

Abstract
Deficiency of acid alpha glucosidase (GAA) causes Pompe disease, which is usually fatal if onset occurs in infancy. Patients synthesize a non-functional form of GAA or are unable to form native enzyme. Enzyme replacement therapy with recombinant human GAA (rhGAA) prolongs survival in infantile Pompe patients but may be less effective in cross-reactive immunologic material (CRIM)-negative patients. We retrospectively analyzed the influence of CRIM status on outcome in 21 CRIM-positive and 11 CRIM-negative infantile Pompe patients receiving rhGAA. Patients were from the clinical setting and from clinical trials of rhGAA, were ≤6 months of age, were not invasively ventilated, and were treated with IV rhGAA at a cumulative or total dose of 20 or 40 mg/kg/2 weeks. Outcome measures included survival, invasive ventilator-free survival, cardiac status, gross motor development, development of antibodies to rhGAA, and levels of urinary Glc4.
Following 52 weeks of treatment, 6/11 (54.5%) CRIM-negative and 1/21 (4.8%) CRIM-positive patients were deceased or invasively ventilated (p < 0.0001). By age 27.1 months, all CRIM-negative patients and 4/21 (19.0%) CRIM-positive patients were deceased or invasively ventilated. Cardiac function and gross motor development improved significantly more in the CRIM-positive group. IgG antibodies to rhGAA developed earlier and serotiters were higher and more sustained in the CRIM-negative group. CRIM-negative status predicted reduced overall survival and invasive ventilator-free survival and poorer clinical outcomes in infants with Pompe disease treated with rhGAA. The effect of CRIM status on outcome appears to be mediated by antibody responses to the exogenous protein.
Keywords: Pompe disease, Glycogen storage disease, Enzyme replacement therapy, Lysosomal storage disease, Acid alpha glucosidase, GAA, Cross-reactive immunologic material, CRIM, Antibody
Introduction
Pompe disease (glycogen storage disease type II) is a lysosomal storage disorder resulting from deficiency of acid alpha glucosidase (GAA). This deficiency causes lysosomal accumulation of glycogen in cardiac, skeletal, and smooth muscle tissue, resulting in progressive cardiac, motor, and respiratory failure. The infantile phenotype is characterized by rapidly progressive muscle weakness, hypertrophic cardiomyopathy, feeding difficulties, and eventual respiratory insufficiency. Patients usually present with disease symptoms at approximately 3 months of age and death occurs at a median age of 6.0–8.7 months [1,2]. The reported frequency of infantile-onset Pompe disease ranges from 1 in 33,333 in Taiwanese populations to 1 in 138,000 in Dutch populations [3,4]. Development of Chinese hamster ovary- (CHO) and transgenic rabbit milk-derived enzyme replacement therapy (ERT) paved the way for the first available targeted therapy for Pompe disease [5,6]. Clinical trials of ERT showed a positive response in infantile Pompe patients [7–12]. CHO-derived rhGAA (alglucosidase alfa; Myozyme®) was approved in the USA, Europe, and Canada in 2006, with subsequent approvals in numerous countries worldwide.
Timely intervention with ERT is essential to halt motor disease progression and reverse cardiomyopathy in infantile Pompe patients; response is more attenuated if ERT is initiated when the disease is more advanced [12]. Despite early treatment, however, some patients with infantile Pompe disease respond poorly to ERT. Factors that are important in determining outcome include extent of pathology and muscle damage at the time of start of ERT [9,11,13]. However, these factors do not sufficiently predict disease course or response to ERT.
In the first clinical trial using CHO cell-derived rhGAA, we suggested that the presence or absence of cross-reactive immunological material (CRIM) may affect prognosis [8]. Patients with two deleterious GAA mutations who are completely unable to form native enzyme are CRIM-negative; patients with presence of some residual, functioning or non-functioning enzyme are CRIM-positive. In this pilot trial the two patients who were CRIM-negative died, while the single patient who was CRIM-positive had a very good motor response and is currently alive and ambulatory [8]. Concomitant with clinical decline, persistent high anti-rhGAA IgG antibody titers were found in the two CRIM-negative patients while titers for the CRIM-positive patient remained low. Similar observations from our group were reported in a subsequent clinical trial [9]. However, this correlation was not confirmed by others [7,14]. CRIM-positive patients tend to have low antibody titers, which are associated with the best response to ERT.
To gain a better understanding of the role of CRIM status as a prognostic factor, we conducted a retrospective analysis comparing outcomes of CRIM-positive with outcomes of CRIM-negative patients with Pompe disease from the clinical setting and from clinical trials of rhGAA. Subjects were age 6 months or younger at the initiation of ERT. Inclusion and exclusion criteria and endpoints were similar to those used in the pivotal trial of alglucosidase alfa [11]. We hypothesized that CRIM-negative patients would have a poorer clinical outcome compared to CRIM-positive patients. Primary clinical outcomes selected were survival, ventilator-free survival, and cardiac and motor response, corresponding with the key endpoints in the pivotal trial of alglucosidase alfa [11]. Additional analyses included urinary Glc4 levels and antibody titers.
Methods
Study design
Retrospective data were collected from clinical trials of CHO-derived rhGAA conducted between 1999 and 2006 [8,9,11,12] and from a retrospective chart review of several clinical patients who met inclusion and exclusion criteria and were part of expanded access programs at two sites. Inclusion and exclusion criteria were chosen to resemble those for the pivotal trial of alglucosidase alfa [11]. Principal inclusion criteria were: confirmed diagnosis of Pompe disease; age at enrollment ≤6 months by adjusted gestational age; <1% of normal GAA activity (in skin fibroblasts and/or muscle biopsy); and cardiomyopathy (left ventricular mass index [LVMI] ≥65 g/m2 by echocardiogram). Exclusion criteria included respiratory insufficiency, major congenital anomaly, or any prior rhGAA treatment. Parents or guardians of all patients in these studies gave consent to institutional IRB- or ethics committee-approved protocols. All patients received rhGAA supplied by Genzyme Corporation (Cambridge, MA) at a cumulative or total dose of 20 or 40 mg/kg every 2 weeks according to previously published reports [8,9,11,12].
Clinical outcomes
Survival and invasive ventilator-free survival were analyzed in the original clinical trials; we summarize these data and present updated survival data through June 2006, at which time the database was locked. Patients included in this analysis from the clinical setting were followed until age 18 months, as done for the pivotal trial of alglucosidase alfa [11]. Two-dimensional, M-mode, and Doppler echocardiography were used to assess LVM index at baseline, 26, and 52 weeks after the initiation of ERT. Motor function evaluation was performed using the Alberta Infant Motor Scale (AIMS) [15] by an experienced physical therapist at baseline, 26, and 52 weeks after the initiation of ERT.
Laboratory methods
Cultured skin fibroblasts obtained from Pompe patients were assayed for GAA enzyme activity using 4-methyl-umbelliferyl-α-D-glucopyranoside (4MUG) as an artificial substrate described in Kishnani et al. [9].
CRIM status was determined as described previously [9,16] based on reactivity of a pool of monoclonal or polyclonal antibodies generated against GAA that could recognize both native and recombinant forms of GAA. Briefly, cell lysates derived from patient fibroblast cells were subjected to Western blot analysis using a 4–12% gradient gel and a pool of monoclonal antibodies (generated against rhGAA) and a polyclonal antibody that was generated against human placental GAA, which recognizes both precursor and processed forms of GAA protein. A patient was designated as CRIM-positive if any of the GAA protein forms (unprocessed precursor band at 110 kDa or any of the processed forms) were detectable on the Western blot analysis; a patient was designated as CRIM-negative if none of these protein forms was detectable on Western blots (processed and unprocessed). Results were initially generated by one laboratory and were confirmed by a second, independent laboratory (one using monoclonal antibodies, the other polyclonal antibodies) in a blinded fashion. Representative Western blot showing CRIM-positive and CRIM-negative patients is shown in Fig. 1.
Fig. 1.

Representative gel showing CRIM-positive and CRIM-negative samples run on the gel with known positive and negative control samples.
In patients whose parents provided informed consent for genotyping, DNA was isolated from peripheral blood and sequenced. GAA mutation analysis was determined by Genzyme Corporation, as described previously [9,11].
Anti-rhGAA IgG antibodies were assessed at baseline and at weeks 4, 8, 12, 24, 38, and 52. In surviving infants, serotiters were followed up to 80 weeks. Antibody status was ascertained using enzyme-linked immunosorbent assays and confirmed using radio-immunoprecipitation, as previously described [9]. Patients from the first trial of three patients [8] were excluded from antibody analyses. Additional testing to determine the presence of inhibitory antibodies towards enzyme uptake or enzyme activity was performed in patients according to the respective clinical trial protocols and the requirements of the Genzyme Pharmacovigilance department. An inhibitory antibody assay (enzyme activity) was used to measure inhibition of rhGAA enzyme activity by antibodies present in patient serum and a flow cytometry-based assay (enzyme uptake) was used to evaluate whether patient antibodies interfere with uptake of rhGAA by human fibroblast cells in culture.
Urine oligosaccharides were obtained to assess and follow Glc4, a biomarker for Pompe disease, at baseline and at weeks 4, 12, 26, 38, 52, 64, and 78. Urinary Glc4 was measured by HPLC–UV and tandem mass spectrometry (ESI-MS/MS) as previously described [17,18].
Statistical analysis
Survival data were analyzed using the Kaplan–Meier method [19] with two-tailed p-values generated using the log-rank test. Other reported p-values were generated by the Wilcoxon rank sum test for continuous variables and Fisher’s exact test for categorical variables. Analyses were performed with STATA version 9.0 (StataCorp LP, College Station, Texas). Due to the limited sample size, all group outcome variable data are presented as medians.
Results
A total of 32 patients from the source clinical trials and from a subset of infantile patients at 2 sites who met inclusion and exclusion criteria for this analysis: 11 CRIM-negative patients and 21 CRIM-positive patients. Baseline demographics and disease-related characteristics (age, LVM index, AIMS score, and urinary Hex 4 levels) were comparable between the two groups (Table 1). Genotypes of CRIM-negative patients who consented to genotyping are presented in Table 2.
Table 1.
Baseline demographics.
| CRIM-negative N = 11 | CRIM-positive N = 21 | |
|---|---|---|
| Gender | ||
| Male | 5 (45%) | 15 (71%) |
| Female | 6 (55%) | 6 (29%) |
| Race | ||
| White | 4 (36%) | 10 (48%) |
| Black | 3 (27%) | 2 (10%) |
| Hispanic | 1 (9%) | 3 (14%) |
| Asian | 1 (9%) | 5 (24%) |
| Other | 2 (18%) | 1 (5%) |
| Age at Symptom Onset (months) | ||
| Mean (SD) | 1.23 (0.9) | 1.67 (1.60) |
| Median | 1.45 | 1.50 |
| Min, Max | 0, 2.4 | 0, 5.4 |
| Age at First ERT (months) | ||
| Mean (SD) | 3.55 (1.93) | 4.59 (2.05) |
| Median | 3.00 | 5.10 |
| Min, Max | 0.25, 7.0 | 0.25, 7.3 |
| Adjusted Age at First ERT (months) | ||
| Mean (SD) | 3.19 (1.81) | 4.20 (1.81) |
| Median | 3.15 | 4.8 |
| Min, Max | 0, 6.1 | 0, 6.1 |
Table 2.
GAA gene mutations in 9/11 CRIM-negative patients.
| Race | Maternal allele | Paternal allele |
|---|---|---|
| Caucasian | c.722–723 del TT (frameshift) | c.1687C > T (p.Gln563Stop) |
| African American | c. 2560C > T (p.Arg854Stop) | c.2560C > T (p.Arg854Stop) |
| Asian | c.148_859-11del (p.Glu50 fs, del exons 2–4) null allele c.1726G > A (p.Gly576Ser) | 686insCGGC (p.Arg229fs) |
| Arab | c.2560C > T (p.Arg854Stop) | c.2560C > T (p.Arg854Stop) |
| Israeli | c.1075G>A (p.Gly359Stop) | c.1075G>A (p.Gly359Stop) |
| African American | c.2560C > T (p.Arg854Stop) | c.2560C > T (p.Arg854Stop) |
| Caucasian | c.1754 IVS12 + 1G > A (splicing) | c.722_723delTT(p.Phe241Cys fs-Ter 88) |
| Arab | c.1209delC (p.Asn403Lysfs-Ter37) | c.1209delC (p.Asn403Lysfs-Ter37) |
| Hispanic | c.1496 G > A (p. Trp498Stop) | c.1496 G > A (p.Trp498Stop) |
Kroos et al. [42].
Survival
Ventilator-free survival was significantly better for the CRIMpositive group than the CRIM-negative group [Fig. 2; p < 0.001]. After 52 weeks of ERT, 6/11 (54.5%) CRIM-negative patients were either deceased (n = 1) or on invasive ventilation (n = 5) as compared to only 1/21 (4.8%) CRIM-positive patient, who was ventilator dependent. By 27.1 months of age, all 11 CRIM-negative patients were deceased (n = 5) or on a ventilator (n = 6), compared to 4/21 (19.0%) CRIM-positive patients (n = 1 deceased, n = 3 invasively ventilated).
Fig. 2.

Kaplan–Meier curve of ventilator-free survival of the CRIM-negative (n = 11) and CRIM-positive (n = 21) patients.
Cardiac function: left ventricular mass index
The upper limit of normal LVMI for infants is 64 g/m2 [20]. In this study, both groups showed similarly elevated LVMI at baseline [median LVMI 202.1 g/m2 for CRIM-negative patients (n = 10) and 207.8 in CRIM-positive patients (n = 19) (p = 0.96)]. After 26 weeks of rhGAA, both groups had a net decrease in LVMI (Fig. 3A). In marked contrast, at 52 weeks, CRIM-positive patients (n = 18) demonstrated additional reduction of median LVMI to near-normal levels (LVMI = 63.9 g/m2), while median LVMI in surviving CRIMnegative patients (n = 9) increased to 129 g/m2 (p = 0.0005).
Fig. 3.
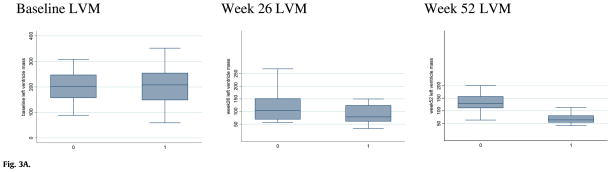
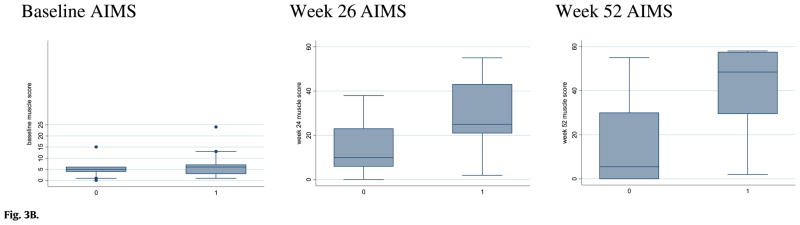
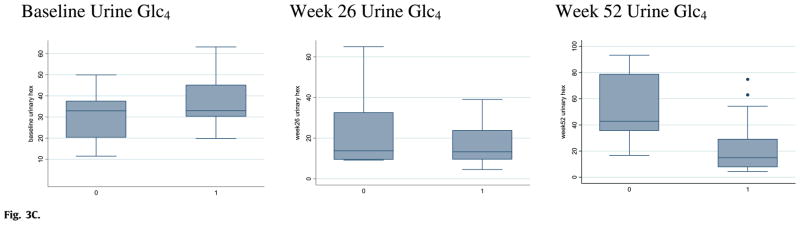
Fig. 3A. Left ventricular mass index (LVMI) at baseline and after 24 and 52 weeks of rhGAA treatment in CRIM-positive and CRIM-negative patients (labeled as 1 and 0 on X-axis, respectively).
Fig. 3B. Alberta Infant Motor Scale (AIMS) scores at baseline and after 26 and 52 weeks of rhGAA treatment in CRIM-positive and CRIM-negative patients (Labeled as 1 and 0 on the x-axis, respectively).
Fig. 3C. Urine Glc4 levels at baseline and after 26 and 52 weeks of rhGAA treatment in CRIM-positive and CRIM-negative patients (Labeled as 1 and 0 on the x-axis, respectively).
Gross motor development
At baseline, gross motor development was delayed and essentially the same for both groups. The average age of CRIM-negative (n = 10) and CRIM-positive (n = 21) patients at ERT initiation was 3.6 months and 4.6 months, respectively; the average score on the Alberta Infant Motor Scales was 5.2 (age equivalent [AE] = 3 weeks) and 7 (AE = 4 weeks), respectively. After 26 weeks of ERT, gross motor function improved in both groups, but the median AIMS score for the CRIM-negative group (10) was below that for the CRIM-positive group (25; p = 0.07) (Fig. 3B). After 52 weeks of ERT, the median AIMS score for CRIM-negative patients (5.5; AE = 3 weeks) was less than the median score for the CRIM-positive patients (48.5; AE = 10 months; p = 0.006).
Anti-rhGAA antibody determination
All CRIM-negative patients (n = 8) and 18/20 (90%) CRIM-positive patients who were tested developed IgG antibodies to rhGAA. All CRIM-negative patients seroconverted by 4 weeks of ERT. The average time of seroconversion in the CRIM-positive group following ERT initiation was 12.7 weeks (Table 3). Serotiters were significantly different at 24 weeks (median titer of 1:51,200 CRIM-negative and 1:600 CRIM-positive; p = 0.0010) and remained significantly different at 52 weeks (median titer of 1:153,600 CRIM-negative and 1:200 CRIM-positive; p = 0.0010).
Table 3.
Antibody data and clinical status of infantile Pompe patients at database lock.
| Median (range) antibody titers
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Baseline | Week 24 | Week 52 | Week 64 | Week 78 | ||||||
| CRIM- | negative | 0 (0–100) n = 8 |
51,200 (12,800– 102,400) n = 7 |
153,600 (3,200– 1,638,400) n = 6 |
409,600 (204,800– 1,638,400) n = 4 |
12,800 (12,800) n = 1 |
||||
| CRIM- | positive | 0 (0–100) n = 19 |
600 (0–51,200) n = 20 |
200 (0–51,200) n = 19 |
300 (0– 51,200) n = 14 |
400 (0–25,600) n = 14 |
||||
| Patient | Inhibition of uptake | Inhibition of activity | Serocon-version (week) | Peak antibody titer | Week of peak titer | Last antibody titer | Week of last titer | Age at invasive ventilation (months) | Age at death (months) | Age at June 2006 database lock (months) |
|
| ||||||||||
| CRIM-negative | ||||||||||
| 1 | ** | ** | ** | ** | ** | ** | ** | 8.2 | 50.2 | |
| 2 | ** | ** | ** | ** | ** | ** | ** | 8.3 | 44.8 | |
| 5 | * | * | 0 | 1:102,400 | 24 | 1:51,200 | 36 | 13.8 | 14.7 | |
| 8 | * | * | 0 | 1:204,800 | 64 | 1:204,800 | 64 | 11.3 | 23.3 | |
| 11 | y | n | 0 | 1:409,600 | 64 | 1:409,600 | 82 | 24.5 | 25.5 | |
| 20 | y | y | 4 | 1:1,638,400 | 52 | 1:1,638,400 | 64 | 18.5 | 32.0 | |
| 24 | y | y | 4 | 1:204,800 | 38 | 1:102,400 | 52 | 9.2 | 31 | |
| 26 | n | n | 4 | 1:51,200 | 24 | 1:25,600 | 38 | 9.1 | 26 | |
| 27 | * | * | 4 | 1:25,600 | 16 | 1:6,400 | 90 | 27.1 | ||
| 28 | n | n | 0 | 1:409,600 | 64 | 1:409,600 | 64 | 18.0 | *** | |
| 31 | ** | ** | ** | ** | ** | ** | ** | 25.3 | 51.4 | |
| CRIM-positive | ||||||||||
| 3 | ** | ** | ** | ** | ** | ** | ** | 83 | ||
| 4 | n | n | 4 | 1:51,200 | 38 | 1:25,600 | 78 | 15.0 | 41.0 | |
| 6 | n | n | 4 | 1:51,200 | 12 | 1:51,200 | 52 | 30 | ||
| 7 | * | * | 0 | 1:25,600 | 12 | 1:800 | 78 | 65 | ||
| 9 | n | n | 4 | 1:25,600 | 24 | 1:1,600 | 78 | 37 | ||
| 10 | * | * | 0 | 1:25,600 | 8 | 1:12,800 | 78 | 32.1 | ||
| 12 | n | n | 4 | 1:12,800 | 12 | 1:200 | 78 | 41 | ||
| 13 | n | n | 4 | 1:12,800 | 16 | 1:3,200 | 80 | 24.4 | 30 | |
| 14 | n | n | 4 | 1:6,400 | 20 | 1:200 | 52 | 19.4 | 19.8 | |
| 15 | n | n | 8 | 1:3,200 | 8 | 1:400 | 78 | 38 | ||
| 16 | n | n | 12 | 1:400 | 20 | 1:100 | 78 | 35 | ||
| 17 | n | n | 8 | 1:400 | 78 | 1:400 | 78 | 36 | ||
| 18 | n | n | 8 | 1:400 | 8 | 1:400 | 78 | 36 | ||
| 19 | n | n | 8 | 1:400 | 38 | 1:100 | 52 | 33 | ||
| 21 | * | n | 64 | 1:400 | 78 | 1:400 | 78 | 22 | ||
| 22 | * | n | 8 | 1:400 | 20 | 1:400 | 38 | *** | ||
| 23 | n | n | 64 | 1:200 | 64 | 1:200 | 78 | 19.7 | 39 | |
| 25 | * | n | 24 | 1:200 | 24 | 1:200 | 52 | *** | ||
| 29 | n | n | Never | 0 | 0 | 0 | 78 | 33 | ||
| 30 | n | n | Never | 0 | 0 | 0 | 78 | 28 | ||
| 32 | * | * | 0 | 1:51,200 | 24 | 1:3,200 | 78 | 62 | ||
Not done.
Patients 1, 2, and 31 had different methods of antibody determination and are excluded from this analysis.
Patients were alive and younger than 18 months on June 2006; data were collected on these patients through age 18 months.
Titers were generally higher in the CRIM-negative patients, whose median peak titer level across subjects was 1:204,800 compared with a peak titer of 1:1800 in CRIM-positive patients. Of the 15 CRIM-positive patients who had seroconverted and for whom antibody data were available at 52 weeks, 9 (60%) had titers less than or equal to 1:800 for that time point and all subsequent time points, while 6 (40%) had titers of 1:3200 or higher at 52 weeks (range 1:3200–1:51,200), with subsequent titers staying at or above 1:1600. Conversely, the CRIM-negative group showed a continued increase in serotiters, effectively tripling median titers from 1:51,200 at 24 weeks to 1:153,600 at 52 weeks of ERT. No CRIMnegative patient showed a consistent decline in titer levels and all CRIM-negative patients developed titers of at least 1:25,600.
Additional testing to detect inhibition of enzyme uptake or inhibition of enzyme activity was conducted on samples from most patients who developed anti-rhGAA antibodies and experienced infusion-associated reactions (IARs) or showed signs of clinical decline. Twenty two patients (5 CRIM-negative, 17 CRIM-positive) were tested for activity inhibition and 19 patients (5 CRIM-negative, 14 CRIM-positive) were tested for uptake inhibition. None of the CRIM-positive patients tested showed detectable levels of uptake or activity inhibition. Three of the five CRIM-negative patients tested positive for uptake inhibition; two of those patients also tested positive for activity inhibition. Five CRIM-negative and 4 CRIM-positive patients in the current study were never tested for activity or uptake inhibition, and an additional 3 CRIM-positive patients were never tested for uptake inhibition.
Biomarker Glc4
There was no significant difference in urine Glc4 levels between the two groups at baseline (z = −0.9032; p = 0.3667). After 52 weeks of ERT, Glc4 levels for CRIM-positive patients remained low (median Glc4 level = 15.0 mmol/mol creatinine), while Glc4 levels in the CRIM-negative group increased (median Glc4 level = 42.9); the difference at 52 weeks was significant (z = 2.829; p = 0.0047; Fig. 3C).
Discussion
ERT with alglucosidase alfa has been shown to be effective in improving survival and motor outcomes in infants with Pompe disease [11,12]. Age at disease symptom onset and stage of disease at treatment initiation have been noted as important factors in determining outcome; however, previous studies demonstrated that not all infants have favorable outcomes even when treatment is begun early. We have reported poor outcome in CRIM-negative patients [8], but no analysis of CRIM status across a large number of patients has been performed.
In the present analysis, CRIM-negative patients clearly show an attenuated response to enzyme in all outcome measures compared to CRIM-positive patients: significantly decreased survival, invasive ventilation-free survival, less improvement in cardiac response, and regression of motor milestones.
This analysis is necessarily limited by the small number of patients for whom robust, long-term clinical data are available. To control for confounding limitations of ERT in Pompe disease, we limited our patient group to those less than 6 months of age at onset of therapy [9]. In patients older than 6 months the muscle is likely in a more advanced stage of disease.
Poor outcome in the CRIM-negative patients could be due to those patients having generally more severe GAA gene mutations— that is, two deleterious mutations, which would be expected to result in complete absence of native enzyme and no band on Western blot. Interestingly, the CRIM-negative patients in this study were observed generally to have a period of improvement in the first 6 months of enzyme replacement followed by a period of decline. Given the coincident rise in antibody response, this suggests that the initial response to treatment with the exogenous enzyme is terminated by the increased antibody activity. Animal studies have suggested that antibody formation to rhGAA reduces the efficacy of ERT [21]. More recently, immune tolerance induction with an adeno-associated virus vector containing a liver-specific promoter in a Pompe knockout mouse model further supports the impact of antibodies in response to ERT [22]. We recently reported the successful induction of immune modulation in a CRIM-negative patient with Pompe disease using the anti- CD20 monoclonal antibody rituximab plus methotrexate and intravenous gamma globulin [23]. In this patient, ERT was started at 7 weeks of age, anti-rhGAA antibodies developed at 23 weeks, and immune modulation therapy was started at 25 weeks; at 24 months of age, the patient continued to be antibody free and to gain motor milestones on ERT.
The significant differences in clinical outcomes between the two CRIM groups in response to ERT were coincident with significant differences in serotiter levels of antibodies against the enzyme. In the CRIM-negative group antibody titers were higher, seroconversion occurred earlier, and titers were sustained at higher levels. It appears that serotiter levels play a role in the clinical decline in these patients. Three of the 5 CRIM-positive patients who did poorly (i.e., died or were invasively ventilated) had peak titers of 12,800, 25,600, or 51,200; titers stayed elevated with no downward trend ever noted in two patients, while the third patient had a slight downward trend. Two of the five CRIM-positive patients with poor outcome had lower peak titers: one had a peak titer of 6400, which dropped to 200 at week 52, while one had zero titers until week 64 when titers increased to 200. Both of these patients had very poor AIMS scores at baseline with no apparent gains in motor function so it is likely that other factors were responsible for their poor outcomes.
Antibody interference has been described as a complicating factor in patients with other lysosomal storage disorders and with other conditions for which a foreign protein is administered, such as hemophilia A and B [24–28]. Seroconversion rates in other LSDs vary; studies suggest that this variation is linked to the relative severity of gene mutations (and therefore potentially CRIM status) commonly seen in each disease [29,30]. More severe mutations are associated with CRIM negativity and a more marked immune response to the replacement enzyme, which appears to the immune system as a foreign protein. Resistance to factor replacement in patients with hemophilia A and B associated with the development of antibodies has been well described and is associated with mutations that produce absent or truncated factors [31]. The present analysis includes genotype data on 9 of 11 CRIM-negative patients; these patients all show severe mutations consistent with CRIM-negative status and a strong, sustained immune response to rhGAA.
Data from clinical trials [8,9,11] suggest that approximately 20% of all infantile Pompe cases are CRIM-negative, accounting for 6–7 cases of infantile Pompe disease in the US per year. Patients of African American descent who are homozygous for the p.Arg854X mutation account for at least half the CRIM-negative infantile patients identified in the US. Patients of Arabic Muslim descent who are homozygous for p.Lys114fsX32 are also identified to be CRIM-negative. Although only a small number of patients are expected to be CRIM-negative, investigation of CRIM status may assist in risk stratification of infantile Pompe patients and in selection of treatment interventions that have the potential to be successful in ablating antibodies, such as immune modulation, prior to start of or early in the course of ERT.
Resistance to therapeutic protein may be susceptible to modulation via tolerance-inducing protocols [32–34]. In many LSDs, although CRIM-positive patients generate antibody responses initially, such responses tolerize over a 1- to 2-year period on continued therapy [30,35]. Successful immunomodulation has been reported in animal models and in some patients with various factor or enzyme deficiencies for whom routine treatment was ineffective [8,34,36–38]. For CRIM-negative patients with Pompe disease or severe Factor IX deficiency, reversing the antibody response once entrenched has generally not been successful, even with relatively strenuous pharmacologic approaches. Indeed, some patients developed nephrotic syndrome from immune complex-mediated nephritis on such therapies [39]. Additional tolerance-inducing strategies, such as the one described above [23], may include preventive tolerance induction at initiation of therapy or use of more novel B and T cell targeting agents to induce tolerance after the development of inhibitory antibodies. Immune tolerance via a gene therapy approach has also been shown to be efficacious in a GAAKO mouse model of Pompe disease [22]. Clinical trials of immunomodulation must be conducted, as the outlook is otherwise dismal for CRIM-negative patients. Moreover, elucidating the immunologic mechanism of antibody development, especially with respect to the necessity for T cell help, may further define additional prophylactic or therapeutic targets for tolerance induction.
The potential benefit of enzyme replacement therapy and need for efficacious early intervention in Pompe disease has led to the development of dried blood spot GAA testing [40] and interest in incorporating GAA testing into newborn screening protocols. Blood-based assays of CRIM status are currently under development to rapidly determine CRIM status and drive treatment-related decisions (personal communication). Pompe disease is now becoming a treatable disease of infancy. Successful enzyme replacement therapies for other lysosomal storage disorders have become the standard of care, and similarly GAA replacement infusions are standard therapy for infantile Pompe disease, in conjunction with attentive supportive care for comorbidities [41]. CRIMnegative status currently means patients may have limited benefit from this expensive treatment, raising a difficult ethical dilemma: whether to withhold treatment from these patients. The treatment of CRIM-negative patients with ERT is therefore challenging; however, efforts are currently underway to develop tolerance-inducing therapies, which may offer a treatment path for such patients. This study also challenges our understanding of the immune system’s role in resistance to ERT and heightens the need for immunomodulation to improve ERT.
Acknowledgments
The authors would like to thank the patients, their families, and the health care providers who participated in the clinical studies of rhGAA whose outcomes are summarized here. We would like to thank the GCRC staff at Duke University for the excellent care provided to patients at the Duke site. The clinical trials were supported by Genzyme Corporation and Grant 1UL1RR024128 from the Duke Clinical Research Unit Program, National Center for Research Resources, and the National Institutes of Health. Dr. Deya Corzo of Genzyme Corporation provided input and critical review of the paper. Katherine Lewis of Genzyme Corporation provided editorial assistance. The original clinical studies from which patients were culled for this analysis were sponsored by Synpac, Inc. (Durham, NC, USA) and Genzyme Corporation (Cambridge, MA, USA). We thank Susan Richards and her clinical science laboratory at Genzyme Corporation for providing antibody analyses. All data included in this paper were verified and analyzed independently by the authors. P.S. Kishnani had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Trial Registration. NCT00025896, NCT00074919, NCT00059280, NCT00125879, NCT00053573.
Footnotes
Disclosures
P Kishnani, J Li, D Bali, S Young, D Koeberl, and H Mandel have received research/grant support from Genzyme Corporation. P Kishnani, S DeArmey, J Li, D Bali, and YT Chen have received honoraria from Genzyme Corporation. P Goldenberg received a fellowship grant from Genzyme Corporation. P Kishnani is a member of the Pompe and Gaucher Registry Boards of Advisors.
rhGAA, in the form of Genzyme’s product, Myozyme, has been approved by the US FDA and the European Union as therapy for Pompe disease. Duke University and inventors for the method of treatment and predecessors of the cell lines used to generate the enzyme (rhGAA) used in this clinical trial receive royalty payments pursuant to the University’s Policy on Inventions, Patents and Technology Transfer.
References
- 1.Kishnani PS, Hwu WL, Mandel H, Nicolino M, Yong F, Corzo D. A retrospective, multinational, multicenter study on the natural history of infantile-onset Pompe disease. J Pediatr. 2006;148:671–676. doi: 10.1016/j.jpeds.2005.11.033. [DOI] [PubMed] [Google Scholar]
- 2.van den Hout HM, Hop W, van Diggelen OP, Smeitink JA, Smit GP, Poll-The BT, Bakker HD, Loonen MC, de Klerk JB, Reuser AJ, van der Ploeg AT. The natural course of infantile Pompe’s disease: 20 original cases compared with 133 cases from the literature. Pediatrics. 2003;112:332–340. doi: 10.1542/peds.112.2.332. [DOI] [PubMed] [Google Scholar]
- 3.Ausems MG, Verbiest J, Hermans MP, Kroos MA, Beemer FA, Wokke JH, Sandkuijl LA, Reuser AJ, van der Ploeg AT. Frequency of glycogen storage disease type II in The Netherlands: implications for diagnosis and genetic counselling. Eur J Hum Genet. 1999;7:713–716. doi: 10.1038/sj.ejhg.5200367. [DOI] [PubMed] [Google Scholar]
- 4.Chien YH, Chiang SC, Zhang XK, Keutzer J, Lee NC, Huang AC, Chen CA, Wu MH, Huang PH, Tsai FJ, Chen YT, Hwu WL. Early detection of Pompe Disease by newborn screening is feasible: results from the Taiwan screening program. Pediatrics. 2008 doi: 10.1542/peds.2007-2222. [DOI] [PubMed] [Google Scholar]
- 5.Fuller M, Van der Ploeg A, Reuser AJ, Anson DS, Hopwood JJ. Isolation and characterisation of a recombinant, precursor form of lysosomal acid alpha-glucosidase. Eur J Biochem. 1995;234:903–909. doi: 10.1111/j.1432-1033.1995.903_a.x. [DOI] [PubMed] [Google Scholar]
- 6.Van Hove JL, Yang HW, Wu JY, Brady RO, Chen YT. High-level production of recombinant human lysosomal acid alpha-glucosidase in Chinese hamster ovary cells which targets to heart muscle and corrects glycogen accumulation in fibroblasts from patients with Pompe disease. Proc Natl Acad Sci USA. 1996;93:65–70. doi: 10.1073/pnas.93.1.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Van den Hout H, Reuser AJ, Vulto AG, Loonen MC, Cromme-Dijkhuis A, Van der Ploeg AT. Recombinant human alpha-glucosidase from rabbit milk in Pompe patients. Lancet. 2000;356:397–398. doi: 10.1016/s0140-6736(00)02533-2. [DOI] [PubMed] [Google Scholar]
- 8.Amalfitano A, Bengur AR, Morse RP, Majure JM, Case LE, Veerling DL, Mackey J, Kishnani P, Smith W, McVie-Wylie A, Sullivan JA, Hoganson GE, Phillips JA, 3rd, Schaefer GB, Charrow J, Ware RE, Bossen EH, Chen YT. Recombinant human acid alpha-glucosidase enzyme therapy for infantile glycogen storage disease type II: results of a phase I/II clinical trial. Genet Med. 2001;3:132–138. doi: 10.109700125817-200103000-00007. [DOI] [PubMed] [Google Scholar]
- 9.Kishnani PS, Nicolino M, Voit T, Rogers RC, Tsai AC, Waterson J, Herman GE, Amalfitano A, Thurberg BL, Richards S, Davison M, Corzo D, Chen YT. Chinese hamster ovary cell-derived recombinant human acid alpha-glucosidase in infantile-onset Pompe disease. J Pediatr. 2006;149:89–97. doi: 10.1016/j.jpeds.2006.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ansong AK, Li JS, Nozik-Grayck E, Ing R, Kravitz RM, Idriss SF, Kanter RJ, Rice H, Chen YT, Kishnani PS. Electrocardiographic response to enzyme replacement therapy for Pompe disease. Genet Med. 2006;8:297–301. doi: 10.1097/01.gim.0000195896.04069.5f. [DOI] [PubMed] [Google Scholar]
- 11.Kishnani PS, Corzo D, Nicolino M, Byrne B, Mandel H, Hwu WL, Leslie N, Levine J, Spencer C, McDonald M, Li J, Dumontier J, Halberthal M, Chien YH, Hopkin R, Vijayaraghavan S, Gruskin D, Bartholomew D, van der Ploeg A, Clancy JP, Parini R, Morin G, Beck M, De la Gastine GS, Jokic M, Thurberg B, Richards S, Bali D, Davison M, Worden MA, Chen YT, Wraith JE. Recombinant human acid [alpha]-glucosidase: major clinical benefits in infantile-onset Pompe disease. Neurology. 2007;68:99–109. doi: 10.1212/01.wnl.0000251268.41188.04. [DOI] [PubMed] [Google Scholar]
- 12.Nicolino M, Byrne B, Wraith JE, Leslie N, Mandel H, Freyer DR, Arnold GL, Pivnick EK, Ottinger CJ, Robinson PH, Loo JC, Smitka M, Jardine P, Tato L, Chabrol B, McCandless S, Kimura S, Mehta L, Bali D, Skrinar A, Morgan C, Rangachari L, Corzo D, Kishnani PS. Clinical outcomes after long-term treatment with alglucosidase alfa in infants and children with advanced Pompe disease. Genet Med. 2009;11:210–219. doi: 10.1097/GIM.0b013e31819d0996. [DOI] [PubMed] [Google Scholar]
- 13.Hawes ML, Kennedy W, O’Callaghan MW, Thurberg BL. Differential muscular glycogen clearance after enzyme replacement therapy in a mouse model of Pompe disease. Mol Genet Metab. 2007;91:343– 351. doi: 10.1016/j.ymgme.2007.04.018. [DOI] [PubMed] [Google Scholar]
- 14.Van den Hout JM, Kamphoven JH, Winkel LP, Arts WF, De Klerk JB, Loonen MC, Vulto AG, Cromme-Dijkhuis A, Weisglas-Kuperus N, Hop W, Van Hirtum H, Van Diggelen OP, Boer M, Kroos MA, Van Doorn PA, Van der Voort E, Sibbles B, Van Corven EJ, Brakenhoff JP, Van Hove J, Smeitink JA, de Jong G, Reuser AJ, Van der Ploeg AT. Long-term intravenous treatment of Pompe disease with recombinant human alpha-glucosidase from milk. Pediatrics. 2004;113:e448–457. doi: 10.1542/peds.113.5.e448. [DOI] [PubMed] [Google Scholar]
- 15.Piper M. Motor assessment of the developing infant. Saunders; Philadelphia: 1994. [Google Scholar]
- 16.Klinge L, Straub V, Neudorf U, Schaper J, Bosbach T, Gorlinger K, Wallot M, Richards S, Voit T. Safety and efficacy of recombinant acid alpha-glucosidase (rhGAA) in patients with classical infantile Pompe disease: results of a phase II clinical trial. Neuromuscul Disord. 2005;15:24–31. doi: 10.1016/j.nmd.2004.10.009. [DOI] [PubMed] [Google Scholar]
- 17.An Y, Young SP, Hillman SL, Van Hove JL, Chen YT, Millington DS. Liquid chromatographic assay for a glucose tetrasaccharide, a putative biomarker for the diagnosis of Pompe disease. Anal Biochem. 2000;287:136–143. doi: 10.1006/abio.2000.4838. [DOI] [PubMed] [Google Scholar]
- 18.Young SP, Stevens RD, An Y, Chen YT, Millington DS. Analysis of a glucose tetrasaccharide elevated in Pompe disease by stable isotope dilution-electrospray ionization tandem mass spectrometry. Anal Biochem. 2003;316:175–180. doi: 10.1016/s0003-2697(03)00056-3. [DOI] [PubMed] [Google Scholar]
- 19.Kaplan EL, Meier P. Nonparametric estimation from incomplete observations. J Am Stat Assoc. 1958;53:457–481. [Google Scholar]
- 20.Vogel M, Staller W, Buhlmeyer K. Left ventricular myocardial mass determined by cross-sectional echocardiography in normal newborns, infants, and children. Pediatr Cardiol. 1991;12:143–149. doi: 10.1007/BF02238520. [DOI] [PubMed] [Google Scholar]
- 21.Raben N, Nagaraju K, Lee A, Lu N, Rivera Y, Jatkar T, Hopwood JJ, Plotz PH. Induction of tolerance to a recombinant human enzyme, acid alpha-glucosidase, in enzyme deficient knockout mice. Transgenic Res. 2003;12:171–178. doi: 10.1023/a:1022998010833. [DOI] [PubMed] [Google Scholar]
- 22.Sun B, Bird A, Young SP, Kishnani PS, Chen YT, Koeberl DD. Enhanced response to enzyme replacement therapy in Pompe disease after the induction of immune tolerance. Am J Hum Genet. 2007;81:1042–1049. doi: 10.1086/522236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mendelsohn NJ, Messinger YH, Rosenberg AS, Kishnani PS. Elimination of antibodies to recombinant enzyme in Pompe’s disease. N Engl J Med. 2009;360:194–195. doi: 10.1056/NEJMc0806809. [DOI] [PubMed] [Google Scholar]
- 24.Starzyk K, Richards S, Yee J, Smith SE, Kingma W. The long-term international safety experience of imiglucerase therapy for Gaucher disease. Mol Genet Metab. 2007;90:157–163. doi: 10.1016/j.ymgme.2006.09.003. [DOI] [PubMed] [Google Scholar]
- 25.Antonarakis SE, Rossiter JP, Young M, Horst J, de Moerloose P, Sommer SS, Ketterling RP, Kazazian HH, Jr, Negrier C, Vinciguerra C, Gitschier J, Goossens M, Girodon E, Ghanem N, Plassa F, Lavergne JM, Vidaud M, Costa JM, Laurian Y, Lin SW, Lin SR, Shen MC, Lillicrap D, Taylor SA, Windsor S, Valleix SV, Nafa K, Sultan Y, Delpech M, Vnencak-Jones CL, Phillips JA, 3rd, Ljung RC, Koumbarelis E, Gialeraki A, Mandalaki T, Jenkins PV, Collins PW, Pasi KJ, Goodeve A, Peake I, Preston FE, Schwartz M, Scheibel E, Ingerslev J, Cooper DN, Millar DS, Kakkar VV, Giannelli F, Naylor JA, Tizzano EF, Baiget M, Domenech M, Altisent C, Tusell J, Beneyto M, Lorenzo JI, Gaucher C, Mazurier C, Peerlinck K, Matthijs G, Cassiman JJ, Vermylen J, Mori PG, Acquila M, Caprino D, Inaba H. Factor VIII gene inversions in severe hemophilia A: results of an international consortium study. Blood. 1995;86:2206–2212. [PubMed] [Google Scholar]
- 26.Warrier I, Ewenstein BM, Koerper MA, Shapiro A, Key N, DiMichele D, Miller RT, Pasi J, Rivard GE, Sommer SS, Katz J, Bergmann F, Ljung R, Petrini P, Lusher JM. Factor IX inhibitors and anaphylaxis in hemophilia B. J Pediatr Hematol Oncol. 1997;19:23–27. doi: 10.1097/00043426-199701000-00003. [DOI] [PubMed] [Google Scholar]
- 27.Germain DP, Kaneski CR, Brady RO. Mutation analysis of the acid beta-glucosidase gene in a patient with type 3 Gaucher disease and neutralizing antibody to alglucerase. Mutat Res. 2001;483:89–94. doi: 10.1016/s0027-5107(01)00232-9. [DOI] [PubMed] [Google Scholar]
- 28.Linthorst GE, Hollak CE, Donker-Koopman WE, Strijland A, Aerts JM. Enzyme therapy for Fabry disease: neutralizing antibodies toward agalsidase alpha and beta. Kidney Int. 2004;66:1589–1595. doi: 10.1111/j.1523-1755.2004.00924.x. [DOI] [PubMed] [Google Scholar]
- 29.Wilcox WR, Banikazemi M, Guffon N, Waldek S, Lee P, Linthorst GE, Desnick RJ, Germain DP. Long-term safety and efficacy of enzyme replacement therapy for Fabry disease. Am J Hum Genet. 2004;75:65–74. doi: 10.1086/422366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wraith JE, Beck M, Lane R, van der Ploeg A, Shapiro E, Xue Y, Kakkis ED, Guffon N. Enzyme replacement therapy in patients who have mucopolysaccharidosis I and are younger than 5 years: results of a multinational study of recombinant human alpha-L-iduronidase (laronidase) Pediatrics. 2007;120:37–46. doi: 10.1542/peds.2006-2156. [DOI] [PubMed] [Google Scholar]
- 31.Oldenburg J, Pavlova A. Genetic risk factors for inhibitors to factors VIII and IX. Haemophilia. 2006;12(Suppl 6):15–22. doi: 10.1111/j.1365-2516.2006.01361.x. [DOI] [PubMed] [Google Scholar]
- 32.Kakkis E, Lester T, Yang R, Tanaka C, Anand V, Lemontt J, Peinovich M, Passage M. Successful induction of immune tolerance to enzyme replacement therapy in canine mucopolysaccharidosis I. Proc Natl Acad Sci USA. 2004;101:829–834. doi: 10.1073/pnas.0305480101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Winsor-Hines D, Merrill C, O’Mahony M, Rao PE, Cobbold SP, Waldmann H, Ringler DJ, Ponath PD. Induction of immunological tolerance/hyporesponsiveness in baboons with a nondepleting CD4 antibody. J Immunol. 2004;173:4715–4723. doi: 10.4049/jimmunol.173.7.4715. [DOI] [PubMed] [Google Scholar]
- 34.Garman RD, Munroe K, Richards SM. Methotrexate reduces antibody responses to recombinant human alpha-galactosidase A therapy in a mouse model of Fabry disease. Clin Exp Immunol. 2004;137:496–502. doi: 10.1111/j.1365-2249.2004.02567.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wraith JE, Clarke LA, Beck M, Kolodny EH, Pastores GM, Muenzer J, Rapoport DM, Berger KI, Swiedler SJ, Kakkis ED, Braakman T, Chadbourne E, Walton-Bowen K, Cox GF. Enzyme replacement therapy for mucopolysaccharidosis I: a randomized, double-blinded, placebo-controlled, multinational study of recombinant human alpha-L-iduronidase (laronidase) J Pediatr. 2004;144:581–588. doi: 10.1016/j.jpeds.2004.01.046. [DOI] [PubMed] [Google Scholar]
- 36.Chemnitz J, Draube A, Scheid C, Staib P, Schulz A, Diehl V, Sohngen D. Successful treatment of severe thrombotic thrombocytopenic purpura with the monoclonal antibody rituximab. Am J Hematol. 2002;71:105–108. doi: 10.1002/ajh.10204. [DOI] [PubMed] [Google Scholar]
- 37.Darabi K, Berg AH. Rituximab can be combined with daily plasma exchange to achieve effective B-cell depletion and clinical improvement in acute autoimmune TTP. Am J Clin Pathol. 2006;125:592–597. doi: 10.1309/RLNM-J01W-BJRN-LH03. [DOI] [PubMed] [Google Scholar]
- 38.Joseph A, Munroe K, Housman M, Garman R, Richards S. Immune tolerance induction to enzyme-replacement therapy by co-administration of short-term, low-dose methotrexate in a murine Pompe disease model. Clin Exp Immunol. 2008;152:138–146. doi: 10.1111/j.1365-2249.2008.03602.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hunley TE, Corzo D, Dudek M, Kishnani P, Amalfitano A, Chen YT, Richards SM, Phillips JA, 3rd, Fogo AB, Tiller GE. Nephrotic syndrome complicating alpha-glucosidase replacement therapy for Pompe disease. Pediatrics. 2004;114:e532–535. doi: 10.1542/peds.2003-0988-L. [DOI] [PubMed] [Google Scholar]
- 40.Zhang H, Kallwass H, Young SP, Carr C, Dai J, Kishnani PS, Millington DS, Keutzer J, Chen YT, Bali D. Comparison of maltose and acarbose as inhibitors of maltase-glucoamylase activity in assaying acid alpha-glucosidase activity in dried blood spots for the diagnosis of infantile Pompe disease. Genet Med. 2006;8:302–306. doi: 10.1097/01.gim.0000217781.66786.9b. [DOI] [PubMed] [Google Scholar]
- 41.Kishnani PS, Steiner RD, Bali D, Berger K, Byrne BJ, Case LE, Crowley JF, Downs S, Howell RR, Kravitz RM, Mackey J, Marsden D, Martins AM, Millington DS, Nicolino M, O’Grady G, Patterson MC, Rapoport DM, Slonim A, Spencer CT, Tifft CJ, Watson MS. Pompe disease diagnosis and management guideline. Genet Med. 2006;8:267–288. doi: 10.1097/01.gim.0000218152.87434.f3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kroos M, Pomponio RJ, Vliet LV, Palmer RE, Phipps M, Helm R, Halley D, Reuser A. Update of the Pompe Disease mutation database with 107 sequence variants and a format for severity rating. Hum Mutat. 2008;29:E13–E26. doi: 10.1002/humu.20745. [DOI] [PubMed] [Google Scholar]