

Abstract
Using a murine model, we demonstrated that endobronchial administration of antibodies (Abs) to MHC class I results in cellular infiltration, epithelial metaplasia, fibrosis and obstruction of the small airways (Obliterative Airway Disease (OAD)) mediated predominantly by Th17 responses to self-antigens. This resembles bronchiolitis obliterans syndrome developed following human lung transplantation. Since B cells play a crucial role in induction of autoimmune responses, we defined the role of B cells and its antigen presenting properties in induction of OAD in this study. Anti-MHC class I was administered endobronchially in B−/− and wild type mice. In contrast to wild type, B−/− animals did not demonstrate cellular infiltration, epithelial metaplasia and obstruction of airways following anti-MHC. Frequency of Kα1-tubulin and CollagenV specific IL-17 cells was significantly decreased in B−/− mice. As expected, Abs against self-antigens and germinal center formation were not developed in B−/− mice. Thus we conclude that B cells and its antigen presenting capacity play an important role in induction of immune responses to self-antigens and immunopathogenesis of OAD following administration of anti-MHC. Therefore, strategies to block B cell and its antigen presenting functions should be considered for preventing the development of chronic rejection.
Keywords: B cell, autoimmunity, anti-MHC antibodies, OAD, cytokines, IL-17
INTRODUCTION
Chronic rejection following human lung transplantation (LTx) clinically diagnosed, as Bronchiolitis Obliterans Syndrome (BOS) remains a major detriment to long-term lung function. BOS was initially described in 1901 (1), however considering its impact, there has been a renewed interest in understanding its immunopathogenesis. Studies from two large centers, including ours, have clearly shown that the prevalence and mortality of BOS is 60% to 80% in less than 7 years following LTx (2–5), making it the most significant long term cause of morbidity and mortality after LTx. Current evidence for the pathogenesis of BOS suggests that it is an immunologically mediated process with injury directed toward pulmonary epithelial or the endothelial cells or both (6). BOS histopathologically comprises of a fibroproliferation leading to obliteration of tubular structures in the lung (7, 8).
Recent reports support that de novo production of antibodies (Abs) to donor MHC post-transplant correlates with development of BOS following human LTx (9, 10). Based on this, we developed a unique murine model in which administration of anti-H2Kd class I MHC mAb endobronchially directly into the lung resulted in epithelial metaplasia, endotheliitis and obliterative airway disease (OAD) of distal airways very similar to the pathology of BOS seen following human LTx (11). We also demonstrated that immune responses to self-antigens, K-α1 tubulin (K-α1T) and Collagen V (ColV), mediated predominantly by Th17 cells play a crucial role in the development of OAD (11). Neutralization of Th17 responses by administration of an anti-IL-17 resulted in abrogation of immune responses to self-antigens and prevention of epithelial metaplasia, cellular infiltration and development of fibrosis as well as OAD (11). Although these mice demonstrated IFN-γ responses to self-antigens, considering that anti-IL-17 reduced OAD lesions (11) and previous studies demonstrating a significant role for IL-17 in tissue inflammation and fibrosis (12), our results led us to conclude that predominant Th17 mediated immune responses to self-antigens lead to development of OAD.
B cells, through production of Abs, play a crucial role in humoral responses and have been implicated in chronic allograft rejection. B cells have also been shown to present antigens and can therefore augment adaptive immune responses (13). Recent studies have shown that there is a significant increase in B cells that infiltrate the lungs following injury (14). Studies using B−/− mice have also shown that the absence of B cells results in decreased T cell responses and lung injury. Moreover, it has been proposed that IL-17 through the recruitment of B cells leads to development of auto-immunity - in particular Abs to self-antigens and autoimmune diseases (15). Based on these findings, we postulated that injury to the lung by the administration of anti-MHC may also lead to recruitment of B cells that are crucial for the development of immune responses to self-antigens and pathogenesis of OAD.
In this study we demonstrate that endobronchial administration of anti-MHC class I in B−/− mice results in a significant reduction in the T cell infiltration to the lungs along with the loss of induction of the Th17 responses against K-α1T and ColV and decreased germinal center formation in the spleen. The decreased T cell infiltration, lack of Th17 responses and the resulting absence of Abs to self-antigens results in the prevention of OAD following administration of anti-MHC in B−/− mice.
METHODS
Anti-MHC class I administration in native lungs of wild type and B-cell knock-out (KO) mice
All experiments were performed in compliance with the guidelines of the Institutional Laboratory Animal Care and Use Committee of Washington University School of Medicine. Murine mAb to H2Kb (C57BL/6, 6week, male, IgG2a), which has no detectable endotoxin, as measured by LAL assay was given into C57/BL6 or B6.129S2-Igh-6tm1Cgn/J as described earlier (11). Ab (200 μg/dose) was administered into the lung on day 1, 2, 3, 6 and then weekly thereafter. C1.18.4 (isotype control) was similarly administered as control.
Histological analysis
Lungs were fixed in 10% formaldehyde and sections cut at 5 μm thickness and stained with Masson's trichrome and hematoxylin & eosin (H&E). Lesions that displayed cellular infiltration, epithelial abnormalities, and fibro-proliferation were analyzed by random sampling. Morphometric analysis for fibrosis and cellular infiltration was performed. Fibrosis was calculated using Optimas software version 6.5.172 (Media Cybernetics), as a percentage of total area enclosed by basement membrane. Cellular infiltration and epithelial abnormalities were similarly calculated as a percentage of the total bronchiole and vessels visualized, respectively.
Isolation of lung infiltrating lymphocytes
Lung infiltrating lymphocytes were isolated as described previously (11). Briefly, lung tissue sections were stirred in a suspension of RPMI-1640 medium (Invitrogen, Carlsbad, CA) supplemented with 0.1% collagenase type XI (Sigma, St. Louis, MO) and 0.002% DNAse (Sigma, St Louis, MO) for enzymatic digestion O/N at RT. The suspension was filtered thought a cell strainer and washed with PBS twice. Lymphocytes were separated using Ficoll-Paque density centrifugation. Lymphocytes isolated from lung tissues were suspended in RPMI medium.
ELISPOT assay
ELISPOT assays were performed as described previously (10). Briefly, 3×105 cells/well were cultured in triplicate in the presence of ColV (20 μg/ml) or human K-α1T (1 μg/ml) and irradiated feeder autologous splenocytes (1:1 ratio) in cytokine Ab coated 96 well plates for 48–72h. The plates were developed as per manufacturers instruction and spots were analyzed in an ImmunoSpot Series I analyzer (Cellular Technology), and the results were expressed as spots per million cells (spm) ± standard error. Any spots obtained by culturing T-cell lines with antigen presenting cells (APCs) alone without ColV or human K-α1T were subtracted from number of spots in test cultures.
ELISA for Auto Abs
ELISA plates were coated with pure ColV (BDBiosciences) or recombinant human K-α1T (1μg/ml) in PBS O/N at 4°C. Serum samples were tested (1:250 and 1:500) and detection was done with anti-mouse IgG, IgM–HRP and developed using TMB substrate and read at 450nm. Concentration of Abs was calculated based on a standard curve using the binding of known concentration of anti-ColV Abs or anti-K-α1T Abs (Santa Cruz Biotech).
Staining for Germinal centers
To determine development of germinal centers in the spleen following administration anti-MHC class I C57BL/6 mice and B cell KO mice were treated with anti-MHC class I or Isotype control Abs as described, and spleens analyzed at day 30 for germinal center formation by immunohistochemical staining using anti-PNA Ab (BDBiosciences).
RESULTS
Endobronchial administration of anti-MHC class I results in a significant increase in B cell infiltration into the lungs
In order to determine whether there is an increase in infiltrating B cells following lung injury by administration of anti-MHC we administered anti-H2Kb or control Abs endobronchially in C57BL/6 mice on days 1, 2 and 3. The lungs were harvested on day 4 and cells obtained by collagenase digestion. B cells in lung were analyzed by flow cytometry using anti-CD19 stain. Lungs harvested from anti-H2Kb administered mice as shown in Figure 1 demonstrated significantly increased B cells when compared to mice treated with control Ab (2.3±0.6×106 vs. 0.9±0.4×106, P<0.05). These indicate that B cells infiltrate the lungs at early time points following administration of anti-MHC class I.
Figure 1. Increased infiltration of B cells in lungs of mice administered anti-MHC Abs endobronchially.
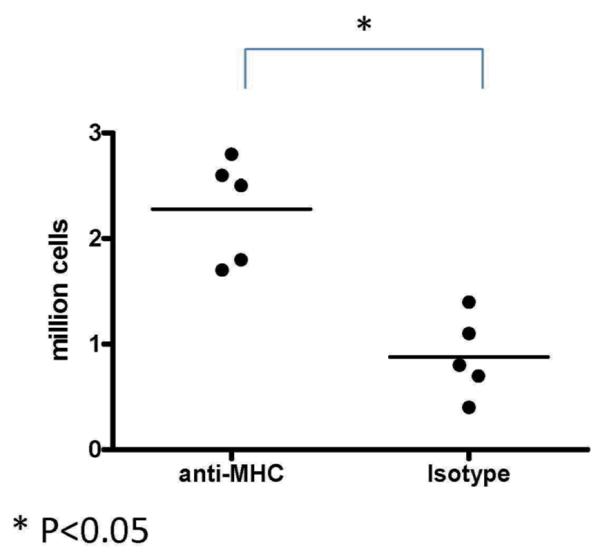
Anti-H2kb or control (C1.18.4) Ab was administered endobronchially in C57BL/6 mice on days 1, 2, 3. The lungs were harvested on day 4 and analyzed for the number of infiltrating cells by flow cytometry using anti-CD19 Abs. There was a significant increase in the number of B cells in the lung following anti-MHC Ab administration. Represented as mean number of B cells with 5 mice in each group. *Denotes p<0.05
B cells play a role in the induction of IL-17 secreting cells following exposure to lower concentrations of self-antigens
B cells have been suggested to play an important role in the processing and presentation of lower threshold of self-antigens, which are thought to be involved in various autoimmune responses (16, 17). In order to test this we stimulated the same number of splenocytes (1×106 cells) from wild type mice and B cell KO mice (5 mice each) with 1μg, 250ng or 100ng of K-α1T or ColV and analyzed the frequency of IL-17 secreting T cells by ELISPOT. The results demonstrated that there was a 90% reduction (3 vs. 48 spm for K-α1T and 8 vs. 72 spm for ColV, p<0.05) in the frequency of IL-17-secreting cells in the B cell KO mice when compared to wild type controls at an antigen concentration of 100ng (Figure 2). Moreover, addition of B cells to the splenocytes from the B cell KO mice resulted in significant increases in the frequency of IL-17 secreting K-α1T and ColV specific cells whereas depletion of the B cells (using CD19 magnetic beads in two consecutive depletions) from the splenocytes from wild type animals before stimulation with the self-antigens resulted in a 2 fold reduction (26 vs. 48 spm, p<0.05) of the IL-17 secreting cells (Figure 2). However, stimulation with 1μg, or 250ng of self-antigens did not demonstrate any difference in the frequency of IL-17 secreting T cells among the groups (p>0.05). This indicates that B cells play an important role in the induction of IL-17 secreting T cells involved in the immune responses to self-antigens and this effect is more profound at a lower concentration of antigens.
Figure 2. B cell depletion results in decreased IL-17 secreting cells when stimulated with low concentrations of self-antigens.

Splenocytes (3×105 per well) from B6.129S2-Igh-6tm1Cgn/J (B−/−) or control C57BL/6 mice (Wild type – WT) were stimulated with 1μg, 250ng and 100 ng of K-α1T or ColV (cells cultured in triplicate for each concentration of antigen) and the frequency of IL-17 secreting cells analyzed using ELISPOT. In another set of experiment B cells from wild type mice were added to the T cells from B6.129S2-Igh-6tm1Cgn/J or B cells were depleted from the splenocytes of wild type mice and stimulated with self-antigens to determine the frequency of IL-17 secreting T cells. The frequency of IL-17 secreting T cells were lower in splenocytes from B6.129S2-Igh-6tm1Cgn/J and wild type mice depleted of B cells (at 100ng antigen concentration) when compared to wild type mice as well as B cells added splenocytes from B6.129S2-Igh-6tm1Cgn/J mice. Frequency of IL-17 secreting cells following stimulation at higher antigen concentration (250ng and 1μg) did not differ between the groups. Results are expressed as mean ± SD spots per million cells observed in 5 mice per group. * Denotes P<0.05
B cell deficiency leads to decreased production of pro-inflammatory cytokines and chemokines following administration of Abs to MHC class I
B cells through the production of TNF-α, IL-6, IFN-γ and CXCL12 has been shown to influence the immunogenicity of the infiltrating T cells (18). In order to determine the role of the B cells in the induction of cytokines and chemokines that affect T cell responses following administration of anti-MHC, we analyzed the expression of cytokines and chemokines in the lungs by quantitative real-time PCR. Analysis of the lungs on day 4 demonstrated that there was a significant reduction in the expression of TNF-α (4.6 folds), IL-6 (6.8 folds), IL-2 (2.7 folds), IFN-g (8.3 folds) and CXCL12 (14.4 folds) in the lungs of B−/− mice when compared to the wild type controls (Figure 3). These support a crucial role for B cells in the induction of immune responses to self-antigens following administration of anti-MHC.
Figure 3. Significant reduction in the pro-inflammatory cytokines and chemokines in B cell deficient mice following anti-MHC Ab administration.

B6.129S2-Igh-6tm1Cgn/J (B−/−) or control C57BL/6 mice (Wild type) were treated with anti-MHC class I Abs day 1, 2 and 3. We analyzed the expression levels of cytokines and chemokines in the lungs by quantitative real-time PCR on Day 4. There was a significant reduction in the expression of TNF-α (4.6 folds), IL-6 (6.8 folds), IL-2 (2.7 folds), IFN-γ (8.3 folds) and CXCL12 (14.4 folds) in the lungs of B cell deficient mice when compared to the wild type controls. Data representative of mean ± SD fold change observed, with 5 mice per group.
B cell deficiency results in decrease in the frequency of lung infiltrating T lymphocytes secreting IFN-γ and IL-17 specific for K-α1T or ColV
In order to determine the role of B cells in the induction of autoimmune responses following administration of anti-MHC, T cells were isolated from lung tissue at 30 days after anti-MHC class I administration and stimulated with K-α1T or ColV (3×105 T-cells/well in triplicate) in the presence of autologous splenocytes and the frequency of IL-17 and IFN-γ secreting cells was determined by ELISPOT. B cell deficiency led to a significant reduction in cells secreting IL-17 and IFN-γ in response to K-α1T or ColV, when compared to the wild type mice and was similar to those observed in the control isotype (C1.18.4) treated mice (p<0.05) (Figure 4). This demonstrates that absence of B cells leads to decreased lung infiltrating T-cell response to K-α1T or ColV following administration of anti-MHC.
Figure 4. B cell deficiency results in a significant reduction in the frequency of IFN-γ and IL-17 secreting T cells against K-α1T and ColV in the lung following anti-MHC Ab administration.

B6.129S2-Igh-6tm1Cgn/J (B cell KO) or control C57BL/6 mice were treated with anti-MHC class I Abs on day 1, 2, 3, 6 and weekly thereafter. T cells infiltrating the lungs were harvested by collagenase digestion and the frequency of T cells secreting IFN-γ, IL-4 and IL-17 on stimulation with K-α1T and ColV (3×105 cells/well cultured in triplicate for each antigen) were analyzed by ELISPOT. A significant reduction in the frequency of T cells against K-α1T and ColV was observed in B cell deficient mice when compared to controls. IFN-g secreting cells to LPS in vitro did not differ among the groups. Data represented as mean ± SD spots per million cells (spm) of 5 mice per group.
Alloimmune responses are induced in B cell KO mice albeit of a lower magnitude
In order to determine whether an alloimmune response could be induced in B−/− mice, we immunized B6.129S2-Igh-6tm1Cgn/J or control C57BL/6J mice twice with 106 BALB/c splenocytes intraperitoneally at weekly intervals and analyzed the frequency of IFN-γ secreting T cells that respond to the BALB/c splenocytes. Analysis of T cell responses by ELISPOT (using 3×105 cells in triplicate) demonstrated that cells from the B−/− mice immunized with BALB/c splenocytes mounted T cell response against the mismatched MHC antigens albeit of a lower magnitude when compared to their wild type controls (67.6±13.8 spm vs. 135.6±26.2 spm, Figure 5). Also, B cell KO mice demonstrated IFN-γ responses following LPS stimulation in vitro (Figure 4), indicating that B−/− mice are capable of mounting effective T cell responses.
Figure 5. Alloimmune responses are preserved in B cell KO mice albeit of a lower magnitude compared to wild type mice.

B6.129S2-Igh-6tm1Cgn/J (B cell KO) or control C57BL/6 (Wild type) mice were immunized twice with 1 million BALB/c splenocytes intraperitoneally at weekly intervals. Frequency of IFN-γ secreting cells that respond to the BALB/c splenocytes were analyzed by ELISPOT. B cell deficient mice immunized with BALB/c splenocytes mounted an IFN-γ response against the mismatched MHC antigens albeit of a lower magnitude when compared to their wild type controls. Represented as mean spots per million cells of 5 mice per group.
Production of Abs to self-antigens and germinal center formation is absent in B cell KO mice following administration of anti-MHC class I Abs
Abs against ColV and K-α1T have been demonstrated to develop in LTx recipients with chronic rejection as well as in animal model of OAD induced by anti-MHC class me (11, 19). Sera from the B−/− mice and wild type controls (day 30 post anti-MHC administration) were analyzed for Abs against K-α1T and ColV. Following administration of the anti-MHC, in B−/− mice as expected, there was no development of auto-Abs whereas the wild type controls developed Abs against K-α1T and ColV (Figure 6A).
Figure 6.

A: Decreased auto-Ab production in B cell deficient mice following anti-MHC Ab administration. B6.129S2-Igh-6tm1Cgn/J (B cell KO) or control C57BL/6 (wild type) mice were treated with anti-MHC class I Abs day1, 2, 3, 6 and weekly thereafter. Serum collected at day 30 was analyzed for auto-Abs against K-α1T and ColV by ELISA. Abs to K-α1T and ColV were significantly reduced in B cell deficient mice when compared to wild type following administration of anti-MHC Abs. Represented as mean ± SD μg/mL of 5 mice per group. * Denotes p<0.05
B: Formation of germinal centers is significantly reduced in the B cell deficient mice following administration of anti-MHC Abs when compared to wild type controls. B6.129S2-Igh-6tm1Cgn/J (B cell KO) or control C57BL/6 (wild type) mice were treated with anti-MHC class I Abs day 1, 2, 3, 6 and weekly thereafter. Spleens from the mice were harvested on day 30 and analyzed for the germinal center formation by staining with anti-PNA Abs. Immunohistochemical analysis of the spleen from the wild type mice demonstrated significantly increased number of germinal centers in comparison to B cell deficient mice.
Activation of the immune responses following antigen stimulation leads to the formation of germinal centers in the spleen. In order to define the role of B cells in the formation of germinal centers following administration of anti-MHC Abs, the spleens from the wild type controls and B−/− animals were analyzed on day 30 for germinal center formation by staining with anti-PNA Abs. Wild type mice demonstrated a fourfold higher number of germinal centers in comparison to B−/− mice (Figure 6B). The number and the sizes of germinal centers in the B−/− mice were similar to those observed in the wild type mice treated with control Abs (data not shown).
B cell deficiency results in decreased cellular infiltration, endotheliitis, epithelial hyperplasia and fibrosis following administration of anti-MHC class I
In order to define the role of B cells in the induction of OAD following administration of anti-MHC, B cell KO or control C57BL/6 mice (5 each) were treated with anti-MHC class I on day 1, 2, 3, 6 and weekly thereafter. Following anti-MHC class I, all B cell KO mice treated with anti-MHC class I showed significant reduction in the cellular infiltration near vessels and bronchioles when compared to control mice at day 30 (Figure 7 b&g vs. c&h). Morphometric analysis demonstrated a 10-fold reduction in fibro-proliferation and epithelial damage in B cell KO mice when compared to wild type mice treated with anti-MHC class I (Figure 8). None of the B cell KO mice treated with C1.18.4 control Ab or wild type mice treated with nonspecific anti-MHC (anti H2kd) demonstrated any lesions (Figure 7 a&f and d&i). For further confirmation of the role of B cells, we passively transferred 2.5 million B cells from wild type mice into B cell KO mice on day 0 of the anti-MHC administration. These mice developed significant increase in cellular infiltration, epithelial metaplasia and development of fibrosis similar to those observed in the wild type mice (Figure 7 e&j and Figure 8). These demonstrate that B cells play an important role in the immunopathogenesis of OAD.
Figure 7. B cell deficiency results in a significant reduction in cellular infiltration around vessel and bronchiole, hyperplasia of the bronchial epithelium and fibrosis.

B6.129S2-Igh-6tm1Cgn/J (B cell KO), control C57BL/6 (wild type), and B cell KO mice with passive transfer of 2.5×106 B cells were treated with either anti-MHC class I (anti-H2kb), isotype (C1.184) or nonspecific anti-MHC class I (anti H2kd) Abs on day 1, 2, 3, 6 and weekly thereafter (5 mice in each group). The lungs were harvested on day 30. Formalin preserved tissues were sectioned randomly and analyzed by H&E (a – e) and Masson's trichrome staining (d – i). Images were captured and representative area depicted in the figure. At least 4 out of 5 mice from each group demonstrated similar lesions in various sections of the lung. None of the B cell KO mice treated with anti-H2kb (b, g) demonstrated any significant fibrosis or airway obliteration, with minimal cellular infiltration and epithelial hyperplasia compared to wild type mice treated with anti-H2kd (c, h). B cell KO mice with passive transfer of B cells (e, j) demonstrated lesions similar to wild type mice. Wild type mice treated with non-specific anti-MHC class I (anti H2kd) (d, i) demonstrated no lesions. a,f: B cell KO with isotype control Abs; b,g: B cell KO with anti H2kb; c,h: wild type with anti-H2kb; d,i: wild type with anti-H2kd (non specific anti-MHC); e,j: B cell KO with passive transfer of B cells. B: bronchiole and V: vessels. Blue staining in trichrome stains denotes collagen deposition in the lungs.
Figure 8. Morphometric analysis showing reduced cellular infiltration, epithelial hyperplasia, fibrosis and luminal occlusion in B cell KO mice and in B cell KO mice with passive transfer of auto-Abs.

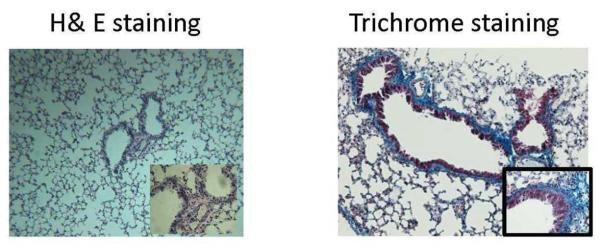

A: B6.129S2-Igh-6tm1Cgn/J (B cell KO), control C57BL/6 (wild type), B cell KO mice with passive transfer of 2.5×106 B cells (purified from splenocytes using B cell enrichment kit, Miltenyi Biotech) and B cell KO mice with passive transfer of 250ng each of Abs to ColV and K-α1T (on day 1) were treated with anti-MHC class I (anti-H2kb Abs on day 1, 2, 3, 6 and weekly thereafter (5 mice in each group). The lungs were harvested on day 30. Formalin preserved tissues were sectioned randomly and analyzed by H&E and Masson's trichrome staining. Images were captured and sections morphometric analysis performed using Optimas software (Media cybernetics). B-cell KO mice demonstrated significantly lesser bronchioles and vessels with cellular infiltration and epithelial hyperplasia. On passively transferring auto-Abs, only some bronchioles and vessels had epithelial hyperplasia or cellular infiltration with absence of any fibrosis when compared to wild type mice or B cell KO mice with passively transferred B cells. % occurrence refers to the percentage of bronchioles/vessels with cellular infiltration, epithelial hyperplasia, fibrosis or luminal occlusion per total number of bronchioles/vessels visualized in the lung sections. Data is mean ± SD of that observed in 5 mice per group. B: H&E and trichrome staining of the lungs. C: ELISPOT analysis of the lung infiltrating T cells specific for K-α1T and ColV in wild type mice, B−/− mice and B−/− mice passively transferred B cells from wild type mice.
Passive transfer of Abs specific to self-antigens to B−/− mice does not result in OAD
In order to determine whether the lack of auto-Ab development is the reason for absence of lesions in B cell KO mice, we administered B cell KO mice with Abs to both K-α1T and ColV (250μg each) following anti-MHC endobronchially. Passive administration of Abs to both K-α1T and ColV in B cell KO resulted in the development of mild lesions in the lungs, with epithelial hyperplasia affecting significantly lesser bronchioles and vessels compared to wild type mice following administration of anti-MHC (19.1 % vs. 38 %, Figure 8A). However, no luminal occlusion or fibrosis was observed in these mice (Fig 8b). On the other hand, B cell KO animals when passively transferred B cells, demonstrated fibrosis, luminal occlusion epithelial hyperplasia and cellular infiltration similar to the wild type mice (Figure 8). This indicates that just the absence of auto-Abs produced by B-cells weren't alone important for B cell KO mice to develop lesions. Analysis of lung infiltrating T cells in the B cell KO mice transferred with B cells by ELISPOT showed a significant increase in the frequency of K-α1T and ColV specific T cells in B−/− mice with passive transfer of B cells when compared to B−/− mice (fig. 8c).
DISCUSSION
Long term survival and function of human lung allografts is limited by the development of chronic rejection clinically diagnosed as BOS, an irreversible and often fatal condition that is unresponsive to therapy. Multiple hypotheses have been proposed for the etiological factors for BOS; however definitive mechanisms for the pathogenesis of BOS remain unclear. Studies from our laboratory, and others, have indicated a predominant role of cellular and humoral alloimmune mediated injury in the development of BOS (20–22). We recently characterized a unique murine model by administration of Abs to MHC class I resulting in induction of autoimmunity to lung specific self-antigens K-α1T and ColV and the development of OAD in the small airways of native murine lungs. This model strongly corroborates previous findings of development of Abs to donor HLA preceding and predisposing to BOS following human LTx, since the OAD developed only with anti-MHC and administration of nonspecific anti-MHC (anti-H2kd in C57BL/6 mice, Figure 7 d&i), isotype control (C1.184) or anti-keratin Abs did not result in OAD (11). In this study, we demonstrate that B cells play a crucial role in the induction of T cell responses especially to self-antigens following administration of anti-MHC class I. B cell deficiency results in decreased production of inflammatory cytokines which we propose to abrogate the development of immune responses against the self-antigens. These results further indicate that development of immune responses to self-antigens is essential for development of OAD.
Recent studies have demonstrated that besides Abs production, B cells play a role in induction of cellular immune responses through production of chemokines and cytokines, antigen presentation and immunoregulation (14). Analysis of cells infiltrating the lungs following administration of anti-MHC endobronchially at early time point (4 days) demonstrated a significant increase in the number of B cells in the lungs (Figure 1). These results are similar to those observed in other models where injury or inflammation in the lungs results in an increase in B cell infiltration (14). It has also been reported that B cells play an important role in the induction of T-cell mediated autoimmune responses in a murine model of autoimmune encephalitis (23). These studies as well as our results following administration of anti-MHC into lungs demonstrate that following inflammation there is a significant increase in the infiltration of B cells into the lungs that is essential for the development of T cell responses against self-antigens.
Our studies using B−/− mice have demonstrated that lack of B cells results in a loss of induction of self-antigen reactive T cells (Figure 4). ELISPOT analysis of lung infiltrating T cells in the B−/− mice following administration of anti-MHC demonstrated diminished Th17 cells that respond to K-α1T and ColV (Figure 4). Keeping in mind that B cell KO mice can have diminished T cells in circulation leading to lower immune responses, we used the same number of T-cells from all animals (3×105 cells per well in triplicate) for our ELISPOT. In addition, previous studies (24, 25) as well as our results presented clearly demonstrate T-cells in B cell KO animals are capable of mounting effective antigenic responses following immunization with cellular antigens since B cell KO animals have an effective IFN-γ response to LPS in vitro (Figure 4) and against allo-antigens (Figure 5). Also, wild type mice following depletion of B cells demonstrated IL-17 responses following stimulation with K-α1T and ColV (Figure 2). B cells are also known to control T cell immune responses through their production of cytokines and chemokines (26). Analysis of the cytokine expression profile in the lungs following anti-MHC administration demonstrated a significant reduction in the expression of TNF-α (4.6 folds), IL-6 (6.8 folds), IL-2 (2.7 folds), IFN-g (8.3 folds) and CXCL12 (14.4 folds) in the lungs of B−/− mice (Figure 3). Since the cytokine and chemokine milieu at the site of injury or infection tightly regulate T cell infiltration and activation (26), we postulate that the absence of B cells at the early time points following administration of anti-MHC may result in decreased production of pro-inflammatory cytokines and chemokines that are essential for self-antigen reactive T cell infiltration and activation in the lungs.
IL-6 is a critical cytokine in the induction and development of Th17 cells (27, 28). IL-6 through its effect on MIP1a and MIP-2 has also been shown to regulate neutrophil infiltration (29) and promote its survival (30, 31). Increases in neutrophil infiltration have been observed in lung injury as well as following LTx and has been postulated to be predictive of BOS development (32). Decreased IL-6 observed in the lung tissue following administration of anti-MHC in the B−/− mice along with a significant reduction in the mononuclear cells that infiltrate the lungs support the hypothesis that lack of B cells during early period following tissue injury can result in decreased neutrophil infiltration leading to decreased exposure of the self-antigens to immune system. This reduces responses to self-antigens and prevents development of OAD. The impact of B cells on IL-17 can also explain the prevention of OAD. IL-17 signaling has been shown to activate MAPK pathways (ERK1/ERK2 and p38 MAPK) trough IκB kinase and stress-activated protein kinase through ubiquitination of TARF 6 that are essential for neutrophil infiltration at the site of inflammation as well as induction of fibrosis (33–35).
It has been proposed that B cells play an important role in presentation of self-antigens in autoimmune diseases and this action of B cells is most pronounced at a lower threshold of the antigen (14, 23) (Figure 2). B cells can recruit professional APCs to the site of inflammation through the production of cytokines and chemokines and augment adaptive immune responses (18). The absence of the immune responses to self-antigens and abrogation of development of OAD lesions including fibrosis following administration of anti-MHC in the B−/− mice demonstrate that B cells are important in the early stages of immune activation following administration of anti-MHC. Moreover, passive administration of the Abs to K-α1T and ColV in the B−/− animals along with anti-MHC administration only resulted in a mild cellular infiltration and hyperplasia that did not progress to fibrosis and OAD. The presence of cellular infiltration not amounting the fibrosis in B cell KO mice that were administered anti-MHC (Figure 7, 8) with decreased IL-17 and IFN-γ secreting cells (Figure 4), suggests that besides activated T-cells or there may be infiltration with other mononuclear cells such as monocytes and macrophages as well as non activated T-cells that may also occur following administration of anti-MHC. Administration of anti-MHC results in increased interstitial inflammation, perivascular infiltrates, and lymphocytic bronchiolitis with peribronchiolar fibrosis, the hallmark features of autoimmune lung disease or chronic lung allograft rejection. However, due to the absence of significant activation of IL-17 there is lack of sufficient tissue injury and fibrosis amounting to OAD, which is also supported by our previous study when administration of anti-IL-17 significantly reduced OAD lesions (11).
Overall, these findings strongly suggest that B cells are necessary for the induction of T cell responses through either antigen presentation or through secretion of cytokines that can lead to the induction of immune responses to self-antigens and development of OAD following administration of anti-MHC. Based on these results, we propose that targeting of the B cells during the early stages of immune activation can potentially inhibit the development of immune responses to self-antigens, which can result in prevention of chronic rejection following human LTx.
ACKNOWLEDGEMENTS
This work is supported by NIH HL092514 and the BJC Foundation (TM). The authors would like to thank Ms. Billie Glasscock for her assistance in submitting this paper.
Footnotes
DISCLOSURE
The authors of the manuscript have no conflicts of interest to disclose as described by the American Journal of Transplantation.
REFERENCES
- 1.Lange W. Uber eine eigenthumbiche erkankung der kleinen broncheim and bronchiolen. Deutsche Arch Klin Med. 1901;70:342–364. [Google Scholar]
- 2.Jain R, Hachem RR, Morrell MR, Trulock EP, Chakinala MM, Yusen RD, et al. Azithromycin is associated with increased survival in lung transplant recipients with bronchiolitis obliterans syndrome. J Heart Lung Transplant. doi: 10.1016/j.healun.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sundaresan RS, Trulock EP, Mohanakumar T, Cooper JD, Patterson GA. Prevalence and outcome of bronchiolitis obliterans syndrome after lung transplantation. Washington University Lung Transplant Group. Annals of Thoracic Surgery. 1995;60:1341–1347. doi: 10.1016/0003-4975(95)00751-6. [DOI] [PubMed] [Google Scholar]
- 4.Valentine VG, Robbins RC, Berry GJ, Patel HR, Reichenspurner H, Reitz BA, et al. Actuarial survival of heart-lung and bilateral sequential lung transplant recipients with obliterative bronchiolitis. J Heart Lung Transplant. 1996;15:371–383. [PubMed] [Google Scholar]
- 5.Smith MA, Sundaresan S, Mohanakumar T, Trulock EP, Lynch JP, Phelan DL, et al. Effect of development of antibodies to HLA and cytomegalovirus mismatch on lung transplantation survival and development of bronchiolitis obliterans syndrome. J Thorac Cardiovasc Surg. 1998;116(5):812–820. doi: 10.1016/S0022-5223(98)00444-9. [DOI] [PubMed] [Google Scholar]
- 6.Griffith BP, Paradis IL, Zeevi A, Rabinowich H, Yousem SA, Duquesnoy RJ, et al. Immunologically mediated disease of the airways after pulmonary transplantation. Annals of Surgery. 1988;208:371–378. doi: 10.1097/00000658-198809000-00015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Boehler A, Kesten S, Weder W, Speich R. Bronchiolitis obliterans after lung transplantation: a review. Chest. 1998;114:1411–1426. doi: 10.1378/chest.114.5.1411. [DOI] [PubMed] [Google Scholar]
- 8.Estenne M, Hertz MI. Bronchiolitis obliterans after human lung transplantation. Am J Resp Crit Care Med. 2003;166:440–444. doi: 10.1164/rccm.200201-003pp. [DOI] [PubMed] [Google Scholar]
- 9.Reznik SI, Jaramillo A, Zhang L, Patterson GA, Cooper JD, Mohanakumar T. Anti-HLA antibody binding to hla class I molecules induces proliferation of airway epithelial cells: a potential mechanism for bronchiolitis obliterans syndrome. J Thorac Cardiovasc Surg. 2000;119(1):39–45. doi: 10.1016/s0022-5223(00)70215-7. [DOI] [PubMed] [Google Scholar]
- 10.Bharat A, Narayanan K, Street T, Fields RC, Steward N, Aloush A, et al. Early posttransplant inflammation promotes the development of alloimmunity and chronic human lung allograft rejection. Transplantation. 2007;83(2):150–158. doi: 10.1097/01.tp.0000250579.08042.b6. [DOI] [PubMed] [Google Scholar]
- 11.Fukami N, Ramachandran S, Saini D, Walter M, Chapman W, Patterson GA, et al. Antibodies to MHC class I induce autoimmunity: role in the pathogenesis of chronic rejection. J Immunol. 2009;182(1):309–318. doi: 10.4049/jimmunol.182.1.309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bettelli E, Oukka M, Kuchroo VK. T(H)-17 cells in the circle of immunity and autoimmunity. Nature immunology. 2007;8(4):345–350. doi: 10.1038/ni0407-345. [DOI] [PubMed] [Google Scholar]
- 13.Noorchashm H, Reed AJ, Rostami SY, Mozaffari R, Zekavat G, Koeberlein B, et al. B cell-mediated antigen presentation is required for the pathogenesis of acute cardiac allograft rejection. J Immunol. 2006;177(11):7715–7722. doi: 10.4049/jimmunol.177.11.7715. [DOI] [PubMed] [Google Scholar]
- 14.Lindell DM, Berlin AA, Schaller MA, Lukacs NW. B cell antigen presentation promotes Th2 responses and immunopathology during chronic allergic lung disease. PLoS One. 2008;3(9):e3129. doi: 10.1371/journal.pone.0003129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hsu HC, Yang P, Wang J, Wu Q, Myers R, Chen J, et al. Interleukin 17-producing T helper cells and interleukin 17 orchestrate autoreactive germinal center development in autoimmune BXD2 mice. Nat Immunol. 2008;9(2):166–175. doi: 10.1038/ni1552. [DOI] [PubMed] [Google Scholar]
- 16.Tian J, Zekzer D, Lu Y, Dang H, Kaufman DL. B cells are crucial for determinant spreading of T cell autoimmunity among beta cell antigens in diabetes-prone nonobese diabetic mice. J Immunol. 2006;176(4):2654–2661. doi: 10.4049/jimmunol.176.4.2654. [DOI] [PubMed] [Google Scholar]
- 17.Yan J, Harvey BP, Gee RJ, Shlomchik MJ, Mamula MJ. B cells drive early T cell autoimmunity in vivo prior to dendritic cell-mediated autoantigen presentation. J Immunol. 2006;177(7):4481–4487. doi: 10.4049/jimmunol.177.7.4481. [DOI] [PubMed] [Google Scholar]
- 18.Lund FE. Cytokine-producing B lymphocytes-key regulators of immunity. Curr Opin Immunol. 2008;20(3):332–338. doi: 10.1016/j.coi.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Goers TA, Ramachandran S, Aloush A, Trulock E, Patterson GA, Mohanakumar T. De novo production of K-alpha 1 tubulin-specific Abs: role in chronic lung allograft rejection. J Immunol. 2008;180(7):4487–4494. doi: 10.4049/jimmunol.180.7.4487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jaramillo A, Smith MA, Phelan D, Sundaresan S, Trulock E, Lynch J, et al. Temporal relationship between the development of anti-HLA antibodies and the development of bronchiolitis obliterans syndrome after lung transplantation. Transplant Proc. 1999;31(1–2):185–186. doi: 10.1016/s0041-1345(98)01495-x. [DOI] [PubMed] [Google Scholar]
- 21.Hachem RR. Lung allograft rejection: diagnosis and management. Curr Opin Organ Transplant. 2009;14(5):477–482. doi: 10.1097/MOT.0b013e32832fb981. [DOI] [PubMed] [Google Scholar]
- 22.Grossman EJ, Shilling RA. Bronchiolitis obliterans in lung transplantation: the good, the bad, and the future. Transl Res. 2009;153(4):153–165. doi: 10.1016/j.trsl.2009.01.005. [DOI] [PubMed] [Google Scholar]
- 23.Fillatreau S, Sweenie CH, McGeachy MJ, Gray D, Anderton SM. B cells regulate autoimmunity by provision of IL-10. Nat Immunol. 2002;3(10):944–950. doi: 10.1038/ni833. [DOI] [PubMed] [Google Scholar]
- 24.Nozaki T, Rosenblum JM, Ishii D, Tanabe K, Fairchild RL. CD4 T cell-mediated rejection of cardiac allografts in B cell-deficient mice. J Immunol. 2008;181(8):5257–5263. doi: 10.4049/jimmunol.181.8.5257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bosio CM, Elkins KL. Susceptibility to secondary Francisella tularensis live vaccine strain infection in B-cell-deficient mice is associated with neutrophilia but not with defects in specific T-cell-mediated immunity. Infect Immun. 2001;69(1):194–203. doi: 10.1128/IAI.69.1.194-203.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Weaver CT, Hatton RD, Mangan PR, Harrington LE. IL-17 family cytokines and the expanding diversity of effector T cell lineages. Annu Rev Immunol. 2007;25:821–852. doi: 10.1146/annurev.immunol.25.022106.141557. [DOI] [PubMed] [Google Scholar]
- 27.Korn T, Mitsdoerffer M, Croxford AL, Awasthi A, Dardalhon VA, Galileos G, et al. IL-6 controls Th17 immunity in vivo by inhibiting the conversion of conventional T cells into Foxp3+ regulatory T cells. Proc Natl Acad Sci U S A. 2008;105(47):18460–18465. doi: 10.1073/pnas.0809850105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Serada S, Fujimoto M, Mihara M, Koike N, Ohsugi Y, Nomura S, et al. IL-6 blockade inhibits the induction of myelin antigen-specific Th17 cells and Th1 cells in experimental autoimmune encephalomyelitis. Proc Natl Acad Sci U S A. 2008;105(26):9041–9046. doi: 10.1073/pnas.0802218105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fenton RR. Molesworth-Kenyon S, Oakes JE, Lausch RN. Linkage of IL-6 with neutrophil chemoattractant expression in virus-induced ocular inflammation. Invest Ophthalmol Vis Sci. 2002;43(3):737–743. [PubMed] [Google Scholar]
- 30.Wolters PJ, Wray C, Sutherland RE, Kim SS, Koff J, Mao Y, et al. Neutrophil-derived IL-6 limits alveolar barrier disruption in experimental ventilator-induced lung injury. J Immunol. 2009;182(12):8056–8062. doi: 10.4049/jimmunol.0801323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fielding CA, McLoughlin RM, McLeod L, Colmont CS, Najdovska M, Grail D, et al. IL-6 regulates neutrophil trafficking during acute inflammation via STAT3. J Immunol. 2008;181(3):2189–2195. doi: 10.4049/jimmunol.181.3.2189. [DOI] [PubMed] [Google Scholar]
- 32.Zheng L, Walters EH, Ward C, Wang N, Orsida B, Whitford H, et al. Airway neutrophilia in stable and bronchiolitis obliterans syndrome patients following lung transplantation. Thorax. 2000;55(1):53–59. doi: 10.1136/thorax.55.1.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McAllister F, Henry A, Kreindler JL, Dubin PJ, Ulrich L, Steele C, et al. Role of IL-17A, IL-17F, and the IL-17 receptor in regulating growth-related oncogene-alpha and granulocyte colony-stimulating factor in bronchial epithelium: implications for airway inflammation in cystic fibrosis. Journal of immunology. 2005;175(1):404–412. doi: 10.4049/jimmunol.175.1.404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huang F, Kao CY, Wachi S, Thai P, Ryu J, Wu R. Requirement for both JAK-mediated PI3K signaling and ACT1/TRAF6/TAK1-dependent NF-kappaB activation by IL-17A in enhancing cytokine expression in human airway epithelial cells. Journal of immunology. 2007;179(10):6504–6513. doi: 10.4049/jimmunol.179.10.6504. [DOI] [PubMed] [Google Scholar]
- 35.Roussel L, Houle F, Chan C, Yao Y, Berube J, Olivenstein R, et al. IL-17 promotes p38 MAPK-dependent endothelial activation enhancing neutrophil recruitment to sites of inflammation. Journal of immunology. 2010;184(8):4531–4537. doi: 10.4049/jimmunol.0903162. [DOI] [PubMed] [Google Scholar]