

Abstract
Sera of patients with cancer contain membraneous microvesicles (MV) able to induce apoptosis of activated T cells by activating the Fas/Fas ligand pathway. However, the cellular origin of MV found in cancer patients’ sera varies as do their molecular and cellular profiles. To distinguish tumor-derived MV in cancer patients’ sera, we used MAGE 3/6+ present in tumors and MV. Molecular profiles of MAGE 3/6+ MV were compared in Western blots or by flow cytometry with those of MV secreted by dendritic cells or activated T cells. These profiles were found to be distinct for each cell type. Only tumor-derived MV were MAGE 3/6+ and were variably enriched in 42-kDa Fas ligand and MHC class I but not class II molecules. Effects of MV on signaling via the TCR and IL-2R and proliferation or apoptosis of activated primary T cells and T cell subsets were also assessed. Functions of activated CD8+ and CD4+ T lymphocytes were differentially modulated by tumor-derived MV. These MV inhibited signaling and proliferation of activated CD8+ but not CD4+ T cells and induced apoptosis of CD8+ T cells, including tumor-reactive, tetramer+CD8+ T cells as detected by flow cytometry for caspase activation and annexin V binding or by DNA fragmentation. Tumor-derived but not dendritic cell-derived MV induced the in vitro expansion of CD4+CD25+FOXP3+ T regulatory cells and enhanced their suppressor activity. The data suggest that tumor-derived MV induce immune suppression by promoting T regulatory cell expansion and the demise of antitumor CD8+ effector T cells, thus contributing to tumor escape.
Microvesicles (MV)3are membrane vesicles that form within late endocytic compartments or multivesicular bodies and are secreted upon fusion of multivesicular bodies with the plasma membrane (1–3). Various normal cells secrete MV, including hemopoietic cells such as reticulocytes, B lymphocytes, platelets, mastocytes, T lymphocytes, and dendritic cells (DC), as well as nonhemopoietic cells, e.g., epithelial cells or fibroblasts (2–5). In addition, tumor cells of various histologies are known to produce and secrete MV (6–10).
MV originating from different cell types have certain features in common, including an outer lipid bilayer, the size (50–100 nm in diameter), the ability to float at the density of 1.13–1.20 g/ml on sucrose gradients, and a characteristic molecular profile (2, 9–11). MV are characterized by the presence of members of the tetraspanin family: CD9, CD63, and CD81, integral and membrane-associated adhesion molecules, e.g., MFG-E8, integrins, and ICAM, and various housekeeping proteins such as tsg 101, several annexins, and Rab GTPases (2, 8). MV with these characteristics have also been referred to as exosomes (3, 8–11). In addition, MV are often enriched in molecules characteristic of their parent cell. Thus, DC-derived exosomes (termed “dexosomes”) accumulate proteins involved in T cell costimulation, the MHC class I and II molecules, as well as CD80 and CD86 (9). In contrast, tumor-derived MV carry proteins unique to or enriched in parental tumor cells, including tumor-associated Ags (TAA) or heat-shock proteins (10).
MV perform multiple cellular functions, including the elimination of unnecessary proteins, exchange of proteins and lipids among cells, and delivery of molecular signals from the parent cell to distantly situated cellular targets (4, 12–14). MV can also drive antitumor immune responses by delivering TAA to APC. In tumor-bearing mice, MV purified from DC, which had been pulsed with TAA, promoted antitumor T cell responses and induced tumor regression (15–17). However, MV can also inhibit T cell responses. Thus, MV secreted by intestinal epithelial cells can induce Ag-specific oral tolerance (18); MV secreted by human trophoblasts maintain tolerance at the maternal-fetal interface during pregnancy (19, 20); and MV derived from tumor cells expressing MHC class I molecules and bioactive ligands such as Fas ligand (FasL) or TRAIL suppress functions of antitumor effector T cells (7, 21–24). We have previously reported that MV expressing 42-kDa FasL were detected in the sera of patients with oral carcinoma and that MV-associated FasL levels correlated with the patients’ tumor burden and nodal involvement (25). These MV induced caspase-3 cleavage, cytochrome c release, and loss of mitochondrial membrane potential and reduced TCRζ-chain expression in activated T lymphocytes (25). On the basis of these data, we have suggested that by inducing effector T cell apoptosis, tumor-derived MV facilitate tumor progression and its immune escape (26).
Recent evidence (27) suggests that tumor-derived MV present in cancer patients’ body fluids might be prognostically useful. Because the MV found in the sera of cancer patients could originate from tumor cells and/or various nonmalignant cells, it is important to be able to distinguish those derived from the tumor and confirm their role in effector T cell apoptosis and their prognostic significance. In this study, we report that tumor-derived MV are distinguishable from DC- or T cell-derived MV by a unique molecular profile and immunosuppressive functions, including the promotion of regulatory T cell (Treg) expansion, which contribute to tumor escape from the host immune system.
Materials and Methods
Cells and cell lines
The head and neck squamous cell carcinoma (SCCHN) cell line, PCI-13, was established in our laboratory and maintained as described previously (28). It was retrovirally transfected with the human FasL gene obtained from Dr. S. Nagata (Osaka Biosciences Institute, Osaka, Japan) as reported previously (29). Melanoma cell lines, Mel-SW and SLM-2, obtained from Dr. J. M. Kirkwood (University of Pittsburgh, Pittsburgh, PA), were established from primary tumors and cultured in RPMI 1640 medium supplemented with 10% (v/v) FBS, L-glutamine, and antibiotics (all from Invitrogen). Jurkat cells were obtained from American Type Culture Collection and were also cultured in a complete RPMI 1640 medium. Cultures in the log phase of growth were used for all experiments. In some experiments, Jurkat cells transfected with the CD8 gene were used. The transfected cells were obtained from Dr. H. Rabinowich (University of Pittsburgh). Human fibroblasts were cultured from skin explants as described previously (30). Human epidermal keratinocytes were purchased from Cascade Biologic and cultured according to the manufacturer’s suggestions.
Human monocytes and T lymphocytes were isolated from PBMC obtained from consented normal controls (NC) under the Institutional Review Board (IRB)-approved protocol (IRB no. 980633). PBMC were isolated by centrifugation on Ficoll-Hypaque gradients (GE Healthcare Bio-Sciences). Monocytes were isolated via plastic adherence for 1 h at 37°C in an atmosphere of 5% CO2 in air, and the nonadherent PBMC were collected as a T lymphocyte fraction. Lymphocytes were immediately used for experiments or cryopreserved. CD3+ T cells were purified by negative selection using the Pan T Cell Isolation Kit II (Miltenyi Biotec) on an AutoMACS system according to the manufacturer’s instructions. In some experiments, CD4+ T cells were negatively selected from PBMC as described previously (31). Purified CD3+ T cells or negatively selected T cell subsets (CD4+ or CD8+) were then cultured for 3–5 days in AIM V medium (Invitrogen) supplemented with 10% AB human serum (Gemini) in the presence of beads coated with anti-CD3 and anti-CD28 Abs (T Cell Activation/Expansion Kit; Miltenyi Biotec).
To generate DC, monocytes were cultured in AIM V medium (Invitrogen) supplemented with 10% FBS, 10 ng/ml recombinant human IL-4 (Cellgenix), and 103 IU/ml recombinant human GM-CSF (Berlex) at 37°C in an atmosphere of 5% CO2 in air for 6 days.
All cell lines, DC, and T cells were cultured in media containing FBS depleted of MV by ultracentrifugation.
Serum samples
Venous blood was collected from consented patients with SCCHN or melanoma (IRB no. 960279). All sera were stored at −80°C until use.
Antibodies
Anti-FasL Ab Ab-3 (Oncogene/EMD Biosciences) was purchased for FasL detection by Western blots. Anti-Fas (CH-11) agonistic mAb, IgM isotype control for CH-11, anti-Fas blocking mAb (clone ZB4), and isotype IgG1 control for ZB4 were all purchased from Upstate Biotechnology. Abs used for flow cytometry included anti-CD3, -CD4, -CD8, -CD25, -CD63, -CD80, -CD83, and -CD86 and were all purchased from Beckman Coulter. Anti-Foxp3 Ab was obtained from eBioscience, and annexin V was purchased from Beckman Coulter. mAb W6/32 was produced by Dr. A. De-Leo (University of Pittsburgh), using a hybridoma obtained from American Type Culture Collection. Anti-MHC class I Ab HC10 and anti-MHC class II Ab LGIII 612.14 were provided by Dr. S. Ferrone (University of Pitts-burgh). Anti-MAGE 3/6 mAb was a gift from Dr. G. C. Spagnoli (Institute for Surgical Research and Hospital Management, Basel, Switzerland). Polyclonal rabbit Ab to the C terminus of human MAGE 3/6 peptide273–284 (CYEFLWGPRALV) was used for immunostaining. Antiserum was generated by Covance Research Products and purified by Dr. A. DeLeo.
Isolation of MV
MV were isolated from culture supernatants of the parental or FasL-transduced PCI-13 cell lines, melanoma cell lines, normal human fibroblasts or keratinocytes, DC, and in vitro-activated T cells. MV were also isolated from the sera of melanoma and SCCHN patients whose tumors were in culture and the sera of NC. MV were isolated as described previously (25). Briefly, sera or concentrated (10×) cell culture supernatants were fractionated by a two-step procedure (size-exclusion chromatography and ultracentrifugation). Aliquots (10 ml) of sera or supernatants were applied to a Sepharose 2B (Amersham Biosciences) column (1.5 × 35 cm) equilibrated with PBS. The protein content in 1-ml fractions was monitored by measuring absorbance at 280 nm. The exclusion peak material was centrifuged at 105,000 × g for 1 h at 4°C, and the pellet was resuspended in 0.3 ml of sterile PBS. Protein concentrations were estimated using Lowry’s assay (Bio-Rad), with BSA used as standard.
MAGE 3/6 expression
RT-PCR analysis was used to determine expression of MAGE-6 mRNA by tumors. RNA was isolated from tumor cells using TRIzol reagent (Invitrogen) following the manufacturer’s recommendations. RT-PCR was performed with a GeneAmp RNA PCR Kit (Applied Biosystems). The following primer set was used: MAGE-6, forward, TGGAGGAC CAGAGGCCCCC, and reverse, CAGGATGATTATCAGGAAGCCT GT; product size 728 bp with cycles: melting at 94°C for 45 s, annealing at 68°C for 45 s, and extension at 72°C for 1 min.
MAGE 3/6 expression in tumor cells was also determined by immunohistochemistry, using cytospins of SW-mel and PCI-13 cells (30,000 cells/field). The smears were fixed in 2% (w/v) paraformaldehyde, rehydrated with PBS containing 0.5% BSA, and stained with polyclonal rabbit anti-human MAGE 3/6 Ab (1/200 in 0.5% BSA). Donkey anti-rabbit FITC-labeled Ig (Santa Cruz Biotechnology) was used as a secondary Ab (1/200 in 0.5% BSA). Smears were blocked with 2% BSA (w/v) for 45 min before staining. Secondary Ab alone was used for control slides. The mounting medium contained 4′,6-diamidino-2-phenylindole (Vector Laboratories) to trace cell nuclei. Slides were evaluated in the Olympus Provis (Olympus) fluorescence microscope. For digital image analysis, Adobe Photoshop 6.0 version software was used.
Western blot assays
For Western blots, samples equivalent to 25 μg of protein were used. After separation by 12% SDS-PAGE, proteins were transferred to polyvinylidene difluoride membranes and incubated overnight at 4°C with appropriate Abs. After washing, membranes were incubated with HRP-conjugated secondary Ab at 1/150,000 dilution (Pierce). The signal was detected with a SuperSignal detection system (Pierce). The anti-FasL Abs, Ab-3 and G247–4, previously shown to detect the 42-kDa form of FasL in addition to the soluble 27-kDa form in immunoblots (29), were selected to study FasL expression in MV.
Analysis of MV by flow cytometry
MV (50 μl) were incubated with 20 μl of aldehyde/sulfate latex beads (4 μm in diameter; Interfacial Dynamics) for 15 min at room temperature. MV-coated beads were incubated with BSA suspended in PBS, washed in 100 mM glycine for 30 min, and finally resuspended in 500 μl of flow solution. Aliquots of beads (50 μl) were then incubated with 10 μl of appropriate Abs for 30 min at 4°C, washed with flow solution, and analyzed on Coulter Epics XL-MCL flow cytometer.
Generation of human Treg
Peripheral blood CD4+ T cells were negatively selected using Mitenyi Biotec beads (31). To separate CD4+CD25+ from CD4+CD25− cells, positive selection on anti-CD25 Ab-coated magnetic beads (Miltenyi Biotec) was then applied (31). In some cases, CD4+CD25high T cells were single-cell sorted and used fresh as described previously (31). The selected CD4+CD25+ T cells were cultured in wells of 96-well plates (1 × 105 cells/well) with autologous-irradiated PBMC (20 × 103 cells/well), OKT3 (1 μg/ml), anti-CD28 Ab (1 μg/ml), IL-15 (10 ng/ml), and IL-2 (150 IU/ml). On day 5 or 6 of culture, rapamycin was added (at the final concentration of 50 nM) together with anti CD3/CD28 mAb-coated beads and IL-2 (500 IU/ml). The cells were cultured in a 37°C incubator in the atmosphere of 5% CO2 in air, split every 3–4 days, and supplemented with fresh medium. On day 20 of culture, Treg were phenotyped and tested for proliferation ± MV in CFSE-based assays as described previously (31).
Coincubation of primary T cells or Jurkat cells with MV
T cells were activated in flasks precoated with OKT3 Ab (1 μg/ml; University of Pittsburgh Cancer Institute Pharmacy, Pittsburgh, PA) and cultured in AIM V medium plus soluble anti-CD28Ab or were activated with microbeads coated with anti-CD3 and anti-CD28 Abs (Miltenyi Biotec) at 37°C in 5% CO2 in air for 48 h. MV were coincubated with activated CD3+, CD4+, or CD8+ T cells or Jurkat cells. [3H]Thymidine (5 μCi/ml) was added for the last 12 h of culture to cells that were to be used in JAM assay. T cells for other assays were cultured for 72 h, harvested, and counted in a trypan blue dye. In some experiments, CFSE-labeled responder cells were used to measure proliferation (31). Jurkat T cells were in the log phase of growth and had 100% viability on the day of experiment. Jurkat cells or primary-activated T cells were harvested after coincubation with MV, washed, and used in biological assays. In initial cocultures, MV obtained from various sera or cell supernatants were titrated (from 10 to 200 μg of protein per 1 × 106 T cells), and 30 μg was sufficient to induce Jurkat cell apoptosis, but because MV of various origins had very different biologic activities per protein content, we generally used an excess of MV (100–200 μg of protein per 1 × 106 T cells) unless otherwise specified.
DNA fragmentation (JAM) assay
Jurkat cells or activated T cells were coincubated with MV, and DNA fragmentation in lymphocytes was measured using JAM assays as described previously (32). Percent apoptosis was quantitated using the following formula: percent-specific apoptosis = [(CPMspontaneous − CPMexperimental)/(CPMspontaneous − CPMmaximum) × 100]. CPMspontaneous indicates spontaneous apoptosis of Jurkat T cells, and CPMmaximal indicates maximal apoptosis after treatment of Jurkat cells with 5% (v/v) Triton X-100 in buffer.
Blocking with mAbs
Before JAM assays, anti-Fas-neutralizing mAb, ZB4 (1–10 μg/ml), or IgG1 isotype control Ab was added to Jurkat cells or primary-activated T cells, and the cells were then incubated with MV-containing fractions. The percent inhibition in the presence of Abs was calculated. Pan-caspase inhibitor Z-VAD-FMK (BD Pharmingen) was used in selected blocking experiments at the concentration of 20 μM.
In vitro stimulation (IVS) and apoptosis of tumor-specific T cells
PBMC obtained form HLA-A2+ melanoma patients (n = 5) were used for preparation of immature DC (iDC) as described above. DC were matured using a mixture of IL-1β, TNF-α, IL-6 (all at 10 ng/ml), and PGE2 (1 μg/ml) for 24 h. Mature DC (mDC) were then pulsed with melanoma peptides: tyrosinase368 –376, MART-127–35, gp100209 –217, and MAGE 3/6271–279 (10 μg/ml) and used for IVS of autologous CD3+ T cells at the ratio 1:20 in the presence of IL-2 and IL-7 (10 ng/ml) as described previously (33). CD3+ T cells in 7-day IVS cultures were coincubated with tumor-derived MV for 24 h and analyzed in ELISPOT for IFN-γ secretion upon restimulation with T2 cells pulsed with the same melanoma peptides used for priming (33). The frequency of MART-1- and gp100-positive T cells after IVS ± MV treatment was also analyzed by flow cytometry after MART-1 and gp100 tetramer staining. Apoptosis of effector cells ± MV was determined by annexin V binding as described previously (25).
Caspase activation and annexin V binding to tetramer+ T cells
Jurkat cells or primary-activated T cells or T cell subsets coincubated with MV were tested for activation of intracellular caspases using CaspACE FITC-VAD-FMK In Situ Marker (Promega). T cells were labeled with 5 μM VAD-FITC, washed, incubated with Abs to CD3, CD4 or CD8, and analyzed by flow cytometry. To test for annexin V binding, T cells coin-cubated with MV were first stained with PE-conjugated, HLA-A2-restricted tetramers: MART-1 (AAGIGILTV) and gp100 (IMDQVPFSV) produced by the Tetramer Facility, National Institute of Allergy and Infectious Diseases. After tetramer staining for 30 min in the dark, T cells were incubated for 15 min with ECD-CD3 and PC7-CD8 Abs and with 7-aminoactinomycin D to discriminate dead cells. After washing with PBS (2% FBS, 0.2% azide), cells were washed once with annexin-binding buffer and stained with Annexin VFITC for 15 min on ice and immediately analyzed for five parameters in a FC500 cytometer (Beckman Coulter).
ELISPOT assay for IFN-γ release
The ELISPOT IFN-γ assay was performed as previously described in 96-well flat-bottom nitrocellulose plates (MAHAS4510; Millipore) using the anti-IFN-γ mAb, 1-D1K, as the capture mAb and the biotinylated anti-IFN-γ mAb, 7-B6-1, as the detection mAb (both from Mabtech) (33). Plates were developed with avidin-peroxidase (Vectastain Elite kit; Vector Laboratories) followed by 3-amino-9-ethyl-carbazole (Sigma-Aldrich). The spots were automatically counted by computer-assisted video image analysis (ELISPOT 4. 14.3; Zeiss).
Transmission electron microscopy
Transmission electron microscopy of MV was done in the Center for Biologic Imaging at the University of Pittsburgh Medical School. For transmission electron microscopy, the exosome pellets were fixed in 2.5% (w/v) glutaraldehyde in PBS, dehydrated, and embedded in Epon. Ultrathin sections (65 nm) were cut and stained with uranyl acetate and Reynold’s lead citrate. The sections were examined in a JEOL 1210 transmission electron microscope.
Results
MV in tumor cell supernatants and sera
MV were isolated from the sera of normal individuals (n = 25), the sera of cancer patients (SCCHN and melanoma; n = 35), tumor culture supernatants and supernatants of ex vivo-differentiated DC, primary-activated T cells, fibroblasts, and keratinocytes using a procedure summarized in the supplemental Fig. 1A.4 MV isolated from supernatants of transfected PCI-13 cells, which contained both soluble FasL and the 42-kDa membranous form of FasL, were used as positive controls. All MV fractions analyzed by transmission electron microscopy contained characteristic MV bound by a double membrane (supplemental Fig. 1B).4 MV varied in size from 50 to 100 nm in diameter. MV were present in the sera of >75% cancer patients, but few, if any, MV were observed in the sera of NC. Protein content in MV isolated from 10-ml aliquots of serum was much higher for cancer patients than for the normal individuals: 15–321 vs 0 – 65 μg, respectively.
MV purified from the sera of cancer patients contain TAA
We had available sera of the two melanoma and one SCCHN patients, whose tumors had been previously established as cell lines. These three tumor cell lines (PCI- 13, Mel-SW, and SLM-2) were tested for expression of MAGE 3/6, a cancer testis Ag, by immunohistochemistry and by RT- PCR. The tumor cells, including PCI-13, showed intracytoplasmic staining for MAGE 3/6 (Fig. 1A). All three cell lines, but not Jurkat cells used as controls, were positive for MAGE 3/6 mRNA (Fig. 1B) and MAGE 3/6 protein in the lysates of isolated MV by Western blots (Fig. 1C). MV purified from supernatants of these cells as well as MV purified from the banked sera of the patients from whom these tumors originated were all positive for MAGE 3/6 (Fig. 1C). MAGE 3/6 was not detected on MV purified from normal cells (DC, T cells, fibroblasts, and keratin-ocytes) or MV fractions purified from the sera obtained from NC. These results suggest that at least a fraction of MV present in the sera of cancer patients originate from the tumor. MAGE 3/6 was also detected on 6 of 27 MV fractions purified from the sera of randomly selected SCCHN patients (data not shown).
FIGURE 1.

MAGE 3/6 is expressed by tumor cells and is present on tumor-derived MV. A, PCI 13 tumor cells immunostained for MAGE 3/6: 1) isotype control; and 2) a confocal microscopy image showing intracytoplasmic staining (green) for MAGE 3/6; cell nuclei are blue (4′,6-diamidino-2-phenylindole (DAPI+)); magnification, ×200. Inset, magnification, ×600. B, Expression of MAGE 3/6 by tumor cells as tested by RT-PCR. A representative of three experiments performed. C, Western blot analysis of MAGE 3/6 expression in serum-derived or cell culture-derived MV. A representative of five Western blot experiments performed is shown.
Molecular profile of tumor-derived MV in the sera of cancer patients
To further explore the cellular profile of MV recovered from cancer patients’ sera, expression of membrane-bound FasL, MHC class I, and costimulatory molecules on these MV and also on MV obtained from supernatants of cultured tumor and normal cells was evaluated. As shown in Fig. 2A, MV purified from the sera of the three cancer patients and supernatants of their tumor cells all contained 42-kDa FasL as did MV purified from supernatants of primary-activated T cells whereas MV originating from DC, keratinocytes, or normal fibroblasts and from the sera of NC did not. Fig. 2, A and B, also show that all MV, regardless of their origin, contained lysosomal-associated membrane protein 1 (albeit at different levels), a marker of the endosomal compartment, and tetraspanin CD63 (data not shown), confirming their endosomal origin. All analyzed MV also contained MHC class I but not always MHC class II molecules. Tumor-derived MV had MHC class II levels similar to those on tumor cells from which they originated. Thus, PCI-13 cells with low MHC class II expression (34) produced MV with undetectable MHC class II, whereas SLM-2 and Mel-SW, which are rich in MHC class II, produced MV, which were MHC class II+ (data not shown).
FIGURE 2.

Western blot and flow cytometry analyses of MV derived from tumor cells, sera of patients with cancer, activated T cells, mDC, and cultured tumor or normal cells. A, Western blots of serum derived or culture-derived MV. Each lane was loaded with 25 μg of protein. A representative of 10 Western blot experiments performed is shown. B, Flow analysis of MV bound to latex beads. Aliquots of bead-bound MV were incubated with the PE-labeled Abs as indicated. Dotted lines indicate controls (i.e., bead-bound MV incubated with isotype control). A representative experiment of six performed is shown.
MV were also analyzed for the presence of B7 costimulatory molecules. Only DC-derived MV expressed CD86. Tumor-derived MV were characterized by the paucity of CD86 (Fig. 2B) and CD80 (data not shown). Only MV originating from activated T cells expressed CD3. Surprisingly, T cell-derived MV expressed little CD95 on their surface (data not shown).
Effects of MV on T cell proliferation
CD3+ T cells purified from PBMC of NC by negative selection were separated into CD4+ and CD8+ T cells and used as responder cells in cocultures with purified MV originating from different cells. Relative to anti-CD3 Ab, DC- or T cell-derived MV only moderately supported proliferation of resting CD4+ and CD8+ cells (Fig. 3A). MV originating from tumor cell lines (PCI-13 and SW-mel) did not support proliferation of T cells. Surprisingly, tumor-derived MV added to preactivated (anti-CD3 Ab-treated) T cell subsets induced expansion of CD4+ T cells while they were consistently and significantly inhibitory with primary-activated CD8+ T cells (p < 0.01; Fig. 3B). These data showed that MV can regulate T cell expansion and that resting vs activated T cell subsets are differentially regulated by tumor-derived MV.
FIGURE 3.

Effects of MV on proliferation of CD4+ or CD8+ T lymphocyte subsets. A, Resting CD4+ and CD8+ T cells were purified and coin-cubated with MV of various origins. Proliferation was measured by [3H]thy-midine incorporation. Responder cells (0.5 × 106) were cultured in the presence of MV (200 μg of protein). MV were isolated from activated T cells, mDC, SW-mel, or PCI-13 cells. As a positive control, OKT-3 Ab was used. B, Effects of MV on proliferation of activated CD4+ or CD8+ T cells. MV of different origins were coincubated with CFSE-labeled, OKT-3 Ab-activated T cell subsets, and proliferation was assayed by flow cytometry. The proliferation index was calculated using Modfit software. The data are mean values ± SD. The asterisks indicate p < 0.001 for CD4+ T cells and p < 0.01 for CD8+ T cells. C, Four-color flow cytometry analysis of activated CD4+ T cell populations cultured in the presence or absence of tumor-derived (PCI-13) or DC-derived MV. On day 7 of culture, cells were tested for CD25 and FOXP3 expression. The percentages of FOXP3+ cells within the CD3+CD4+ CD25+ subset of cells are indicated in each panel. The data shown in A–C are from individual representative experiments of five performed with T cells obtained from different normal donors.
Tumor-derived MV promote proliferation of Treg
To further investigate the observed increases in proliferation of activated CD4+ T cells upon coincubation with MV, CD4+ T cells were preactivated with anti-CD3 Ab and cultured ± MV derived from tumor cells or DC for 7 days. Gating on CD3+CD4+CD25+ cells in these cultures, we determined the percentages of FOXP3+ cells present, because this marker is generally associated with Treg (31, 35). As shown in Fig. 3C, tumor-derived MV, but not DC-derived MV, induced substantial expansion of CD4+CD25+ FOXP3+ Treg. Titration experiments (1–60 μg of MV protein) showed that the dose of MV equal to 5 μg of protein/0.5 × 106 cells was optimal for expansion of Treg (M. Szajnik, M. Czystowska, M. J. Szczepanski, and T. L. Whiteside, manuscript in preparation). These cells also expressed CTLA-4 and other Treg markers (data not shown).
Treg were also obtained from normal PBMC by expansion of the sorted CD4+CD25+ subset in the presence of IL-2, OKT3/CD28 Ab-coated beads, and rapamycin as previously described by us (35). The expanded cell population consisted entirely of CD4+CD25highFOXP3+ cells (Fig. 4A). All of these cells also expressed GITR and CTLA-4 (data not shown) and mediated strong suppression of activated autologous CD4+CD25− responder cells (35). Coincubation of these Treg in the presence of tumor-derived of MV for 6 days resulted in a significant Treg expansion compared with controls, as determined in CFSE assays or by cell counts (Fig. 4B). In contrast, MV isolated from supernatants of human iDC did not enhance Treg proliferation (Fig. 4C). When freshly isolated CD4+CD25high Treg were used in place of those cultured with rapamycin, the same results were observed (Fig. 4, B and C, right).
FIGURE 4.

Tumor-derived MV expand Treg. Isolated CD4+CD25+ T cells were cultured in the presence of rapamycin as described in Materials and Methods. A, Phenotypic characteristics of rapamycin-expanded Treg tested in day 20 of culture. All expanding cells were CD4+CD25highFOXP3+. B, Proliferation of CFSE-labeled, ex vivo-generated Treg ± MV in 7-day cultures. MV were either tumor-derived (PCI-13) or were isolated from supernatants of human iDC. Proliferation index (PI) for each culture was obtained using Modfit software. C, Cell numbers (means ± SD of 4 wells) in cultures of rapamycin (Rapa)-expanded (left) or freshly isolated (right) Treg incubated ± MV derived from PCI-13 or iDC supernatants. The data shown in C are from three independent experiments.
In addition to increasing Treg expansion, tumor-derived MV also induced TGF-β production by Treg (Table I) and up-regulated SMAD 2/3 phosphorylation in Treg (1).
Table I.
Level of TGF-β1 in the supernatant of Treg coincubated with or without MVa
| TGF-β1 (pg/ml) | |
|---|---|
| Treg alone | 20 ± 3 |
| Treg coincubated with: | |
| MVSKOV3 | 144 ± 34 |
| MVHeLa | 136 ± 27 |
| MVAD10 | 68 ± 22 |
| Medium plus MV | BLDb |
Rapamycin-expanded Treg (1 × 106 cells/well) were cultured in the presence or absence of MV for 72 h. Supernatants were tested for TGF-β1 level by ELISA.
BLD, below level of detection.
Tumor-derived MV induce apoptosis in Jurkat T cells
As indicated above, tumor-derived MV promoted expansion of total activated CD4+ T cells and of Treg but inhibited that of activated CD8+ T cells, consistent with the previously reported sensitivity of primary CD8+ T cells to MV-induced apoptosis (36). Therefore, we confirmed using CD8+ Jurkat cells that tumor-derived MV induced death in these T cells. First, DNA fragmentation induced by tumor-derived MV was measured in CD8+ Jurkat cells using JAM assays, and it was evident in ~ 20% of these cells (Fig. 5A) as also reported previously (25). This DNA fragmentation was significantly inhibited by zVAD (Fig. 5A). In contrast to tumor-derived MV, those derived from DC (Fig. 5A) or T cells (data not shown) did not induce any DNA fragmentation in Jurkat cells. The CH-11 Ab used as a positive control induced DNA fragmentation in ~60% of these cells. To confirm these data, we counted the viable and dead Jurkat cells in cocultures with MV using a trypan blue dye. As shown in Table II, only tumor-derived MV induced significant death of CD8+ Jurkat cells. Furthermore, the percentages of CD8+ Jurkat cells binding annexin V or positive for pan-caspase activity were comparable to those observed with a positive control Ab (CH-11) as shown in Fig. 5, B and C.
FIGURE 5.
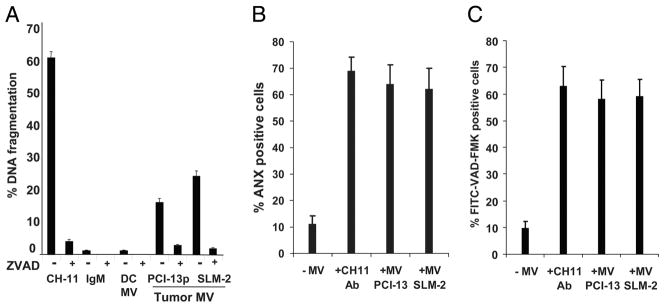
Tumor-derived MV induce apoptosis and signaling defects in Jurkat cells. A, Apoptosis of CD8+ Jurkat cells after coincubation with MV measured in JAM assay as DNA fragmentation. Apoptosis was blocked by preincubation of T cells with pan-caspase inhibitor Z-VAD-FMK. Data are means ± SD of quadruplicate wells obtained in 10 experiments performed is shown. B, A representative experiment of caspase activation in CD8+ Jurkat cells coincubated with tumor-derived MV or CH-11A or Ab isotype control for 24 h. C, Annexin V (ANX) binding to CD8+ Jurkat cells coincubated with tumor-derived MV as in B. The data in B and C are mean values ± SD from one representative experiment of five performed.
Table II.
Death of CD8+ Jurkat cells coincubated with MV derived from normal cells or from tumor cellsa
| Treatment | No. of Cells ×104
|
Percentage of Dead Cells | ||
|---|---|---|---|---|
| Live Cells | Dead Cells | Total Cells | ||
| Untreated Jurkat | 159 | 17 | 176 | 10 |
| + MV (keratinocytes) | 137 | 32 | 169 | 19 |
| + MV (fibroblasts) | 175 | 42 | 217 | 19 |
| + MV (DC) | 173 | 29 | 202 | 14 |
| + MV (SW-mel) | 67 | 123 | 190 | 65b |
| + MV (PCI 13p) | 51 | 130 | 181 | 72b |
CD8+ Jurkat cells in the log phase of growth were coincubated with MV (100 μg/ml) for 72 h. Cells stained with a trypan blue dye were counted per well in triplicate wells. The data are mean values.
Significant differences from untreated Jurkat cells at p < 0.001.
Signaling in activated T cells coincubated with tumor-derived MV
To evaluate effects of tumor-derived MV on signaling in activated primary T cells, expression of the TCR-associated ζ-chain as well as JAK3 and phosphorylated STAT5 was studied by Western immunoblots. Primary CD8+ and CD4+ T cells were activated with OKT3/anti-CD28 Ab-coated microbeads before the addition of MV. MV derived from normal cells (DC and fibroblasts) did not down-regulate expression of the ζ-chain or JAK 3 in T cells, whereas tumor-derived MV consistently significantly and inhibited expression of these signaling molecules (Fig. 6A). Interestingly, time course experiments showed that CD3 ζ-chain expression was dramatically decreased upon the addition of MV only in resting CD8+ T and not resting CD4+ T cells (Fig. 6B). Activated CD8+ T cells had lower ζ-chain expression than activated CD4+ T cells, and they were delayed in recovering its expression after coincubation with MV (Fig. 6C). The data indicate that MV induce down-regulation of ζ in resting as well as activated CD8+ T cells, but not CD4+ T cells, and that CD8+ T cells recover ζ expression very slowly after MV intervention.
FIGURE 6.

MV induce down-regulation of CD3ζ and JAK3 expression in primary-activated CD8+ T cells. A, Western blot analysis of CD3ζ and JAK3 expression in activated T cells after coincubation with MV. The CD3ζ/actin and JAK3/actin ratios for the Western blot analysis are shown on the right. The data are representative of five experiments performed with primary T cells of different donors. B, CD3ζ expression in nonactivated (resting) T cells ± MV. C, CD3ζ expression in the T cell subsets activated with microbeads coated with OKT3 and anti-CD28 mAb for 48 h before the addition of MV. The data in B and C are ratios of CD3ζ/actin derived from Western blot analyses performed at different times after tumor-derived MV were added to activated T cells. D, Phosphorylated STAT5/STAT5 ratios in T cell subsets activated as indicated above and incubated ± MV. B–D, Representative experiments of three performed from each condition is shown.
STAT5 phosphorylation was higher in activated CD8+ than CD4+ T cells (Fig. 6D). The addition of MV to activated CD8+ T cells dramatically decreased phosphorylated STAT5 expression (Fig. 6D). In contrast, MV increased phosphorylated STAT5 expression in activated CD4+ T cells.
In aggregate, these experiments are consistent with the conclusion that activated primary CD8+ T cells are selectively targeted by tumor-derived MV, which interfere with TCR- and IL-2R-mediated signaling. In contrast, DC-, T cell-, or fibroblast-derived MV did not cause similar signaling defects in activated primary T cells.
Tumor-derived MV eliminate tumor-reactive T cells
To determine whether tumor-reactive activated T cells were especially sensitive to inhibitory signals and/or apoptosis mediated by tumor-derived MV, T cells obtained from HLA-A2+ melanoma patients were in vitro-sensitized using autologous mDC pulsed with the melanoma peptides tyrosinase368–376, MART-127–35, gp100209–217, and MAGE 3/6271–279. After 7 days of stimulation, during which expansion of melanoma-specific T cells occurred (as determined by tetramer-based analysis; data not shown), the expanded CD3+ T cells were coincubated with melanoma-derived MV for 4–24 h. These T cells were then analyzed in ELISPOT for IFN-γ secretion upon restimulation with T2 cells pulsed with the same melanoma peptides. As shown in Fig. 7A, MV induced a significant decrease in the number of peptide-responsive T cells. Thus, peptide-specific T cells were sensitive to MV-mediated apoptosis, although the elimination by MV of melanoma peptide-specific T cells was not complete.
FIGURE 7.

Effects of MV on tumor peptide-reactive T cells. A, IFN-γ ELISPOT analysis of an HLA-A2+ melanoma patient’s T cells generated by IVS with melanoma peptides presented by autologous mDC. These T cells were then coincubated with tumor-derived MV as described in Materials and Methods. The data are mean spot numbers ± SD from quadruplicate wells in a representative ELISPOT assay out of five performed. The numbers of IFN-γ spots produced by T cells not treated with MV (□) and (■) the numbers of spots produced by MV-treated T cells. MV-treated or MV-untreated T cells were stimulated with T2 cells loaded with the indicated peptides in ELISPOT assays. Asterisks indicate p < 0.01. B, Annexin V binding to melanoma-specific (MART-1 tetramer+ or gp100 tetramer+) T cells coincubated with tumor-derived MV or control MART-1 tetramer+ or gp100 tetramer+CD8+ T cells incubated in the absence of MV. The last right panel shows inhibition annexin V binding in the presence of ZB4 mAb. A representative experiment of three performed is shown.
To further document that MV induced apoptosis of the peptide-responsive CD8+ T cells generated in IVS, annexin V binding to MART-1 and gp100 tetramer+CD8+ T cells ± MV was measured by flow cytometry. As shown in Fig. 7B, in the absence of MV, only a small fraction of tetramer+CD8+ T cells bound annexin V. After the coincubation of melanoma peptide-activated T cells with MV, a 2- to 4-fold increase in the proportion of annexin V +tetramer+CD8+ T cells was observed. However, when ZB4 Ab was added, annexin V binding to tetramer+CD8+ T cells was inhibited (Fig. 7B, right panels). The data are consistent with the interpretation that peptide-specific CD8+ T cells are sensitive to MV-mediated apoptosis, which is mediated by Fas/FasL interactions.
Discussion
The ability to produce and release MV is an attribute of activated cells. MV found in the sera of cancer patients can originate not only from the tumor but also from clinically activated immune or nonimmune cells. MV that are tumor-derived contain TAA, and their molecular profile is distinct from that of DC- or T cell-derived MV. We demonstrate that MV originating from the tumor in culture and those derived from the serum of the patient bearing the same tumor have the same molecular profile. This confirms that cancer patients’ sera can be a reliable source of tumor-derived MV. Nevertheless, to be able to segregate tumor-derived MV and those originating from other cells in the sera of patients, it will be necessary to capture and concentrate them for further analysis. Strategies using Ab-coated beads are being currently developed to isolate tumor-derived MV from patients’ body fluids to be able to evaluate their prognostic potential relative to that of other serum markers.
Not only the phenotype, but also functions of MV differed depending on their origin. Although T cell-derived MV or DC-derived MV were immunostimulatory and promoted ex vivo proliferation of resting T cells, tumor-derived MV did not. In fact, they consistently impaired signaling and induced apoptosis of activated primary CD8+ T cells, thus inhibiting their proliferation. Also, tumor-derived MV significantly promoted proliferation of activated CD4+ T cells despite the low level or absence of MHC class II Ags associated with these MV, as shown by flow cytometry. A substantial fraction of these proliferating CD4+ T cells was represented by CD4+CD25highFOXP3+ Treg. The stimulatory effects of tumor-derived MV on CD4+ T cells, especially Treg, contrast with inhibitory activities these MV exert on CD8+ T lymphocytes. These features of tumor-derived MV suggest that in vivo they could be involved in facilitating tumor escape from the host immune system.
Recent studies have identified various molecular and cellular mechanisms responsible for tumor escape, including accumulations of Treg at the tumor site and in the peripheral circulation of cancer patients and local as well as systemic apoptosis of CD8+ T lymphocytes (37). With respect to Treg, we and others (35, 38) have reported that cancer progression is associated with expansion of Treg in tumor-infiltrating lymphocytes and the peripheral blood of patients. These increased frequencies of Treg have been linked to poor prognosis and shorter survival in patients with cancer (39). The mechanisms involved in the recruitment of Treg to tumors or in Treg expansion in situ are not well defined. One such mechanism could involve tumor-derived MV, which are present in serum and other body fluids and thus exert not only local but also systemic effects on immune cells. These MV preferentially stimulate expansion of CD4+ T cells by mechanisms that are yet unknown. Our preliminary data indicate that PCI-13-derived MV, as with PCI-13 tumor cells, bear ICOSL (B7-H2; our unpublished data), which could interact with ICOS+CD4+ T cells inducing their transition to Treg. We have previously reported that the ICOS-ICOSL costimulation is a key step for differentiation of nonactivated CD4+CD25+ precursor cells into Treg and for promoting their expansion (40). Therefore, our ex vivo results showing that tumor-derived, but not DC-derived, MV promote Treg expansion from precursors in the peripheral blood, induce TGF-β reduction by Treg, and enhance their suppressor activity (1) provide yet another indication that the tumor actively participates in down-regulating host immune responses.
The data reported by Clayton et al. (41) show that tumor-derived MV selectively interfere with proliferation of T cells in response to IL-2. This inhibitory effect was mediated by membrane-associated TGF-β present in MV (41). The same authors further reported that tumor-derived exosomes down-regulate NKG2D expression on NK or CD8+ T cells via membrane-associated TGF-β1 (42). As TGF-β is involved in the differentiation of Treg in the tumor microenvironment (43) and is produced by Treg, representing one mechanism of Treg-mediated suppression, it is reasonable to assume that MV could induce immune suppression via favoring Treg expansion. For example, MV present in human breast milk have been reported to increase the number of CD25+FOXP3+CD4+ Treg upon coincubation with normal PBMC (44).
The presence of CD8+ antitumor effector cells in the circulation of cancer patients has been demonstrated in many studies (e.g., Ref. 45). However, these antitumor effector cells are largely ineffective in controlling tumor progression. We and others (46) have suggested that tumor-driven untimely death of antitumor effector cells is one of the mechanisms responsible for this lack of antitumor activity. In this study, we used an IVS model to show that lymphocytes obtained from a patient with melanoma and activated ex vivo with cognate melanoma peptides were highly susceptible to apoptosis induced by tumor-derived MV, which effectively eliminated a significant fraction of IFN-γ producing peptide-specific CD8+ T cells. In the IVS cultures enriched in CD8+ tetramer+ T cells, MV derived from the autologous tumor or allogeneic HLA-A2+ tumors other than melanoma effectively eliminated tumor peptide-specific (tetramer+CD8+) effector cells. The mechanism(s) responsible for T effector demise might involve the biologically active membrane form of FasL present on tumor-derived MV (24, 25, 46–48). Levels of FasL expression on MV isolated from cancer patients’ sera correlated with their apoptosis-inducing activity and with disease activity in patients with head and neck cancer (27). However, not all MV derived from the sera of cancer patients express FasL. Their death-inducing, growth inhibitory effects could be mediated by other death ligands such as programmed death ligand 1 or TRAIL known to be present in tumor-derived MV and able to engage the extrinsic pathway of apoptosis (22). We have recently shown (34) that these MV also activate the intrinsic (mitochondrial) death pathway in CD8+ T cells. The subsequent activation of the executor caspases leads to the demise of effector T cells, which are normally responsible for immune defense.
Previous studies have shown that tumor-derived factors are responsible for down-regulation of CD3ζ expression as well as the JAK3 and p-STAT5 signaling pathway in T cells coincubated with tumor cells (49, 50). In this study, tumor-derived MV, despite low expression levels of FasL, were able to suppress expression of CD3ζ and JAK3 proteins in activated CD8+ T cells whereas DC-or fibroblasts-derived MV did not. Modulation via TCR engagement of the JAK3 pathway is critical for type 1 cytokine receptors, which use the common γ-chain and can modulate CD3ζ-mediated activation upon exposure to IL-2, IL-7, or IL-15. Down-regulation by MV of the key components (the ζ-chain and JAK3) of this signaling pathway appears to lead to T cell anergy (49–51).
In aggregate, our ex vivo experiments indicate that tumor-derived MV ubiquitous in cancer patients’ sera or supernatants of tumor cells have the capacity to eliminate antitumor effector cells and, in parallel, to support expansion of CD4+CD25highFOXP3+ Treg. This coordinated double insult to cellular immunity appears to represent an important mechanism contributing to immune evasion in cancer.
Supplementary Material
Footnotes
This work is supported in part by the National Institutes of Health Grant P01 CA109688 (to T.L.W.). M.S. was supported by the National Heart, Lung, and Blood Institute-sponsored Production Assistance for Cellular Therapies Program N01-HB-37165.
Abbreviations used in this paper: MV, microvesicle; DC, dendritic cell; FasL, Fas ligand; IVS, in vitro stimulation; NC, normal control; SCCHN, head and neck squamous cell carcinoma; TAA, tumor-associated Ag; Treg, regulatory T cell; iDC, immature DC.
The online version of this article contains supplemental material.
Disclosures
The authors have no financial conflict of interest.
References
- 1.Denzer K, Kleijmeer MJ, Heijnen HF, Stoorvogel W, Gellze HJ. Exosome: from internal vesicle of the multivesicular body to intercellular signaling device. J Cell Sci. 2000;113:3365–3374. doi: 10.1242/jcs.113.19.3365. [DOI] [PubMed] [Google Scholar]
- 2.Johnstone RM. Exosomes biological significance: a concise review. Blood Cells Mol Dis. 2006;36:315–321. doi: 10.1016/j.bcmd.2005.12.001. [DOI] [PubMed] [Google Scholar]
- 3.van Niel G, Porto-Carreiro I, Simors S, Raposo G. Exosomes: a common pathway for a specialized function. J Biochem. 2006;140:13–21. doi: 10.1093/jb/mvj128. [DOI] [PubMed] [Google Scholar]
- 4.Ratajczak J, Wysoczynski M, Hayek F, Janowska-Wieczorek A, Ratajczak MZ. Membrane-derived microvesicles: important and underappreciated mediators of cell-to-cell communication. Leukemia. 2006;20:1487–1495. doi: 10.1038/sj.leu.2404296. [DOI] [PubMed] [Google Scholar]
- 5.Martinez-Lorenzo MJ, Anel A, Gamen S, Monle NI, Lasierra P, Larrad L, Pineiro A, Alava MA, Naval J. Activated human T cells release bioactive Fas ligand and APO2 lignad in microvesicles. J Immunol. 1999;163:1274–1281. [PubMed] [Google Scholar]
- 6.Martinez-Lorenzo MJ, Anel A, Alava MA, Pineiro A, Naval J, Lasierra P, Larrad L. The human melanoma cell line MelJuSo secretes bioactive FasL and APO2L/TRAIL on the surface of microvesicles: possible contribution to tumor counterattack. Exp Cell Res. 2004;295:315–329. doi: 10.1016/j.yexcr.2003.12.024. [DOI] [PubMed] [Google Scholar]
- 7.Valenti R, Huber V, Iero M, Filipazzi P, Parmiani G, Rivoltini L. Tumor-released microvesicles as vehicles of immunosuppression. Cancer Res. 2007;67:2912–2915. doi: 10.1158/0008-5472.CAN-07-0520. [DOI] [PubMed] [Google Scholar]
- 8.Keller S, Sanderson MP, Stoeck A, Altevogt P. Exosomes: from biogenesis and secretion to biological function. Immunol Lett. 2006;107:102–108. doi: 10.1016/j.imlet.2006.09.005. [DOI] [PubMed] [Google Scholar]
- 9.Mears R, Craven RA, Hanrahan S, Totty N, Upton C, Young SL, Patel P, Selby PJ, Banks RE. Proteomic analysis of melanoma-derived exosomes by two-dimensional polyacrylamide gel electrophoresis and mass spectrometry. Proteomics. 2004;4:4019–4031. doi: 10.1002/pmic.200400876. [DOI] [PubMed] [Google Scholar]
- 10.Hegmans JP, Bard MP, Hemmes A, Luider TM, Kleijmeer MJ, Prins JB, Zitvogel L, Burgers SA, Hoogsteden HC, Lambrecht BN. Proteomic analysis of exosomes secreted by human mesothelioma cells. Am J Pathol. 2004;164:1807–1815. doi: 10.1016/S0002-9440(10)63739-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bard MP, Hegmans JP, Hemmes A, Luider TM, Willemsen R, Severijnen LA, van Meerbeeck JP, Burgers SA, Hoogsteden HC, Lambrecht BN. Proteomic analysis of exosomes isolated from human malignant pleural effusions. Am J Respir Cell Mol Biol. 2004;31:114–121. doi: 10.1165/rcmb.2003-0238OC. [DOI] [PubMed] [Google Scholar]
- 12.Al-Nedawi K, Meehan B, Micallef J, Lhotak V, May L, Guha A, Rak J. Intercellular transfer of the oncogenic receptor EGFRvIII by microvesicles derived from tumour cells. Nat Cell Biol. 2008;10:619–624. doi: 10.1038/ncb1725. [DOI] [PubMed] [Google Scholar]
- 13.Cocucci E, Racchetti G, Meldolesi J. Shedding microvesicles: artefacts no more. Cell. 2008;19:43–51. doi: 10.1016/j.tcb.2008.11.003. [DOI] [PubMed] [Google Scholar]
- 14.Fader CM, Savina A, Sanchez D, Colombo MI. Exosome secretion and red cell maturation: exploring molecular components involved in the docking and fusion of multivesicular bodies in K562 cells. Blood Cells Mol Dis. 2005;35:153–157. doi: 10.1016/j.bcmd.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 15.Yang Y, Xiu F, Cai Z, Wang J, Wang Q, Fu Y, Cao X. Increased induction of antitumor response by exosomes derived from interleukin-2 gene-modified tumor cells. J Cancer Res Clin Oncol. 2007;133:389–399. doi: 10.1007/s00432-006-0184-7. [DOI] [PubMed] [Google Scholar]
- 16.Segura E, Amigorena S, Thery C. Mature dendritic cells secrete exosomes with strong ability to induce antigen-specific effector immune responses. Blood Cells Mol Dis. 2005;35:89–93. doi: 10.1016/j.bcmd.2005.05.003. [DOI] [PubMed] [Google Scholar]
- 17.Kovar M, Boyman O, Shen X, Hwang I, Kohler R, Sprent J. Direct stimulation of T cells by membrane vesicles from antigen-presenting cells. Proc Natl Acad Sci USA. 2006;103:11671–11676. doi: 10.1073/pnas.0603466103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lin XP, Almqvist N, Telemo E. Human small intestinal epithelial cells constitutively express the key elements for antigen processing and the production of exosomes. Blood Cells Mol Dis. 2005;35:122–128. doi: 10.1016/j.bcmd.2005.05.011. [DOI] [PubMed] [Google Scholar]
- 19.Taylor DD, Akyol S, Gercel-Taylor C. Pregnancy-associated exosomes and their modulation of T cell signaling. J Immunol. 2006;176:1534–1542. doi: 10.4049/jimmunol.176.3.1534. [DOI] [PubMed] [Google Scholar]
- 20.Taylor DD, Bohler HC, Gercel-Taylor C. Pregnancy-linked suppression of TcR signaling pathways by a circulating factor absent in recurrent spontaneous pregnancy loss (RPL) Mol Immunol. 2006;43:1872–1880. doi: 10.1016/j.molimm.2005.10.008. [DOI] [PubMed] [Google Scholar]
- 21.Taylor DD, Gercel-Taylor C, Lyons KS, Stanson J, Whiteside TL. T cell apoptosis and suppression of T cell receptor/CD3-ζ by Fas ligand-containing membrane vesicles shed from ovarian tumors. Clin Cancer Res. 2003;9:5113–5119. [PubMed] [Google Scholar]
- 22.Valenti R, Huber V, Filipazzi P, Pilla L, Sovena G, Villa A, Corbelli A, Fais S, Parmiani G, Rivoltini L. Human tumor-released microvesicles promote the differentiation of myeloid cells with transforming growth factor β-mediated suppressive activity on T lymphocytes. Cancer Res. 2006;66:9290–9298. doi: 10.1158/0008-5472.CAN-06-1819. [DOI] [PubMed] [Google Scholar]
- 23.Taylor DD, Gercel-Taylor C. Tumour-derived exosomes and their role in cancer-associated T cell signalling defects. Br J Cancer. 2005;92:305–311. doi: 10.1038/sj.bjc.6602316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Andreola G, Rivoltini L, Castelli C, Huber V, Perego P, Deho P, Squarcina P, Accornero P, Lozupone F, Lugini L, et al. Induction of lymphocyte apoptosis by tumor cell secretion of FasL-bearing microvesicles. J Exp Med. 2002;195:1303–1316. doi: 10.1084/jem.20011624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim JW, Wieckowski E, Taylor DD, Reichert TE, Watkins S, Whiteside TL. Fas ligand-positive membranous vesicles isolated from sera of patients with oral cancer induce apoptosis of activated T lymphocytes. Clin Cancer Res. 2005;11:1010–1020. [PubMed] [Google Scholar]
- 26.Whiteside TL. Tumour-derived exosomes or microvesicles: another mechanism of tumour escape from the host immune system? Br J Cancer. 2005;92:209–211. doi: 10.1038/sj.bjc.6602360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bergmann C, Strauss L, Wieckowski E, Czystowska M, Albers A, Wang Y, Zeidler R, Lang S, Whiteside TL. Tumor-derived microvesicles in sera of patients with head and neck cancer and their role in tumor progression. Head Neck. 2008;31:371–380. doi: 10.1002/hed.20968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Heo DS, Synderman C, Gollin SM, Pan S, Walker E, Deka R, Barnes EL, Johnson JT, Herberman RB, Whiteside TL. Biology, cytogenetics, and sensitivity to immunological effector cells of new head and neck squamous cell carcinoma lines. Cancer Res. 1989;49:5167–5175. [PubMed] [Google Scholar]
- 29.Gastman BR, Atarshi Y, Reichert TE, Saito T, Balkir L, Rabinowich H, Whiteside TL. Fas ligand is expressed on human squamous cell carcinomas of the head and neck and it promotes apoptosis of T lymphocytes. Cancer Res. 1999;59:5356–5364. [PubMed] [Google Scholar]
- 30.Elder EM, Lotze MT, Whiteside TL. Successful culture and selection of cytokine gene-modified human dermal fibroblasts for the biologic therapy of patients with cancer. Hum Gene Ther. 1996;7:479–487. doi: 10.1089/hum.1996.7.4-479. [DOI] [PubMed] [Google Scholar]
- 31.Strauss L, Whiteside TL, Knights A, Bergmann C, Knuth A, Zippelius A. Selective survival of naturally occurring human CD4+CD25+Foxp3+ regulatory T cells cultured with rapamycin. J Immunol. 2007;178:320–329. doi: 10.4049/jimmunol.178.1.320. [DOI] [PubMed] [Google Scholar]
- 32.Matzinger P. The JAM test: a simple assay for DNA fragmentation and cell death. J Immunol Methods. 1991;45:185–192. doi: 10.1016/0022-1759(91)90325-a. [DOI] [PubMed] [Google Scholar]
- 33.Schultes BC, Whiteside TL. Monitoring of immune responses to CA125 with an IFN-γ ELISPOT assay. J Immunol Methods. 2003;279:1–15. doi: 10.1016/s0022-1759(03)00253-9. [DOI] [PubMed] [Google Scholar]
- 34.Ferris RL, Whiteside TL, Ferrone S. Immune escape associated with functional defects in antigen processing machinery in head and neck cancer. Clin Cancer Res. 2006;12:3890–3895. doi: 10.1158/1078-0432.CCR-05-2750. [DOI] [PubMed] [Google Scholar]
- 35.Strauss L, Bergmann C, Gooding W, Johnson JT, Whiteside TL. The frequency and suppressor function of CD4+CD25highFoxp3+ T cells in the peripheral circulation of patients with squamous cell carcinoma of the head and neck. Clin Cancer Res. 2007;13:6301–6311. doi: 10.1158/1078-0432.CCR-07-1403. [DOI] [PubMed] [Google Scholar]
- 36.Czystowska M, Szczepanski MJ, Szajnik M, Quadrini K, Brandwein H, Hadden JW, Signorelli K, Whiteside TL. IRX-2, a novel immunotherapeutic, protects human T cells from tumor-induced cell death. Cell Death Diff. 2009;16:708–718. doi: 10.1038/cdd.2008.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Whiteside TL. Immune suppression in cancer: effects on immune cells, mechanisms and future therapeutic intervention. Semin Cancer Biol. 2006;16:3–15. doi: 10.1016/j.semcancer.2005.07.008. [DOI] [PubMed] [Google Scholar]
- 38.Wolf AM, Wolf D, Stearer M, Gastl G, Gunsilius E, Grubeck-Loebenstein B. Increase of regulatory T cells in the peripheral blood of cancer patients. Clin Cancer Res. 2003;9:606–612. [PubMed] [Google Scholar]
- 39.Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, Evdemon-Hogan M, Conejo-Garcia JR, Zhang L, Burow M, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. 2004;10:942–949. doi: 10.1038/nm1093. [DOI] [PubMed] [Google Scholar]
- 40.Strauss L, Bergmann C, Szczepanski MJ, Lang S, Kirkwood JM, Whiteside TL. Expression of ICOS on human melanoma-infiltrating CD4+CD25highFoxp3+ T regulatory cells: Implications and impact on tumor-mediated immune suppression. J Immunol. 2008;180:2967–2980. doi: 10.4049/jimmunol.180.5.2967. [DOI] [PubMed] [Google Scholar]
- 41.Clayton A, Mitchell JP, Court J, Mason MD, Tabi Z. Human tumor-derived exosomes selectively impair lymphocyte responses to interleu-kin-2. Cancer Res. 2007;67:7458–7466. doi: 10.1158/0008-5472.CAN-06-3456. [DOI] [PubMed] [Google Scholar]
- 42.Clayton A, Mitchell JP, Court J, Linnane S, Mason MD, Tabi Z. Human tumor-derived exosomes down-modulate NKG2D expression. J Immunol. 2008;180:7249–7258. doi: 10.4049/jimmunol.180.11.7249. [DOI] [PubMed] [Google Scholar]
- 43.Bergmann C, Strauss L, Wang Y, Szczepanski MJ, Lang S, Johnson JT, Whiteside TL. T regulatory Type 1 cells (Tr1) in squamous cell carcinoma of the head and neck: mechanisms of suppression and expansion in advanced disease. Clin Cancer Res. 2008;14:3706–3715. doi: 10.1158/1078-0432.CCR-07-5126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Admyre C, Johansson SM, Qazi KR, Filen JJ, Lahesmaa R, Norman M, Neve EP, Scheynius A, Gabrielsson S. Exosomes with immune modulatory features are present in human breast milk. J Immunol. 2007;179:1969–1978. doi: 10.4049/jimmunol.179.3.1969. [DOI] [PubMed] [Google Scholar]
- 45.Romero P, Cerottini JC, Speiser DE. The human T cell response to melanoma antigens. Adv Immunol. 2006;92:187–224. doi: 10.1016/S0065-2776(06)92005-7. [DOI] [PubMed] [Google Scholar]
- 46.Krammer PH, Arnold R, Lavrik IN. Life and death in peripheral T cells. Nat Rev Immunol. 2007;7:532–542. doi: 10.1038/nri2115. [DOI] [PubMed] [Google Scholar]
- 47.Abrahams VM, Straszewski SL, Kamsteeg M, Hanczaruk B, Schwartz PE, Rutherford TJ, Mor G. Epithelial ovarian cancer cells secrete functional Fas ligand. Cancer Res. 2003;63:5573–5581. [PubMed] [Google Scholar]
- 48.Gastman BR, Johnson DE, Whiteside TL, Rabinowich H. Caspase-mediated degradation of T-cell receptor ζ-chain. Cancer Res. 1999;59:1422–1427. [PubMed] [Google Scholar]
- 49.Chen M, Cheng A, Chen YQ, Hymel A, Hanson EP, Kimmel L, Minami Y, Taniguchi T, Changelian PS, O’Shea JJ. The amino terminus of JAK3 is necessary and sufficient for binding to the common γ-chain and confers the ability to transmit interleukin 2-mediated signals. Proc Natl Acad Sci USA. 1997;94:6910–6915. doi: 10.1073/pnas.94.13.6910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stepkowski SM, Kirken RA. Janus tyrosine kinases and signal transducers and activators of transcription regulate critical functions of T cells in allograft rejection and transplantation tolerance. Transplantation. 2006;82:295–303. doi: 10.1097/01.tp.0000228903.03118.be. [DOI] [PubMed] [Google Scholar]
- 51.Bhattacharyya S, Mandal D, Saha B, Sen GS, Das T, Sa G. Curcumin prevents tumor-induced T cell apoptosis through Stat-5a-mediated Bcl-2 induction. J Bio Chem. 2007;282:15954–15964. doi: 10.1074/jbc.M608189200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.