

Abstract
Background
Cell motility is essential for embryonic development and physiological processes such as the immune response, but also contributes to pathological conditions such as tumor progression and inflammation. However, our understanding of the mechanisms underlying migratory processes is incomplete. Drosophila border cells provide a powerful genetic model to identify the roles of genes that contribute to cell migration.
Results
Members of the Hedgehog signaling pathway were uncovered in two independent screens for interactions with the small GTPase Rac and the polarity protein Par-1 in border cell migration. Consistent with a role in migration, multiple Hh signaling components were enriched in the migratory border cells. Interference with Hh signaling by several different methods resulted in incomplete cell migration. Moreover, the polarized distribution of E-Cadherin and a marker of tyrosine kinase activity were altered when Hh signaling was disrupted. Conservation of Hh-Rac and Hh-Par-1 signaling was illustrated in the wing, in which Hh-dependent phenotypes were enhanced by loss of Rac or par-1.
Conclusions
We identified a pathway by which Hh signaling connects to Rac and Par-1 in cell migration. These results further highlight the importance of modifier screens in the identification of new genes that function in developmental pathways.
Keywords: cell migration, border cells, Drosophila, Hedgehog, Par-1, Rac
Introduction
Cell migration is integral to normal processes such as the formation and remodeling of tissues during development. Because misregulated migration contributes to birth defects and cancer, gaining a better understanding of the mechanisms that control normal migration is vital. While more is known about single cell motility, cells frequently migrate in small to large collectives throughout development and in tumor metastasis (Friedl and Gilmour, 2009; Friedl et al., 2012). Our knowledge of how individual cells receive and integrate outside signals to coordinate group level migration within their native tissue environment is still incomplete. Studies using the Drosophila border cell model have provided recent insight into the cellular and molecular mechanisms governing in vivo collective cell migration (Yilmaz and Christofori, 2010; He et al., 2011). Border cells are a specialized group of cells that migrate during ovarian development (He et al., 2011; Montell et al., 2012). The Drosophila ovary is composed of strings of progressively developing egg chambers, each of which contains an oocyte and 15 supportive nurse cells surrounded by a monolayer follicle cell epithelium (Spradling, 1993; He et al., 2011). Border cells are specified in the epithelium at early stage 9 of oogenesis (Fig. 1A). A specialized pair of follicle cells at the anterior end of the egg chamber, the polar cells, recruit 4 to 8 additional cells surrounding the polar cells to become migratory border cells. Border cells detach from the epithelium as a cohesive cluster, migrate between the germline-derived nurse cells over a distance of ~150 μm and stop at the oocyte border by stage 10 (Fig. 1A).
Fig. 1. Overexpression of hh rescues RacN17 border cell migration defects.
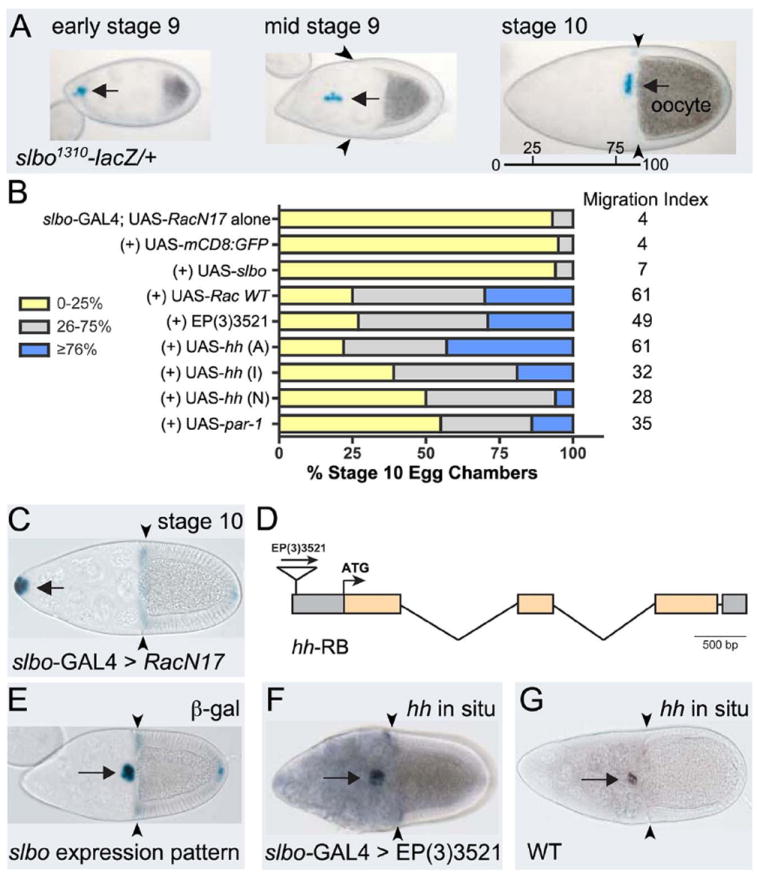
(A) slbo1310-lacZ/+ egg chambers at indicated stages stained for β-galactosidase activity (blue) to show border cells at beginning (left), during (middle) and end of migration when they reach the oocyte (right); migration distance indicated (bar). (B) Quantification of border cell migration at stage 10, with relative MI, following overexpression of indicated genes in a slbo-Gal4; UAS-RacN17 background (controls from (Geisbrecht and Montell, 2004)). Migration is shown as the percentage of border cells that migrated 0-25% (yellow), 26-75% (gray), or ≥76% (blue) of the distance to the oocyte. N > 100 egg chambers for each genotype. (C) Stage 10 slbo-Gal4; UAS-RacN17 egg chamber stained for slbo1310-lacZ; border cells did not migrate. (D) Diagram of the hh gene with coding exons (orange). EP(3)3521 is inserted 424 bp upstream of the ATG. (E) Stage 10 slbo1310/+ egg chamber stained to show slbo expression pattern (blue). (F, G) RNA in situ hybridization to detect hh expression at stage 10. (F) slbo-GAL4 driving EP(3)3521 shows increased hh expression in the slbo pattern. (G) Wild-type egg chamber. Arrows indicate border cells; arrowheads show extent of follicle cell rearrangement. Anterior to the left in this and subsequent figures.
The stereotypical migration of border cells is precisely regulated by the action of at least four known signaling pathways. Signaling through the JAK/STAT pathway specifies border cell identity and regulates border cell motility (Silver and Montell, 2001; Beccari et al., 2002; Ghiglione et al., 2002; Silver et al., 2005). In parallel, steroid hormone signaling through the Ecydsone receptor coordinates the timing of migration (Bai et al., 2000; Jang et al., 2009). JNK signaling promotes proper levels of cell adhesion between border cells to maintain the cluster during migration (Llense and Martin-Blanco, 2008; Melani et al., 2008). Finally, multiple growth factor ligands secreted by the oocyte activate two receptor tyrosine kinases (RTKs) expressed on border cells, Platelet Derived Growth Factor Receptor/Vascular Endothelial Growth Factor Receptor-related (PVR) and Epidermal Growth Factor Receptor (EGFR), to direct border cells to the oocyte (Duchek et al., 2001; McDonald et al., 2003; McDonald et al., 2006). A current challenge is to understand how all of these signals are integrated at the spatial-temporal level to promote border cell detachment, to maintain cluster cohesiveness, and to establish directional guidance.
Like most migrating cells, border cells require the small GTPase Rac for movement. Rac regulates the actin cytoskeleton to promote cellular protrusions at the leading edge of single cells or sheets of cells. These protrusions allow cells to grip their migratory substrate and/or enable cells to sense directional signals in the environment for navigation within tissues. In border cells, RTK signaling promotes the formation of actin-rich cellular protrusions at the leading edge of the migrating cluster through a localized increase in the activation of Rac (Bianco et al., 2007; Prasad and Montell, 2007; Wang et al., 2010). Moreover, activation of Rac in one border cell is sufficient to polarize the cluster and direct their migration (Wang et al., 2010). However, the mechanism by which asymmetric Rac activation is interpreted between cells in the cluster to promote organized forward movement is still unclear. To help elucidate these and other mechanisms of border cell migration, we performed a screen to identify genes required for Rac-dependent cell motility. We report here that hedgehog (hh) is a strong suppressor of dominant-negative Rac-induced migration defects.
Hh was originally identified in a mutant screen designed to identify genes required for Drosophila embryonic patterning (Nusslein-Volhard and Wieschaus, 1980). Homologs are found in many species and play vital roles in a wide range of developmental processes, including cell fate determination, patterning, and cell proliferation (Ingham et al., 2011). Hh is a secreted morphogen that is capable of both short- and long-range signaling. Because Hh is a potent signaling molecule, its production, secretion, movement, and reception are highly regulated. The binding of Hh to its receptor, Patched (Ptc), alleviates Ptc-dependent repression of the protein Smoothened (Smo) to activate downstream signaling events. Thus, Hh and Ptc act antagonistically and hh loss-of-function phenotypes are similar to those observed when ptc is overexpressed (Ingham et al., 2011).
The known transcriptional effects of Hh signaling are mediated by the zinc-finger transcription factor Cubitus interruptus (Ci). This protein is a member of the vertebrate Gli family of transcription factors that can either activate or repress target gene transcription (Huangfu and Anderson, 2006). Upon Hh stimulation, the full-length activated form of Ci, known as Ci-155, translocates to the nucleus to activate transcription of downstream targets such as ptc and decapentaplegic (dpp). However, in the absence of Hh signaling, the C-terminal region of Ci undergoes a series of phosphorylation events that inhibits Ci activity (Aza-Blanc et al., 1997). After initial phosphorylation by cAMP-dependent protein kinase (PKA), both glycogen synthase kinase 3 (GSK-3) and casein kinase 1 (CK1) are capable of phosphorylating Ci (Aza-Blanc et al., 1997; Price and Kalderon, 1999; Price and Kalderon, 2002). These sequential modifications result in protein cleavage to generate a short form called Ci-75 which when ectopically overexpressed represses Hh activity and target genes (Aza-Blanc et al., 1997). Thus, modulation of Hh signaling at the level of signal pathway initiation or the control of post-translational modifications of downstream components is important to maintain proper gene expression in a wide variety of tissues.
While Hh functions as a chemoattractant of migrating cells and axons (Lin et al., 2012), the role (if any) of hh and its vertebrate homologs in cell motility apart from chemotactic responses is less understood. Moreover, the extent to which Hh signaling is generally required for cell migration during Drosophila and/or vertebrate development is unclear. Among its many biological functions, Hh promotes follicle cell differentiation during Drosophila oogenesis (Forbes et al., 1996a; Forbes et al., 1996b). Mutations in the negative regulators ptc, cos-2 and the Pka catalytic subunit (Pka-C1) result in ectopic polar cells, which in some cases recruits cells to form additional border cell clusters (Liu and Montell, 1999; Zhang and Kalderon, 2000; Bai and Montell, 2002). Although a few of these extra border cell clusters complete their migration to the oocyte/nurse cell boundary, other clusters exhibit delayed migration. Whether this is due to a direct role for the Hh pathway in cell migration or is a consequence of altered cell fate remains to be determined. In this study, we present data that highlight a role for Hh signaling in border cell migration apart from border cell fate specification. Based upon the identification of Hh pathway components in two independent genetic screens, we propose that Hh signaling intersects with both Rac and the polarity protein Par-1 to mediate proper migration. Moreover, Hh may be required to maintain the subcellular localization of E-cadherin and polarized tyrosine phosphorylation, a marker of active tyrosine kinase signaling, to facilitate cell migration.
Results
Identification of hh in a Gain-of-Function Screen for Genes that Modify Rac-Dependent Border Cell Motility
Rac is a major regulator of border cell migration and promotes the actin-rich protrusions needed for their efficient collective motility (Murphy and Montell, 1996; Geisbrecht and Montell, 2004; Wang et al., 2010). However, relatively little is known about the genetic and molecular pathways that interact with Rac in this highly regulated process. We previously described a screen to identify genes that when overexpressed, modify the Rac-dependent cell migration defect (Geisbrecht and Montell, 2004). We report here the complete results of the screen and describe one candidate, hh, in more detail (Table 1; Fig. 1B-G). An inactive form of Rac (RacN17) was expressed in border cells using slow border cells- (slbo-) GAL4, which drives expression in all border cells of the cluster, but not the central polar cells, prior to and throughout their migration (Fig. 1A, 1E). Expression of UAS-RacN17 driven by slbo-GAL4 resulted in a complete block in border cell migration and significantly reduced female sterility (Fig. 1B, C) (Murphy and Montell, 1996; Geisbrecht and Montell, 2004). Therefore, we performed a screen to identify genes that when overexpressed have the ability to overcome this collective cell migration defect.
Table 1.
EP lines that suppress RacN17 border cell migration defects
| EP line | Map position | Rescue strength | Gene over-expressed | EST/cDNA | Orientationa/molecular informationb | Rescue RacV12c | Rescue RasV12c | Rescue slbo1310 c |
|---|---|---|---|---|---|---|---|---|
| EP(X)1451 | 11A1 | I/S | CG1806 (novel) | LP03706 | (+/+) -4300 bp of ATG | − | − | − |
| EP(X)1569 | 5C7 | S | actin5Cd, e | (+/+) -828 bp of ATG | − | − | + | |
| (X)1604 | 5C7 | S | actin5Cd, e | (+/+) -833 bp of ATG | − | − | + | |
| EP(2)2172 | 23B7 | S | CG3059 (NTPase) | LD11641 | (+/+) -6508 bp of ATG | − | − | − |
| EP(2)2604 | 49B10 | S | spt4 | LD44495 | (+/+) +84 bp of ATG | − | − | + |
| EP(2)2633 | 60C5-6 | S | CG3394 | GH22220 | (+/+) -163 bp of ATG | − | + | ND |
| EP(3)3203 | 98A14 | W | RpS10a | LD32148 | (+/+) -508 bp of ATG | +/- | − | − |
| (3)3597 | 98A14 | S | RpS10a | LD32148 | (+/+) -497 bp of ATG | − | ND | − |
| EP(3)3279 | 72D1 | S | diap1d, e | GH15335 | (+/+) -8737 bp of ATG | − | − | − |
| (3)3308 | 72D1 | I/S | diap1d, e | GH15335 | (+/+) -8154 bp of ATG | − | − | − |
| EP(3)3521 | 94E2 | S | hedgehogd | hh FL cDNAf | (+/+) -424 bp of ATG | − | − | − |
| EP(3)3556 | 66F1 | I | smaug | LD07551 | (+/+) -4237 bp of ATG | − | − | − |
Orientation of EP line with respect to coding strand (+/+ or +/-).
Distances calculated from genomic sequence.
EP lines were crossed to slbo-GAL4;UAS-RacV12, slbo-GAL4;UAS-RasV12 or slbo1310/slbo1310 to determine specificity of RacN17 suppression. The ability to suppress (+) or to not suppress (−) the respective border cell migration defects was determined.
Enhancer traps show expression in border cells: G(1)0177 (actin5C); thj5C8 (diap1); hh90E (hh).
Data from Geisbrecht and Montell (2004).
hh full length cDNA obtained from P. Beachy.
Abbreviations: S, strong; I, intermediate; W, weak; ND, not determined.
We crossed flies of the genotype slbo-GAL4; UAS-RacN17 to an available collection of 2273 randomly inserted EP (enhancer/promoter) “target” P-element lines designed to drive overexpression of genes located near the insertion site (Rorth et al., 1998; Geisbrecht and Montell, 2004). Ovaries from the progeny of misexpressed EP lines that significantly rescued fertility were dissected and immunostained to directly examine the extent of border cell migration in stage 10 egg chambers. We then determined the migration index (MI), which is calculated from the weighted average migration distance of each border cell cluster from the anterior end of the egg chamber to the nurse cell/oocyte border (Fig. 1A; 0 = no migration; 100 = complete migration). The MI allowed us to determine which EP lines significantly rescued the RacN17-mediated border cell migration defects (Fig. 1B; Table 1). We verified that expression of additional UAS constructs did not non-specifically titrate out the amount of GAL4 protein required to maintain expression of inactive Rac. Expression of UAS-mCD8:GFP or UAS-slbo resulted in a MI of 4 and 7, respectively, which was not significantly different from slbo-GAL4, UAS-RacN17 alone with a MI of 4 (Fig. 1B) (Geisbrecht and Montell, 2004).
Twenty-nine EP lines that exhibited a MI ≥ 20 were kept for further study. Of these, nine genes were confirmed to be overexpressed by the upstream EP-line in a GAL4-dependent manner (Table 1). All of these lines were located either upstream or immediately downstream of the predicted ATG in the correct orientation. The roles of two genes identified in this screen, Drosophila inihibitor of apoptosis 1 (diap1) and actin 5C, were previously described (Geisbrecht and Montell, 2004). To address specificity of the EP lines for rescue of RacN17 migration defects, we further tested their ability to rescue the border cell migration defects of other known mutants (Table 1). The overall levels and spatial activity of Rac are important for border cell migration; uniform expression of constitutively active Rac (RacV12) in all border cells completely inhibits their migration (Murphy and Montell, 1996; Duchek et al., 2001; Geisbrecht and Montell, 2004). Notably, however, none of the EP lines significantly suppressed RacV12. Constitutively active Ras (RasV12), a different GTPase independently involved in border cell migration, also strongly disrupts border cell migration (Lee et al., 1996). However, only one EP line (CG3394) rescued RasV12. Slbo is a C/EBP transcription factor that regulates the expression of genes required for border cell motility (Montell et al., 1992; Borghese et al., 2006; Wang et al., 2006). Similar to RacN17, slbo mutant border cells fail to migrate and remain at the anterior tip of the egg chamber (Montell et al., 1992). Only overexpression of the EP lines inserted into the actin5C and spt4 genes suppressed the slbo migration defects. Thus, while a few EP lines were able to rescue other border cell mutants, the majority of the EP lines identified in this screen specifically suppressed the RacN17 phenotype.
The strongest suppressor identified in the screen was EP(3)3521 (Fig. 1B), which rescued the RacN17 migration defects almost as effectively as the positive control, UAS-RacWT (Fig. 1B). This EP line is inserted in the 5’UTR of the hh gene (Fig. 1D). We performed in situ hybridizations to confirm that EP(3)3521 specifically overexpressed hh transcript in a slbo-GAL4 specific pattern (Fig. 1E-G). Moreover, expression of Hh using three different, independent UAS-hh transgenes rescued the RacN17 migration defects, although to varying degrees. The least level of suppression occurred with UAS-hh (N), a truncated active form of Hh (Porter et al., 1996) possibly because too much Hh signal was induced (see below). This Hh-mediated rescue of border cell migration was specific for RacN17, because overexpression of EP(3)3521/Hh did not suppress the RacV12, RasV12, or slbo mutant migration defects (Table 1).
Identification of ptc in a Screen for Genes that Interact with par-1 in border cell migration
Dominant genetic interaction screens often reveal components of genetic pathways or cellular processes that may be missed in other types of screens (St Johnston, 2002). We recently identified a critical role for the serine-threonine kinase and cell polarity protein Par-1 in border cell detachment and motility (McDonald et al., 2008). Heterozygotes of a strong loss-of-function allele of par-1, par-127C1, exhibit normal border cell migration (Table 2). However, par-127C1 exhibits strong border cell migration defects in transheterozygous combination with mutant alleles of several genes required for border cell migration (Liu and Montell, 1999; McDonald et al., 2003). We reasoned that other genes required for border cell migration, and potentially those that function with par-1, might dominantly interact with par-1 in a similar manner. Therefore, we performed an EMS mutagenesis screen of the second chromosome for alleles that caused border cell migration defects in heterozygous combination with par-127C1.
Table 2.
Complementation groups isolated in par-1 dominant interaction screen
| Gene | Allele(s) | par-127C1 interaction (% migration defects)a | Chromosome locationb | par-1 interaction with known allele | Synonyms/homologs | Role in border cell migration |
|---|---|---|---|---|---|---|
| par-127C1/+ (control) | – | 3 | – | – | – | – |
| taiman (tai; pdig1) | 4A2 | 27 | 30A2-30A6 | Yes (tai61G1) | DAIB1/AIB1 | Bai et al., 2001 |
| pdig2 | 11H1 | 11 | 35E1-35F7 | – | Unknown | – |
| pdig3 | 25G8 | 10 | 59B1-59B3c | – | Unknown | – |
| pdig4 (sec5)d | 30E5 | 19 | 23F2-3e | NDf | Sec5 | Assaker et al., 2010 |
| patched (ptc; pdig5) | 31A7 | 13 | 44D5-44E1 | Yes (ptc9) | Ptc1 | This study |
| shotgun (shg; pdig6) | 31D8 | 24 | 57B15-57B16 | Yes (shgR69) | DE-cadherin/E-cadherin | Niewiadomska et al., 1999 |
| 46G1 | 15 |
Dominant interaction with par-1 (pdig, +/+, par-127C1) scored as percentage of egg chambers in which border cells did not complete their migration to the oocyte by stage 10; n ≥ 100 egg chambers for each genotype. Alleles exhibited similar but milder interaction with par-1Δ16.
Chromosomal location based on lethal complementation with mutant alleles and/or overlapping deficiencies from the Bloomington Stock Center. In most cases the non-complementing deficiency exhibited a comparable interaction with par-1.
Second lethal mutation mapped to 40A5-40D3 (Df(2L)Exel6049), but par-1 did not interact with this deficiency.
Missense point mutation mapped to the sec5 open reading frame: Q393K (nucleotide 1178 C to A).
Based on lethality; par-1 had a milder interaction with Df(2L)BSC31, which removes this region (23E5;23F4-5).
ND, not determined.
Out of 2,198 chromosomes screened, six complementation groups (seven mutant alleles) disrupted border cell migration when crossed to par-127C1 (Table 2). These genes were termed pdig, for par-1 dominant interacting genes. Two pdig complementation groups were allelic to known border cell migration genes: the pdig1 allele disrupts taiman (tai) and the pdig6 group is allelic to the Drosophila E-cadherin gene shotgun (shg). Since tai and shg are both essential for border cell migration (Niewiadomska et al., 1999; Bai et al., 2000), the isolation of mutations in these two genes validated the screen approach. The four other pdig genes (pdig2, pdig3, pdig4 and pdig5) were subsequently mapped to specific chromosomal regions using the cytologically defined Bloomington deficiency kit (Table 2). A mutation in pdig4 maps to sec5, which encodes a member of the exocyst complex and has recently been implicated in border cell migration (Assaker et al., 2010). The other three pdig alleles map to regions that do not contain genes known to regulate border cells, suggesting we have identified mutations in new genes involved in border cell migration.
To identify the gene mutated by pdig531A7, the allele was mapped to the cytological region 44D5-E3 by complementation with overlapping deficiencies (Table 2). Available lethal mutant alleles for this region were tested for complementation with pdig531A7. Two strong loss of function alleles of the Hh receptor Ptc, ptc7 and ptc9, were lethal in combination with pdig5, suggesting that they are allelic. Moreover, par-127C1 exhibited comparable border cell migration defects in heterozygous combination with ptc9 (12% of egg chambers; n = 396). Therefore, pdig531A7 is an allele of ptc and we identified a genetic interaction between par-1 and ptc. Surprisingly, we also observed a genetic interaction between par-1 and a positive regulator of the Hh pathway, smo. In egg chambers heterozygous for both par-127C1 and smo3, 11% of border cells did not complete their migration (n = 254 egg chambers). We observed a weaker interaction with hh; 7% of egg chambers heterozygous for par-127C1 and hhAC had a border cell migration defect (n = 338 egg chambers). These results suggest that reduction of par-1 sensitizes border cells to changes in the overall levels of Hh signaling. Since Hh was identified as a suppressor of dominant-negative Rac, we also tested the ability of Par-1 to modify RacN17 phenotypes in this assay (Fig. 1B). Overexpression of par-1 suppressed Rac-mediated border cell migration defects, although to a lesser extent than Hh.
Members of the Hh Pathway are Expressed in Border Cells During Late Oogenesis
The identification of members of the Hh pathway in two independent genetic screens strongly suggested a role for Hh signaling in border cells. While the expression and function of the Hh pathway has been examined at earlier stages of oogenesis (Forbes et al., 1996a; Forbes et al., 1996b; Zhang and Kalderon, 2000; Zhang and Kalderon, 2001; Hartman et al., 2010), it has not been described during later oogenesis. Thus, we determined the expression pattern of key members of the Hh pathway during stages 8 to 10 of oogenesis when border cells are recruited and undergo migration. We first investigated the expression of a nuclear-lacZ enhancer trap in the hh gene locus, which has been shown to report the endogenous localization of hh in various tissues including the early ovary (Forbes et al., 1996a). hh-lacZ is expressed in the germarium (Forbes et al., 1996a) and then turns off until later oogenesis, when it specifically labels border cells but not central polar cells (Fig. 2A). We confirmed the expression of hh in border cells via in situ hybridization using an anti-sense probe (Fig. 1G).
Fig. 2. Expression of Hh pathway components in wild-type border cells.
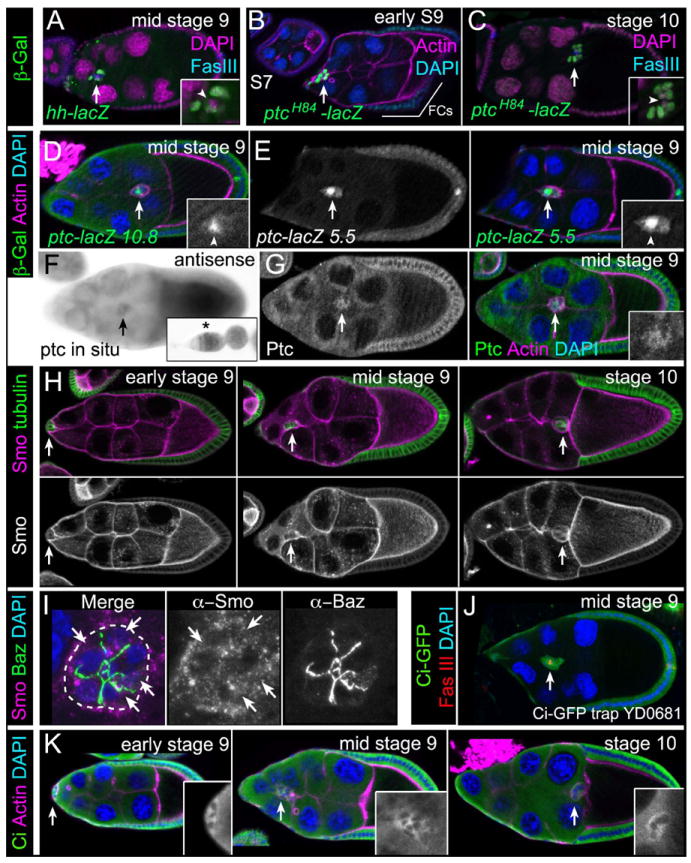
Representative images of egg chambers. Arrows indicate border cells. (A-E) Immunostaining for β-galactosidase (green) to detect hh-lacZ (A) or ptc-lacZ (B-E) enhancer trap (A-C) and promoter (D, E) reporters. Co-stains are Fas III (polar cells; blue in [A, C]), DAPI (nuclei; magenta in [A, C], blue in [B, D, E]) and F-actin (magenta [B, D, E]). Insets, magnified views of border cells (lacZ and DAPI in [A, C]); arrowheads, polar cells; FCs, follicle cells. (F) ptc mRNA expression. Inset, ptc expression in germarium. (G) Egg chamber immunostained for Ptc (green) and co-stained for F-actin (magenta). Inset, magnified view of border cells (Ptc). (H, I) Immunostaining for Smo (magenta). (H) Smo protein localization before, during, and after migration. α-Tubulin (green) labels all follicle cells. (I) Merged z-stack of border cells stained for Smo (magenta) and Bazooka (green). Smo localizes to cytoplasmic punctae (arrows); cluster outer border is outlined. (J, K) Ci protein (green) is present in border cells as detected by a GFP protein trap in ci (J) and using an antibody to Ci-FL protein (K; insets). Egg chambers were co-stained for Fas III (red [J]), F-actin (magenta [K]) and DAPI (blue).
We next determined the expression of the Hh receptor Ptc, which was assayed first with a nuclear-lacZ enhancer trap in the ptc gene. ptcH84-lacZ reflects ptc endogenous expression and reports Hh activity (Forbes et al., 1996b). ptcH84-lacZ was expressed at very low levels at stage 7 in all cells but was expressed at high levels in border cells starting when they form at late stage 8 (Fig. 2B). At stages 9 and 10, border cells had detectable ptcH84-lacZ with low expression in other follicle cells including the polar cells (Fig. 2B, 2C). These data were confirmed with two lacZ reporter constructs encompassing 10.8 kb (Fig. 2D) and 5.5 kb (Fig. 2E), respectively, of the ptc 5’ upstream regulatory region. These constructs are expressed in the normal ptc RNA and protein patterns in the wing disc (data not shown) (Chen and Struhl, 1996). ptc-lacZ 10.8 was highly expressed in polar cells and at low levels in border cells and follicle cells (Fig. 2D). ptc-lacZ 5.5 was expressed at similarly high levels in the polar cells but at higher levels in border cells and follicle cells beginning at late stage 8 and continuing through stage 10 (Fig. 2E). We observed slight differences in the expression levels of the ptc reporters in border cells, which could be due to insertion site variability and/or difference in the constructs themselves. Nonetheless, the observed patterns of ptc-lacZ are consistent with ptc expression turning on within border cells prior to and during their migration. We confirmed the ptc expression pattern using in situ hybridization; signal was observed early in the germarium, as previously reported (Forbes et al., 1996b), as well as in border cells (Fig. 2F). Ptc protein is normally found in intracellular vesicles and undergoes active trafficking within the cell (Zhu et al., 2003; Torroja et al., 2004). Using an antibody to Ptc, we detected uniform but low Ptc protein in cytoplasmic punctae in all follicle cells including border cells (Fig. 2G). Ptc protein was found in a broader pattern than that of ptc-lacZ, but agrees with previously published results and the RNA expression pattern (Fig. 2F) (Forbes et al., 1996b).
Next, we examined the localization of Smo, the obligate transducer of Hh signal (Fig. 2H, 2I). Smo protein was broadly expressed in egg chambers, with high levels in the germline and lower levels in follicle cells. When we examined the localization of Smo in border cells in further detail, we observed Smo in cytoplasmic-associated punctae within border cells (Fig 2I). This subcellular localization of Smo is consistent with its known association with intracellular vesicles in Hh signaling-responsive cells (Zhu et al., 2003; Torroja et al., 2004).
Finally, we analyzed the expression of the transcription factor Ci in later-staged egg chambers. A GFP protein trap in the first intron of the ci gene (Quinones-Coello et al., 2007) showed high expression in follicle cells and border cells (Fig. 2J). To confirm this expression, we used an antibody to detect full-length Ci (Ci-FL), which is the form of Ci that translocates to the nucleus and activates the Hh pathway (Aza-Blanc et al., 1997). Ci-FL undergoes shuttling between the cytoplasm and the nucleus and under normal antibody staining conditions appears cytoplasmic (Aza-Blanc et al., 1997; Hooper, 2003). Notably, high levels of Ci-FL were detected in the cytoplasm of follicle cells with lower levels in border cells (Fig. 2K), in agreement with a report by Sun and Deng (Sun and Deng, 2007).
STAT Regulates hh and ptc Expression in Border Cells
The transcription factor Stat92E regulates expression of multiple downstream targets specifically required for border cell migration, including slbo (Silver and Montell, 2001; Beccari et al., 2002). Since hh and ptc are both expressed at relatively high levels in border cells, we wanted to know whether JAK/STAT signaling was important for their expression. In egg chambers homozygous for a hypomorphic allele of unpaired (upd), the activating ligand for the JAK/STAT pathway, hh-lacZ expression was decreased in border cells compared to wild-type (Fig. 3A-B’). The expression of ptcH84-lacZ was also decreased in egg chambers mutant for a viable combination of stat alleles, stat397/stat3391 (Fig. 3C-D’). These data suggest that STAT signaling is necessary for hh and ptc expression in migratory border cells. In agreement with this idea, we observed genetic interactions between stat and members of the hh pathway (Fig. 3E). Consistent with previous results, loss of one copy of stat revealed mild border cell migration defects (Silver and Montell, 2001). Viable females that were trans-heterozygous for stat397 and a known interactor, slbo1310, showed border cell migration defects in approximately 30% of egg chambers (Fig. 3E) (Silver and Montell, 2001). Similarly, we observed genetic interactions between stat and mutations in several genes in the hh pathway, including Pka-C1, cos-2 and hh itself; the border cell migration defects ranged from 25-60% when these mutant alleles were trans-heterozygous with stat397. We did not observe a genetic interaction between smo and stat, which may indicate that this genetic combination is not dosage sensitive or the alleles chosen for this analysis did not reveal an interaction. No border cell migration defects were observed in egg chambers trans-heterozygous for hhAC and slbo1310. In addition, overexpression of hh was unable to suppress the slbo mutant phenotype (Table 1). Therefore, hh may be a slbo-independent downstream target of STAT.
Fig. 3. JAK/STAT signaling regulates expression of hh and ptc in border cells.
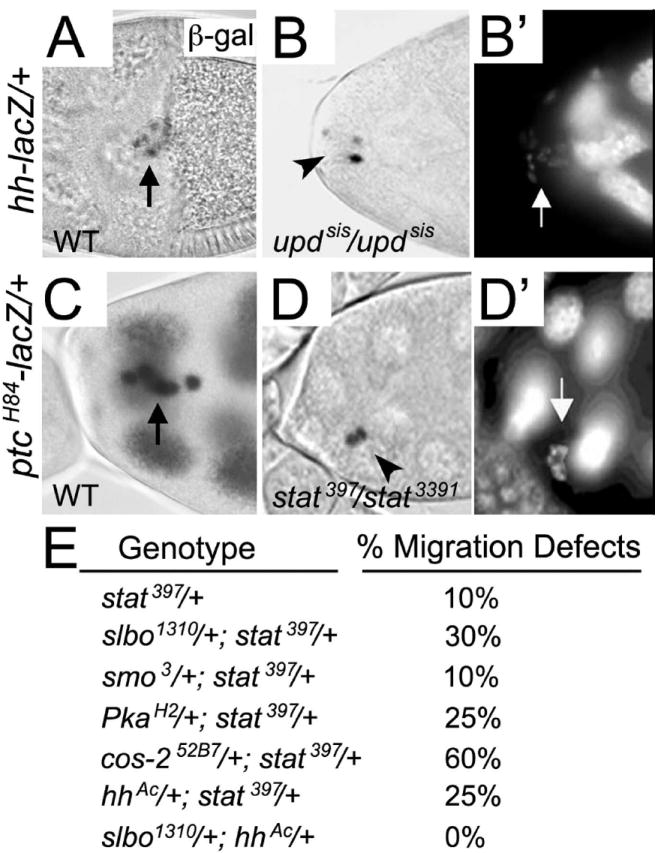
(A-D’) Stage 10 egg chambers stained for nuclear lacZ activity in the border cells (arrows) and counterstained with DAPI (B’, D’) to mark nuclei. (A) LacZ reporter activity is present in all border cells in hh-lacZ egg chambers. (B, B’) Homozygous updsis mutant egg chamber in which the border cell cluster remained at the anterior tip of the egg chamber. Few border cell nuclei show hh-lacZ expression (arrowhead in B) even though border cells are present (arrow in B’). (C) LacZ reporter activity is present in all border cells in ptc-lacZ egg chambers. This egg chamber was slightly overexposed and thus staining in the nurse cells is more apparent. (D, D’) stat397/stat3391 mutant egg chamber in which border cells failed to migrate (arrowhead in D). ptc-lacZ expression is only present in two border cells, but all cells are visible by nuclear DAPI staining (arrow in D’). (E) Table showing results of dominant stat genetic interaction tests, expressed as the percentage of border cell migration defects found in trans-heterozygous flies of the indicated genotypes; 42 ≤ n ≤ 190 egg chambers for each genotype.
Hh Signaling is Required for Efficient Border Cell Migration
The expression of Hh pathway members in border cells along with the observed genetic interactions with known border cell migration genes suggested that Hh signaling may regulate border cell migration. To test this directly, we employed multiple genetic approaches to knock down the function of multiple members of the Hh pathway in border cells. To test if the loss of Hh signaling proteins results in defective border cell migration, we obtained transgenic RNAi lines for smo, ci and hh that have been used previously to knock down the respective genes (Hartman et al., 2010; Su et al., 2011). Specific knockdown of these genes in all border cells using slbo-GAL4 caused migration defects in 10-22% of egg chambers (Fig. 4A-C). This is similar to the results achieved by knockdown of par-1 using RNAi with the same GAL4 driver (Fig. 4A) (McDonald et al., 2008). RNAi approaches can lead to incompletely penetrant phenotypes due to partial knockdown. Therefore, to confirm these results we took several complementary approaches to disrupt Hh activity. Expression of a smo mutant in which all PKA serine phosphorylation sites are mutated to alanines (smoPKA-SA) competes with endogenous Smo and thus reduces Hh signaling in vivo (Apionishev et al., 2005; Su et al., 2011). Expression of this phosphorylation-defective UAS-smoPKA-SA in border cells driven by slbo-GAL4 inhibited their migration to a similar extent as smo RNAi (Fig. 4A). Overexpression of wild-type ptc greatly reduces Hh signaling (Johnson et al., 1995). We found that overexpression of ptc B1 (wild-type ptc) in border cells disrupted migration in 32% of egg chambers (Fig. 4A, 4D). The stronger phenotypes caused by ptc overexpression compared to RNAi knockdown of ci, smo and hh likely reflects the uniformly high inhibition of Hh activity by ptc; RNAi, though useful, commonly results in variable efficiency of gene knockdown (Perrimon et al., 2010). Together, these results indicate that knockdown of the Hh pathway disrupts border cell migration.
Fig. 4. Migration defects upon loss of Hh pathway members in border cells.
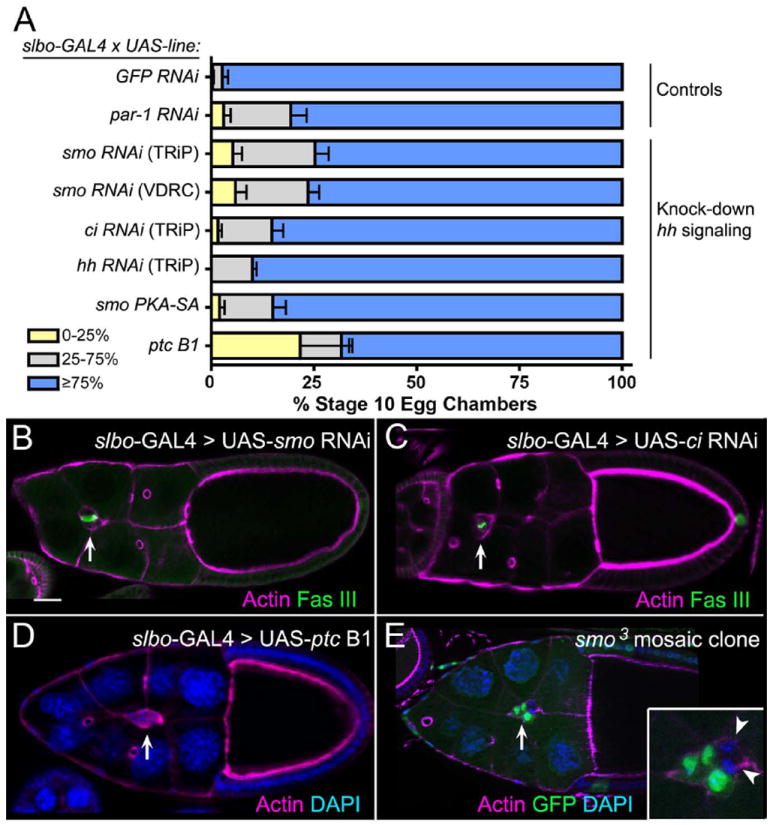
(A) Quantification of border cell migration at stage 10 upon knockdown of Hh pathway components using RNAi or overexpression approaches. Migration is shown as the percentage of border cells that migrated 0-25% (yellow), 26-75% (gray), or ≥76% (blue) of the distance to the oocyte. N ≥ 193 egg chambers for each genotype; at least 3 experiments were performed. Error bars represent standard error of the mean (SEM). (B-E) Fixed stage 10 egg chambers stained for phalloidin to mark F-actin (magenta) at cell membranes and DAPI (blue) to mark nuclei. GFP (green) marks wild-type cells in (E) and Fas III (green) marks polar cell membranes in (B, C). Arrows point to border cells. (B-D) Examples of egg chambers in which RNAi knockdown of smo (B) or ci (C) or in which ptc overexpression (D) is driven by slbo-GAL4; all show incomplete border cell migration. (E) Loss of smo by mosaic clonal analysis reveals a migration defect in a stage 10 egg chamber. Inset shows that two cells in the border cell cluster are mutant (GFP-negative; arrowheads). Scale bar in (B) represents 20 μm.
We next utilized available mutant alleles to confirm the role of Hh signaling components in border cell migration. We first tested the function of this pathway in border cells by mosaic clonal analysis using alleles of smo. We only obtained small clones with a strong loss-of-function allele, smo2. Therefore, we induced clones of the cold-sensitive smo allele, smo3. We incubated the flies at 25°C, which has been used previously to recover smo clones in the imaginal wing disc (Rodriguez and Basler, 1997). Border cells homozygous mutant for smo3 exhibited defective migration, although the phenotype was incompletely penetrant (Fig. 4E). Clusters with one to four smo3 mutant border cells exhibited migration defects in 28% of egg chambers (n = 43); border cells that did not reach the oocyte stalled between 50-75% of the normal migration distance (Fig. 4E). We did not observe completely mutant smo border cell clusters, possibly due to reduced proliferation of mutant cells in the clones (Duman-Scheel et al., 2002). Similar results were obtained when we analyzed additional Hh pathway members using mosaic clone analysis. Clusters that contained at least one border cell mutant for hh (hhAC allele), dispatched (disp; dispS037707 allele), which is required for release of Hh from signaling cells (Burke et al., 1999), or the transcription factor ci (ci94 allele) also exhibited migration defects (n ≥ 8 migration-defective egg chambers for each genotype). We note that the incompletely penetrant phenotypes suggest that Hh may function in parallel to one or more other pathways that regulate border cell migration.
We next examined the effects of increasing the levels of Ci, the downstream transcription factor for Hh signaling, in border cells. Overexpression of either wild-type ci, or an activated version (ci m1-4), severely disrupted the ability of border cells to complete their migration (Fig. 5). Most border cells expressing high levels of wild-type ci either did not migrate or only migrated a short distance away from the anterior tip of the egg chamber (Fig. 5A, 5C), in agreement with Sun and Deng (Sun and Deng, 2007). However, border cells overexpressing activated ci were more severely affected (Fig. 5B, 5C). These results suggest that having the correct spatial-temporal organization and/or level of Hh signaling is important for migratory border cells, similar to Rac, Slbo, Upd and other genes required for border cell migration (Rorth et al., 2000; Duchek et al., 2001; Silver and Montell, 2001). Alternatively, overexpression of Ci could disrupt the normal Hh signaling circuit in border cells or indirectly affect other signaling pathways, thus accounting for the observed migration defects.
Fig. 5. Overexpression of ci disrupts border cell migration.
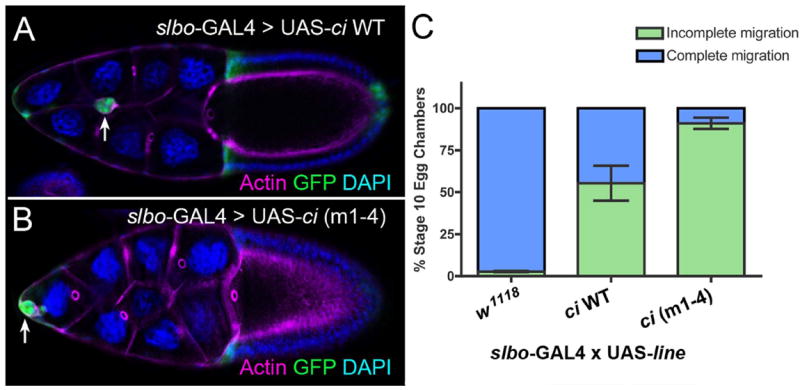
(A, B) Stage 10 egg chambers stained for phalloidin to mark F-actin (magenta) at cell membranes. GFP (green) marks the GAL4 expression pattern and DAPI (blue) marks the nuclei. Overexpression of WT ci (A) or an activated version of ci (B), ci (m1-4), driven by slbo-GAL4 results in a strong migration defect. (C) Quantification of stage 10 border cells that did not reach the oocyte (incomplete; green) versus those that reached the oocyte (complete; blue) upon overexpression of ci in the border cells. N ≥ 200 egg chambers for each genotype; at least 3 experiments were performed. Error bars represent SEM.
Loss of Hh Signaling Disrupts Distribution of E-cadherin and Phosphorylated Tyrosine
To determine which aspect of border cell migration was disrupted in the absence of Hh signaling, we analyzed several key markers of the cytoskeleton, border cell identity and membrane organization. We overexpressed ptc to consistently and maximally decrease the levels of Hh signaling in all border cells. Two markers of the cytoskeleton, α-tubulin to detect microtubules and phalloidin to detect F-actin, were each normal in border cells lacking Hh activation (Fig. 6A-D). We next analyzed the distribution of STAT in border cells to determine whether decreased Hh signaling affects STAT localization or protein levels. STAT localizes to the nucleus when STAT is active (Meyer and Vinkemeier, 2004) and is a marker of differentiated border cells (Silver et al., 2005). Both control and UAS-ptc border cells had similar levels of nuclear-localized STAT (Fig. 6E, 6F), consistent with Hh activity being downstream of STAT signaling (Fig. 3).
Fig. 6. Loss of Hh signaling affects Ecad and p-Tyr distribution within border cells.
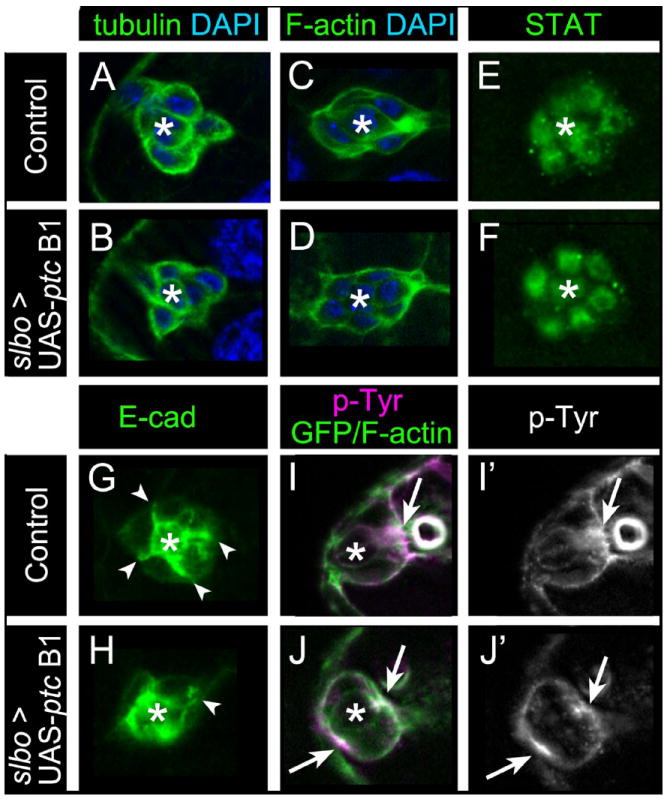
Representative images of stage 9 control slbo-GAL4/+ (A, C, E, G, I) or slbo-GAL4/UAS-ptc B1 (B, D, F, H, J) egg chambers stained for border cell markers; asterisks mark polar cells. (A, B) Border cells labeled for α-tubulin (green) to mark microtubules and DAPI (blue) to label nuclei. Microtubule organization is normal. (C, D) Border cells stained for phalloidin to mark F-actin (green) and DAPI (blue). F-actin localization is normal. (E, F) Border cell nuclei stained for STAT (green). Nuclear STAT is normal. (G, H) Border cells stained for E-cad (green). (G) In control, E-cad is enriched at membranes between border cells (arrowheads) and polar cells (asterisk). (H) ptc B1 cluster in which E-cad distribution is disrupted at border cell membranes; one cell has normal E-cad (arrowhead). (I-J’) Border cells prior to migration stained for p-Tyr (magenta [I, J]; white [I’, J’]) and F-actin/GFP (green in I, J). (I, I’) p-Tyr is enriched at the front of control border cell clusters (arrow). (J, J’) ptc B1 border cell cluster in which p-Tyr is enriched at the front and rear (arrows). N ≥ 10 egg chambers for each genotype.
In contrast, the distribution/levels of several membrane-associated markers were altered when hh signaling was disrupted. Wild-type border cells have enriched levels of the cell adhesion protein E-cadherin (E-cad) at the membrane interfaces between border cells and at polar cell membranes (Fig. 6G) (Niewiadomska et al., 1999; Bai et al., 2000). Overexpression of ptc disrupted the normally organized membrane E-cad localization; 64% of clusters had increased localization around the polar cells and uneven localization between border cells (Fig. 6H; n = 11). Finally, we examined the localization of phosphorylated tyrosine (p-Tyr), which has been used to analyze levels of tyrosine kinase activity, including the RTKs PVR and EGFR, in border cells (Jekely et al., 2005; Assaker et al., 2010). Wild-type border cells normally have membrane-enriched p-Tyr staining at the front of the cluster at stage 9 prior to their detachment from the follicular epithelium and subsequent migration (Fig. 6I, 6I’; 91%, n = 11 egg chambers). However, when ptc was overexpressed we observed aberrant p-Tyr staining localization (Fig. 6J, 6J’). Border cells overexpressing ptc had either membrane-enriched p-Tyr at both the front and back of the cluster (Fig. 6J, J’; 32%, n = 19 egg chambers) or p-Tyr was no longer enriched at either the front or back (16%). Thus, a marker of overall border cell identity and the organization of the cytoskeleton appear to be normal when Hh signaling is disrupted, but the localization of specific membrane-enriched proteins are altered.
Rac and par-1 Interact with hh in the Wing
To determine the extent to which the hh, Rac and par-1 pathways interact in other tissues, we chose to focus on the patterning of the adult Drosophila wing blade. The role of Hh signaling in this tissue is well-defined and genetic interactions are commonly used to ascertain whether mutants enhance or suppress pathway-specific wing phenotypes. Compared to wild-type wing blades (Fig. 7A), expression of ptc under control of the developing wing 71B-Gal4 driver at 25°C resulted in a reduction in wing blade size, a slight narrowing between the L3-L4 longitudinal veins and a fusion of the L3-L4 veins proximal to the anterior crossvein (ACV) (Fig. 7B). This is a mild ptc overexpression phenotype (Johnson et al., 1995). At higher temperatures (29°C), 71B-GAL4 confers a higher level of ptc overexpression (and disruption of Hh signal), resulting in a more severe phenotype (Fig. 7C) (Johnson et al., 1995). Removal of one copy of par-1 at 25°C enhances the mild veination phenotype, further reducing the space proximal to the ACV (Fig. 7D; compare to Fig. 7B). Furthermore, loss of one copy each of the three Drosophila Rac genes (Rac1, Rac2 and Mtl) upon ptc overexpression at 25°C abolished the ACV and partially deleted L3 (Fig. 7E, 7F), which closely resembles the 29°C ptc overexpression phenotype (Fig. 7C). These data demonstrate that both par-1 and Rac interact genetically with the Hh signaling pathway in the developing wing. This suggests that there is a conserved relationship amongst these pathways in the wing and ovary, although the precise biochemical functions may differ in the two tissues. Moreover, the observation that Rac mutations severely enhance the wing vein phenotypes associated with decreased Hh signaling is consistent with overexpression of Hh suppressing the RacN17 phenotypes in border cells.
Fig. 7. Loss of one copy of par-1 or the Rac genes enhances adult wing defects caused by reduced Hh signaling.
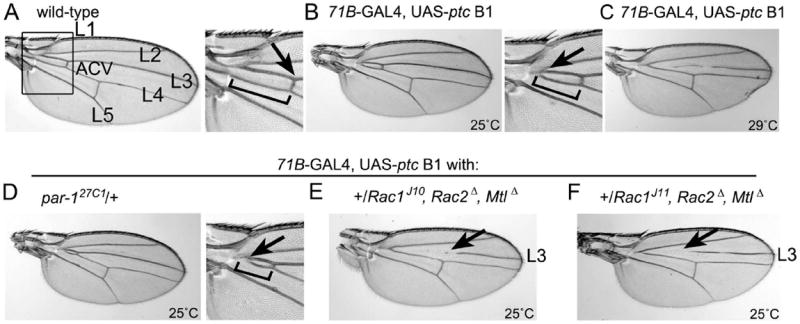
(A) Wild-type wing in which the longitudinal veins L1-L5 and anterior crossvein (ACV) are indicated (left panel). The boxed region is expanded (right panel) to show the normal length of veins L3-L4 (bracket) on the proximal side of the ACV (arrow) located between L3 and L4. (B) Reduced Hh signaling via overexpression of ptc at 25°C results in mild wing phenotypes (Johnson et al., 1995); reduced length of the L3-L4 longitudinal veins proximal to the ACV (bracket) and fusion of the proximal end (arrow) are indicated. (C) Severe wing vein phenotype caused by overexpression of ptc at 29°C. (D) Enhancement of the mild ptc overexpression vein fusion phenotype (arrow, bracket) upon loss of one copy of par-1 at 25°C. (E, F) Removal of one copy of all three Rac genes, using two different Rac1 alleles, at 25°C strongly enhances the mild ptc overexpression phenotype. The ACV and portions of the L3 longitudinal vein (arrows) are eliminated, which phenocopies severe ptc overexpression at 29°C (compare to [C]). Note that overexpression of ptc reduces overall wing size (Johnson et al., 1995).
Discussion
In this work, a role for the Hh signaling pathway in collective migration of the border cells was uncovered in two independent genetic screens. Previous genetic mosaic screens in border cells identified a role for cos2 in polar cell differentiation (Liu and Montell, 1999), but had yet to reveal a role for Hh signaling components in border cell migration. It has long been recognized that alternative screening methods are advantageous in uncovering genes that may be required earlier in development and/or for those genes with redundant functions. Both of these explanations are supported by published literature and the data presented here. First, ectopic Hh signaling, either by overexpression of hh itself or loss of the downstream components ptc, cos2, or Pka-C1, produces early ovarian phenotypes that include oocyte mis-positioning and excess polar cells (Forbes et al., 1996a; Liu and Montell, 1999; Zhang and Kalderon, 2000; Bai and Montell, 2002). These events occur prior to border cell recruitment and migration and thus may complicate analyses of Hh signaling in subsequent oogenic processes. We bypassed this potential issue by inducing downregulation of the Hh pathway specifically in border cells just prior to their migration. Second, the migration defects due to loss of Hh pathway components appear to be incompletely penetrant. Despite considerable reduction of Hh signaling due to overexpression of ptc, most border cells were able to complete their migration. However, the significant suppression of RacN17 motility defects by overexpression of Hh particularly indicates an important functional role for this pathway in border cells. Our data are thus consistent with other, at present unknown, signaling pathways functioning in concert with Hh for proper cell migration.
Function of Hh Signaling in Border Cell Migration
The Hh pathway is capable of regulating a wide variety of cellular responses through transcriptional regulation of downstream target genes (Ogden et al., 2004). In most tissues, Hh is secreted from a local source, but the downstream effects occur only in ptc-receiving cells that may reside up to ten cell diameters away (Ingham and McMahon, 2001). In migrating border cells, the results presented here suggest an autocrine mechanism where Hh is both produced and received by the same cells. Both the hh-lacZ and multiple ptc-lacZ enhancer traps/reporters reveal transcriptional activity in the outer, migratory cells of the cluster. Furthermore, we show that hh expression is regulated by JAK/STAT signaling and is independent of Slbo regulation (Fig. 8). It remains a distinct possibility that the Hh signal is relayed between border cells within the cluster. Nonetheless, our data favor a role for Hh specifically in border cells rather than receiving Hh signal from other cells in the ovary. This idea is supported by our findings that migration was impaired when Hh and proteins required for its signal reception and transduction were knocked down by RNAi selectively in the border cells using slbo-GAL4. Furthermore, border cells mutant for disp did not complete their migration. As Disp is required in Hh-secreting cells for release of lipid-modified active Hh (Burke et al., 1999) this further indicates that border cells produce Hh signal. It is still unclear whether paracrine versus autocrine Hh signaling is biologically important. However, a number of studies have reported roles for autocrine Hh activity in the Drosophila wing disc and optic primordium in the embryo and the salivary gland in the larva, as well as autocrine Sonic hedgehog (Shh), a vertebrate Hh homolog, in neural stem cells, B-cell lymphoma, and interferon-stimulated cerebellar dysplasia during brain development (Zhu et al., 2003; Wang et al., 2004; Cai et al., 2008; Biehs et al., 2010; Singh et al., 2010). This type of Hh signaling mechanism also occurs in a variety of human tumors, where abnormal Hh pathway activation in an autocrine fashion increases cell proliferation and invasion (Katoh and Katoh, 2005; Datta and Datta, 2006).
Fig. 8. Model for Hh signaling in border cells.
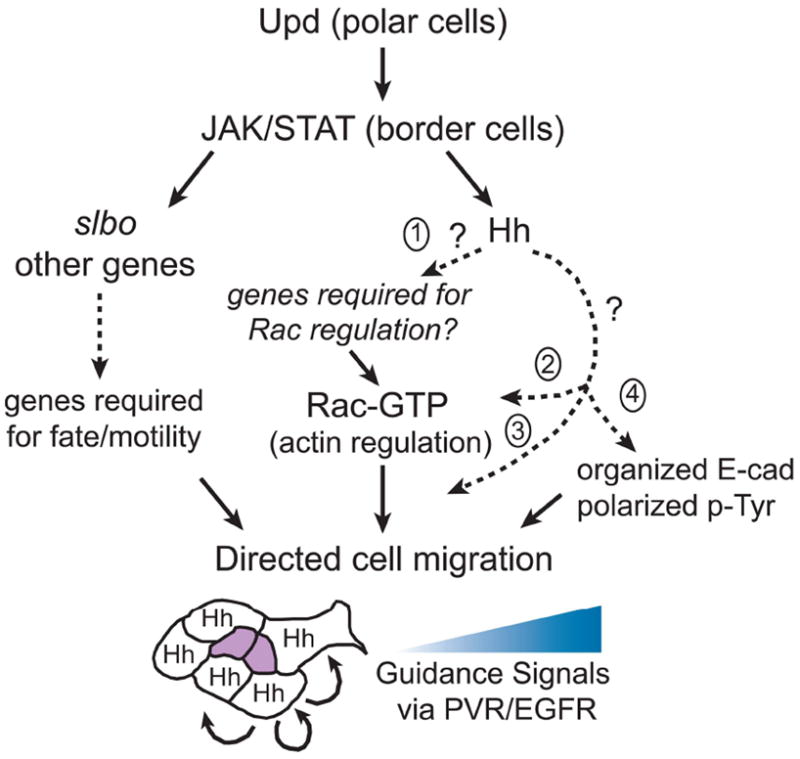
Upd signaling from the polar cells activates JAK/STAT in surrounding border cells (reviewed in Montell et al., 2012). JAK/STAT triggers expression of slbo and other genes required for border cell fate and motility. Hh is a downstream target of JAK/STAT but is independent of Slbo. Hh may be upstream (1) of active Rac (Rac-GTP), for example if it regulates the expression of genes required for Rac regulation such as a guanine-nucleotide exchange factor (GEF). Alternatively, the genetic data are consistent with Hh functioning with (2, 3), or in parallel to (4) Rac-GTP, possibly through Par-1 (for simplicity not shown). Hh could upregulate Rac levels in border cells (2), act downstream of Rac on presently unknown targets (3) and/or regulate E-cad/p-Tyr localization in parallel to Rac (4). Together, the border cell signaling pathways converge to direct collective border cell migration in response to a gradient of guidance signals secreted from the oocyte via the RTKs, PVR/EGFR. Hh is active in each border cell and likely received by the same cell, although autocrine signaling between cells in the cluster could also occur (arrows, border cell schematic). See text for additional details.
A question raised by our study is what role the Hh signaling pathway plays in border cell migration. As depicted in our proposed model (Fig. 8), border cells with reduced Hh activation exhibited altered localization of E-cad and depolarized p-Tyr, either of which could affect border cell motility. E-cad is required for border cell migration by promoting proper adhesion with the nurse cell substrate. Importantly, disruption of E-cad localization contributes to migration defects caused by loss of steroid hormone signaling and cell polarity genes (Niewiadomska et al., 1999; Bai et al., 2000; Pinheiro and Montell, 2004). Loss of one of the guidance ligands, pvf1, disrupts E-cad localization in border cells similar to what we observed when Hh activity was impaired (McDonald et al., 2003). Given that PVF1 signals to the receptor PVR on border cells, this suggests a connection between Hh and RTK signaling. Indeed, wild-type border cells exhibit polarized activation of the RTKs PVR and EGFR prior to migration as assayed by global tyrosine phosphorylation and specific phosphorylation of PVR (Tyr-1428) (Jekely et al., 2005; Janssens et al., 2010). Disruption of this polarized RTK by several mechanisms, as shown by reduced or mislocalized p-Tyr, impairs border cell migration (Jekely et al., 2005; Assaker et al., 2010; Quinones et al., 2010). Our data suggest that Hh signaling restricts polarized p-Tyr to the front of the border cell cluster. Interestingly, overexpression of vertebrate Shh in keratinocytes increased activation of EGFR and invasion through matrix (Bigelow et al., 2005). Moreover, there is evidence for synergism between Hh and EGFR signaling to activate Gli (Ci homolog) transcription targets in human cells (Kasper et al., 2006). Nonetheless, proteins other than RTKs (and/or their targets) can be phosphorylated on tyrosines and thus recognized by the p-Tyr antibody; thus, the role for Hh signaling in border cells may be independent of the RTK pathways. Regulators of endocytosis are also required for localized, high levels of p-Tyr in border cells (Jekely et al., 2005; Assaker et al., 2010). However, in contrast to loss of hh, loss of endocytic pathway members do not significantly impair E-cad levels or localization (Assaker et al., 2010). Thus, Hh likely regulates border cell migration by a distinct mechanism. Further experiments will be needed to determine if Hh signaling is required for the proper levels or distribution of an unknown protein(s) that affects tyrosine phosphorylation and E-cad localization during border cell migration.
Connection(s) between Rac, Par-1, and Hh Pathways
The data presented here suggest a link between the Rac, Hh, and Par-1 signaling pathways. However, our understanding of how these proteins function together to modulate migration remain a mystery. The results from our suppression screen indicate that overexpression of Hh can overcome Rac-dependent migration defects. In fact, Hh was the strongest suppressor obtained from our screen. A simple explanation for this observation is that Ci induces transcription of one or more as yet unknown downstream target genes required in the migratory process (Fig. 8). However, the entire repertoire of Ci targets has yet to be elucidated and specific targets (apart from ptc itself) in border cells are unknown. Interestingly, Rac2 was recently uncovered as a potential target of the Hh signaling pathway in the Drosophila embryo (Biehs et al., 2010). Both Rac1 and Rac2 are essential for border cell migration (Geisbrecht and Montell, 2004). Thus, it is intriguing to speculate that upregulation of Rac2 by Hh is a possible mechanism to overcome the RacN17 migration phenotype. Alternatively, the Hh pathway may directly or indirectly affect regulation of Rac protein activity either by increasing the amount of active Rac-GTP or by regulating the subcellular localization of activated Rac within the border cell cluster (Fig. 8). Hek 293 cells exposed to Shh had increased levels of the active form of the small GTPase RhoA (Kasai et al., 2004). Similarly, Shh stimulated RhoA and Rac via phosphoinositide 3-kinase (PI3K) signaling during chemotaxis of fibroblasts (Polizio et al., 2011). Because border cells do not rely on PI3K activity (Fulga and Rorth, 2002), another mechanism is likely involved.
What is the connection between Par-1 and the Hh signaling pathway? One possibility is the emerging requirement for microtubules in Hh signaling. Disruption of microtubules with the drug nocodazole prevents downstream transcriptional responses, possibly due to nuclear translocation of Gli proteins (Tukachinsky et al., 2010). Furthermore, Cos2-mediated subcellular motility and translocation of its cargo Ci requires microtubules in Drosophila (Farzan et al., 2008). Par-1 is a central player in mediating microtubule polymerization and dynamics (Hayashi et al., 2011). More specifically, phosphorylation of microtubule-associated proteins (MAPs) by the Par-1 kinase induces detachment of MAPs from microtubules. It is interesting to speculate that the kinase activity of Par-1 is essential in the Hh pathway to regulate Cos2 or MAP proteins for Ci mobility. Another possibility is that Par-1 functions through Rac. Overexpression of Par-1 partially suppressed the RacN17 migration defect, similar to overexpression of Hh. Although it is unclear why Par-1 rescued the Rac phenotype, it is possible that Par-1 acts in parallel to Hh signaling to promote Rac-mediated border cell motility. Notably, mammalian MARK2 (Par-1 homolog) promotes microtubule growth downstream of Rac1 at the leading edge of migrating cells (Nishimura et al., 2012). Further studies, however, are needed to determine the precise molecular relationships amongst the Rac, Hh and Par-1 pathways in collectively migrating border cells.
A Conserved Role for Hh in Other Morphogenic Processes
Cell shape changes are important for most aspects of morphogenic processes, including cell contractility and cell migration. Hh signaling induces cell shape changes in the developing Drosophila eye via regulation of non-muscle myosin II (Corrigall et al., 2007; Escudero et al., 2007). Thus, the role of Hh in border cells may be to regulate cell shape during migration. Accumulating evidence points to a non-canonical role for Hh in mediating mammalian cell migration. Shh can function as a chemoattractant in migrating cells and guidance of axons independent of Gli-induced gene transcription (Jenkins, 2009; Lin et al., 2012). In axon guidance, Shh stimulates phosphorylation and activation of Src kinase and thereby facilitates axon turning through regulation of the actin cytoskeleton (Yam et al., 2009). Specifically, Shh induced phosphorylation of Src at Tyr-418, an activating site, and polarization of Src family kinases within the axon. This appears to be consistent with our finding that loss of Hh activity depolarized global p-Tyr distribution. In other migratory cell types, Shh also acts as a chemoattractant that induces cytoskeletal rearrangements and migration independent of the canonical transcriptional response (Hochman et al., 2006; Bijlsma et al., 2007; Bijlsma et al., 2012). However, the mechanism of Hh function in border cell migration is likely to be different for several reasons. First, Hh is unlikely to function as a long-range chemoattractant, because border cells are the likely source of Hh signal (as discussed above). Second, a role for Src in border cell migration is unknown at present, so Hh may mediate tyrosine phosphorylation of other substrates in border cells. Third, we have evidence that Ci is involved in border cell migration and therefore canonical Hh-induced transcription is predicted to be important. Nonetheless, our results are consistent with a conserved role for the Hh pathway in regulating cytoskeletal-mediated events in migrating cells, which in border cells likely functions through the Rac GTPase.
Experimental Procedures
Drosophila Genetics and Mosaic Analysis
Fly culture and crosses were performed according to standard procedure at 25° C, except where indicated. The following mutant and transgenic fly stocks were used for the initial analysis of RacN17 overexpression and specificity controls: slbo-Gal4; UAS-RacN17 (Geisbrecht and Montell, 2004); UAS-mCD8-GFP (Bloomington Stock Center); UAS-slbo (Geisbrecht and Montell, 2004); UAS-RacWT (Bloomington Stock Center); UAS-RacV12 (Bloomington Stock Center); UAS-RasV12 (Lee et al., 1996). Additional lines utilized: slbo1310/CyO (slbo-lacZ; (Montell et al., 1992)); hh-lacZ/TM6B (Forbes et al., 1996a); ptc-lacZ reporter lines encompassing the upstream regulatory region of ptc: ptc-lacZ 5.5 and ptc-lacZ 10.8 (gifts of Joan Hooper); ptcH84lacZ/Cyo (P(A92)ptcH84 enhancer trap; (Ingham et al., 1991)); Ci-GFPYD0681 (Quinones-Coello et al., 2007); UAS-hh N (N-terminus active form of hh; (Porter et al., 1996)); UAS-hh A (full-length hh; (Porter et al., 1996)); UAS-hh I (full-length hh; (Ingham and Fietz, 1995)); updsisC5 (Sefton et al., 2000); stat3391 (Stat92EEP3391; (Silver and Montell, 2001)); UAS-ptc B1 (wild-type Ptc; (Johnson et al., 1995)); UAS-smoPKA-SA (Apionishev et al., 2005); UAS-ci WT (HA-tagged; (Chen et al., 1999)); UAS-ci (m1-4) (HA-tagged; (Chen et al., 1999)); par-127C1 (McDonald et al., 2008); par-1Δ16 (Cox et al., 2001); 71B-GAL4 (wing GAL4 driver; (Brand et al., 1994)); RacJ10, Rac2Δ, MtlΔ and RacJ11, Rac2Δ, MtlΔ (Bloomington Stock Center; (Hakeda-Suzuki et al., 2002)). Other alleles used for genetic interactions: stat397 (Silver and Montell, 2001); smo3 (from D. Kalderon; (Quirk et al., 1997)); Pka-C1H2 (Zhang and Kalderon, 2000); cos-252B7 (Liu and Montell, 1999); hhAC (null allele from P. Beachy; (Lee et al., 1992)); ptc7 (ptc9B; (Phillips et al., 1990)); ptc9 (ptcIN108; (Schuske et al., 1994)); tai61G1 (Bai et al., 2000); shgR69 (Godt and Tepass, 1998). w1118 was used as the wild-type strain.
UAS-RNAi lines were obtained from the VDRC and Bloomington Stock Center (TRiP Harvard collection): UAS-dsRNA to GFP (GFP RNAi); UAS-par-1 RNAi (McDonald et al., 2008); UAS-smo RNAi (VDRC line v9542); UAS-smo RNAi (TRiP line JF02363); UAS-ci RNAi (TRiP line JF01715); UAS-hh RNAi (TRiP line JF01804). GAL4-UAS overexpression and transgenic RNAi was performed after overnight incubation at 29°C. slbo-Gal4 (recombined with UAS-mCD8:GFP) was used throughout these studies (Rorth et al., 1998). slbo-GAL4, UAS-mCD8:GFP was outcrossed to w1118 and used as a control. The more widely-expressed c306-GAL4 (Murphy and Montell, 1996) produced similar phenotypes, but was not used further since its expression early in oogenesis could interfere with early Hh functions.
Mutant clones were generated by mitotic recombination using the FLP/FRT system using an X-chromosome hs-flp (Xu and Rubin, 1993). Adult flies of the correct genotype were heat shocked for 1 hour, twice a day at 37°C and incubated at 25°C to enable recovery of clones. Females were fattened and dissected at 25°C 4-7 days after the last heat shock. Clones were marked by loss of the ubi-nlsGFP marker. The following alleles were used: FRT82B, hhAC; smo3, FRT40A; and FRT82B, dispS037707. ci mutant clones (marked by loss of GFP) were generated by analyzing females of the genotype hs-flp; FRT42D P[ci+] hsp70-GFP/FRT42D; ci94/ci94 (Methot and Basler, 1999).
For the wing genetic interaction studies, 71B-GAL4 was recombined with UAS-ptc B1 and crossed to the relevant mutant alleles at 25°C. Female progeny were collected and analyzed as described (Johnson et al., 1995; Zhu et al., 2003). Wing phenotypes were 100% penetrant (n > 30 for each genotype).
Rac Modifier Screen
The slbo-GAL4; UAS-RacN17 stock was crossed to 2273 EP lines (Bloomington Stock Center) (Rorth et al., 1998). The progeny from these crosses were initially screened for rescue of female fertility and secondarily screened for the extent of border cell migration. Lines that exhibited a MI ≥ 20 were kept for further analysis and specificity tests as described in the Results section. Nine lines were confirmed to be in the correct orientation for gene induction using sequence annotation information available through FlyBase. In situ hybridization was performed for a subset of the candidates to confirm that the respective EP line driven by slbo-GAL4 overexpressed the downstream gene.
par-1 Dominant Genetic Interaction Screen
Mutant alleles that exhibited dominant genetic interaction in viable heterozygous combination with par-127C1 were identified as part of a previous mutagenesis screen for new par-1 alleles (McDonald et al., 2008). Briefly, isogenized dp, cn, bw males were fed 22 mM EMS (ethyl methanesulfonate; Sigma) in 1% sucrose for 24 hours (Bokel, 2008). Mutagenized males were allowed to recover for 24 hours and then crossed to virgin w; Sco/CyO females for 3-4 days. 2198 mutagenized mutant male dp, cn, bw, */Sco progeny (where the asterisk indicates the mutagenized chromosome) were crossed to virgin PZ6356, par-127C1/CyO females to test for genetic interaction. Female dp, cn, bw, */PZ6356, par-127C1 progeny were fattened overnight. Ovaries were dissected, fixed in 96-well plates and stained for β-galactosidase activity to detect PZ6356, an enhancer trap that labels border cells and the oocyte nucleus (Liu and Montell, 1999). Lines with a putative phenotype were identified first using a stereo microscope and then confirmed at higher magnification. F2 generation dp, cn, bw, */Sco males were backcrossed to virgin w; Sco/CyO females to establish a balanced stock. Inter se crosses were used to establish complementation groups. Once lines were rescreened and the phenotype confirmed, they were crossed to the cytologically defined Bloomington chromosome 2 deficiency kit to map the location of homozygous lethal mutations. Those deficiencies that were lethal in combination with the mutant allele were then crossed to par-127C1 to determine whether the deficiency had a comparable dominant interaction with par-1.
In Situ Hybridization, Immunofluorescence and Microscopy Analysis
Ovary dissections and in situ hybridizations were carried out as described in Wang, et al. (Wang et al., 2006) and S. Zimmerman, N. Peters, A. Altaras, and C. Berg (in preparation). Probes for in situ hybridization were made from hh and ptc cDNA clones (Porter et al., 1996; Su et al., 2011) by in vitro transcription; the ptc anti-sense probe was previously used to analyze ptc in the wing disc (Su et al., 2011).
Antibody staining was performed as described (McDonald and Montell, 2005). Fixation was generally performed in 4% formaldehyde/0.1 M potassium phosphate buffer, pH 7.4 for 10 min at room temperature; except fixation for anti-Ci and anti-Smo antibody staining was performed for 15 min on ice followed by 15 min at room temperature. Antibodies used were: rabbit anti-β-galactosidase at 1:2000 (Cappel); mouse anti-Fas III at 1:10 (7G10, Developmental Studies Hybridoma Bank, DSHB); concentrated rat anti-Ci at 1:150 (2A1; DSHB) (Motzny and Holmgren, 1995); mouse anti-Smo at 1:10 (20C6; DSHB) (Lum et al., 2003); concentrated mouse concentrated anti-Ptc at 1:600 (Apa1; DSHB) (Torroja et al., 2004); mouse anti-α-Tubulin at 1:800 (DM1A, Sigma); rat anti-E-Cadherin at 1:25 (DCAD2; DSHB) (Uemura et al., 1996); rabbit anti-STAT at 1:1000 (a gift from S. Hou); and mouse anti-phosphotyrosine at 1:1000 (p-Tyr-100; Cell Signaling Technology). Secondary antibodies or phalloidin (to label F-actin) conjugated to Alexa Fluor 488 or Alexa Fluor 568 (1:400; Invitrogen) were used. DAPI (0.05 μg/mL; Sigma) was used to stain nuclei. Samples were mounted on slides in Vectashield (Vector Labs) or Aqua-Poly/Mount (Polysciences, Inc.). Imaging was performed on a Zeiss AxioImager Z1 motorized epifluorescent microscope using the ApoTome System and a MRm CCD camera. Either a Plan-Apochromat 20x 0.75 numerical aperture (NA) objective or a Plan-Neofluar 40x 1.3 NA oil objective was used. Image adjustments (e.g. brightness, contrast and/or color) were performed using Photoshop CS4 or ImageJ. Figures were assembled in Adobe Photoshop CS4 or Illustrator CS4. GraphPad Prism 4 was used to compile graphs and calculate SEM.
Bullet points.
Two independent genetic screens uncover a role for Hh signaling in cell migration.
Overexpression of Hh rescues Rac-dependent cell migration defects.
Disruption of Hh signaling affects E-cadherin and phosphorylated-tyrosine localization in the migrating border cells.
The Rac, Par-1 and Hh signaling pathways intersect in wing vein development.
Acknowledgments
We would like to thank Lynn Cooley/FlyTrap, DSHB, NIG-Fly, Bloomington Stock Center, TRiP, VDRC, Daniel Kalderon, Phil Beachy, Celeste Berg, Joan Hooper and Denise Montell for kindly sharing flies, antibodies and protocols. Sean Karpen, Tina Bridges, Colleen Dudley, Jinyan Bo, Tanika Harris and Poornima Vanguri provided essential technical assistance at various stages of this study. Special thanks to Denise Montell in whose lab the genetic interaction screens and initial experiments were performed.
Grant Sponsor: National Institutes of Health; Grant Numbers: R01AR060788 (E.R.G.), R01GM085175 (A.J.Z.), R01GM078526 (J.A.M.); Grant Sponsor: March of Dimes Basil O’Connor Award; Grant Number: 5-FY07-41 (A.J.Z.); Grant Sponsor: American Heart Association; Grant Number: 12SDG8870002 (Y.S.)
References
- Apionishev S, Katanayeva NM, Marks SA, Kalderon D, Tomlinson A. Drosophila Smoothened phosphorylation sites essential for Hedgehog signal transduction. Nat Cell Biol. 2005;7:86–92. doi: 10.1038/ncb1210. [DOI] [PubMed] [Google Scholar]
- Assaker G, Ramel D, Wculek SK, Gonzalez-Gaitan M, Emery G. Spatial restriction of receptor tyrosine kinase activity through a polarized endocytic cycle controls border cell migration. Proc Natl Acad Sci U S A. 2010;107:22558–22563. doi: 10.1073/pnas.1010795108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aza-Blanc P, Ramirez-Weber FA, Laget MP, Schwartz C, Kornberg TB. Proteolysis that is inhibited by hedgehog targets Cubitus interruptus protein to the nucleus and converts it to a repressor. Cell. 1997;89:1043–1053. doi: 10.1016/s0092-8674(00)80292-5. [DOI] [PubMed] [Google Scholar]
- Bai J, Montell D. Eyes absent, a key repressor of polar cell fate during Drosophila oogenesis. Development. 2002;129:5377–5388. doi: 10.1242/dev.00115. [DOI] [PubMed] [Google Scholar]
- Bai J, Uehara Y, Montell DJ. Regulation of invasive cell behavior by taiman, a Drosophila protein related to AIB1, a steroid receptor coactivator amplified in breast cancer. Cell. 2000;103:1047–1058. doi: 10.1016/s0092-8674(00)00208-7. [DOI] [PubMed] [Google Scholar]
- Beccari S, Teixeira L, Rorth P. The JAK/STAT pathway is required for border cell migration during Drosophila oogenesis. Mech Dev. 2002;111:115–123. doi: 10.1016/s0925-4773(01)00615-3. [DOI] [PubMed] [Google Scholar]
- Bianco A, Poukkula M, Cliffe A, Mathieu J, Luque CM, Fulga TA, Rorth P. Two distinct modes of guidance signalling during collective migration of border cells. Nature. 2007;448:362–365. doi: 10.1038/nature05965. [DOI] [PubMed] [Google Scholar]
- Biehs B, Kechris K, Liu S, Kornberg TB. Hedgehog targets in the Drosophila embryo and the mechanisms that generate tissue-specific outputs of Hedgehog signaling. Development. 2010;137:3887–3898. doi: 10.1242/dev.055871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bigelow RL, Jen EY, Delehedde M, Chari NS, McDonnell TJ. Sonic hedgehog induces epidermal growth factor dependent matrix infiltration in HaCaT keratinocytes. J Invest Dermatol. 2005;124:457–465. doi: 10.1111/j.0022-202X.2004.23590.x. [DOI] [PubMed] [Google Scholar]
- Bijlsma MF, Borensztajn KS, Roelink H, Peppelenbosch MP, Spek CA. Sonic hedgehog induces transcription-independent cytoskeletal rearrangement and migration regulated by arachidonate metabolites. Cell Signal. 2007;19:2596–2604. doi: 10.1016/j.cellsig.2007.08.011. [DOI] [PubMed] [Google Scholar]
- Bijlsma MF, Damhofer H, Roelink H. Hedgehog-stimulated chemotaxis is mediated by smoothened located outside the primary cilium. Sci Signal. 2012;5:ra60. doi: 10.1126/scisignal.2002798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bokel C. EMS screens: from mutagenesis to screening and mapping. Methods Mol Biol. 2008;420:119–138. doi: 10.1007/978-1-59745-583-1_7. [DOI] [PubMed] [Google Scholar]
- Borghese L, Fletcher G, Mathieu J, Atzberger A, Eades WC, Cagan RL, Rorth P. Systematic analysis of the transcriptional switch inducing migration of border cells. Dev Cell. 2006;10:497–508. doi: 10.1016/j.devcel.2006.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand AH, Manoukian AS, Perrimon N. Ectopic expression in Drosophila. Methods Cell Biol. 1994;44:635–654. doi: 10.1016/s0091-679x(08)60936-x. [DOI] [PubMed] [Google Scholar]
- Burke R, Nellen D, Bellotto M, Hafen E, Senti KA, Dickson BJ, Basler K. Dispatched, a novel sterol-sensing domain protein dedicated to the release of cholesterol-modified hedgehog from signaling cells. Cell. 1999;99:803–815. doi: 10.1016/s0092-8674(00)81677-3. [DOI] [PubMed] [Google Scholar]
- Cai C, Thorne J, Grabel L. Hedgehog serves as a mitogen and survival factor during embryonic stem cell neurogenesis. Stem Cells. 2008;26:1097–1108. doi: 10.1634/stemcells.2007-0684. [DOI] [PubMed] [Google Scholar]
- Chen Y, Cardinaux JR, Goodman RH, Smolik SM. Mutants of cubitus interruptus that are independent of PKA regulation are independent of hedgehog signaling. Development. 1999;126:3607–3616. doi: 10.1242/dev.126.16.3607. [DOI] [PubMed] [Google Scholar]
- Chen Y, Struhl G. Dual roles for patched in sequestering and transducing Hedgehog. Cell. 1996;87:553–563. doi: 10.1016/s0092-8674(00)81374-4. [DOI] [PubMed] [Google Scholar]
- Corrigall D, Walther RF, Rodriguez L, Fichelson P, Pichaud F. Hedgehog signaling is a principal inducer of Myosin-II-driven cell ingression in Drosophila epithelia. Dev Cell. 2007;13:730–742. doi: 10.1016/j.devcel.2007.09.015. [DOI] [PubMed] [Google Scholar]
- Cox DN, Lu B, Sun TQ, Williams LT, Jan YN. Drosophila par-1 is required for oocyte differentiation and microtubule organization. Curr Biol. 2001;11:75–87. doi: 10.1016/s0960-9822(01)00027-6. [DOI] [PubMed] [Google Scholar]
- Datta S, Datta MW. Sonic Hedgehog signaling in advanced prostate cancer. Cell Mol Life Sci. 2006;63:435–448. doi: 10.1007/s00018-005-5389-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duchek P, Somogyi K, Jekely G, Beccari S, Rorth P. Guidance of cell migration by the Drosophila PDGF/VEGF receptor. Cell. 2001;107:17–26. doi: 10.1016/s0092-8674(01)00502-5. [DOI] [PubMed] [Google Scholar]
- Duman-Scheel M, Weng L, Xin S, Du W. Hedgehog regulates cell growth and proliferation by inducing Cyclin D and Cyclin E. Nature. 2002;417:299–304. doi: 10.1038/417299a. [DOI] [PubMed] [Google Scholar]
- Escudero LM, Bischoff M, Freeman M. Myosin II regulates complex cellular arrangement and epithelial architecture in Drosophila. Dev Cell. 2007;13:717–729. doi: 10.1016/j.devcel.2007.09.002. [DOI] [PubMed] [Google Scholar]
- Farzan SF, Ascano M, Jr, Ogden SK, Sanial M, Brigui A, Plessis A, Robbins DJ. Costal2 functions as a kinesin-like protein in the hedgehog signal transduction pathway. Curr Biol. 2008;18:1215–1220. doi: 10.1016/j.cub.2008.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forbes AJ, Lin H, Ingham PW, Spradling AC. hedgehog is required for the proliferation and specification of ovarian somatic cells prior to egg chamber formation in Drosophila. Development. 1996a;122:1125–1135. doi: 10.1242/dev.122.4.1125. [DOI] [PubMed] [Google Scholar]
- Forbes AJ, Spradling AC, Ingham PW, Lin H. The role of segment polarity genes during early oogenesis in Drosophila. Development. 1996b;122:3283–3294. doi: 10.1242/dev.122.10.3283. [DOI] [PubMed] [Google Scholar]
- Friedl P, Gilmour D. Collective cell migration in morphogenesis, regeneration and cancer. Nat Rev Mol Cell Biol. 2009;10:445–457. doi: 10.1038/nrm2720. [DOI] [PubMed] [Google Scholar]
- Friedl P, Locker J, Sahai E, Segall JE. Classifying collective cancer cell invasion. Nat Cell Biol. 2012;14:777–783. doi: 10.1038/ncb2548. [DOI] [PubMed] [Google Scholar]
- Fulga TA, Rorth P. Invasive cell migration is initiated by guided growth of long cellular extensions. Nat Cell Biol. 2002;4:715–719. doi: 10.1038/ncb848. [DOI] [PubMed] [Google Scholar]
- Geisbrecht ER, Montell DJ. A role for Drosophila IAP1-mediated caspase inhibition in Rac-dependent cell migration. Cell. 2004;118:111–125. doi: 10.1016/j.cell.2004.06.020. [DOI] [PubMed] [Google Scholar]
- Ghiglione C, Devergne O, Georgenthum E, Carballes F, Medioni C, Cerezo D, Noselli S. The Drosophila cytokine receptor Domeless controls border cell migration and epithelial polarization during oogenesis. Development. 2002;129:5437–5447. doi: 10.1242/dev.00116. [DOI] [PubMed] [Google Scholar]
- Godt D, Tepass U. Drosophila oocyte localization is mediated by differential cadherin-based adhesion. Nature. 1998;395:387–391. doi: 10.1038/26493. [DOI] [PubMed] [Google Scholar]
- Hakeda-Suzuki S, Ng J, Tzu J, Dietzl G, Sun Y, Harms M, Nardine T, Luo L, Dickson BJ. Rac function and regulation during Drosophila development. Nature. 2002;416:438–442. doi: 10.1038/416438a. [DOI] [PubMed] [Google Scholar]
- Hartman TR, Zinshteyn D, Schofield HK, Nicolas E, Okada A, O’Reilly AM. Drosophila Boi limits Hedgehog levels to suppress follicle stem cell proliferation. J Cell Biol. 2010;191:943–952. doi: 10.1083/jcb.201007142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi K, Suzuki A, Ohno S. A novel function of the cell polarity-regulating kinase PAR-1/MARK in dendritic spines. Bioarchitecture. 2011;1:261–266. doi: 10.4161/bioa.1.6.19199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He L, Wang X, Montell DJ. Shining light on Drosophila oogenesis: live imaging of egg development. Curr Opin Genet Dev. 2011;21:612–619. doi: 10.1016/j.gde.2011.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochman E, Castiel A, Jacob-Hirsch J, Amariglio N, Izraeli S. Molecular pathways regulating pro-migratory effects of Hedgehog signaling. J Biol Chem. 2006;281:33860–33870. doi: 10.1074/jbc.M605905200. [DOI] [PubMed] [Google Scholar]
- Hooper JE. Smoothened translates Hedgehog levels into distinct responses. Development. 2003;130:3951–3963. doi: 10.1242/dev.00594. [DOI] [PubMed] [Google Scholar]
- Huangfu D, Anderson KV. Signaling from Smo to Ci/Gli: conservation and divergence of Hedgehog pathways from Drosophila to vertebrates. Development. 2006;133:3–14. doi: 10.1242/dev.02169. [DOI] [PubMed] [Google Scholar]
- Ingham PW, Fietz MJ. Quantitative effects of hedgehog and decapentaplegic activity on the patterning of the Drosophila wing. Curr Biol. 1995;5:432–440. doi: 10.1016/s0960-9822(95)00084-4. [DOI] [PubMed] [Google Scholar]
- Ingham PW, McMahon AP. Hedgehog signaling in animal development: paradigms and principles. Genes Dev. 2001;15:3059–3087. doi: 10.1101/gad.938601. [DOI] [PubMed] [Google Scholar]
- Ingham PW, Nakano Y, Seger C. Mechanisms and functions of Hedgehog signalling across the metazoa. Nat Rev Genet. 2011;12:393–406. doi: 10.1038/nrg2984. [DOI] [PubMed] [Google Scholar]
- Ingham PW, Taylor AM, Nakano Y. Role of the Drosophila patched gene in positional signalling. Nature. 1991;353:184–187. doi: 10.1038/353184a0. [DOI] [PubMed] [Google Scholar]
- Jang AC, Chang YC, Bai J, Montell D. Border-cell migration requires integration of spatial and temporal signals by the BTB protein Abrupt. Nat Cell Biol. 2009;11:569–579. doi: 10.1038/ncb1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssens K, Sung HH, Rorth P. Direct detection of guidance receptor activity during border cell migration. Proc Natl Acad Sci U S A. 2010;107:7323–7328. doi: 10.1073/pnas.0915075107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jekely G, Sung HH, Luque CM, Rorth P. Regulators of endocytosis maintain localized receptor tyrosine kinase signaling in guided migration. Dev Cell. 2005;9:197–207. doi: 10.1016/j.devcel.2005.06.004. [DOI] [PubMed] [Google Scholar]
- Jenkins D. Hedgehog signalling: emerging evidence for non-canonical pathways. Cell Signal. 2009;21:1023–1034. doi: 10.1016/j.cellsig.2009.01.033. [DOI] [PubMed] [Google Scholar]
- Johnson RL, Grenier JK, Scott MP. patched overexpression alters wing disc size and pattern: transcriptional and post-transcriptional effects on hedgehog targets. Development. 1995;121:4161–4170. doi: 10.1242/dev.121.12.4161. [DOI] [PubMed] [Google Scholar]
- Kasai K, Takahashi M, Osumi N, Sinnarajah S, Takeo T, Ikeda H, Kehrl JH, Itoh G, Arnheiter H. The G12 family of heterotrimeric G proteins and Rho GTPase mediate Sonic hedgehog signalling. Genes Cells. 2004;9:49–58. doi: 10.1111/j.1356-9597.2004.00701.x. [DOI] [PubMed] [Google Scholar]
- Kasper M, Schnidar H, Neill GW, Hanneder M, Klingler S, Blaas L, Schmid C, Hauser-Kronberger C, Regl G, Philpott MP, Aberger F. Selective modulation of Hedgehog/GLI target gene expression by epidermal growth factor signaling in human keratinocytes. Mol Cell Biol. 2006;26:6283–6298. doi: 10.1128/MCB.02317-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh Y, Katoh M. Hedgehog signaling pathway and gastric cancer. Cancer Biol Ther. 2005;4:1050–1054. doi: 10.4161/cbt.4.10.2184. [DOI] [PubMed] [Google Scholar]
- Lee JJ, von Kessler DP, Parks S, Beachy PA. Secretion and localized transcription suggest a role in positional signaling for products of the segmentation gene hedgehog. Cell. 1992;71:33–50. doi: 10.1016/0092-8674(92)90264-d. [DOI] [PubMed] [Google Scholar]
- Lee T, Feig L, Montell DJ. Two distinct roles for Ras in a developmentally regulated cell migration. Development. 1996;122:409–418. doi: 10.1242/dev.122.2.409. [DOI] [PubMed] [Google Scholar]
- Lin C, Nozawa YI, Chuang PT. The path to chemotaxis and transcription is smoothened. Sci Signal. 2012;5:pe35. doi: 10.1126/scisignal.2003423. [DOI] [PubMed] [Google Scholar]
- Liu Y, Montell DJ. Identification of mutations that cause cell migration defects in mosaic clones. Development. 1999;126:1869–1878. doi: 10.1242/dev.126.9.1869. [DOI] [PubMed] [Google Scholar]
- Llense F, Martin-Blanco E. JNK signaling controls border cell cluster integrity and collective cell migration. Curr Biol. 2008;18:538–544. doi: 10.1016/j.cub.2008.03.029. [DOI] [PubMed] [Google Scholar]
- Lum L, Zhang C, Oh S, Mann RK, von Kessler DP, Taipale J, Weis-Garcia F, Gong R, Wang B, Beachy PA. Hedgehog signal transduction via Smoothened association with a cytoplasmic complex scaffolded by the atypical kinesin, Costal-2. Mol Cell. 2003;12:1261–1274. doi: 10.1016/s1097-2765(03)00426-x. [DOI] [PubMed] [Google Scholar]
- McDonald JA, Khodyakova A, Aranjuez G, Dudley C, Montell DJ. PAR-1 kinase regulates epithelial detachment and directional protrusion of migrating border cells. Curr Biol. 2008;18:1659–1667. doi: 10.1016/j.cub.2008.09.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald JA, Montell DJ. Analysis of cell migration using Drosophila as a model system. Methods Mol Biol. 2005;294:175–202. doi: 10.1385/1-59259-860-9:175. [DOI] [PubMed] [Google Scholar]
- McDonald JA, Pinheiro EM, Kadlec L, Schupbach T, Montell DJ. Multiple EGFR ligands participate in guiding migrating border cells. Dev Biol. 2006;296:94–103. doi: 10.1016/j.ydbio.2006.04.438. [DOI] [PubMed] [Google Scholar]
- McDonald JA, Pinheiro EM, Montell DJ. PVF1, a PDGF/VEGF homolog, is sufficient to guide border cells and interacts genetically with Taiman. Development. 2003;130:3469–3478. doi: 10.1242/dev.00574. [DOI] [PubMed] [Google Scholar]
- Melani M, Simpson KJ, Brugge JS, Montell D. Regulation of cell adhesion and collective cell migration by hindsight and its human homolog RREB1. Curr Biol. 2008;18:532–537. doi: 10.1016/j.cub.2008.03.024. [DOI] [PubMed] [Google Scholar]
- Methot N, Basler K. Hedgehog controls limb development by regulating the activities of distinct transcriptional activator and repressor forms of Cubitus interruptus. Cell. 1999;96:819–831. doi: 10.1016/s0092-8674(00)80592-9. [DOI] [PubMed] [Google Scholar]
- Meyer T, Vinkemeier U. Nucleocytoplasmic shuttling of STAT transcription factors. Eur J Biochem. 2004;271:4606–4612. doi: 10.1111/j.1432-1033.2004.04423.x. [DOI] [PubMed] [Google Scholar]
- Montell DJ, Rorth P, Spradling AC. slow border cells, a locus required for a developmentally regulated cell migration during oogenesis, encodes Drosophila C/EBP. Cell. 1992;71:51–62. doi: 10.1016/0092-8674(92)90265-e. [DOI] [PubMed] [Google Scholar]
- Montell DJ, Yoon WH, Starz-Gaiano M. Group choreography: mechanisms orchestrating the collective movement of border cells. Nat Rev Mol Cell Biol. 2012;13:631–645. doi: 10.1038/nrm3433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motzny CK, Holmgren R. The Drosophila cubitus interruptus protein and its role in the wingless and hedgehog signal transduction pathways. Mech Dev. 1995;52:137–150. doi: 10.1016/0925-4773(95)00397-j. [DOI] [PubMed] [Google Scholar]
- Murphy AM, Montell DJ. Cell type-specific roles for Cdc42, Rac, and RhoL in Drosophila oogenesis. J Cell Biol. 1996;133:617–630. doi: 10.1083/jcb.133.3.617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niewiadomska P, Godt D, Tepass U. DE-Cadherin is required for intercellular motility during Drosophila oogenesis. J Cell Biol. 1999;144:533–547. doi: 10.1083/jcb.144.3.533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura Y, Applegate K, Davidson MW, Danuser G, Waterman CM. Automated screening of microtubule growth dynamics identifies MARK2 as a regulator of leading edge microtubules downstream of Rac1 in migrating cells. PLoS One. 2012;7:e41413. doi: 10.1371/journal.pone.0041413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nusslein-Volhard C, Wieschaus E. Mutations affecting segment number and polarity in Drosophila. Nature. 1980;287:795–801. doi: 10.1038/287795a0. [DOI] [PubMed] [Google Scholar]
- Ogden SK, Ascano M, Jr, Stegman MA, Robbins DJ. Regulation of Hedgehog signaling: a complex story. Biochem Pharmacol. 2004;67:805–814. doi: 10.1016/j.bcp.2004.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrimon N, Ni JQ, Perkins L. In vivo RNAi: today and tomorrow. Cold Spring Harb Perspect Biol. 2010;2:a003640. doi: 10.1101/cshperspect.a003640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips RG, Roberts IJ, Ingham PW, Whittle JR. The Drosophila segment polarity gene patched is involved in a position-signalling mechanism in imaginal discs. Development. 1990;110:105–114. doi: 10.1242/dev.110.1.105. [DOI] [PubMed] [Google Scholar]
- Pinheiro EM, Montell DJ. Requirement for Par-6 and Bazooka in Drosophila border cell migration. Development. 2004;131:5243–5251. doi: 10.1242/dev.01412. [DOI] [PubMed] [Google Scholar]
- Polizio AH, Chinchilla P, Chen X, Kim S, Manning DR, Riobo NA. Heterotrimeric Gi proteins link Hedgehog signaling to activation of Rho small GTPases to promote fibroblast migration. J Biol Chem. 2011;286:19589–19596. doi: 10.1074/jbc.M110.197111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter JA, Ekker SC, Park WJ, von Kessler DP, Young KE, Chen CH, Ma Y, Woods AS, Cotter RJ, Koonin EV, Beachy PA. Hedgehog patterning activity: role of a lipophilic modification mediated by the carboxy-terminal autoprocessing domain. Cell. 1996;86:21–34. doi: 10.1016/s0092-8674(00)80074-4. [DOI] [PubMed] [Google Scholar]
- Prasad M, Montell DJ. Cellular and molecular mechanisms of border cell migration analyzed using time-lapse live-cell imaging. Dev Cell. 2007;12:997–1005. doi: 10.1016/j.devcel.2007.03.021. [DOI] [PubMed] [Google Scholar]
- Price MA, Kalderon D. Proteolysis of cubitus interruptus in Drosophila requires phosphorylation by protein kinase A. Development. 1999;126:4331–4339. doi: 10.1242/dev.126.19.4331. [DOI] [PubMed] [Google Scholar]
- Price MA, Kalderon D. Proteolysis of the Hedgehog signaling effector Cubitus interruptus requires phosphorylation by Glycogen Synthase Kinase 3 and Casein Kinase 1. Cell. 2002;108:823–835. doi: 10.1016/s0092-8674(02)00664-5. [DOI] [PubMed] [Google Scholar]
- Quinones-Coello AT, Petrella LN, Ayers K, Melillo A, Mazzalupo S, Hudson AM, Wang S, Castiblanco C, Buszczak M, Hoskins RA, Cooley L. Exploring strategies for protein trapping in Drosophila. Genetics. 2007;175:1089–1104. doi: 10.1534/genetics.106.065995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinones GA, Jin J, Oro AE. I-BAR protein antagonism of endocytosis mediates directional sensing during guided cell migration. J Cell Biol. 2010;189:353–367. doi: 10.1083/jcb.200910136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quirk J, van den Heuvel M, Henrique D, Marigo V, Jones TA, Tabin C, Ingham PW. The smoothened gene and hedgehog signal transduction in Drosophila and vertebrate development. Cold Spring Harb Symp Quant Biol. 1997;62:217–226. [PubMed] [Google Scholar]
- Rodriguez I, Basler K. Control of compartmental affinity boundaries by hedgehog. Nature. 1997;389:614–618. doi: 10.1038/39343. [DOI] [PubMed] [Google Scholar]
- Rorth P, Szabo K, Bailey A, Laverty T, Rehm J, Rubin GM, Weigmann K, Milan M, Benes V, Ansorge W, Cohen SM. Systematic gain-of-function genetics in Drosophila. Development. 1998;125:1049–1057. doi: 10.1242/dev.125.6.1049. [DOI] [PubMed] [Google Scholar]
- Rorth P, Szabo K, Texido G. The level of C/EBP protein is critical for cell migration during Drosophila oogenesis and is tightly controlled by regulated degradation. Mol Cell. 2000;6:23–30. doi: 10.1016/s1097-2765(05)00008-0. [DOI] [PubMed] [Google Scholar]
- Schuske K, Hooper JE, Scott MP. patched overexpression causes loss of wingless expression in Drosophila embryos. Dev Biol. 1994;164:300–311. doi: 10.1006/dbio.1994.1200. [DOI] [PubMed] [Google Scholar]
- Sefton L, Timmer JR, Zhang Y, Beranger F, Cline TW. An extracellular activator of the Drosophila JAK/STAT pathway is a sex-determination signal element. Nature. 2000;405:970–973. doi: 10.1038/35016119. [DOI] [PubMed] [Google Scholar]
- Silver DL, Geisbrecht ER, Montell DJ. Requirement for JAK/STAT signaling throughout border cell migration in Drosophila. Development. 2005;132:3483–3492. doi: 10.1242/dev.01910. [DOI] [PubMed] [Google Scholar]
- Silver DL, Montell DJ. Paracrine signaling through the JAK/STAT pathway activates invasive behavior of ovarian epithelial cells in Drosophila. Cell. 2001;107:831–841. doi: 10.1016/s0092-8674(01)00607-9. [DOI] [PubMed] [Google Scholar]
- Singh RR, Kim JE, Davuluri Y, Drakos E, Cho-Vega JH, Amin HM, Vega F. Hedgehog signaling pathway is activated in diffuse large B-cell lymphoma and contributes to tumor cell survival and proliferation. Leukemia. 2010;24:1025–1036. doi: 10.1038/leu.2010.35. [DOI] [PubMed] [Google Scholar]
- Spradling AC. Germline cysts: communes that work. Cell. 1993;72:649–651. doi: 10.1016/0092-8674(93)90393-5. [DOI] [PubMed] [Google Scholar]
- St Johnston D. The art and design of genetic screens: Drosophila melanogaster. Nat Rev Genet. 2002;3:176–188. doi: 10.1038/nrg751. [DOI] [PubMed] [Google Scholar]
- Su Y, Ospina JK, Zhang J, Michelson AP, Schoen AM, Zhu AJ. Sequential phosphorylation of smoothened transduces graded hedgehog signaling. Sci Signal. 2011;4:ra43. doi: 10.1126/scisignal.2001747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J, Deng WM. Hindsight mediates the role of notch in suppressing hedgehog signaling and cell proliferation. Dev Cell. 2007;12:431–442. doi: 10.1016/j.devcel.2007.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torroja C, Gorfinkiel N, Guerrero I. Patched controls the Hedgehog gradient by endocytosis in a dynamin-dependent manner, but this internalization does not play a major role in signal transduction. Development. 2004;131:2395–2408. doi: 10.1242/dev.01102. [DOI] [PubMed] [Google Scholar]
- Tukachinsky H, Lopez LV, Salic A. A mechanism for vertebrate Hedgehog signaling: recruitment to cilia and dissociation of SuFu-Gli protein complexes. J Cell Biol. 2010;191:415–428. doi: 10.1083/jcb.201004108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uemura T, Oda H, Kraut R, Hayashi S, Kotaoka Y, Takeichi M. Zygotic Drosophila E-cadherin expression is required for processes of dynamic epithelial cell rearrangement in the Drosophila embryo. Genes & development. 1996;10:659–671. doi: 10.1101/gad.10.6.659. [DOI] [PubMed] [Google Scholar]
- Wang J, Lin W, Popko B, Campbell IL. Inducible production of interferon-gamma in the developing brain causes cerebellar dysplasia with activation of the Sonic hedgehog pathway. Mol Cell Neurosci. 2004;27:489–496. doi: 10.1016/j.mcn.2004.08.004. [DOI] [PubMed] [Google Scholar]
- Wang X, Bo J, Bridges T, Dugan KD, Pan TC, Chodosh LA, Montell DJ. Analysis of cell migration using whole-genome expression profiling of migratory cells in the Drosophila ovary. Developmental cell. 2006;10:483–495. doi: 10.1016/j.devcel.2006.02.003. [DOI] [PubMed] [Google Scholar]
- Wang X, He L, Wu YI, Hahn KM, Montell DJ. Light-mediated activation reveals a key role for Rac in collective guidance of cell movement in vivo. Nat Cell Biol. 2010;12:591–597. doi: 10.1038/ncb2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu T, Rubin GM. Analysis of genetic mosaics in developing and adult Drosophila tissues. Development. 1993;117:1223–1237. doi: 10.1242/dev.117.4.1223. [DOI] [PubMed] [Google Scholar]
- Yam PT, Langlois SD, Morin S, Charron F. Sonic hedgehog guides axons through a noncanonical, Src-family-kinase-dependent signaling pathway. Neuron. 2009;62:349–362. doi: 10.1016/j.neuron.2009.03.022. [DOI] [PubMed] [Google Scholar]
- Yilmaz M, Christofori G. Mechanisms of motility in metastasizing cells. Mol Cancer Res. 2010;8:629–642. doi: 10.1158/1541-7786.MCR-10-0139. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Kalderon D. Regulation of cell proliferation and patterning in Drosophila oogenesis by Hedgehog signaling. Development. 2000;127:2165–2176. doi: 10.1242/dev.127.10.2165. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Kalderon D. Hedgehog acts as a somatic stem cell factor in the Drosophila ovary. Nature. 2001;410:599–604. doi: 10.1038/35069099. [DOI] [PubMed] [Google Scholar]
- Zhu AJ, Zheng L, Suyama K, Scott MP. Altered localization of Drosophila Smoothened protein activates Hedgehog signal transduction. Genes Dev. 2003;17:1240–1252. doi: 10.1101/gad.1080803. [DOI] [PMC free article] [PubMed] [Google Scholar]