-cell death.
Figure 3.

Preincubation with IRX-2 protects CD8+ Jurkat cells from MV-induced DNA fragmentation. (A) Fluorescent microscopy of CD8+ Jurkat cells that were either untreated (a), incubated for 24 h with IRX-2 alone (b) or MV alone for 24 h (c) or preincubated with IRX-2 for 24 h and subsequently treated with MV for 24 h (d) and then stained by the TUNEL method to reveal DNA strand breaks (red nuclei) indicative of apoptosis. (B) Percentage of TUNEL-positive CD8+ Jurkat cells in the above cocultures. A minimum of 300 CD8+ Jurkat cells were counted for each treatment group. Results are expressed as the mean percentages±S.D. of two independent experiments (*P<0.0002 compared to MV-treated sample). (C) Antiapoptotic effects of IRX-2 require a pretreatment of T cells. CD8+ Jurkat cells were untreated, treated with IRX-2 (1 : 3 dilution) for 24 h (+ IRX), MV for 3 h (+ MV), preincubated with IRX-2 for 24 h and then treated with MV (10 μg) for 3 h (+ IRX→MV) or first incubated with MV and then treated with IRX-2 (+ MV→IRX-2) or incubated with both agents simultaneously (+ MV and IRX) for 3 h 24 + + or h, respectively. Activation of caspases was analyzed by flow cytometry. Results are mean percentages±S.D. from a representative experiment out of three experiments performed (*P<0.002 compared to MV-treated sample)
IRX-2 does not reverse MV-induced apoptosis
To determine whether IRX-2 could protect T cells from apoptosis once the apoptotic cascade had been initiated, CD8+ Jurkat cells were treated first with MV and then incubated with IRX-2. Alternatively, Jurkat cells were simultaneously treated with MV and IRX-2. In comparison to the effects of IRX-2 treatment before the addition of MV, apoptosis was reduced by about 50% after simultaneous co-incubation of T cells with MV + IRX-2 (Figure 3C). When IRX-2 was added 3 h after treatment with MV, the protective effect of IRX-2 was completely abrogated. IRX-2 was not able to inhibit the apoptotic cascade already initiated in T cells by MV. Thus, it appears that IRX-2 acts through a protective mechanism and cannot induce a reversal of apoptosis once it is initiated by MV.
IRX-2 protects T cells from both apoptotic pathways
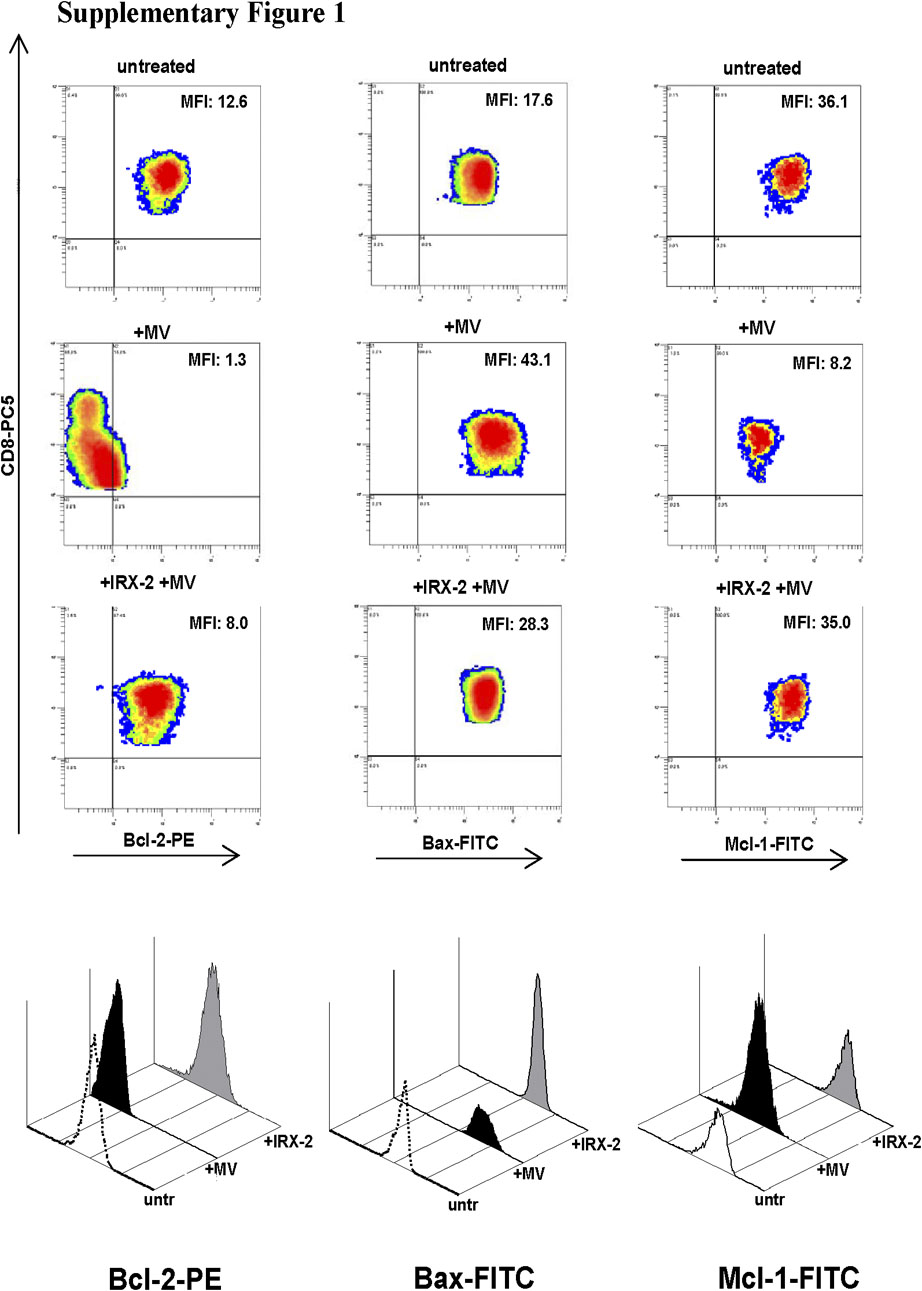
To further examine the mechanisms responsible for IRX-2-mediated protection of T cells from MV-induced apoptosis, we performed western blots using cytosolic and heavy membrane (mainly mitochondrial) cell fractions. As shown in Figure 4, MV-induced mitochondrial release of intermembrane apoptogenic proteins (cytochrome c, SMAC and AIF). The appearance of cytochrome c and SMAC in the cytosol was associated with their reduced levels in the mitochondria-containing fraction. IRX-2 effectively inhibited mitochondrial release of these proteins. Cleavage of Bid and mitochondrial translocation of tBid were also inhibited in cells pretreated with IRX-2. Moreover, cleavage of caspase-8 by MV into p43/41 and p18 fragments in the cytosol was also inhibited by IRX-2. Antiapoptotic proteins, Bcl-2 and Bcl-XL, were not decreased by short-term MV treatment in mitochondria, although pretreatment with IRX-2 appeared to increase their expression. In contrast, when primary activated CD8+ T cells were used, MV induced significant downregulation in Bcl-2, Bcl-XL and FLIP expression, whereas IRX-2 pretreatment in part prevented their loss, as demonstrated by quantitative flow cytometry (Table 2). The difference between flow and western blot results in antiapoptotic protein expression may be due to the use of different methods applied to different cell types. In any case, MV caused a significant upregulation of the proapoptotic proteins Bax and Bim, which was prevented by IRX-2 pretreatment (Table 2; Supplementary Figure 1). Although MV alone increased Fas expression on Jurkat or primary T cells, IRX pretreatment decreased (P<0.002) Fas expression on the surface of these cells (data not shown). MV and IRX had little or no effect on the expression of proapoptotic FasL (data not shown). Similar results were obtained after IRX-2 incubation and MV treatment of activated primary CD4+ cells and CD8+ Jurkat cells (data not shown).
Figure 4.

Expression of proapoptotic and antiapoptotic proteins in the cytosol and mitochondrial fractions of T cells treated with MV or IRX-2 + MV. Jurkat cells were treated with MV and IRX-2 as indicated. The cells and were then subjected to subcellular fractionation to obtain cytosol and heavy membrane (HM) fractions. The subcellular fractions were resolved by SDS-PAGE, and the indicated proteins were detected by immunoblotting. β-Actin and Cox IV served as controls for equal loading for the cytosol and HM fractions, respectively
Table 2.
MV and IRX-2 modulate the expression of pro- and antiapoptotic proteins in primary T cellsa
| Untreated | +MV | P-valueb | +IRX-2+MV | P-valueb | |
|---|---|---|---|---|---|
| Bcl-2 | 7.7±0.4 | 1.9±0.1 | 0.0008 | 4.8±0.6 | 0.0049 |
| Bax | 17.9±1.2 | 40.0±1.5 | 0.0001 | 26.8±2.3 | 0.0003 |
| Bcl-xL | 20.1±0.1 | 8.2±0.4 | 0.0001 | 11.0±0.8 | 0.0019 |
| FLIP | 42.4±0.6 | 17.5±0.6 | 0.0002 | 25.5±2.0 | 0.0030 |
| Bim | 8.1±0.3 | 16.7±1.6 | 0.0016 | 9.2±0.3 | 0.0020 |
| Mcl-1 | 37.5±3.8 | 7.1±1.1 | 0.0004 | 35.1±1.6 | 0.0003 |
Activated peripheral blood CD8+ cells were preincubated with IRX-2 (at 1 : 3 dilution) for 24 h and then treated with 10 μg MV for additional 24 h. Expression levels (mean fluorescence intensity) of different pro- and antiapoptotic protein were measured by quantitative flow cytometry. The data are the MFI±S.D. of pro- and antiapoptotic proteins in MV- and IRX-2-treated activated primary CD8+ cells and were obtained in three independent experiments.
The P-values indicate changes in MFI between untreated and MV-treated or IRX-2+MV-treated cells
Experiments were also performed to evaluate effects of cycloheximide on IRX-2-mediated protection of T cells. IRX-2 protection was significantly reduced in the presence of cycloheximide (P<0.002), and this reduction was accompanied by downregulation in expression levels of FLIP, Mcl-1 and Bcl-2 (data not shown).
IRX-2 protects T cells from MV-induced downregulation of JAK3 and STAT5 expression
It has been previously observed that MV derived from sera of patients with cancer downregulate expression of molecules mediating the common γ-chain cytokine receptor signaling pathway, including JAK3 and STAT5.14 Similar here, MV caused a significant downregulation of JAK3 in T cells (Figure 5a, panel 1, compare lanes 1 and 3), which intensified with the extended time of co-incubation (Figure 5a, panel 1, lanes 3 and 4). Although IRX-2 alone did not increase JAK3 expression (Figure 5a, panel 1, compare lanes 1 and 2), it was able to reverse completely the MV-induced JAK3 downregulation and restore its expression (Figure 5a, panel 1, compare lanes 3 and 4 with lanes 5 and 6). Furthermore, IRX-2 caused a strong activation of STAT5, a JAK3 signal transducer, as indicated by phosphorylation of this protein (Figure 5a, panel 2, lanes 2 and 5). This dramatic activation of STAT5 was sustained even after prolonged (24 h) incubation with MV (Figure 5a, panel 2, lane 6). In addition, we observed a loss in CD3ζ expression in T cells after MV treatment. Here again, preincubation with IRX-2 protected T lymphocytes from MV-mediated CD3ζ downregulation (Figure 5a, panel 4, compare lanes 3 and 4 with lanes 5 and 6).
Figure 5.

MV and IRX-2 modulate the expression of signaling molecules in the cytokine and TCR pathway and the activation of the Akt/PI3K pathway. CD8+ Jurkat cells were untreated or treated with IRX-2 (1 : 3 dilution) and MV in different combinations. Whole-cell lysates of cells from each treatment group were separated on SDS-PAGE and transferred to PVDF membranes for subsequent western blotting. (a) The expression levels of JAK3, phosphorylated and total STAT5, CD3ζ and FLIP was analyzed by probing the membrane with the specific Abs. Reprobing with β-actin Ab confirmed equal protein loading. The results shown are representative of four independent experiments. (b) The activation of Akt-1/-2 was analyzed by immunoblotting with Ser473-specific anti-phospho Akt mAb. Reprobing with a total Akt Ab confirmed equal protein loading. Results shown are from a representative experiment out of three experiments performed. (c) CD8+ Jurkat cells were preincubated with IRX-2 for 24 h or left untreated. Cells were then treated with the Akt inhibitor, Akti-1/2, at different concentrations (0–5 μM) for 1 h before the addition of MV for additional 3 h. The level of apoptosis was measured by FITC-VAD-FMK staining and flow cytometry. Results are mean percentages of positive cells±S.D. obtained in three independent experiments (*P<0.05; **P<0.01 compared to MV-treated sample without IRX-2 and Akt inhibitor)
The Akt/PI3K pathway is the main downstream target of antiapoptotic activity of IRX-2
Because our findings showed a substantial influence of IRX-2 on several key proteins of the Bcl-2 family, which could be regulated by the Akt/PKB signaling pathway, we measured the activation of Akt-1/2 in response to MV and/or IRX-2 by western blots. Preincubation with IRX-2 did not enhance basal Akt phosphorylation (Figure 5b, compare lanes 1 and 2). However, when the cells were treated with MV, a dramatic, time-dependent dephosphorylation of Akt-1/2 was observed (Figure 5b, panel 1, lanes 3 and 4). A time-course study with 10 μg of MV showed that Akt dephosphorylation started at 3 h of incubation and intensified over time (data not shown). Pretreatment of Jurkat cells with IRX-2 completely inhibited MV-induced dephosphorylation of Akt-1/2 at both 3 and 24 h of treatment (Figure 5b, panel 1, lanes 5 and 6).
This pronounced prosurvival effect of IRX-2 on CD8+ Jurkat cells, which clearly counteracted the MV-induced Akt inactivation, indicated that Akt might serve as the main downstream target of IRX-2 signaling. As shown in Figure 5c, pretreatment of Jurkat cells with the Akti-1/2 inhibitor before IRX-2 and MV treatments resulted in a gradual abrogation of the antiapoptotic effect of IRX-2. At a relatively low inhibitor concentration of 1 μM, the protection from apoptosis provided by IRX-2 was only slightly inhibited. However, it was completely blocked at the inhibitor concentration of 5 μM. At these inhibitor concentrations, cell viability was not affected. In addition, the inhibitor significantly increased Bax levels and decreased expression levels of Bcl-2 and FLIP (P<0.02 for all) in Jurkat cells exposed to MV and IRX-2 (data not shown). In aggregate, these findings strongly suggest that Akt may be the main downstream coordinator of the survival signal provided by IRX-2, by modulating expression levels of anti- and proapoptotic proteins.
The survival signals promoted by IRX-2 are comparable to protective effects of IL-2, IL-7 and IL-15
Because IL-2 is a principal cytokine in IRX-2, the observed antiapoptotic activity of IRX-2 could be largely IL-2 dependent. However, synergy with other cytokines present in IRX-2 could promote survival. Activated primary CD8+ and CD4+ T cells were incubated with IRX-2 or 100 IU/ml of recombinant human IL-2, a dose approximating that present in IRX-2 (at 1 : 3 dilution) and compared for the ability to inhibit MV or CH-11 Ab-induced apoptosis. As shown in Table 3A, IL-2 had a similar protective effect against MV-induced apoptosis as IRX-2 in CD8+ T cells, but had a somewhat lower, although still significant, protective effect in CD4+ T cells. In terms of protection against CH-11 Ab-induced apoptosis, IL-2 was significantly less effective than IRX-2 in enhancing survival of CD4+ T cells and had almost no effect in CD8+ T cells (Table 3B). These findings indicate that the survival-enhancing potential of IRX-2 is greater than that of its main cytokine IL-2 and that support by other cytokines present in IRX-2 at very low physiological concentrations might contribute to these effects.
Table 3.
Antiapoptotic effects of IRX-2 in comparison to IL-2, IL-7 and IL-15 in MV- or CH-11 Ab-induceda primary T cellsa
| CD8+ cells |
CD4+ cells |
|||
|---|---|---|---|---|
| FITC-VAD-FMK+ cells | P-valueb | FITC-VAD-FMK+ cells | P-valueb | |
| (A) MV-induced apoptosis | ||||
| Control | 21.8±3.9 | — | 14.3±5.0 | — |
| No IRX-2+MV | 71.0±8.0 | — | 45.7±1.6 | 0.0222c |
| +IRX-2+MV | 20.6±0.8 | 0.0006 | 10.2±0.4 | 0.0038 |
| +IL-7+MV | 49.5±3.5 | 0.0004 | 34.5±1.9 | 0.0124 |
| +IL-15+MV | 16.8±6.2 | 0.0044 | 16.7±2.1 | 0.0072 |
| +IL-7/IL-15+MV | 11.8±6.2 | 0.0036 | 11.9±0.8 | 0.0031 |
| +IL-2+MV | 21.9±2.7 | 0.0002 | 22.2±2.7 | 0.0113 |
| (B) CH-11 Ab-induced apoptosis | ||||
| Control | 12.3±2.8 | — | 11.4±1.3 | — |
| No IRX-2+MV | 50.0±0.2 | — | 46.8±3.0 | — |
| +IRX-2+MV | 13.4±3.0 | 0.0007 | 12.0±1.7 | 0.0001 |
| +IL-7+MV | 37.7±1.4 | 0.0015 | 31.8±10.6 | — |
| +IL-15+MV | 28.8±3.1 | 0.0020 | 21.1±5.3 | 0.0086 |
| +IL-7/IL-15+MV | 24.2±4.9 | 0.0028 | 20.2±0.1 | 0.0010 |
| +IL-2+MV | 44.2±6.9 | — | 31.7±3.0 | 0.0128 |
Activated primary CD8+ and CD4+ T cells were preincubated with IRX-2 (1 : 3 dilution), recombinant human IL-2 (100 IU/ml), IL-7 (10 ng/ml), IL-15 (10 ng/ml) or IL-7 and IL-15 (both 10 ng/ml) for 24 h and then treated with 10 μg MV or 400 ng/ml CH-11 Ab for an additional 24 h. Activation of caspases was analyzed by flow cytometry. The data are mean percentages of FITC-VAD-FMK+ cells±S.D. obtained in three independent experiments.
The P-values refer to significant differences between cells pretreated with IRX-2 compared to MV alone or cells pretreated with the indicated cytokine compared to those pretreated with IRX-2.
The P-value refers to the difference in sensitivity to MV-mediated apoptosis between CD8+ and CD4+ T cells
In addition, we compared the protective effect of IRX-2 with the activity of recombinant IL-7 and IL-15, both potent survival cytokines for T lymphocytes, which are not present in the IRX-2 mixture. Preincubation of T cells with these cytokines at a concentration of 10 ng/ml, alone or in combination, provided protection from MV- or CH-11 Ab-induced apoptosis in all cases, although to a different extent (Table 3A and B). A combination of both cytokines blocked apoptosis in the CD8+ and CD4+ cell subsets, and the level of apoptosis inhibition was similar to that mediated by IRX-2, but only in case of MV-induced apoptosis (Table 3A and B). Thus, the protective effects of IRX-2 were comparable to or in some cases (e.g., protection of CD8+ cells) even stronger than those of the recombinant survival cytokines, IL-7 and IL-15.
Discussion
One of the mechanisms used by human tumors to escape from the host’s immune system involves targeted elimination of CD8+ effector T cells through apoptosis mediated by tumor-derived MV.15,16 Immunosuppressive MV have been found in neoplastic lesions, sera, ascites and pleural effusions obtained from cancer patients and have been linked to apoptosis and TCR alterations in effector T cells in these patients.16 MV-driven elimination of effector T cells, which are necessary for antitumor host defense, might contribute to cancer progression. Therefore, protection of antitumor effector cells from functional impairments and death is a major objective of immune therapy. Clinical and experimental data show that certain cytokines, especially survival cytokines using the common receptor γ-chain, are able to protect activated T cells from tumor-induced death and enhance their antitumor activity.17
A new biologic agent called IRX-2 contains natural cytokines and growth factors produced by ex vivo stimulated human peripheral blood mononuclear cells (PBMC) under cGMP conditions. In the initial clinical studies with HNSCC and cervical carcinoma patients, immunotherapy with IRX-2 led to immune-mediated tumor regression in some of these patients.12,18–20 In HNSCC patients treated with IRX-2, tumor regression was accompanied by extensive leukocyte infiltrates. These local changes were, in turn, accompanied by a delayed tumor recurrence and an increase in overall survival as compared to historical controls.12,21 Another effect of IRX-2 was the restoration and normalization of lymphocyte counts in preclinical studies in mice and in lymphopenic HNSCC patients, respectively, especially in combination with thymosin-α1.20
In the current study, we asked whether IRX-2 is additionally able to protect T cells from apoptosis mediated by tumor-derived MV. Using a previously established in vitro model of tumor-induced apoptosis,11 we demonstrated that IRX-2 provides strong protection to T cells from apoptosis mediated by tumor-derived MV through activation of survival pathways, thereby effectively counteracting cancer-related immuno-suppression. The PI3K/Akt pathway is known to have a key function in T-lymphocyte survival through the regulation of pro- and antiapoptotic proteins including Bcl-2 family members.22 We confirmed that the effects of IRX-2 on some of the Bcl-2 family members as well as cFLIP were mediated by the PI3K/Akt pathway. Tumor-derived MV caused a dramatic time-dependent Akt dephosphorylation and IRX-2 pretreatment completely inhibited the MV-induced inactivation of Akt. Our preliminary data show that the Akti-1/2 inhibitor eliminates protective effects of IRX-2 and this is accompanied by a significant downregulation of Bcl-2 and FLIP expression levels as well as upregulation of Bax expression in Jurkat cells (data not shown). These data support the hypothesis that the PI3K/Akt pathway is a central target of IRX-2 for regulating T-cell survival.
IRX-2-mediated protection of activated T cells extends to apoptotic signals induced by agents other than tumor-derived MV, for example, CH-11 Ab or staurosporine. Because the former induces apoptosis mainly through the death receptor pathway23 and the latter activates only the mitochondrial pathway,24 these findings suggest that IRX-2 can protect T cells from both the extrinsic and the intrinsic death pathways. Indeed, it appears that MV initiate caspase-8-dependent apoptotic cascade in association with the production of a proapoptotic Bid fragment. As a BH3-only protein, tBid translocates to the mitochondria, and either activates Bax directly25 or binds and neutralizes antiapoptotic Bcl-2 family members, Bcl-2, Bcl-XL and Mcl-1.26 Binding of tBid to antiapoptotic Bcl-2 proteins may interfere with their interaction with Bax.27 The MV-mediated activation of caspase-3 could represent the combined effect of direct processing of caspase-3 by caspase-8, and further activation of caspase-3 downstream of the mitochondria following the release of the apoptogenic proteins. The significant loss in Mcl-1 expression level may result from its degradation by caspase-8 and caspase-3.28 Because IRX-2 appears to inhibit the MV-mediated activation of caspase-8, it may indirectly block many of the downstream apoptotic events, including Bid cleavage, release of mitochondrial intermembrane apoptogenic proteins and activation of effector caspases. Our data further suggest that the antiapoptotic effect of IRX-2 is not limited to its inhibition of caspase-8 activation, as it independently increases expression levels of antiapoptotic Bcl-2 proteins. Of note, upregulation of Mcl-1 by IL-2, IL-15 and IL-7 has been previously reported.29 Thus, IRX-2-mediated protection appears to be a generalized phenomenon, which is accompanied by upregulation of the antiapoptotic Bcl-2 family members. These results are consistent with numerous other studies reporting changes in Bcl-2, Bcl-XL or Bax expression in circulating T cells of patients with different malignancies,30,31 potentially implicating tumor-derived MV in this process.
In addition, we noted that pretreatment of T cells with IRX-2 partially counteracted the MV-induced downregulation of cFLIP, a dominant-negative inhibitor of caspase-8, and a central modulator of Fas-induced apoptosis.32 It can block the intrinsic mitochondrial pathway indirectly through induction of NF-κB and TCR-induced and IL-2-mediated downregulation of cFLIP has been correlated with the sensitivity of T cells to activation-induced cell death.33 Our preliminary data indicate that FLIP expression levels have a function in IRX-mediated protection of activated T cells from apoptosis.
A principal cytokine in the IRX-2 immunotherapeutic product is IL-2, and it was possible that IL-2 was primarily responsible for the observed cytoprotective effect of IRX-2. However, IRX-2 was found to be significantly more effective than IL-2 alone in enhancing survival of activated human CD8+ and CD4+ T cells. It is likely that cytoprotective effects of IL-2 in IRX-2 are enhanced by the presence of IFNγ and GM-CSF, which in combination, could mediate immuno-potentiating effects.30 The function of other components of IRX-2 (e.g., IL-1α, IL-6, IL-8, TNFα) in promoting T-cell survival is less clear, although studies have shown that depending on tissue location and concentration some of these proinflammatory cytokines can also support antitumor immune responses.30 It is important to note that a functional synergism among the various components of IRX-2 was previously described, demonstrating, for example, that IRX-2 was able to induce maturation of DCs to a greater extent than comparable levels of TNFα alone.13
Cytoprotective effects of IRX-2 were compared with those mediated by the survival cytokines, IL-7 and IL-15, which, like IL-2, are members of the common cytokine receptor γ-chain family. They are important in the peripheral T-cell homeostasis and maintenance of memory T cells.31 IRX-2 was found to be significantly more effective than recombinant IL-7 in protecting activated CD4+ and CD8+ T cells from MV- and CH-11 Ab-induced apoptosis and had similar protective effects as IL-15. Among the cytokines tested, IRX-2 had the greatest survival potency when CH-11 Ab was used to induce apoptosis, implying protection against receptor-mediated apoptosis.
Because it is acknowledged that the common cytokine receptor γ-chain mediates survival signals through the JAK3/STAT5 pathway in T cells,34 effects of MV and IRX-2 on the expression and activation of JAK3 and STAT5 were also evaluated. As expected, a significant downregulation of JAK3 expression by MV was observed.14 IRX-2 was able to restore MV-induced JAK3 downregulation completely, although it did not increase its baseline expression. IRX-2 also induced strong activation of STAT5 proteins that are important survival factors, required for the protection of mature T cells from apoptosis.34 Further, MV-mediated downregulation of CD3ζ, a key signaling molecule of the TCR pathway, in T cells was prevented in the presence of IRX-2. Together, these data emphasize the key function that cytokines present in IRX-2 have in the regulation of death and survival in activated T cells.
Overall, our study indicates that the pretreatment of T cells with IRX-2 restores the balance between pro- and antiapoptotic Bcl-2 family proteins and normalizes the expression of cFLIP, JAK3 and CD3ζ, using the PI3K/Akt pathway as the key regulator. The mechanism of protection mediated by IRX-2 includes neosynthesis of a survival protein that is under the control of the Akt pathway, as Akti-1/2 inhibitor abolished protection and at the same time, decreased expression levels of antiapoptotic proteins but upregulated that of proapoptotic Bax. This mechanism provides potent protection of activated T cells from tumor-induced death, inhibiting the Fas-mediated as well as the mitochondrial pathway of apoptosis. As effector T cells resistant to immunosuppressive influences of the tumor microenvironment are essential for antitumor host defense, utilization of IRX-2 in cancer immunotherapies is likely to improve their survival and promote antitumor effects significantly.
Materials and Methods
Antibodies and reagents
The following mAbs were used for flow cytometry analysis: anti-CD3-ECD, -CD8-PC5, -CD4-PE (Beckman Coulter, Miami, FL, USA); anti-Bcl-2-FITC, -Bcl-2-PE, -Fas-FITC, -FasL-PE (BD Biosciences, San Jose, CA, USA); anti-Bax-FITC, -Bcl-xL-FITC (Santa Cruz Biotechnology, Santa Cruz, CA, USA) and anti-Bid-mAb (Abcam Inc., Cambridge, MA, USA). Polyclonal antibodies (Abs) were: anti-Bim (Cell Signaling Technology Inc., Danvers, MA, USA), anti-FLIP (GenWay Biotech, San Diego, CA, USA) and anti-Mcl-1 Ab (Santa Cruz Biotechnology). FITC-conjugated Annexin V was purchased from Beckman Coulter. FITC-conjugated anti-rabbit IgG was purchased from Jackson ImmunoResearch Laboratories (West Grove, PA, USA) and the isotype controls (IgG1-FITC, IgG2a-FITC and IgG2b-FITC, and IgG2-PE) were purchased from BD Biosciences. Antibodies purchased for western blot analysis included anti-Cox IV (Molecular Probes Inc.); anti-actin (Sigma-Aldrich Inc.); anti-SMAC (Cell Signaling Technology Inc.); anti-Bid, anti-AIF, anti-Cyt c, anti-Mcl-1, anti-Bcl-2 and anti-Bcl-XL (all from Santa Cruz Inc.); anti-Casp-8 (BD Pharmingen); polyclonal phospho-Akt (Ser473), polyclonal total Akt, monoclonal phospho-STAT5 (Tyr694) and monoclonal total STAT5 (Cell Signaling Technology Inc.), monoclonal Bcl-2, monoclonal CD3ζ, monoclonal JAK3, polyclonal caspase-3 and polyclonal anti-FasL Ab3 (BD Biosciences). Anti-Fas (CH-11) agonistic mAb, IgM isotype control for CH-11, anti-Fas blocking mAb, clone ZB4 and isotype IgG1 control for ZB4 were all purchased from Upstate Biotechnology. All cell culture reagents including AIM V medium, RPMI 1640 medium, phosphate-buffered saline (PBS), heat-inactivated fetal calf serum, streptomycin, penicillin, l-glutamine, recombinant trypsin-like enzyme (TrypLE) and Trypan blue dye were purchased from Gibco/Invitrogen (Grand Island, NY, USA). The human recombinant cytokines, rhIL-2, rhIL-7 and rhIL-15, were purchased from PeproTech Inc. Bovine serum albumin (BSA), saponin and staurosporine were from Sigma-Aldrich. 7-Amino-actinomycin D (7-AAD) and the pan-caspase inhibitor, z-VAD-FMK, were obtained from BD Biosciences. The selective inhibitor of Akt1/Akt2 was purchased from Calbiochem.
IRX-2 description
IRX-2 is a multitargeted biologic currently in clinical trials for HNSCC containing nanogram to picogram quantities of IL-1β, IL-2, IL-6, IL-8, GM-CSF, IFNγ and TNF-α. It is prepared by IRX Therapeutics (Farmingdale, NY, USA) under pharmaceutical cGMP standards from phytohemagglutinin-stimulated normal human PBMC as previously described.12 Quality control tests including a bioassay with CTLL cells and enzyme-linked immunosorbent assay determinations of cytokine levels are routinely performed to guarantee the consistency of various IRX-2 preparations. For all experiments reported here, unless otherwise noted (e.g., in titrations), a 1 : 3 dilution of IRX-2 was used. This dilution yields the following cytokine concentrations (all in ng/ml): IL-1β, 0.3; IL-2, 4.3; IL-6, 0.7; IL-8, 25.2; GM-CSF, 0.6; IFNγ, 2.2; TNFα, 1.6.
Cells and cell lines
The HNSCC cell line PC-13 was established in our laboratory and maintained as previously described.11 It was retrovirally transfected with the human FasL gene obtained from Dr. S Nagata (Osaka Biosciences Institute, Osaka, Japan) as previously reported.5 Supernatants of transfected PCI-13 cells (PCI-13-FasL), which contained both sFasL and the 42 kDa membranous form of FasL, were used as a source of tumor-derived MV. Jurkat cells were obtained from American Tissue Culture Collection (ATCC, Manassas, VA, USA) and were transfected with CD8 cDNA by Dr. H Rabinowich, at the University of Pittsburgh. The CD8+ Jurkat cells were cultured in RPMI 1640 medium supplemented with 10% (v/v) fetal bovine serum (FBS), l-glutamine and antibiotics. Human T lymphocytes were isolated from PBMC obtained from consented normal donors. PBMC were isolated by Ficoll-Hypaque density-gradient centrifugation (GE Healthcare Bio-Sciences Corp.), washed and plated for 1 h at 37°C in T162 culture flasks in an atmosphere of 5% CO2 to remove CD14+ monocytes. The nonadherent T-lymphocyte fraction was collected and immediately used for experiments or cryopreserved. CD8+ or CD4+ T cells were purified by positive selection using CD8 MicroBeads or CD4 MicroBeads, respectively (Miltenyi Biotec), using the AutoMACS system according to the manufacturer’s instructions. Purified CD8+ or CD4+ T cells were then cultured for 2–3 days in AIM V medium supplemented with 10% FBS (previously depleted of MV by ultracentrifugation) in the presence of beads coated with anti-CD3 and anti-CD28 antibodies (T Cell Activation/Expansion Kit; Miltenyi Biotec). All cells used for the above-described experiments were in the log phase of growth.
Isolation of microvesicles
Microvesicles were isolated from culture supernatants of the FasL-transfected PCI-13 cell line as previously described.11 Briefly, the concentrated cell supernatants were fractioned by a two-step procedure, including size exclusion chromatography and ultracentrifugation. PCI-13-FasL supernatants were concentrated at least 10×, using Centriprep Filters (Fisher Scientific, Pittsburgh, PA, USA). Next, 10 ml aliquots of the concentrated supernatants were applied to a Sepharose 2B (Amersham Biosciences, Piscataway, NJ, USA) column (1.5 × 35 cm) equilibrated with PBS. Fractions (1 ml) were collected and the protein content was monitored by measuring absorbance at 280 nm. The exclusion peak material, containing proteins of >50 million kDa, was then centrifuged at 105 000 × g for 2 h at 4°C. The pellet was resuspended in 300–500 μl of sterile PBS. Protein concentration in each MV preparation was estimated by a Lowry’s protein assay (Bio-Rad Laboratories, Hercules, CA, USA) with BSA used as a standard.
Subcellular fractionation
The cells were suspended in digitonin permeabilization buffer (50 mmol/l sucrose, 137 mmol/l NaCl, 70 mmol/l KCl, 4.3 mmol/l Na2HPO4, 1.4 mmol/l K2HPO4, 0.2 mg/ml digitonin and 0.1% Hydorol M), vortexed and incubated on ice for 5 min. The cells were then centrifuged at 1000 × g for 5 min at 4°C. The supernatant contained the cytosolic fraction. The remaining pellet was resuspended in RIPA buffer (50 mmol/l Tris-HCl (pH 7.4), 150 mmol/l NaCl, 1 mmol/l EDTA, 1 mmol/l EGTA, 1% Triton X-100, 1% sodium deoxycholate and 0.1% SDS), vortexed and incubated on ice for 5 min. The lysate was vortexed and centrifuged at 10 000 × g for 10 min at 4°C. The supernatant contained the heavy membrane fraction.
Western blot assays
To determine total or phosphorylated Akt, Bcl-2, CD3ζ, caspase-3, JAK3, STAT5 and FLIP, CD8+ Jurkat cells or purified activated CD8+ or CD4+ primary T cells were co-incubated with MV at the indicated concentration and/or with IRX-2 (1 : 3 final dilution) for the indicated periods of time at 37 °C. The cells were then washed, centrifuged at 4 °C and lysed in equal volumes of ice-cold lysis buffer (50 mM Tris-HCL (pH 7.5), 150 mM NaCl, 0.5% Nonidet P-40) and protease inhibitor cocktail (Pierce Chemical Co., Rockford, IL, USA). After lysis, the homogenates were clarified by centrifugation. The supernatants were isolated, and boiled for 5 min in 5 × Laemmli sample buffer. Equivalent protein quantities, as determined by Lowry, were loaded on each gel. For determination of apoptosis-related proteins subcellular fractions of MV/IRX-2-treated cells were used. Proteins were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and electrotransferred to polyvinylidene difluoride (PVDF) membranes. The membranes were blocked in 5% fat-free milk or 5% BSA in TTBS (0.05% Tween 20 in Tris-buffered saline) for 1 h at room temperature (RT) and then incubated overnight at 4°C with the appropriate antibodies. After washing (3 × 15 min) with TTBS at RT, membranes were incubated with horseradish-peroxidase-conjugated secondary antibody at 1 : 150 000 dilution (Pierce Chemical Co.) for 1 h at RT. After washes, membranes were developed with a SuperSignal chemoluminescent detection system (Pierce Chemical Co.). To reprobe with another primary antibody, membranes were incubated in stripping buffer (0.5 M NaCl, 3% (v/v) glacial acetic acid), washed and then used for further study.
Co-incubation of CD8+ Jurkat cells or activated normal T lymphocytes with MV and IRX-2
CD8+ Jurkat cells or activated primary T lymphocytes were plated at 0.3 × 106 cells per well in a 96-well plate and pretreated or not with IRX-2 or with recombinant human cytokines at a final concentration of 10 ng/ml or 100 IU/ml for 24 h (unless otherwise noted). A 1 : 3 dilution of IRX-2 is defined as containing 90 IU/ml of IL-2. MV (10 μg protein per 0.3 × 106 cells) were then added for 3–24 h. In some experiments, cells were first co-incubated with MV for 3–24 h, then washed and treated further with IRX-2 or cytokines or treated for the indicated time period with MV and IRX-2 added simultaneously. In selected blocking experiments, anti-Fas neutralizing mAb, ZB-4, the pan-caspase inhibitor, Z-VAD-FMK or the specific Akt inhibitor were added at the indicated concentrations before MV co-incubation.
Cell-surface staining
Microvesicles and/or IRX-2 co-incubated CD8+ Jurkat cells or activated primary T lymphocytes (at least 300 000 cells per tube) were washed twice in staining buffer (0.1% w/v BSA and 0.1% w/v NaN3). Cells were stained for cell-surface markers as previously described.4,5 Briefly, cells were incubated with the optimal dilution of each Ab for 20 min at RT in the dark, washed twice with staining buffer and finally fixed in 1% (v/v) paraformaldehyde (PFA) in PBS. The following Abs were used for surface staining: anti-CD3-ECD, anti-CD4-PE, anti-CD8-PC5, anti-Fas-FITC and anti-FasL-PE. The appropriate isotype control Abs were used in all experiments.
Flow cytometry
Four-color flow cytometry was performed using a FACScan flow cytometer (Beckman Coulter) equipped with Expo32 software (Beckman Coulter). Lymphocytes were gated based on morphology, and debris, MV as well as monocytes and granulocytes were excluded, collecting data on at least 105 cells. For the analysis of activated primary T lymphocytes, gates were restricted to the CD3+CD8+ or CD3+CD4+ T-cell subsets. Data were analyzed using Coulter EXPO 32 version l.2 analysis software.
Annexin-V-binding assay
Annexin V binding to MV and/or IRX-2 co-incubated CD8+ Jurkat cells or activated T lymphocytes was measured by flow cytometry to evaluate spontaneous or in vitro induced apoptosis. Following surface staining with antibodies to CD3, CD8 or CD4, the cells were resuspended in ANX-binding buffer and incubated with FITC-conjugated ANX for 15 min on ice. Additional staining with 7-AAD was performed to discriminate dead and live cells. Routinely, fewer than 10% of cells were 7AAD+. The cells were analyzed by flow cytometry within 30 min of staining.
Measurement of caspase activation
Pan-caspase activity was tested by intracellular staining of activated caspases using a pan-caspase inhibitor, CaspACE™ FITC-VAD-FMK In Situ Marker (Promega, Madison, WI, USA). Cells were resuspended in PBS and FITC-VAD-FMK was added at a final concentration of 5 μM. The cells were incubated for 20 min at 37°C, 5% CO2 and washed with PBS. Then cells were stained for cell-surface receptors, fixed with 1% PFA and analyzed by flow cytometry. The specific activation of caspase-3 and caspase-7 was measured using the Vybrant® FAM caspase-3 and -7 Assay Kit from Invitrogen (Carlsbad, CA, USA) according to the manufacturers’ instructions. Briefly, cells were resuspended in PBS and stained with a 150× dilution of the carboxyfluorescein (FAM)-labeled FMK-caspase inhibitor for 60 min at 37°C, 5% CO2. Then the cells were washed in wash buffer and fixed with 1% PFA. The cells were analyzed by flow cytometry with the fluorescein measured on the FL1 channel.
Measurement of the mitochondrial membrane potential
The loss of MMP as a hallmark of apoptosis was measured using the MitoProbet JC-1 Assay Kit from Invitrogen. The cationic dye JC-1 (5,5′, 6,6′-tetrachloro-1,1′, 3, 3′-tetraethylbenzimidazolylcarbocyanine iodide) exists in healthy cells as a green monomer in the cytosol and also accumulates as red aggregates in the mitochondria. In apoptotic and necrotic cells, JC-1 remains only in the cytoplasm due to mitochondrial depolarization, which can be detected by flow cytometry as a decrease in the red/green fluorescence intensity ratio. CD8+ Jurkat cells or activated T lymphocytes were incubated in PBS containing 2 μM of JC-1 for 30 min at 37°C, 5% CO2. An aliquot of the cells was treated with 50 μM of the mitochondrial uncoupler carbonyl cyanide 3-chlorophenylhydrazone (CCCP) during the staining period as a positive control for mitochondrial depolarization. The cells were analyzed using a flow cytometer immediately after staining.
Evaluation of apoptosis-related proteins
Expression of antiapoptotic proteins Bcl-2, Bcl-xL, FLIP and Mcl-1 and the proapoptotic proteins Bax, Bim and Bid was investigated in CD8+ Jurkat cells or activated primary T lymphocytes using multicolor flow cytometry. The cells were first stained for surface T-cell markers as described above. For intracellular staining of apoptosis-related proteins the cells were fixed with 1% (v/v) PFA in PBS at RT for 10 min and then permeabilized with saponin (0.1% v/v in PBS) for 15 min at 4°C. Next, the cells were stained for 30 min at 4°C with FITC- or PE-conjugated antihuman Bcl-2, Bax and Bcl-xL or unconjugated antibodies specific for FLIP, Bim, Bid or Mcl-1, followed by a wash with 0.1% saponin. Samples stained with unconjugated antibodies were further incubated with an FITC-conjugated goat anti-rabbit IgG for 15 min at RT. After washing with 0.1% saponin, cells were fixed in 1% (v/v) PFA. Isotype control-matched antibodies were used for both surface and intracellular controls and all antibodies were pretitered on fresh PBMC. In some experiments, Jurkat cells were preincubated with cycloheximide at the concentration of 1 or 10 μg alone or with MV + IRX + CHX and expression levels of pro- and antiapoptotic proteins as well as Annexin V binding to Jurkat cells were measured.
TUNEL assay
DNA fragmentation was measured using the In Situ Cell Death Detection Kit, TMR red (Roche, Indianapolis, IN, USA). Briefly, cytospin preparations (100 000 cells per slide) of MV- and IRX-2-treated T cells were air-dried and fixed with 4% (v/v) PFA in PBS for 1 h at RT. Slides were rinsed with PBS and incubated with permeabilization solution (01% Triton X-100 in 0.1% sodium citrate) for 2 min on ice. Then the slides were washed twice with PBS and incubated with 20 μl of the TUNEL reaction mixture for 1 h at 37°C in a humidified chamber in the dark. Then the samples were washed extensively with PBS and incubated in a medium with 4′,6-diamidino-2-phenylindole (Vector Laboratories, Burlingame, CA, USA) to trace cell nuclei. Slides were evaluated in a Nikon Eclipse E-800 fluorescence microscope under × 200 magnification. For digital image analysis, Adobe Photoshop 6.0 was used. A minimum of 300 cells were randomly counted in a microscopic field to determine the percentage of cells with DNA fragmentation.
Statistical analysis
Statistical analysis was performed using the Student’s t-test. P-values <0.05 were considered significant.
Supplementary Material
{kind=link}
Acknowledgements
We are grateful to Dr. Hannah Rabinowich for her advice and help with experiment interpretation. This work was supported in part by NIH grant PO1-CA109688 to TLW.
Abbreviations
- MV
microvesicle
- ANX
Annexin
- DC
dendritic cell
- MMP
mitochondrial membrane potential
- HNSCC
head and neck squamous-cell carcinoma
- IFNγ
interferon-γ
- TNFα
tumor necrosis factor-α
- MFI
mean fluorescent intensity
- mAb
monoclonal antibody
- PBMC
peripheral blood mononuclear cell
- PBS
phosphate-buffered saline
- BSA
bovine serum albumin
- PVDF
polyvinylidene difluoride
- PFA
paraformaldehyde
Footnotes
Supplementary Information accompanies the paper on Cell Death and Differentiation website (http://www.nature.com/cdd)
References
- 1.Whiteside TL. Immune suppression in cancer: effects on immune cells, mechanisms and future therapeutic intervention. Semin Cancer Biol. 2006;16:3–15. doi: 10.1016/j.semcancer.2005.07.008. [DOI] [PubMed] [Google Scholar]
- 2.Reichert TE, Strauss L, Wagner EM, Gooding W, Whiteside TL. Signaling abnormalities, apoptosis, and reduced proliferation of circulating and tumor-infiltrating lymphocytes in patients with oral carcinoma. Clin Cancer Res. 2002;8:3137–3145. [PubMed] [Google Scholar]
- 3.Hoffmann TK, Dworacki G, Tsukihiro T, Meidenbauer N, Gooding W, Johnson JT, et al. Spontaneous apoptosis of circulating T lymphocytes in patients with head and neck cancer and its clinical importance. Clin Cancer Res. 2002;8:2553–2562. [PubMed] [Google Scholar]
- 4.Saito T, Kuss I, Dworacki G, Gooding W, Johnson JT, Whiteside TL. Spontaneous ex vivo apoptosis of peripheral blood mononuclear cells in patients with head and neck cancer. Clin Cancer Res. 1999;5:1263–1273. [PubMed] [Google Scholar]
- 5.Gastman BR, Atarashi Y, Reichert TE, Saito T, Balkir L, Rabinowich H, et al. Fas ligand is expressed on human squamous cell carcinomas of the head and neck, and it promotes apoptosis of T lymphocytes. Cancer Res. 1999;59:5356–5364. [PubMed] [Google Scholar]
- 6.O’Connell J, Bennett MW, O’Sullivan GC, Collins JK, Shanahan F. The Fas counterattack: cancer as a site of immune privilege. Immunol Today. 1999;20:46–52. doi: 10.1016/s0167-5699(98)01382-6. [DOI] [PubMed] [Google Scholar]
- 7.Reimer T, Herrnring C, Koczan D, Richter D, Gerber B, Kabelitz D, et al. FasL:Fas ratio – a prognostic factor in breast carcinomas. Cancer Res. 2000;60:822–828. [PubMed] [Google Scholar]
- 8.Soubrane C, Mouawad R, Antoine EC, Verola O, Gil-Delgado M, Khayat D. A comparative study of Fas and Fas-ligand expression during melanoma progression. Br J Dermatol. 2000;143:307–312. doi: 10.1046/j.1365-2133.2000.03655.x. [DOI] [PubMed] [Google Scholar]
- 9.Iero M, Valenti R, Huber V, Filipazzi P, Parmiani G, Fais S, et al. Tumour-released exosomes and their implications in cancer immunity. Cell Death Differ. 2008;15:80–88. doi: 10.1038/sj.cdd.4402237. [DOI] [PubMed] [Google Scholar]
- 10.Taylor DD, Gercel-Taylor C. Tumour-derived exosomes and their role in cancer-associated T-cell signalling defects. Br J Cancer. 2005;92:305–311. doi: 10.1038/sj.bjc.6602316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim JW, Wieckowski E, Taylor DD, Reichert TE, Watkins S, Whiteside TL. Fas ligand-positive membranous vesicles isolated from sera of patients with oral cancer induce apoptosis of activated T lymphocytes. Clin Cancer Res. 2005;11:1010–1020. [PubMed] [Google Scholar]
- 12.Hadden J, Verastegui E, Barrera JL, Kurman M, Meneses A, Zinser JW, et al. A trial of IRX-2 in patients with squamous cell carcinomas of the head and neck. Int Immunopharmacol. 2003;3:1073–1081. doi: 10.1016/S1567-5769(03)00029-8. [DOI] [PubMed] [Google Scholar]
- 13.Egan JE, Quadrini KJ, Santiago-Schwarz F, Hadden JW, Brandwein HJ, et al. IRX-2, a novel in vivo immunotherapeutic, induces maturation and activation of human dendritic cells in vitro. J Immunother. 2007;30:624–633. doi: 10.1097/CJI.0b013e3180691593. [DOI] [PubMed] [Google Scholar]
- 14.Sabapatha A, Gercel-Taylor C, Taylor DD. Specific isolation of placenta-derived exosomes from the circulation of pregnant women and their immunoregulatory consequences. Am J Reprod Immunol. 2006;56:345–355. doi: 10.1111/j.1600-0897.2006.00435.x. [DOI] [PubMed] [Google Scholar]
- 15.Whiteside TL. Tumour-derived exosomes or microvesicles: another mechanism of tumour escape from the host immune system? Br J Cancer. 2005;92:209–211. doi: 10.1038/sj.bjc.6602360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Valenti R, Huber V, Iero M, Filipazzi P, Parmiani G, Rivoltini L. Tumor-released microvesicles as vehicles of immunosuppression. Cancer Res. 2007;67:2912–2915. doi: 10.1158/0008-5472.CAN-07-0520. [DOI] [PubMed] [Google Scholar]
- 17.Smyth MJ, Cretney E, Kershaw MH, Hayakawa Y. Cytokines in cancer immunity and immunotherapy. Immunol Rev. 2004;202:275–293. doi: 10.1111/j.0105-2896.2004.00199.x. [DOI] [PubMed] [Google Scholar]
- 18.Meneses A, Verastegui E, Barrera JL, de la Garza J, Hadden JW. Lymph node histology in head and neck cancer: impact of immunotherapy with IRX-2. Int Immunopharmacol. 2003;3:1083–1091. doi: 10.1016/S1567-5769(03)00017-1. [DOI] [PubMed] [Google Scholar]
- 19.Duenas-Gonzalez A, Verastegui E, Lopez-Graniel C, Gonzalez A, Mota A, Barrera-Franco JL, et al. A pilot study of perilymphatic leukocyte cytokine mixture (IRX-2) as neoadjuvant treatment for early stage cervical carcinoma. Int Immunopharmacol. 2002;2:1007–1016. doi: 10.1016/s1567-5769(02)00048-6. [DOI] [PubMed] [Google Scholar]
- 20.Hadden JW, Verastegui E, Hadden E. IRX-2 and thymosin alpha1 (Zadaxin) increase T lymphocytes in T lymphocytopenic mice and humans. Ann NY Acad Sci. 2007;112:245–255. doi: 10.1196/annals.1415.032. [DOI] [PubMed] [Google Scholar]
- 21.Barrera JL, Verastegui E, Meneses A, Zinser J, de la Garza J, Hadden JW. Combination immunotherapy of squamous cell carcinoma of the head and neck: a phase 2 trial. Arch Otolaryngol Head Neck Surg. 2000;126:345–351. doi: 10.1001/archotol.126.3.345. [DOI] [PubMed] [Google Scholar]
- 22.Song G, Ouyang G, Bao S. The activation of Akt/PKB signaling pathway and cell survival. J Cell Mol Med. 2005;9:59–71. doi: 10.1111/j.1582-4934.2005.tb00337.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wajant H. The Fas signaling pathway: more than a paradigm. Science. 2002;296:1635–1636. doi: 10.1126/science.1071553. [DOI] [PubMed] [Google Scholar]
- 24.Stepczynska A, Lauber K, Engels IH, Janssen O, Kabelitz D, Wesselborg S, et al. Staurosporine and conventional anticancer drugs induce overlapping, yet distinct pathways of apoptosis and caspase activation. Oncogene. 2001;20:1193–1202. doi: 10.1038/sj.onc.1204221. [DOI] [PubMed] [Google Scholar]
- 25.Desagher S, Osen-Sand A, Nichols A, Eskes R, Montessuit S, Lauper S, et al. Bid-induced conformational change of Bax is responsible for mitochondrial cytochrome c release during apoptosis. J Cell Biol. 1999;144:891–901. doi: 10.1083/jcb.144.5.891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Willis SN, Fletcher JI, Kaufmann T, van Delft MF, Chen L, Czabotar PE, et al. Apoptosis initiated when BH3 ligands engage multiple Bcl-2 homologs, not Bax or Bak. Science. 2007;315:856–859. doi: 10.1126/science.1133289. [DOI] [PubMed] [Google Scholar]
- 27.Oltvai ZN, Milliman CL, Korsmeyer SJ. Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that accelerates programmed cell death. Cell. 1993;74:609–619. doi: 10.1016/0092-8674(93)90509-o. [DOI] [PubMed] [Google Scholar]
- 28.Han J, Goldstein LA, Gastman BR, Rabinowich H. Interrelated roles for Mcl-1 and BIM in regulation of TRAIL-mediated mitochondrial apoptosis. J Biol Chem. 2006;281:10153–10163. doi: 10.1074/jbc.M510349200. [DOI] [PubMed] [Google Scholar]
- 29.Opferman JT, Letai A, Beard C, Sorcinelli MD, Ong CC, Korsmeyer SJ. Development and maintenance of B and T lymphocytes requires antiapoptotic MCL-1. Nature. 2003;426:671–676. doi: 10.1038/nature02067. [DOI] [PubMed] [Google Scholar]
- 30.Salazar-Onfray F, Lopez MN, Mendoza-Naranjo A. Paradoxical effects of cytokines in tumor immune surveillance and tumor immune escape. Cytokine Growth Factor Rev. 2007;18:171–182. doi: 10.1016/j.cytogfr.2007.01.015. [DOI] [PubMed] [Google Scholar]
- 31.Ma A, Koka R, Burkett P. Diverse functions of IL-2, IL-15, and IL-7 in lymphoid homeostasis. Annu Rev Immunol. 2006;24:657–679. doi: 10.1146/annurev.immunol.24.021605.090727. [DOI] [PubMed] [Google Scholar]
- 32.Budd RC, Yeh WC, Tschopp J. cFLIP regulation of lymphocyte activation and development. Nat Rev Immunol. 2006;6:196–204. doi: 10.1038/nri1787. [DOI] [PubMed] [Google Scholar]
- 33.Schmitz I, Weyd H, Krueger A, Baumann S, Fas SC, Krammer PH, et al. Resistance of short term activated T cells to CD95-mediated apoptosis correlates with de novo protein synthesis of c-FLIPshort. J Immunol. 2004;172:2194–2200. doi: 10.4049/jimmunol.172.4.2194. [DOI] [PubMed] [Google Scholar]
- 34.Ross JA, Nagy ZS, Cheng H, Stepkowski SM, Kirken RA. Regulation of T cell homeostasis by JAKs and STATs. Arch Immunol Ther Exp (Warsz) 2007;55:231–245. doi: 10.1007/s00005-007-0030-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.