

Abstract
Tumor-induced dysfunction of immune cells is a common problem in cancer. Tumors induce immune suppression by many different mechanisms, including accumulation of regulatory T cells (Treg). Adaptive Treg (Tr1) generated in the tumor microenvironment express CD39 and CD73 ectonucleotidases, produce adenosine and are COX2+PGE2+. Adenosine and PGE2 produced by Tr1 or tumor cells bind to their respective receptors on the surface of T effector cells (Teff) and cooperate in up-regulating cytosolic 3′5′-cAMP levels utilizing adenylyl cyclase isoform 7 (AC-7). In Teff, increased cAMP mediates suppression of anti-tumor functions. Treg, in contrast to Teff, seem to require high cAMP levels for mediating suppression. This differential requirement of Treg and Teff for cAMP offers an opportunity for pharmacologic interventions using selected inhibitors of the adenosine/PGE2 pathways. Blocking of adenosine/PGE2 production by Tr1 or blocking binding of these factors to their receptors on T cells or inhibition of cAMP synthesis in Teff all represent novel therapeutic strategies that used in combination with conventional therapies could restore anti-tumor functions of Teff. At the same time, these inhibitors could disarm Tr1 cells by depriving them of the factors promoting their generation and activity or by down-regulating 3′5′-cAMP levels. Thus, the pharmacologic control of Treg-Teff interactions offers a novel strategy for restoration of anti-tumor Teff functions and silencing of Treg. Used in conjunction with anti-cancer drugs or with immune therapies, this strategy has a potential to improve therapeutic effects by preventing or reversing tumor-induced immune suppression.
Keywords: Adenosine, anti-tumor immunity, effector T cells (Teff), pharmacologic inhibitors, prostaglandin E2 (PGE2), regulatory T cells (Treg)
1. INTRODUCTION
It is well documented that various immunotherapies administered to patients with cancer, including antitumor vaccines, are largely ineffective in inducing clinical responses, i.e., limiting tumor growth, preventing recurrence and prolonging survival [1, 2]. Anti-tumor immune responses generated by these immunetherapies are often weak, short-lived or biased toward Th2 instead of Th1 cellular pathways [3]. Among various explanations for this common situation is the ability of the tumor to disarm the host immune system [4, 5]. Human tumors are known to produce a variety of immunosuppressive factors, including adenosine [6, 7], prostaglandin E2 [8–10], cytokines such as TGF-β [11], tumor-associated gangliosides [12] and others [13, 14]. These factors induce functional abnormalities in those immune cells that are expected to target and eliminate tumor cells [15]. Further, tumor-induced immune dysfunction is not generic but is restricted to tumor-specific immune responses, because only antitumor immune responses are selectively compromised, while anti-viral or antibacterial immune responses are unimpaired in most cancer patients. As a result, cancer patients are unable to mount effective anti-tumor immune responses. Tumor-induced immune cell dysfunction can impact a variety of cellular activities including signal transduction, activation, cytokine production, proliferation, cytotoxicity and cell migration [16]. It can be concomitantly present in a variety of immune cell types, including T cells, natural killer (NK) cells, monocytes, dendritic cells (DC) and B cells [16]. Further, the profile and the severity of dysfunctions demonstrable in immune cells vary broadly between patients [17, 18]. These dysfunctions may be local, involving immune cells in the tumor or tumor-involved lymph nodes, or may be systemic [18, 19]. They are thought to contribute to cancer progression by enabling the tumor to escape from immune control [20]. Human tumors are often infiltrated by immune cells, especially T lymphocytes and myeloid cells, which are recruited to the site by tumor-derived factors and which become subverted by the tumor to promote its growth [21]. In some cases, infiltrating immune cells retain a limited degree of anti-tumor effector functions, although tumors which progress aggressively are strongly immunosuppressive [22, 23]. Among mononuclear cells infiltrating solid tumors are CD3+ T cells, with various proportions of CD4+ and CD8+ lymphocytes. The T cells accumulating in the tumor milieu are often activated, i.e., CD25+ (IL-2R+) [16]. In fact, the role of TIL in tumor progression and prognosis has been debated for many years with more recent views favoring TIL as indicators of better outcome in colorectal carcinoma, melanoma and other cancers [24–27]. Yet, TIL have been reported to contain substantial proportions of CD4+CD25high T cells, which also express FOXP3, a transcription factor necessary for mediating suppression, confirming that they are regulatory T cells (Treg). Accumulations of suppressor cells in human tumors as well as the peripheral blood of patients with cancer have been reported, and their presence has been associated with tumor progression and poor prognosis [28–31]. While tumors are clearly able to orchestrate dysfunction of effector T cells (Teff) in the tumor microenvironment, Treg recruited to the tumor site appear to be resistant to death and mediate higher levels of suppression than Treg in the peripheral circulation [30]. Treg are a small subset of CD4+ T cells (~ 5%) which interact with Teff suppressing their functions [32, 33]. They are functionally heterogeneous, comprising several subsets of phenotypically similar cells which are able to suppress functions of Teff via distinct and often unexpected mechanisms [34–37]. At least two Treg subsets have been recognized in humans: (a) natural Treg (nTreg), which originate in the thymus, mediate suppression by cell contact-dependent mechanisms involving the Granzyme B/perforin or Fas/FasL pathways and constitute the major regulatory T-cell subset responsible for maintaining peripheral tolerance to self [38, 39]; (b) inducible Treg (iTreg) also referred to as type 1 regulatory T cells (Tr1), which are induced in the periphery following chronic antigenic stimulation in the presence of IL-10 derived from tolerogenic antigen-presenting cells [40]. They differentiate into active suppressor cells which mediate suppression via contact-independent mechanisms largely involving TGF-β1, IL-10 or other soluble immunoinhibitory factors [41, 42]. Today, the phenotype of human Treg is not yet firmly defined. Most nTreg are CD3+CD4+CD25highFOXP3+, while Tr1 cells have a somewhat different phenotype characterized by high expression levels of inhibitory cytokines, TGF-β1 and IL-10, and the absence of IL-4 [41]. Considerable controversy exists as to the phenotype and the role of Treg in tumor progression. While they might be largely responsible for tumor escape from the host immune system, it is important to remember that Treg-mediated regulation of activated immune cells represents a physiologically normal response designed to maintain a homeostatic balance and prevent undesirable immune activation. Thus, Treg found in pathologic conditions, such as cancer or chronic infections, may represent suppressor cells recruited and conditioned by the microenvironment to mediate elevated suppression levels and favoring tumor growth. Among tumor-derived soluble immunosuppressive factors found in the tumor microenvironment are adenosine and prostaglandin E2 (PGE2) [41, 43]. Evidence suggests that human Treg are able to utilize adenosine and PGE2 for inducing Teff suppression. In this review, we will describe the evidence for the involvement of adenosine and PGE2 in Treg functions in the tumor microenvironment. Insights into molecular mechanisms driving interactions between Treg and the tumor offer an opportunity to devise novel strategies for silencing Treg-mediated suppression in cancer through pharmacologic or immunologic means. If Treg play a role in tumor progression, then their silencing could eliminate tumor escape from the host immune system. For example, by directly inhibiting adenosine and PGE2 production by the tumor and/or Treg or by protecting Teff from inhibitory effects of these factors, it might be possible to restore effective anti-tumor immunity in patients with malignancies.
2. CD39 ECTONUCLEOTIDASE EXPRESSION BY HUMAN mTreg
Adenosine is an anti-inflammatory product of enzymatic hydrolysis of extracellular ATP by CD39 (ecto- ATPase) and CD73 (ecto-5′-AMP nucleotidase). Fig. (1) illustrates the pathway that results in adenosine generation from extracellular ATP. Adenosine is present in body fluids, although its half-life is very short [44]. The source of adenosine in the tumor microenvironment can be tumor cells, infiltrating immune cells and epithelial cells. The tumor microenvironment is characterized by tissue disruption and numerous dying tumor cells resulting in ATP release, high levels of ectonucleotidase activity and the accumulation of extracellular adenosine (e.g., 10–20 fold increase in tumor vs. normal tissues) [7]. Local hypoxia greatly favors ectonucleotidase activity and adenosine metabolism [37]. Adenosine has well-documented immunosuppressive activity, which contributes to the inhibition of local and systemic anti-tumor immune responses [6, 7]. Overexpression of CD39 and, especially CD73, in human tumor cells, including melanoma, breast carcinoma and HNSCC has been reported [45, 46], suggesting that tumors are a major source of endogenous adenosine. Because of high levels of extracellular ATP generated in the tumor microenvironment, ectonucleotidase activity is important for removing its excess and protecting the tumor from ATP-associated toxicity [47]. At the same time, tumor growth is promoted by adenosine-mediated inhibition of antitumor T-cell immunity [48]. In support of this view, it has been shown that ectonucleotidase expression in tumor cells correlated with tumor progression, invasiveness, neoangiogenesis and metastasis formation [46, 49]. More recently, it has been reported that murine Treg also express CD39 and CD73 proteins which are enzymatically active [50, 51]. CD39 ectonucletidase was found by us and others to be present on the surface of human Treg [52], and we have used CD39 as a marker for the isolation of human Treg [53]. We have initially demonstrated that Treg in human PBMC hydrolyzed exogenous ATP and generated adenosine [54]. On the other hand, isolated and highly purified CD39+ Treg hydrolyze ATP to 5′AMP and not to adenosine (Fig. 1), and only a small subset of CD39+ Treg were shown to co-express CD73. In human lymphocytes, expression of CD39 and CD73 is not limited to Treg, as subsets of effector CD4+ T cells, CD8+ T cells and B cells are positive as well. This probably means that CD39+ Treg require collaboration from other lymphocytes, e.g., B cells which are rich in CD73, or tumor-derived exosomes, which have been recently reported to carry membrane CD73 [55], to produce adenosine. Interestingly, our recent data show that Treg isolated from the peripheral blood based on CD39 expression contain a subset of CD4+CD39+CD25+FOXP3+ cells which mediate suppression and a subset of CD4+CD39+CD25neg FOXP3− cells which do not [53]. These two subsets of CD4+CD39+ T cells cross-talk and influence each other’s functions suggesting plasticity of Treg that may be shaped by events in their milieu [56]. Adenosine binds to A1, A2A, A2B and A3 receptors (R) expressed on the surface of various cell types, including lymphocytes and dendritic cells (DC). In immune cells, suppressive effects of adenosine are largely mediated through A2AR and A2BR signaling [57] with a concomitant activation of adenylyl cyclase (Ac) and upregulation of cAMP resulting in a functional paralysis of responding Teff cells. Human Treg express relatively few A2AR or A2BR as compared to Teff cells [58]. Thus, exogenous adenosine, in part produced by Treg, binds to A2AR or A2BR liberally expressed on CD4+ Teff, which results in the inhibition of proliferation and cytokine production. An elevation of 3′5′-cAMP levels that follows adenosine receptor triggering in these cells leads to activation of protein kinases which mediate protein phosphorylation. Interestingly, studies performed with murine spleen-derived Treg and Teff suggested that 3′5′-cAMP levels are regulated differently in resting vs. IL-2 activated Treg and Teff [59]. Thus, resting Teff have higher Ac and phosphodiesterase (PDE) activity than Treg. Upon IL-2 activation, Ac activity in Treg increases and that of PDE decreases, resulting in elevated 3′5′-cAMP, which these cells apparently need for mediating suppression. In IL-2- activated Teff, on the other hand, Ac activity is down-regulated and PDE activity up-regulated, leading to lower 3′5′-cAMP and favoring Teff proliferation [59]. These provocative data imply that interactions between Treg and Teff are strictly controlled at the level of 3′5′-cAMP and are dependent on cellular activation. There are other differences between Treg and Teff that impact on their biology. For example, Schenk and colleagues recently reported that expression of the ATP receptor, P2XR, on the surface of Treg is increased compared to Teff and that exogeneous ATP inhibits functions of Treg [60]. Therefore, CD39+ Treg have the tools to protect themselves from ATP by hydrolyzing it to 5′AMP and eventually to adenosine.
Fig. 1.
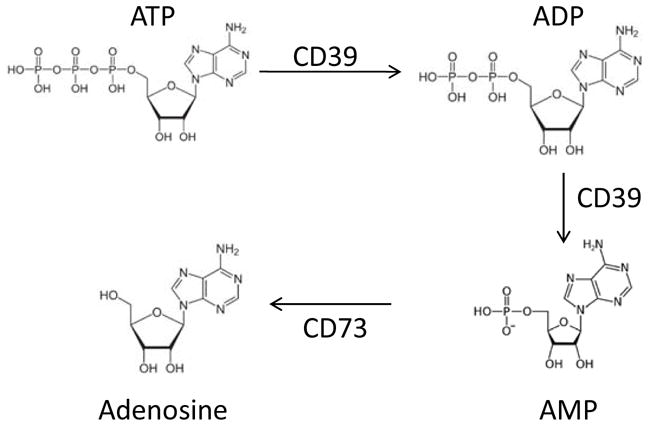
The pathway of ATP hydrolysis by the ectonucleosidases, CD39 and CD73 to adenosine.
3. HUMAN INDUCIBLE Treg (Tr1) AND ADENOSINE PRODUCTION
While adenosine production by human nTreg might require assistance from CD73− expressing neighbors, activated inducible Treg (Tr1) appear to be able to do so alone. We utilized an in vitro assay system to co-culture naive, CD4+CD25neg T cells obtained from the peripheral blood of normal donors with autologous immature dendritic cells (iDC) and irradiated tumor cells in the medium containing low doses of cytokines (IL-2, IL-10 and IL-15) and cross-linked anti-CD3 antibody for 10 days [61]. The T cells outgrowing in these co-cultures gradually acquired the Tr1 phenotype and suppressor activity. By day 10, most of the proliferating T cells were CD3+CD4+CD25+IL-2Rβ+IL2Rγ+ FOXP3+IL-10+TGF-β+IL4−, and they effectively mediated suppression of autologous responder cell proliferation [61]. These cells also hydrolyzed exogenous ATP to adenosine in vitro as demonstrated by ATP consumption assays and by mass spectrometry for adenosine [54]. By flow cytometry and Western blots, Tr1 cells also expressed CD39 and CD73 [54]. These data indicated that Tr1 cells could generate and release adenosine. Using CFSE-based suppression assays, in which naïve autologous CD4+CD25neg T cells were responding to TcR-mediated signals, we showed that in the presence of ARL67156, a selective CD39 antagonist, or α β-methylene ADP, an inhibitor of CD73, as well as an agonist of A2AR, ZM241865, Tr1-mediated suppression of proliferation was significantly inhibited [54]. Furthermore, not only proliferation but also cytokine secretion by T cells responding to TcR-mediated signals was inhibited by Treg-derived adenosine and was relieved in the presence of the above mentioned antagonists. These data suggest that Tr1 producing adenosine can suppress not only the expansion of responder cells but also their effector functions. Although in vitro generated Tr1 appear to express both CD39 and CD73 as shown by flow cytometry and Western blots [54], it was unclear whether Tr1, presumably the major subset of Treg present in cancer patients, also co-expressed these surface enzymes in vivo. In the peripheral circulation of normal donors and patients with cancer, we and others consistently show expression of CD39 on nearly all human Treg and of CD73 on only a small fraction of these cells. However, we expected that inflammatory cells accumulating in human tumor tissues might upregulate expression of ectonucleotidases. We stained tumor tissue sections using anti-CD39, anti-CD73, anti-CD25 and anti-CD4 Abs and searched for expression of these markers by confocal microscopy. We reported that CD4+ T cells expressing CD39 or CD73 were present in the tumor (HNSCC) infiltrating T cells [52], and that at least some Treg (CD4+CD25+) in the infiltrate co-expressed these markers [52]. Regardless of the marker co-expression in the same cells, enzymatically active CD39+ Treg would be able to use CD73 on tumor cells or other infiltrating cells for adenosine production in tumor tissues. We surmise that enzymatically-active CD39 Treg present in the human peripheral blood could similarly utilize CD73 expressed by other lymphocytes [52], exosomes [55], or vascular endothelial cells [62] to produce immunosuppressive adenosine.
Because in vitro generated Tr1 express both ectonucleotidases necessary for adenosine generation from exogenous ATP, it is not unreasonable to speculate that activated Treg induced in the presence of tumor antigens, DC and selected cytokines up-regulate CD73 expression and that they could, therefore, utilize the pathway in mediating suppression of other immune cells.
4. ROLE OF ADENOSINE DEAMINASE (ADA) IN Treg-MEDIATED SUPPRESSION
If Treg are involved in adenosine production and use it for suppression, the question arises as to protective mechanisms these cells use to save themselves from adverse effects of adenosine. Ectonucleotidase, CD39 is present and enzymatically active in human nTreg as well as Tr1, and CD73 either co-expressed or supplied by other cells, catalyzes conversion of AMP to adenosine (Fig. 1). It could, of course, be that the absence of the CD73 co-expression on Treg serves as a protection mechanism from self-produced adenosine. However, more interesting is the observation that another surface marker, CD26, is absent/low in these cells [52]. CD26 is a 110 kD glycoprotein with intrinsic dipeptidyl peptidase IV activity whose extracellular domain is associated with ADA [63]. CD26 is highly expressed on the surface of all conventional CD4+ T cell subsets (Tconv), where it serves as an anchor for ADA and, therefore, localizes ADA to the cell surface. Signaling via CD26 on Tconv cells involves CD45 molecules and links CD26 and ADA to T-cell differentiation into Teff which mediate helper functions [66]. ADA hydrolyzes adenosine to inosine, decreasing its pericellular concentration in CD4+CD25neg Tconv. On the other hand, adenosine-producing Treg, which do not express CD26/ADA, could accumulate adenosine in the pericellular space and use it to inhibit functions of other T cells. The ability of Teff to deaminate adenosine might be a protective mechanism, allowing these cells to in part escape from adenosine-mediated suppression. Activated Teff, which upregulate A2AR expression, are very sensitive to adenosine-mediated inhibition and require ADA for protection from Treg. Because Treg have fewer A2AR relative to Teff, they may be less sensitive to inhibitory activity of adenosine. Due to the increased expression of ectonucleotidases, the absence of the CD26/ADA complex and low ADA activity, Treg are equipped with a set of tools to maintain high levels of extracellular adenosine in their microenvironment and utilize it to induce suppression of Teff activities [52].
5. PGE2 IN THE TUMOR MICROENVIRONMENT
PGE2 is a major product of cyclooxygenase 2 (COX-2) activity [67]. COX-2 is overexpressed by many human tumors, and its expression has been linked to tumor progression and poor patient survival [68]. PGE2 mediates its immunoinhibitory activity via four different G protein-coupled receptors (EP1–EP4) expressed on various cells. PGE2 signaling leads to an intracellular increase and activation of cAMP, with a concomitant decrease in cell proliferation and suppression of other functions in immune cells [69, 70]. PGE2 also induces Tr1 cells and modulates their activity thus contributing to creating a tolerogenic milieu [28]. In a series of in vitro experiments in which tumor cells were engineered to express COX-2 or were either COX-2neg or had COX-2 expression inhibited (the COX-2 gene knock out with siRNA; Diclofenac, a generic COX inhibitor), we demonstrated that the outgrowth of Tr1 cells and their suppressive activity mediated via released IL-10 and TGF-β were dependent on COX-2 expression in the tumor cells [28]. Using the same type of in vitro assays with COX-2+ and COX-2neg tumor cells, we showed that the former induced a significantly greater number of Tr1 cells than the latter. Also, Tr1 generated in co-cultures with COX-2+ tumor cells were significantly more suppressive, hydrolyzed more exogenous ATP, and produced higher levels of adenosine and PGE2 (p<0.05 for all) than Tr1 induced by COX-2neg tumors. Tr1 cells induced in the presence of COX-2+ tumors expressed COX-2 themselves and were able to produce PGE2. These COX-2+ Tr1 co-expressed CD39 and CD73 and in addition to PGE2, they also produced adenosine [54]. Their suppressor function was inhibited in the presence of ectonucleotidase antagonists and also in the presence of indomethacin, confirming that both adenosine and PGE2 contributed to Tr1-mediated suppression.
6. ADENOSINERGIC AND PGE2 PATHWAYS IN THE TUMOR MILIEU
Since tumor cells as well as Tr1 can produce adenosine and PGE2, it has been suggested that these two suppressive factors cooperate in mediating immune suppression in the tumor microenvironment [70]. Both factors activate respective G protein-coupled receptors which mediate intracellular signaling via cAMP (Fig. 2). PGE2 mediates its biologic effects through EP1, EP2, EP3 and EP4 receptors [71], while adenosine binds to A2AR and A2BR expressed on immune cells. By adding AH6809, an EP2R antagonist, to co-cultures of Tr1 and Teff, we determined that PGE2 largely binds to the EP2R on lymphocytes. The addition of AH23848, an EP4R antagonist, or all other EPR antagonists had no effect on Teff proliferation in these co-cultures [54]. Also, studies with an A2AR and A2BR antagonist, ZM241385, showed that Tr1-derived adenosine mediates suppression of Teff proliferation by binding to its receptors on Teff cells. Although it remains to be determined how much each factor contributes to Tr1-mediated suppression, we have determined that the total level of PGE2 produced in co-cultures of Tr1 with Teff cells is approximately 100-fold greater then the level of adenosine. Yet, the use of receptor antagonists in the proliferation inhibition assays showed an almost equal reversal of suppression for both PGE2 and adenosine. The distribution and density of A2AR and EP2R on responding Teff cells as well as receptor affinities for their respective ligands presumably determine the extent of cooperation between Tr1-derived adenosine and PGE2 in mediating suppression of Teff cell proliferation.
Fig. 2.
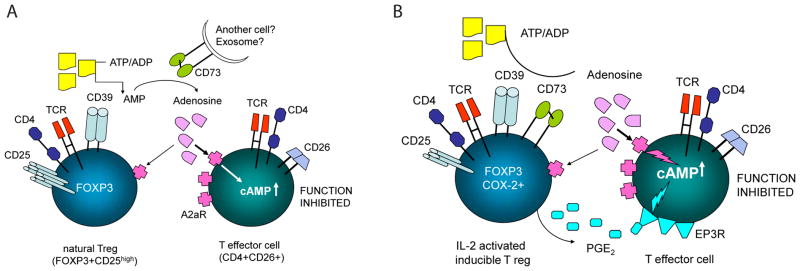
Interactions between nTreg (A) or IL-2 activated inducible Treg (Tr1) with effector T cells (Teff) mediated by the adenosine and PGE2 pathways.
Signals processed by both adenosine and PGE2 receptors down-modulate Teff functions by controlling intracellular 3′5′-cyclicAMP levels [72]. In immune cells, 3′5′-cAMP is produced by the adenylate cyclase-7 (Ac-7) isoform, as suggested by our preliminary data. Consequently, Ac-7 is the point of convergence for A2A receptor and EP2R-mediated signals that regulate 3′5′ cyclic AMP levels (Fig. 2). Downstream from Ac-7, PKA type 1 in Teff cells is involved in inhibitory activities of adenosine and PGE2 [52]. Using Rp-8-Br-cAMPS, an agent which prevents binding of 3′5′-cAMP to the regulatory subunit of PKA type I, we showed that Tr1-mediated suppression of Teff proliferation was significantly inhibited. This suggested that blocking of PKA type I activity in Teff cells protects them from suppression by PGE2- and adenosine-producing Tr1 cells [54].
While the above described in vitro studies suggest that adenosine and PGE2 play a major role in suppression mediated by Tr1 cells, the in vivo involvement of these factors in Treg activities in cancer-related immune suppression has not been documented. We have previously shown that Treg accumulate in the tumor and peripheral blood of patients with HNSCC [31]. These cells could comprise nTreg and Tr1 subsets, although we expect that in cancer patients, Tr1 would be induced and assume a predominant role. Examining PBMC of HNSCC patients for the presence of Tr1 subsets by quantitative multicolor flow cytometry, we observed that the frequency of CD39+ and COX-2+ Treg was significantly increased relative to that in normal controls (NC). We have also reported that the percentage of CD39+ Treg in the patients’ blood was significantly greater in patients with later than early stage disease, suggesting that the increased frequency of CD39+ Treg in the peripheral circulation of HNSCC patients is related to disease progression [73]. Further, we compared the frequency of CD39+ Tr1 cells in the peripheral blood of HNSCC patients prior to and after oncologic treatments and consistently observed a significant increase after oncologic therapy [54]. The frequency of IL-10+ and TGF-β1+ CD4+ T cells was also elevated in the patients’ blood relative to that in NC, and it was significantly reduced after therapy. Also, we found that while CD39 and COX-2 were co-expressed in CD4+ T cells, IL-10 and TGF-β1 were expressed by a non-overlapping, distinct CD4+ T cell subset. These preliminary observations suggest that a Treg subset producing adenosine/PGE2 is distinct from that of comprising cells secreting IL-10 or TGF-β1. Only the former Treg subset expands following radiochemotherapy. Further, we observed co-expression of CD39 and COX-2 in Tr1 cells within the TIL at the tumor site [52]. At the very least, these data support the conclusion that Tr1 cells present in the blood and tumor tissues in cancer patients co-express CD39 and COX-2 and, therefore, have the capability to produce both these immunosuppressive factors.
7. CLINICAL SIGNIFICANCE OF THE ADENOSINERGIC PATHWAY
Adenosine and PGE2 can be produced by many human tumors and are present in the tumor microenvironment [9, 37, 43, 48, 49]. However, the contribution of Treg, a small subset of CD4+ T cells, to adenosine and PGE2 production is a novel and potentially clinically important finding, which has to be viewed in the context of local interactions between immune cells in the tumor microenvironment. If adenosine and PGE2 produced by activated human Treg (Tr1) synergize in mediating suppression of Teff cell functions, the result is a more potent immunosuppressive effect. In vitro, tumor cells expressing COX-2 were shown to drive the development of Tr1 cells that are producers of both adenosine and PGE2 [41, 54]. In patients with cancer, Tr1 cells able to produce both factors appear to be increased in the frequency and may be resistant to conventional oncologic therapies. While these preliminary data suggest that the tumor/tissue microenvironment determines the frequency, quality and mechanisms of suppression inducible Treg employ, more solid evidence is needed to support this hypothesis. As human tumors are often COX-2+ and are rich in extracellular ATP, because of cell death, opportunities exist for ATP-mediated up-regulation of ectonucleotidase activity and COX-2 expression in Tr1 cells generated in or attracted to the tumor microenvironment. In fact, up-regulation of these molecules is known to occur during inflammation that is a frequent component of the tumor development [70, 72]. The adenosine and PGE2 pathways both regulated at the 3′5′-cAMP level represent a powerful and effective combination for suppression of anti-tumor immune responses and thus for tumor progression and tumor escape from immune control. Cooperation between the adenosinergic and PGE2 pathways as exemplified by Treg-Teff interactions also represents a promising target for therapies aimed at the restoration of anti-tumor immune responses that are important for eliminating tumor escape as well as tumor resistance to conventional therapies [73].
8. PHARMACOLOGIC INTERVENTIONS TARGETING THE ADENOSINERGIC PATHWAY
One way to block undesirable immune suppression mediated by adenosine and PGE2 is a pharmacologic intervention utilizing inhibitors or antagonists of either pathway. This may be accomplished by directly interfering with production of adenosine and PGE2 or with their binding to respective receptors on immune cells. A number of clinically applicable pharmacologic strategies exist today for blocking the adenosinergic pathway [73], and many of these strategies have already been utilized in diseases other than cancer [74]. Inhibitors of the PGE2 pathway have been used to treat cancer for many years [75]. Rarely have these pharmacologic strategies been applied to eliminating immune suppression mediated by Treg in cancer. A good example of this approach is a phase II trial of IRX-2, a biologic containing several cytokines, administered to patients with advanced HNSCC in the adjuvant setting in conjunction with daily doses of indomethacin in order to silence suppressor cells [76]. Traditionally, Treg depletion has been used to improve endogenous anti-tumor immunity and the efficacy of immunotherapies usin strategies such as the administration of low-dose cyclophosphamide, daclizumab (anti-CD25 Ab), denileukin diftitox (Ontak) or tyrosine kinase inhibitors (TKIs) such as Sunitinb [77–79]. These anti-Treg regimens transiently reduce Treg numbers and function in the blood of some patients with cancer [79]. Invariably, however, Treg come back, and blocking of their activity becomes again necessary [80].
The use of pharmacologic inhibitors to specifically target the 3′5′-cAMP pathway in order to control excessive Treg function might be a novel therapeutic strategy. As shown here, adenosine binding to A2AR and PGE2 binding to EP2R expressed on Teff cells suppressed their anti-tumor functions in vitro by a common mechanism involving up-regulation of cytosolic 3′5′-cAMP levels and PKA type I activation. Numerous clinically available pharmacologic agents such as inhibitors of ectonucleotidase activity, A2AR and EP2R antagonists or inhibitors of PKA-1 type I can effectively block these pathways and decrease Tr1-mediated suppression of Teff proliferation, as shown in our experiments [52, 54]. Also, Rolipram, a phosphodiesterase 4 (PDE4) inhibitor, increased 3′5′-cAMP levels in Teff and consequently increased their susceptibility to Tr1-mediated suppression [54]. Similarly, many drugs blocking COX-2 activity are in clinical use, including indomethacin, diclofenac, ibuprofen, Colecoxib and others [67, 75]. The inhibitors can be selected to block adenosine or PGE2 production by Treg or block Treg effects in Teff. Specifically, it may be possible to consider blocking A2ARs and EP2Rs on Teff in order to silence signals delivered by Treg while increasing functions of Teff. Alternatively, it can be hypothesized that activating PDEs with, e.g., propanolol, would decrease functions of Treg while simultaneously increasing those of Teff. The feasibility of this type of PDE-directed approach is illustrated by the recently reported studies in mice, where stimulation of CD4+ T cells by allogeneic DC in the presence of cilostomide, an inhibitor of PDE3 resulted in enrichment in numbers and function of Treg [81]. These Treg were functional and blocked allograft rejection in vivo indicating that pharmacological PDE inhibition is a promising strategy for Treg-based therapies [81]. The fact that adenosine- and PGE2-mediated signals received by their respective surface receptors on Teff converge at the level of adenylyl cyclase (Ac), the key enzyme responsible for 3′5′-cAMP synthesis is of special interest for developing pharmacologic inhibitors for future clinical use [82, 83]. The intracellular cAMP concentration is regulated by enzymatic activity of Ac, which catalyzes the formation of 3′5′-cAMP, and by enzymatic activity of PDEs responsible for its degradation [83]. The hypothesis that we entertain is that the blockade of adenosine and PGE2 signaling at the point of their convergence in Teff (i.e., inhibition of Ac activity) will restore anti-tumor immune responses in patients with cancer. Our preliminary data confirms literature reports that Ac-7 isoform is expressed largely, if not exclusively, in hematopoietic cells [84]. This means that the Ac-7 inhibition should selectively downregulate 3′5′-cAMP levels in Teff, up regulate Teff functions, silence Treg and relieve adenosine or PGE2-mediated suppression. An enzyme isoform that is present in lymphocytes and integrates signals generated by both adenosine and PGE2 pathways is potentially a very attractive therapeutic target. Unfortunately, among the currently available pharmacologic inhibitors of Ac, none is specific for the Ac-7 isoform. If experimental data demonstrate that the simultaneous blockade of adenosine and PGE2 signaling at the point of their convergence in Teff can restore anti-tumor immune response in these cells, the development of Ac-7 isoformselective inhibitors would be fully justified. Thus, blocking of adenosine-PGE2 cooperation in Teff potentially leading to the restoration of their anti-tumor competence is a novel application of the “blocking the inhibitor” strategy for therapy of cancer.
9. CONCLUSIONS
Much has been recently said about the role of adenosine and PGE2 in shaping the tumor microenvironment [85, 86]. In this review, we have summarized recent insights into adenosine- and PGE2-mediated suppression utilized by human Treg in patients with cancer. It emerges that this may be the most prominent mechanisms utilized by activated inducible Treg (Tr1) in the microenvironment of solid tumors such as, e.g., HNSCC. In our hands, the frequency of CD4+CD39+ Treg cells in the periphery was related to cancer progression in several different human cancers, an indication that suppressor function mediated by the adenosine or PGE2 pathways may have clinical consequences. The Tr1 subsets present in human tumors or PBMC of cancer patients are COX2+/CD39+ or IL-10+/TGF-β+/CD39+, suggesting that functionally diverse Tr1 subsets may emerge during tumor progression or as a result of therapeutic interventions, both of which might alter the tumor microenvironment. One strategy for overcoming tumor-induced immune suppression and for eliminating tumor escape, involves a pharmacologically mediated blockade of cooperative interactions between inhibitory factors present in the tumor milieu that suppress immune responses. A potentially promising therapeutic target that recently has emerged is Ac-7, an enzyme regulating 3′5′-cAMP synthesis in immune cells. By selectively blocking activity of this enzyme, it may be possible to inhibit adenosine/PGE2 collaboration, disarm Treg and restore anti-tumor immune functions in Teff in cancer patients.
Footnotes
DECLARATION OF INTEREST
Supported in part by NIH grant PO1 CA109688 to TLW.
References
- 1.Eggremont AM. Immunostimulation versus immunosuppression after multiple vaccinations: the woes of therapeutic vaccine development. Clin Cancer Res. 2009;15:6745–6747. doi: 10.1158/1078-0432.CCR-09-2377. [DOI] [PubMed] [Google Scholar]
- 2.Alpizar YA, Chain B, Collins MK, Greenwood J, Katz D, Stauss HJ, Mitchison NA. Ten years of progress in vaccination against cnacer: the need to counteract cancer evasion by dual targeting in future therapies. Cancer Immunol Immunother. 2011;60:1127–1135. doi: 10.1007/s00262-011-0985-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tatsumi T, Kierstead LS, Ranieri E, Gesualdo L, Schena FP, Finke JH, Bukowski RM, Mueller-Berghaus J, Kirkwood JM, Kwok WW, Storkus WJ. Disease-associated bias in T helper type 1 (Th1)/Th2 CD4(+) T cell responses against MAGE-6 in HLA-DRB10401(+) patients with renal cell carcinoma or melanoma. J Exp Med. 2002;196(5):619–628. doi: 10.1084/jem.20012142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Campoli M, Ferrone S. HLA antigen changes in malignant cells: epigenetic mechanisms and biologic significance. Oncogene. 2008;27:5869–5885. doi: 10.1038/onc.2008.273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Frey AB, Monu N. Signaling defects in anti-tumor T cells. Immunol Rev. 2008;222:192–205. doi: 10.1111/j.1600-065X.2008.00606.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hoskin DW, Mader JS, Furlong SJ, Conrad DM, Blay J. Inhibition of T cell and natural killer cell function by adenosine and its contribution to immune evasion by tumor cells (Review) Int J Oncol. 2008;32:527–535. [PubMed] [Google Scholar]
- 7.Blay J, White TD, Hoskin DW. The extracellular fluid of solid carcinomas contains immunosuppressive concentrations of adenosine. Cancer Res. 1997;57:2602–2605. [PubMed] [Google Scholar]
- 8.Sitarz R, Leguit RJ, de Leng WW, Morsink FH, Polkowski WP, Maciejewski R, Offerhaus GJ, Milne AN. Cyclooxygenase-2 mediated regulation of E-cadherin occurs in conventional but not early-onset gastric cancer cell lines. Cell Oncol. 2009;31:475–485. doi: 10.3233/CLO-2009-0496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang D, Dubois RN. Eicosanoids and cancer. Nat Rev Cancer. 2010;10:181–193. doi: 10.1038/nrc2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang Y, Liu Q, Zhang M, Yu Y, Liu X, Cao X. Fas signal promotes lung cancer growth by recruiting myeloid-derived suppressor cells via cancer cell-derived PGE2. J Immunol. 2009;182:3801–3808. doi: 10.4049/jimmunol.0801548. [DOI] [PubMed] [Google Scholar]
- 11.Chen WJ, Konkel JE. TGF-β and “adaptive” FOXP3+ regulatory T cells. J Mol Cell Biol. 2010;2:30–36. doi: 10.1093/jmcb/mjp004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sa G, Das T, Moon C, Hilston CM, Rayman PA, Rini BI, Tannenbaum CS, Finke JH. GD3 an overexpressed tumor-derived ganglioside mediates the apoptosis of activated but not resting T cells. Cancer Res. 2009;69:3095–3104. doi: 10.1158/0008-5472.CAN-08-3776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gabrilovich D, Ishida T, Oyama T, Ran S, Kravtsov V, Nadaf S, Carbone DP. Vascular endothelial growth factor inhibits the development of dendritic cells dramatically affects the differentiation of multiple hematopoietic lineages in vivo. Blood. 1998;92:4150–4166. [PubMed] [Google Scholar]
- 14.Doubrovina ES, Doubrovin MM, Vider E, Sisson RB, O’Reilly RJ, Dupont B, Vyas YM. Evasion from NK cell immunity by MHC class I chain-related molecules expressing colon adenocarcinoma. J Immunol. 2003;171:6891–6899. doi: 10.4049/jimmunol.171.12.6891. [DOI] [PubMed] [Google Scholar]
- 15.Kim R, Emi M, Tanabe K, Arihiro K. Tumor-driven evolution of immunosuppressive networks during malignant progression. Cancer Res. 2006;66:5527–5536. doi: 10.1158/0008-5472.CAN-05-4128. [DOI] [PubMed] [Google Scholar]
- 16.Whiteside TL. Immune responses to malignancies. J Allergy Clin Immunol. 2010;125:S272–S283. doi: 10.1016/j.jaci.2009.09.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Whiteside TL. The role of immune cells in the tumor microenvironment. Cancer Treat Res. 2006;130:103–124. doi: 10.1007/0-387-26283-0_5. [DOI] [PubMed] [Google Scholar]
- 18.Poschke I, Mougiakakos D, Kiessling R. Camouflage and sabotage: tumor escape from the immune system. Cancer Immunol Immunother. 2011;60:1161–1171. doi: 10.1007/s00262-011-1012-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Reichert TE, Strauss L, Wagner EM, Gooding W, Whiteside TL. Signaling abnormalities and reduced proliferation of circulating and tumor-infiltrating lymphocytes in patients with oral carcinoma. Clin Cancer Res. 2002;8:3137–3145. [PubMed] [Google Scholar]
- 20.Ferrone S, Whiteside TL. Tumor microenvironment and immune escape. Surg Oncol Clin North AM. 2007;16:755–774. doi: 10.1016/j.soc.2007.08.004. [DOI] [PubMed] [Google Scholar]
- 21.Whiteside TL. The tumor microenvironment and its role in promoting tumor growth. Oncogene. 2008;27:5904–5912. doi: 10.1038/onc.2008.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Uppaluri R, Dunn GP, Lewis JS., Jr Focus on TILs: prognostic significance of tumor infiltrating lymphocytes in head and neck cancers. Cancer Immunol. 2008;8:16. [PMC free article] [PubMed] [Google Scholar]
- 23.Imai N, Ikeda H, Tawara I, Shiku H. Tumor progression inhibits the induction of multifunctionality in adoptively transferred tumor-specific CD8+ T cells. Eur J Immunol. 2009;39:241–253. doi: 10.1002/eji.200838824. [DOI] [PubMed] [Google Scholar]
- 24.Oble DA, Loewe R, Yu P, Mihm MC., Jr Focus on TILs: prognostic significance of tumor infiltrating lymphocytes in human melanoma. Cancer Immunol. 2009;9:3. [PMC free article] [PubMed] [Google Scholar]
- 25.Fakhry C, Westra WH, Li S, Cmelak A, Ridge JA, Pinto H, Forastiere A, Gillison ML. Improved survival of patients with human papillomavirus-positive head and neck squamous cell carcinoma in a prospective clinical trial. J Natl Cancer Inst. 2008;100:261–269. doi: 10.1093/jnci/djn011. [DOI] [PubMed] [Google Scholar]
- 26.Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pages C, Tosolini M, Camus M, Berger A, Wind P, Zinzindohoue F, Bruneval, Cugnenc PH, Trajanoski Z, Fridman WH, Pages F. Type density and location of immune cells within human colorectal tumors predict clinical outcome. Science. 2006;313(5795):1960–1964. doi: 10.1126/science.1129139. [DOI] [PubMed] [Google Scholar]
- 27.Cupponi A, Wieers G, van Baren N, Coulie PG. Tumor-infiltrating lymphocytes apparently good for melanoma patients. But why? Cancer Immunol Immunother. 2011;60:1153–1160. doi: 10.1007/s00262-011-1026-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bergmann C, Strauss L, Wang Y, Szczepanski MJ, Lang S, Johnson JT, Whiteside TL. T regulatory type 1 cells (Tr1) in squamous cell carcinoma of the head and neck: mechanisms of suppression and expansion in advanced disease. Clin Cancer Res. 2008;14:3706–3715. doi: 10.1158/1078-0432.CCR-07-5126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, Evdemon-Hogan M, Conejo-Garcia JR, Zhang L, Burow M, Zhu Y, Wei S, Kryczek I, Daniel B, Gordon A, Myers L, Lackner A, Disis ML, Knutson KL, Chen L, Zou W. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. 2004;10:942–949. doi: 10.1038/nm1093. [DOI] [PubMed] [Google Scholar]
- 30.Strauss L, Bergmann C, Szczepanski M, Gooding W, Johnson JT, Whiteside TL. A unique subset of CD4+CD25highFoxp3+ T cells secreting IL-10 and TGF-β1 mediates suppression in the tumor microenvironment. Clin Cancer Res. 2007;13:4345–4354. doi: 10.1158/1078-0432.CCR-07-0472. [DOI] [PubMed] [Google Scholar]
- 31.Strauss L, Bergmann C, Gooding W, Johnson JT, Whiteside TL. The frequency and suppressor function of CD4+CD25highFoxp3+ T cells in the circulation of patients with squamous cell carcinoma of the head and neck. Clin Cancer Res. 2007;13:6301–6311. doi: 10.1158/1078-0432.CCR-07-1403. [DOI] [PubMed] [Google Scholar]
- 32.Nishikawa H, Sakaguchi S. Regulatory T cells in tumor immunity. Int J Cancer. 2010;127:759–767. doi: 10.1002/ijc.25429. [DOI] [PubMed] [Google Scholar]
- 33.Mougiakakos D, Choudhury A, Lladser A, Kiessling R, Johansson CC. Regulatory T cells in cancer. Adv Cancer Res. 2010;107:57–117. doi: 10.1016/S0065-230X(10)07003-X. [DOI] [PubMed] [Google Scholar]
- 34.Czystowska M, Strauss L, Bergmann C, Szajnik M, Rabinowich H, Whiteside TL. Reciprocal granzyme/perforin-mediated death of human regulatory and responder T cells is regulated by interleukin-2 (IL-2) J Mol Med. 2010;88:577–588. doi: 10.1007/s00109-010-0602-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Strauss L, Bergmann C, Whiteside TL. Human circulating CD4+CD25highFoxp3+ Treg kill autologous CD8+ but not CD4+ responder cells by Fas-mediated apoptosis. J Immunol. 2009;182:1469–1480. doi: 10.4049/jimmunol.182.3.1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shevach EM. Mechanisms of Foxp3+ T regulatory cell-mediated suppression. Immunity. 2009;30:636–645. doi: 10.1016/j.immuni.2009.04.010. [DOI] [PubMed] [Google Scholar]
- 37.Sitkovsky MV, Kjaergaard J, Lukashev D, Ohta A. Hypoxia-adenosinergic immunosuppression: tumor protection by T regulatory cells and cancerous tissue hypoxia. Clin Cancer Res. 2008;14:5947–5952. doi: 10.1158/1078-0432.CCR-08-0229. [DOI] [PubMed] [Google Scholar]
- 38.Raimondi G, Turner MS, Thomson AW, Morel PA. Naturally occurring regulatory T cells: recent insights in health and disease. Crit Rev Immunol. 2007;27:61–95. doi: 10.1615/critrevimmunol.v27.i1.50. [DOI] [PubMed] [Google Scholar]
- 39.Jonuleit H, Schmitt E, Stassen M, Tuehenberg A, Knop J, Enk AH. Identification and functional characterization of human CD4(+)CD25(+) T cells with regulatory properties isolated from peripheral blood. J Exp Med. 2001;193:1285–1294. doi: 10.1084/jem.193.11.1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Roncarolo MG, Gregori S, Battaglia M, Bacchetta R, Fleischhauer K, Levings MK. Interleukin-10 secreting type 1 regulatory T cells in rodents and humans. Immunol Rev. 2006;212:28–50. doi: 10.1111/j.0105-2896.2006.00420.x. [DOI] [PubMed] [Google Scholar]
- 41.Bergmann C, Strauss L, Zeidler R, Lang S, Whiteside TL. Expansion of human T regulatory type 1 cells in the microenvironment of cyclooxygenase 2 overexpressing head and neck squamous cell carcinoma. Cancer Res. 2007;67:8865–8873. doi: 10.1158/0008-5472.CAN-07-0767. [DOI] [PubMed] [Google Scholar]
- 42.Roncarolo MG, Bacchetta R, Bordignon C, Narula S, Levings MK. Type 1 T regulatory cells. Immunol Rev. 2001;182:68–79. doi: 10.1034/j.1600-065x.2001.1820105.x. [DOI] [PubMed] [Google Scholar]
- 43.Dannenberg AJ, Altorki NK, Boyle JO, Dang C, Howe LR, Weksler BB, Subbaramaialt K. Cyclo-oxygenase 2: a pharmacological target for the prevention of cancer. Lancet Oncol. 2001;2:544–551. doi: 10.1016/S1470-2045(01)00488-0. [DOI] [PubMed] [Google Scholar]
- 44.Sitkovsky MV, Lukashev D, Apasov S, Kojima H, Koshiba M, Caldwell C, Ohta A, Thiel M. Physiological control of immune response and inflammatory tissue damage by hypoxia-inducible factors and adenosine A2A receptors. Annu Rev Immunol. 2004;22:657–682. doi: 10.1146/annurev.immunol.22.012703.104731. [DOI] [PubMed] [Google Scholar]
- 45.Dzhandzhugazyan KN, Kirkin AF, thor Straten P, Zeuthen J. Ecto-ATP diphosphohydrolase/CD39 is overexpressed in differentiated human melanomas. FEBS Lett. 1998;430:227–230. doi: 10.1016/s0014-5793(98)00603-6. [DOI] [PubMed] [Google Scholar]
- 46.Zhang B. CD73: A novel target for cancer immunotherapy. Cancer Res. 2010;70:6407–6411. doi: 10.1158/0008-5472.CAN-10-1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang X, Meng L, He B, Chen J, Liu P, Zhao J, Zhang Y, Li M, An D. The role of P2X7 receptor in ATP-mediated human leukemia cell death: calcium influx-independent. Acta Biochim Biophys Sin, (Shanghai) 2009;41:362–369. doi: 10.1093/abbs/gmp016. [DOI] [PubMed] [Google Scholar]
- 48.Sitkovsky M, Lukashev D, Deaglio S, Dwyer K, Robson SC, Ohta A. Adenosine A2A receptor antagonists: blockade of adenosinergic effects and T regulatory cells. Br J Pharma. 2008;153:S457–S464. doi: 10.1038/bjp.2008.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stagg J, Smyth MJ. Extracellular adenosine triphosphate and adenosine in cancer. Oncogene. 2010;29:5346–5358. doi: 10.1038/onc.2010.292. [DOI] [PubMed] [Google Scholar]
- 50.Borsellino G, Kleinewietfeld M, Di Mitri D, Sternjak A, Diamantini A, Giometto R, Hopner S, Centonze D, Bernardi G, Dell’Acqua ML, Rossini PM, Battistini L, Rotzschke O, Falk K. Expression of ectonucleotidase CD39 by Foxp3+ Treg cells: hydrolysis of extracellular ATP and immune suppression. Blood. 2007;110:1225–1232. doi: 10.1182/blood-2006-12-064527. [DOI] [PubMed] [Google Scholar]
- 51.Deaglio S, Dwyer KM, Gao W, Friedman D, Usheva A, Erat A, Chen JF, Enjyoji K, Linden J, Oukka M, Kuchroo VK, Strom TB, Robson SC. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J Exp Med. 2007;204:1257–1265. doi: 10.1084/jem.20062512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mandapathil M, Hilldorfer B, Szczepanski MJ, Czystowska M, Szajnik M, Ren J, Lang S, Jackson EK, Gorelik E, Whiteside TL. Generation and accumulation of immunosuppressive adenosine by human CD4+CD25highFOXP3+ regulatory T cells. J Biol Chem. 2010;285:7176–7186. doi: 10.1074/jbc.M109.047423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schuler P, Harasymczuk M, Schilling B, Lang S, Whiteside TL. Separation of human CD4+CD39+ T cells by magnetic beads reveals two phenotypically and functionally different subsets. J Immunol Meth. 2011;369:59–68. doi: 10.1016/j.jim.2011.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mandapathil M, Szczepanski MJ, Szajnik M, Ren J, Jackson EK, Johnson JT, Gorelik E, Lang S, Whiteside TL. Adenosine and prostaglandin E2 cooperate in the suppression of immune responses mediated by adaptive regulatory T cells. J Biol Chem. 2010;285:27571–27580. doi: 10.1074/jbc.M110.127100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Clayton A, Al-Taei S, Webber J, Mason MD, Tabi Z. Cancer exosomes express CD39 and CD73, which suppress T cells through adenosine production. J Immunol. 2011;187(2):676–683. doi: 10.4049/jimmunol.1003884. [DOI] [PubMed] [Google Scholar]
- 56.Zhou L, Chong MW, Littman DR. Plasticity of CD4+ T cell lineage differentiation. Immunity. 2009;30:646–655. doi: 10.1016/j.immuni.2009.05.001. [DOI] [PubMed] [Google Scholar]
- 57.Zhang H, Conrad DM, Butler JJ, Zhao C, Blay J, Hoskin DW. Adenosine acts through A2 receptors to inhibit IL-2-induced tyrosine phosphorylation of STAT5 in T lymphocytes: role of cyclic adenosine 3′,5′-monophosphate and phosphatises. J Immunol. 2004;173:932–944. doi: 10.4049/jimmunol.173.2.932. [DOI] [PubMed] [Google Scholar]
- 58.Bynoe MS, Viret C. FoxP3+CD4+ T cell-mediated immunosuppression involves extracellular nucleotide metabolism. Trends Immunol. 2008;29:99–102. doi: 10.1016/j.it.2007.12.005. [DOI] [PubMed] [Google Scholar]
- 59.Bazhin AV, Kahnert S, Kimpfler S, Schadendorf D, Umansky V. Distinct metabolism of cyclic adenosine monophosphate in regulatory and helper CD4+ T cells. Mol Immunol. 2010;47(4):678–684. doi: 10.1016/j.molimm.2009.10.032. [DOI] [PubMed] [Google Scholar]
- 60.Schenk U, Frascoli M, Proietti M, Geffers R, Traggiai E, Buer J, Ricordi C, Westendorf AM, Grassi F. ATP inhibits the generation and function of regulatory T cells through the activation of purinergic P2X receptors. Sci Signal. 2011;4(162):ra12. doi: 10.1126/scisignal.2001270. [DOI] [PubMed] [Google Scholar]
- 61.Bergmann C, Strauss L, Zeidler R, Lang S, Whiteside TL. Expansion and characteristics of human T regulatory type 1 cells in co-cultures simulating tumor microenvironment. Cancer Immunol Immunother. 2007;56:1429–1442. doi: 10.1007/s00262-007-0280-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Algars A, Karikoski M, Yegutkin GG, Stoitzner P, Niemela J, Salmi M, Jalkanen S. Different role of CD73 in leukocyte trafficking via blood and lymph vessels. Blood. 2011;117:4387–4393. doi: 10.1182/blood-2010-11-321646. [DOI] [PubMed] [Google Scholar]
- 63.Kameoka J, Tanaka T, Nojima Y, Schlossman SF, Morimoto C. Direct association of adenosine deaminase with a T cell activation antigen, CD26. Science. 1993;261:466–469. doi: 10.1126/science.8101391. [DOI] [PubMed] [Google Scholar]
- 64.Schrader WP, West CA. Localization of adenosine deaminase and adenosine deaminase complexing protein in rabbit heart. Implications for adenosine metabolism. Circ Res. 1990;66:754–762. doi: 10.1161/01.res.66.3.754. [DOI] [PubMed] [Google Scholar]
- 65.Dong RP, Kameoka J, Hegen M, Tanaka T, Xu Y, Schlossman SF, Morimoto C. Characterization of adenosine deaminase binding to human CD26 on T cells and its biologic role in immune response. J Immunol. 1996;156:1349–1355. [PubMed] [Google Scholar]
- 66.Havre PA, Abe M, Urasaki Y, Ohnuma K, Morimoto C, Dang NH. The role of CD26/dipeptidyl peptidase IV in cancer. Front Biosci. 2008;13:1634–1645. doi: 10.2741/2787. [DOI] [PubMed] [Google Scholar]
- 67.Wang D, Dubois RN. Prostaglandins and cancer. Gut. 2006;55:115–122. doi: 10.1136/gut.2004.047100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Singh B, Cook KR, Vincent L, Hall CS, Berry JA, Multani AS, Lucci A. Cyclooxygenase-2 induces genomic instability, BCL2 expression, doxorubicin resistance, and altered cancer-initiating cell phenotype in MCF7 breast cancer cells. J Surg Res. 2008;147:240–246. doi: 10.1016/j.jss.2008.02.026. [DOI] [PubMed] [Google Scholar]
- 69.Yaqub S, Tasken K. Role for the cAMP-protein kinase A signaling pathway in suppression of antitumor immune responses by regulatory T cells. Crit Rev Oncog. 2008;14:57–77. doi: 10.1615/critrevoncog.v14.i1.40. [DOI] [PubMed] [Google Scholar]
- 70.Su Y, Huang X, Raskovalova T, Zacharia L, Lokshin A, Jackson E, Gorelik E. Cooperation of adenosine and prostaglandin E2 (PGE2) in amplification of cAMP-PKA signaling and immunosuppression. Cancer Immunol Immunother. 2008;57:1611–1623. doi: 10.1007/s00262-008-0494-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sugimoto Y, Narumiya S. Prostaglandin E receptors. J Biol Chem. 2007;282:11613–11617. doi: 10.1074/jbc.R600038200. [DOI] [PubMed] [Google Scholar]
- 72.Su Y, Jackson EK, Gorelik E. Receptor desensitization and blockade of the suppressive effects of prostaglandin E2 and adenosine on the cytotoxic activity of human melanoma-infiltrating T lymphocytes. Cancer Immunol Immunother. 2010 doi: 10.1007/s00262-010-0924-z. epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Whiteside TL. Inhibiting the inhibitors: evaluating agents targeting cancer immunosuppression. Exp Opin Biol Ther. 2010;10:1019–1035. doi: 10.1517/14712598.2010.482207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hasko G, Linden J, Cronstein B, Pacher P. Adenosine receptors: therapeutic aspects for inflammatory and immune disease. Nat Rev Drug Discov. 2008;7:759–770. doi: 10.1038/nrd2638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lev-Ari S, Lichtenberg D, Arber N. Compositions for treatment of cancer and inflammation. Recent Pat Anticancer Drug Discov. 2008;3(1):55–62. doi: 10.2174/157489208783478720. [DOI] [PubMed] [Google Scholar]
- 76.Berinstein NL, Wolf G, Naylor PH, Baltzer L, Eagan JE, et al. Increased lymphocyte infiltration in patients with head and neck cancer treated with the IRX-2 immunotherapy regimen. Cancer Immunol Immunother. 2011 doi: 10.1007/s00262-011-1134-z. submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ghiringhelli F, Menard C, Puig PE, Ladoire S, Roux S, Martin F, Solary E, Lecesne A, Zitvogel L, Chauffert B. Metronomic cyclophosphamide regimen selectively depicts CD4+CD25+ regulatory T cells and restores T and NK effector functions in early stage cancer patients. Cancer Immunol Immunother. 2007;56:641–648. doi: 10.1007/s00262-006-0225-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Morse MA, Hobeika AC, Osada T, Serra D, Niedzwiecki D, Lyerly HK, Clay TM. Depletion of human regulatory T cells specifically enhances antigen specific immune responses to cancer vaccines. Blood. 2008;112:610–618. doi: 10.1182/blood-2008-01-135319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Finke JH, Rini B, Ireland J, Rayman P, Richmond A, Golshayan A, Wood L, Elson P, Garcia J, Dreicer R, Bukowski R. Sunitinib reverses type-1 immune suppression and decreases T-regulatory cells in renal cell carcinoma patients. Clin Cancer Res. 2008;14:6674–6682. doi: 10.1158/1078-0432.CCR-07-5212. [DOI] [PubMed] [Google Scholar]
- 80.DeVries IJ, Castelli C, Huygens C, Jacobs JF, Stockis J, Schuler-Thurner B, Adema GJ, Punt CJ, Rivoltini L, Schuler G, Coulie PG, Luca SS. Frequency of circulating Tregs with demethylated FOXP3 intron 1 in melanoma patients receiving tumor vaccines and potentially Treg-depleting agents. Clin Cancer Res. 2010;17:1–8. doi: 10.1158/1078-0432.CCR-10-2227. [DOI] [PubMed] [Google Scholar]
- 81.Feng G, Nadig SN, Backdahl L, Beck S, Francis RS, Schiopu A, Whatcott A, Wood KJ, Bushell A. Functional regulatory T cells produced by inhibiting cyclic nucleotide phosphodiesterase type 3 prevent allograft rejection. Sci Transl Med. 2011;3(83):83ra40. doi: 10.1126/scitranslmed.3002099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Iwatsubo K, Okumura S, Ishikawa Y. Drug therapy aimed at adenylyl cyclase to regulate cyclic nucleotide signaling. Endocr Metab Immune Disord Drug Targets. 2006;6:239–247. doi: 10.2174/187153006778249994. [DOI] [PubMed] [Google Scholar]
- 83.Pavan B, Biondi C, Dalpiaz A. Adenylyl cyclases as innovative therapeutic goals. Drug Discov Today. 2009;14:982–991. doi: 10.1016/j.drudis.2009.07.007. [DOI] [PubMed] [Google Scholar]
- 84.Duan B, Davis R, Sadat EL, Collins J, Sternweis PC, Yuan D, Jiang LI. Distinct roles of adenylyl cyclase VII in regulating the immune responses in mice. J Immunol. 2010;185:335–344. doi: 10.4049/jimmunol.0903474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ernst PB, Garrison JC, Thompson LF. Much ado about adenosine: adenosine synthesis and function in regulatory T cell biology. J Immunol. 2010;185:1993–1998. doi: 10.4049/jimmunol.1000108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Greenhough A, Smartt HJ, Moore AE, Roberts HR, Williams AC, Paraskeva C, Kaidi A. The COX-2/PGE2 pathway: key roles in the hallmarks of cancer and adaptation to the tumour microenvironment. Carcinogenesis. 2009;30:377–386. doi: 10.1093/carcin/bgp014. [DOI] [PubMed] [Google Scholar]