

Abstract
The juvenile idiopathic inflammatory myopathies (JIIM) are systemic autoimmune diseases characterized by skeletal muscle weakness, characteristic rashes, and other systemic features. In follow-up to our study defining the major clinical subgroup phenotypes of JIIM, we compared demographics, clinical features, laboratory measures, and outcomes among myositis-specific autoantibody (MSA) subgroups, as well as with published data on adult idiopathic inflammatory myopathy patients enrolled in a separate natural history study. In the present study, of 430 patients enrolled in a nationwide registry study who had serum tested for myositis autoantibodies, 374 had either a single specific MSA (n = 253) or no identified MSA (n = 121) and were the subject of the present report. Following univariate analysis, we used random forest classification and exact logistic regression modeling to compare autoantibody subgroups. Anti-p155/140 autoantibodies were the most frequent subgroup, present in 32% of patients with juvenile dermatomyositis (JDM) or overlap myositis with JDM, followed by anti-MJ autoantibodies, which were seen in 20% of JIIM patients, primarily in JDM. Other MSAs, including anti-synthetase, anti-signal recognition particle (SRP), and anti-Mi-2, were present in only 10% of JIIM patients. Features that characterized the anti-p155/140 autoantibody subgroup included Gottron papules, malar rash, “shawl-sign” rash, photosensitivity, cuticular overgrowth, lowest creatine kinase (CK) levels, and a predominantly chronic illness course. The features that differed for patients with anti-MJ antibodies included muscle cramps, dysphonia, intermediate CK levels, a high frequency of hospitalization, and a monocyclic disease course. Patients with anti-synthetase antibodies had higher frequencies of interstitial lung disease, arthralgia, and “mechanic’s hands,” and had an older age at diagnosis. The anti-SRP group, which had exclusively juvenile polymyositis, was characterized by high frequencies of black race, severe onset, distal weakness, falling episodes, Raynaud phenomenon, cardiac involvement, high CK levels, chronic disease course, frequent hospitalization, and wheelchair use. Characteristic features of the anti-Mi-2 subgroup included Hispanic ethnicity, classic dermatomyositis and malar rashes, high CK levels, and very low mortality. Finally, the most common features of patients without any currently defined MSA or myositis-associated autoantibodies included linear extensor erythema, arthralgia, and a monocyclic disease course. Several demographic and clinical features were shared between juvenile and adult idiopathic inflammatory myopathy subgroups, but with several important differences. We conclude that juvenile myositis is a heterogeneous group of illnesses with distinct autoantibody phenotypes defined by varying clinical and demographic characteristics, laboratory features, and outcomes.
INTRODUCTION
The juvenile idiopathic inflammatory myopathies (JIIM) are systemic autoimmune diseases characterized by symmetric proximal weakness, characteristic rashes, and other systemic features.10,27 The adult idiopathic inflammatory myopathies (IIM) and several other autoimmune disorders are recognized as being composed of a number of clinical and serologic phenotypes, each defining relatively homogeneous subsets of patients with common demographic and clinical features, the presence of certain associated autoantibodies, responses to therapy, and outcomes.1,22,25
Recently, a large study of the demographic, clinical, and laboratory features and outcomes of patients with IIM defined homogeneous, distinct clinical phenotypes.29 As a complement to clinical phenotypes, serologic phenotypes, defined by the presence of autoantibodies—which are either relatively specific for myositis (myositis-specific autoantibodies [MSAs]) or are seen in myositis and other autoimmune diseases (myositis-associated autoantibodies [MAAs])—have been recognized in adult patients.20 Recently identified autoantibody specificities have aided in defining additional phenotypes in adult IIM patients.1,22 Although myositis autoantibodies have been described in JIIM patients,12,13,26,28,29,32,39 the full spectrum of demographic, clinical features, and outcomes associated with the myositis autoantibodies has not been fully elucidated in large JIIM populations. The existence of distinct subgroups of patients based on myositis autoantibodies has not been uniformly recognized,5,9 and the degree of similarity between JIIM and adult IIM patients with the same myositis autoantibodies has not been thoroughly investigated.24,27
In follow-up to our prior study of clinical phenotypes,29 we undertook a large study to define the myositis autoantibody phenotypes of JIIM and compare them to adult IIM patients with the same autoantibody specificities.
PATIENTS AND METHODS
Four hundred thirty-six patients with probable or definite juvenile dermatomyositis (JDM) or juvenile polymyositis (JPM)2 were enrolled in the National Institutes of Health Clinical Center or Food and Drug Administration’s investigational review board-approved natural history protocols from March 1989 through August 2010; patients had been diagnosed with myositis between May 1957 and March 2010.29 Of these, 430 patients were tested for myositis autoantibodies using a single serum sample after written consent/assent was obtained according to the Declaration of Helsinki. A final cohort of 374 patients had either a single specific MSA (n = 253) or no identified MSA (n = 121), and they are the primary subjects in the present study. The childhood cohort was compared to an adult IIM study population, which included 148 adult IIM patients enrolled at the National Institutes of Health Clinical Center in Bethesda, MD, between 1983 and 1990 with MSA testing completed; adult IIM patients were diagnosed between January 1965 and January 1990.20
All methods, including patients, autoantibody testing, and statistical approaches, were as described previously,29 with the modified statistical methods detailed here. The Wilcoxon rank sum test was used to compare continuous data, and the chi-square test and, for small sample sizes, Fisher exact test were conducted to determine significant differences between variables of interest and autoantibody subgroups. Following univariate analysis, we again used a 2-staged approach to further identify variables important in the classification of JIIM autoantibody groups. First, for autoantibody groups with more than 15 patients, we used random forest classification tree analysis, a nonlinear, nonparametric algorithm that generates estimates of ranking for predictive importance,21 to identify and validate the predictors from among a large group of candidate variables. Then the top variables from the random forest models, consisting of 500 forests and 1000 trees, were entered into a backward stepwise logistic regression analysis. Variables that were statistically significant from univariable analyses or were clinically significant based on prior reports in the literature were entered into the random forest model (full model). Variables with more than 10% of data missing, such as enzymes, pulmonary function testing, and abnormal cardiac findings, were not entered in the models. Due to these factors, the number of patients with a given autoantibody varied in different random forest models. When the number of patients in the autoantibody groups differed, the data were resampled to ensure balance, using the method of undersampling from the larger group.21 The top variables from the random forest model were selected based on their mean decrease in accuracy (MDA) and their clinical importance in the literature. Variables that were significant in the backward stepwise logistic regression were entered into an exact logistic regression.
For autoantibody groups with n < 15 (anti-signal recognition particle [SRP] and anti-Mi-2 autoantibodies), due to small sample size, random forest classification tree analysis was not performed; thus, these analyses began with a stepwise logistic regression, and then significant variables were entered into the exact logistic regression. Variables in which the frequency was either 100% or 0% in 1 of the 2 groups were not examined by logistic regression, to avoid model instability. Similarly, if a variable was highly correlated with another variable (for example, age at onset and age at diagnosis), the variable that was less significant was removed from the logistic regression analysis. Finally, certain variables led to model instability, and such predictor variables were removed from the logistic regression analysis.
Odds ratios, 95% confidence intervals, and p values to test for significance were calculated. For this analysis, we interpreted a c statistic ≥90% as very good, 90% > c ≥ 80% as good, 80% > c ≥ 70% as fair, and c < 70% as poor. The likelihood ratio test, which compares the log likelihoods of the 2 models and tests whether this difference is statistically significant, also was examined to test the final model’s validity.
RESULTS
Myositis Autoantibody Distributions in JIIM
To assess patterns of MSA and MAA combinations, an autoantibody matrix was created (Table 1). Approximately two-thirds (63%) of JIIM patients had at least 1 MSA, whereas 28% were negative for any currently defined MSA or MAA, and 9% had an isolated MAA. The most frequent MSAs were the anti-p155/140 autoantibody (also called “p155/140” or “anti-p155/140”), present in 32%, and anti-MJ autoantibodies in 20%. The classic MSAs, that is, anti-aminoacyl-tRNA synthetases (anti-synthetase) (4.4%), anti-SRP (1.4%), and anti-Mi-2 (4.4%) autoantibodies, were present in approximately 10% of JIIM patients. Most patients with anti-synthetase autoantibodies had anti-Jo1 (63%), and 5 patients had anti-alanyl, and 1 each had anti-glycyl and anti-asparaginyl-tRNA synthetase autoantibodies.
TABLE 1.
Myositis Autoantibody Matrix for all Patients With Juvenile Idiopathic Inflammatory Myopathies (n = 430)*
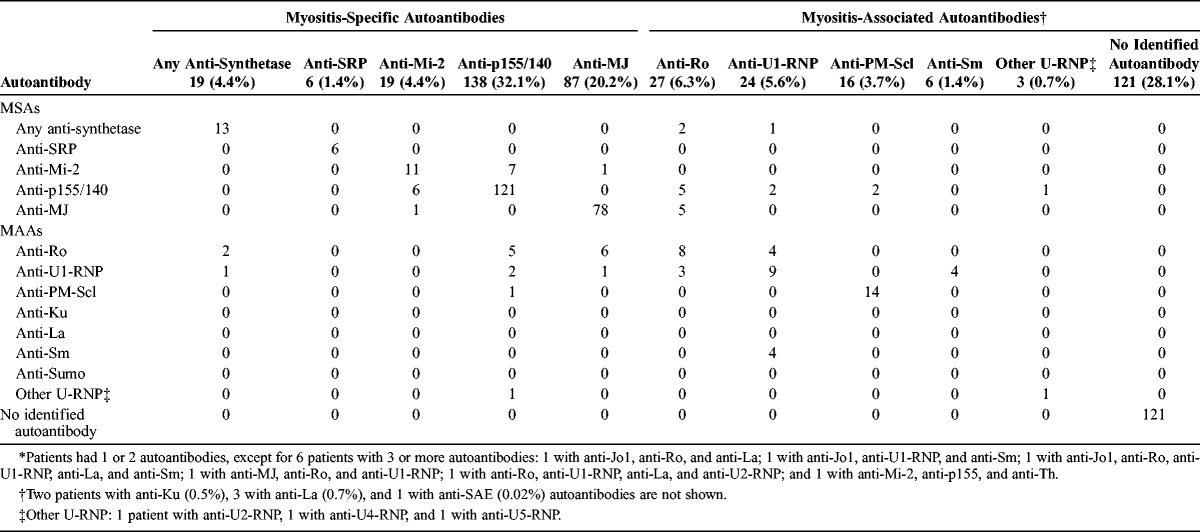
Generally, each MSA was seen in isolation. Several patients had more than 1 MSA: 6 patients had both anti-Mi-2 and anti-p155/140, and 1 patient had both anti-Mi-2 and anti-MJ autoantibodies. These dually positive sera were positive for anti-Mi-2 by protein immunoprecipitation and positive for anti-p155/140 or anti-MJ autoantibodies by the immunoprecipitation-blot assay only. Since the current study of autoantibody phenotypes examined only patients with 1 independent MSA, these 7 patients with more than 1 MSA were excluded from subsequent analyses.
Almost 16% of patients had an MAA. The most frequent MAAs included anti-Ro (6.3%), anti-U1-ribonucleoprotein (RNP) (5.6%), and anti-polymyositis-scleroderma (PM-Scl) (3.7%). As expected, sera from patients with MAA frequently had a concomitant MSA. Frequent combinations of MAA and MSA were anti-Ro and either anti-MJ or anti-p155/140 autoantibodies. Patients with anti-SRP or anti-Mi-2 autoantibodies were not positive for MAA. In terms of combinations of MAAs, all patients with anti-La autoantibodies also had anti-Ro autoantibodies, and many of these also had anti-U1-RNP as well. All patients with the anti-Sm autoantibody were also positive for anti-U1-RNP autoantibody.
We excluded from certain analyses 49 patients with an MAA in isolation who did not also have an MSA. The patients with isolated MAAs who were excluded had anti-La (1 patient), anti-Ku (2 patients), anti-Ro (13 patients), anti-U1-RNP (18 patients), or anti-PM-Scl autoantibodies (15 patients). Three patients were excluded due to missing myositis autoantibody data.
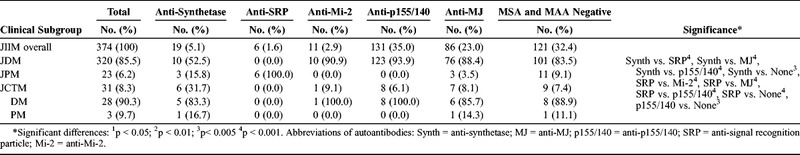
A final 374 patients with either a single specific MSA (n = 253) or no identified MSA (n = 121) were included in the study (Table 2). Over half of the patients with anti-synthetase autoantibodies had JDM, 16% had JPM, and 32% had juvenile myositis overlapping with another autoimmune or connective tissue disease (JCTM). Patients with non-Jo-1 anti-synthetases had JDM more frequently than those with anti-Jo-1 autoantibodies (71% vs. 42%). The JCTM patients with anti-synthetase autoantibodies included 3 patients with JDM overlapping with juvenile rheumatoid arthritis (JRA), 1 patient with JDM overlapping with systemic lupus erythematosus (SLE), 1 patient with JPM and Sjögren syndrome, and 1 patient with JDM, SLE, and JRA. Patients with anti-synthetase autoantibodies had a different distribution of clinical subgroups compared to patients with anti-SRP, anti-MJ, and anti-p155/140 autoantibodies and patients who were negative for an MSA or MAA. All patients with anti-SRP autoantibodies had JPM, which is a significantly different clinical subgroup distribution than patients with anti-Mi-2, anti-p155/140, or anti-MJ autoantibodies and patients who were MSA/MAA negative, who had JDM predominantly. However, 3 patients with anti-MJ had JPM and 1 had JPM overlapping with ulcerative colitis. Of these patients with JPM and anti-MJ autoantibodies, several had features of JDM, but none had the characteristic Gottron papules or heliotrope rash (1 had thigh rash, a muscle biopsy with perifascicular atrophy, and developed calcinosis; 2 had perivascular inflammation on muscle biopsy and periungual capillary changes).
TABLE 2.
Myositis Clinical Subgroups Categorized by the Presence of Myositis-Specific Autoantibodies
The demographics and disease onset among the myositis autoantibody groups are given in Table 3. Patients with anti-synthetase and anti-SRP autoantibodies were significantly older when diagnosed compared with patients with anti-MJ or anti-p155/140 autoantibodies and those without any MSA or MAA. Patients with anti-MJ autoantibodies had a shorter delay to diagnosis than those with anti-p155/140 autoantibodies and those without a defined MSA or MAA. About three-fourths (72%) of the study population were female. Overall, the majority of patients were white, but the racial distribution differed among autoantibody groups. The proportion of white patients was higher in patients with anti-p155/140 (80%) and anti-MJ (72%) autoantibodies than in patients with anti-SRP (17%) and anti-Mi-2 autoantibodies (18%). White patients comprised 53% of the anti-synthetase autoantibody population, which was greater than the anti-Mi-2 autoantibody group. The majority of patients with anti-SRP autoantibodies were black (83%), compared with the percentage of patients with anti-p155/140 and anti-MJ autoantibodies and those who were MSA/MAA negative that were black (5.3%–16%). Six patients (54%) in the anti-Mi-2 group were Hispanic.
TABLE 3.
Demographic and Disease Onset Data for Patients With Juvenile Myositis Categorized by Myositis Autoantibodies
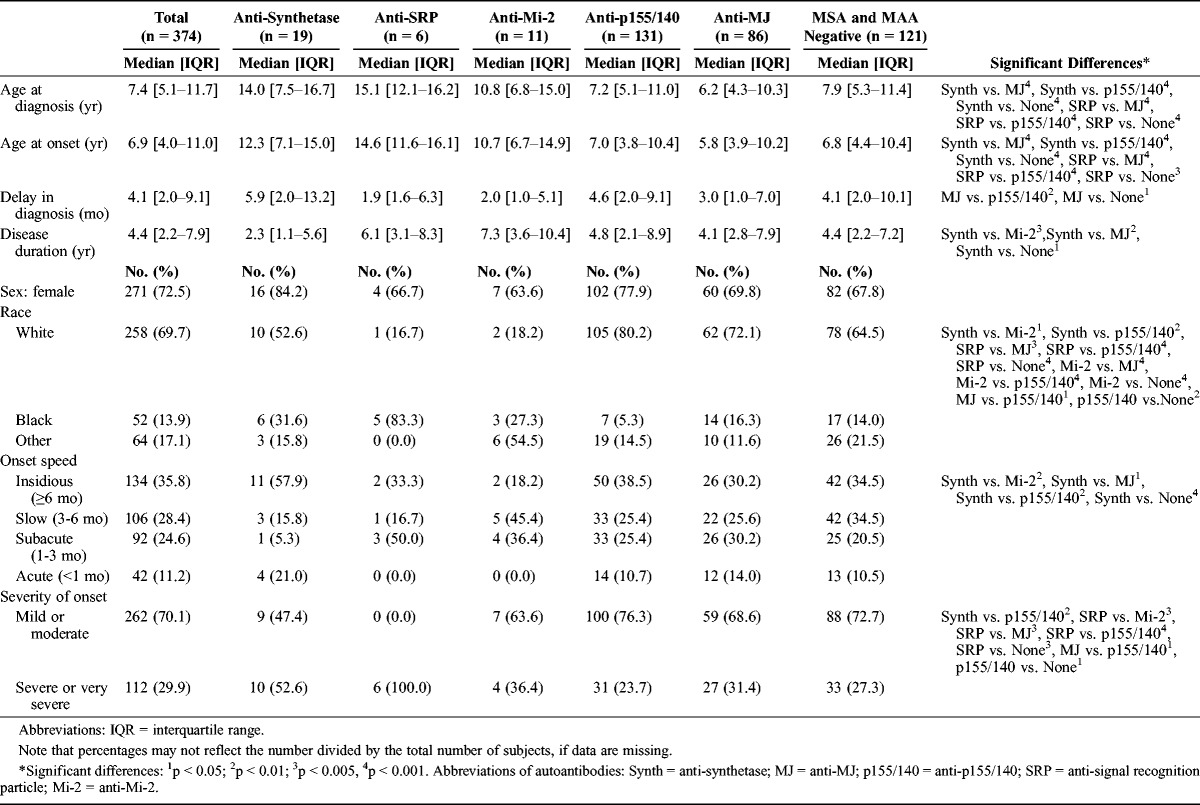
Most patients had either a slow or very slow onset speed. Patients with anti-synthetase autoantibodies (58%) were more likely to have an insidious onset speed compared with the other autoantibody groups. Patients with anti-SRP autoantibodies uniformly had a severe or very severe disease onset in contrast to patients who were MSA/MAA negative or had anti-Mi-2, anti-p155/140, or anti-MJ autoantibodies (24%-39%). Patients with anti-p155/140 autoantibodies more frequently had a mild or moderate illness onset.
Signs and Symptoms Among the Myositis Autoantibody Groups
The frequencies of signs and symptoms (Table 4) showed a number of differences among the myositis autoantibody groups. Almost every patient showed some musculoskeletal system involvement, with proximal muscle weakness being almost universally present, as expected, because it is a cardinal manifestation of the disease. Arthralgia and arthritis were also higher in frequency in patients with anti-synthetase autoantibodies (74%–84%) compared to patients with anti-SRP, anti-Mi-2, anti-p155/140, and anti-MJ autoantibodies (17%–64%). Arthralgia was less common in patients with anti-SRP autoantibodies (17%) compared with patients with anti-MJ autoantibodies or patients with no MSA or MAA (64% and 67%, respectively), and in patients with anti-p155/140 autoantibodies (55%) compared to patients who were MSA/MAA negative (67%). Patients with anti-SRP autoantibodies almost universally experienced falling episodes, distal weakness, and muscle atrophy (83%–100%) as signs of more severe weakness, compared to patients with other myositis autoantibodies.
TABLE 4.
Symptoms and Signs by System Involvement in Patients With Juvenile Myositis Categorized by Myositis Autoantibodies
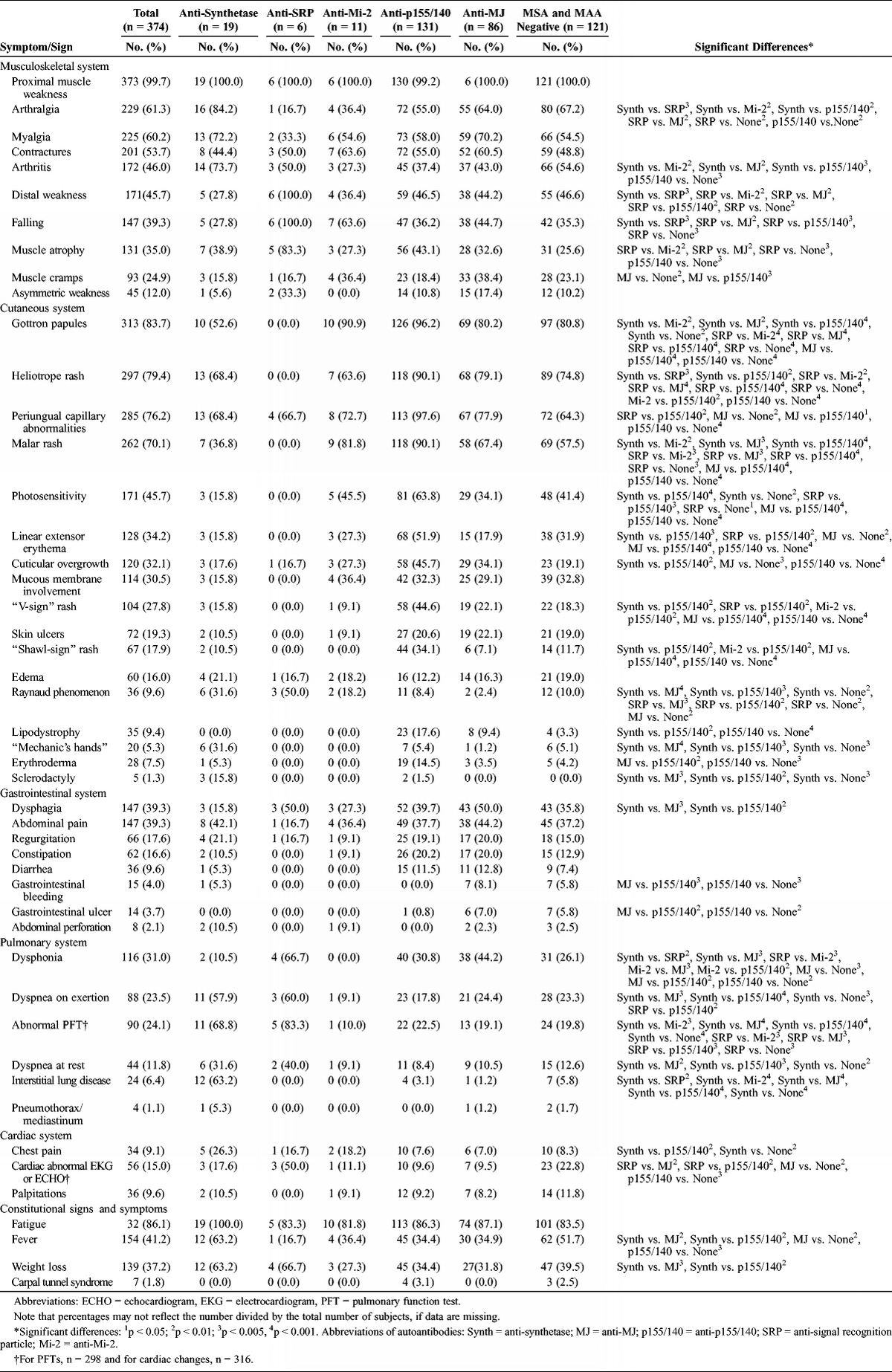
As expected, cutaneous involvement was common in all patients except those with anti-SRP autoantibodies. The most common cutaneous manifestations in patients with anti-p155/140, anti-MJ, and anti-Mi-2 autoantibodies included Gottron papules and heliotrope rash (64%–96%), which are part of the Bohan and Peter classification criteria for dermatomyositis (DM).2 Malar rash was present in most patients with these DM-associated autoantibodies (67%–90%), was less frequent in patients with anti-synthetase autoantibodies (37%), and was absent from patients with anti-SRP autoantibodies, all of whom had JPM. Photosensitivity was also more frequent in patients (34%–64%) with DM-associated autoantibodies (anti-p155/140, anti-MJ, or anti-Mi-2), and was infrequent or absent in patients with anti-synthetase and anti-SRP autoantibodies. Patients with anti-p155/140 autoantibodies also had frequent periungual capillary changes, linear extensor erythema, “V-sign” and “shawl-sign” rashes, and cuticular overgrowth (34%–88%). In contrast, Raynaud phenomenon was more common in patients with anti-SRP (50%) and anti-synthetase autoantibodies (32%) compared with patients with anti-p155/140 and anti-MJ autoantibodies, or with those who were MSA/MAA negative (2%–10%). Sclerodactyly and “mechanic’s hands” were more common in patients with anti-synthetase autoantibodies (16% and 32%, respectively). Lipodystrophy was seen most frequently in patients with anti-p155/140 autoantibodies (18%), whereas it was nearly absent in patients with anti-synthetase autoantibodies and those who were MSA/MAA negative (0% and 3%, respectively). Erythroderma was similarly most frequent in those with anti-p155/140 autoantibodies (14%) and uncommon in those with anti-MJ autoantibodies or those who were MSA/MAA negative (4% each).
Dysphagia was most frequent in patients with anti-MJ and anti-SRP autoantibodies (50% each) and was less common in patients with anti-synthetase autoantibodies (16%). Gastrointestinal bleeding and ulcers, infrequent but severe complications, were most frequent in patients with anti-MJ autoantibodies and in those who were MSA/MAA negative (present in 6%–8%), but were absent in patients with anti-p155/140 autoantibodies.
Dysphonia was reported in 67% of patients with anti-SRP autoantibodies, in 44% of patients with anti-MJ autoantibodies, and less frequently in other antibody phenotypes (10%–31%), and was absent from patients with anti-Mi-2 autoantibodies. Dyspnea on exertion was more prevalent in patients with anti-SRP and anti-synthetase autoantibodies (60% and 58%, each) compared with patients with anti-p155/140 and anti-MJ autoantibodies and those who were MSA/MAA negative (18%–24%). Interstitial lung disease was much more common in patients with anti-synthetase autoantibodies (63%) than in the other antibody phenotypes (0–6%). Dyspnea at rest was also more common in anti-synthetase-positive patients (32%) than in those with anti-p155/140 or anti-MJ autoantibodies or those who were MSA/MAA negative (8%–13%). Chest pain was more frequent in patients with anti-synthetase autoantibodies (26%) compared to those with anti-p155/140 autoantibodies or those who were MSA/MAA negative (8%). Cardiac abnormalities on electrocardiogram or echocardiogram were most frequent in those with anti-SRP autoantibodies (50%) and less common in those with anti-MJ and anti-p155/140 autoantibodies (10% each).
Fatigue was a common symptom in all clinical groups, occurring in 82%–100% of patients and not differing among autoantibody groups. Fever and weight loss (63% each) were more common in the anti-synthetase autoantibody-positive patients than in those with anti-p155/140 or anti-MJ autoantibodies (32%–35%).
Enzyme Levels and Antinuclear Antibody Titers Among Myositis Autoantibody Groups
There were significant differences in the percentage of patients who had elevated serum levels of muscle-derived enzymes among the myositis autoantibody groups (Table 5). Only 71% of patients with anti-p155/140 autoantibodies had an elevated creatine kinase (CK) level compared with 92%–95% of patients with anti-MJ autoantibodies or patients who were MSA/MAA negative. Patients with anti-MJ and anti-synthetase autoantibodies were more likely to have elevated CK levels than patients with anti-p155/140 autoantibodies.
TABLE 5.
Abnormal Enzyme Levels in Patients With Juvenile Myositis Categorized by Myositis Autoantibodies
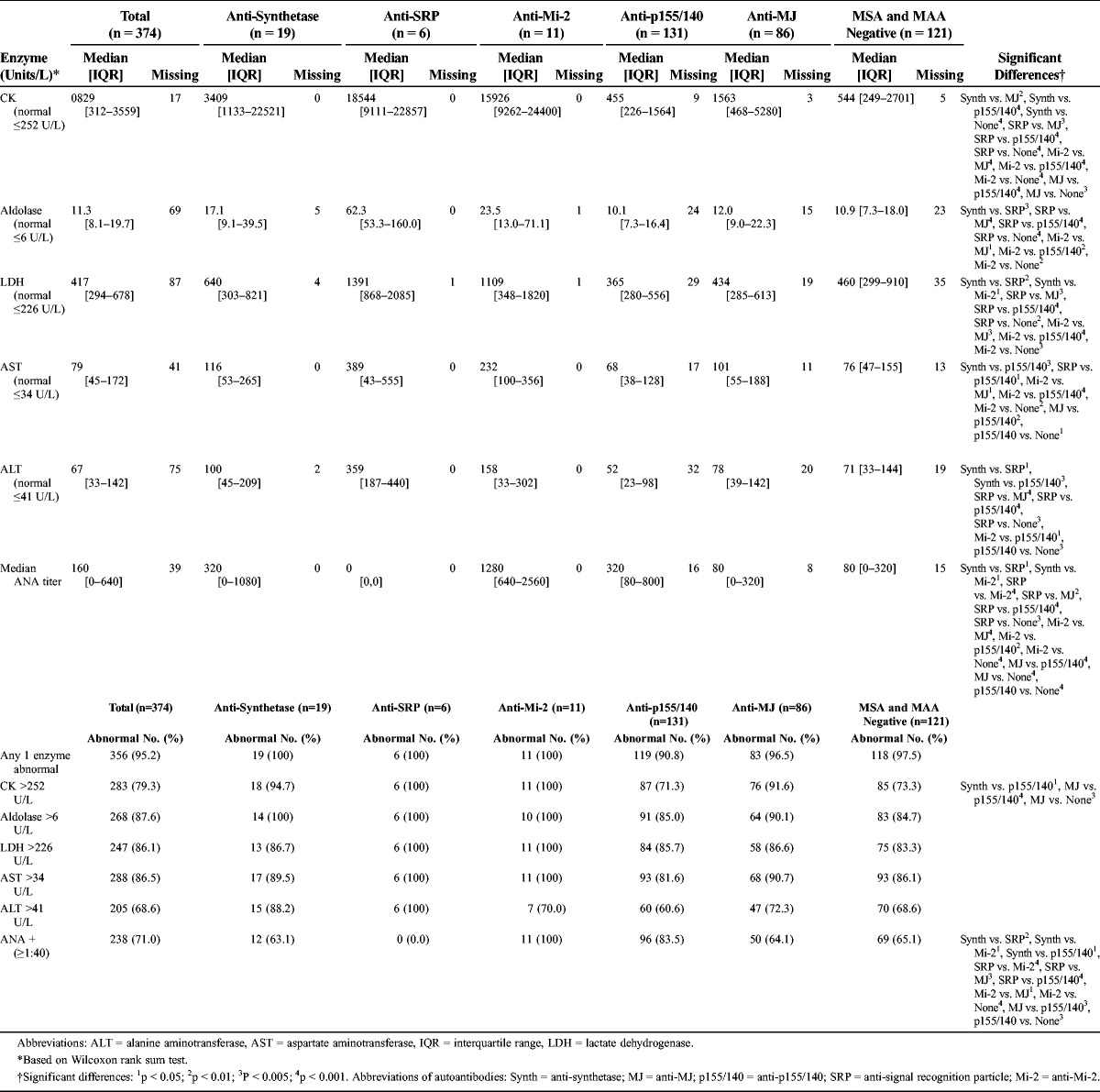
Patients with anti-SRP and anti-Mi-2 autoantibodies had significantly higher median levels of CK (10- to 40-fold greater) compared with patients with anti-MJ and anti-p155/140 autoantibodies and patients who were MSA/MAA negative. Patients with anti-synthetase autoantibodies had intermediate CK levels. Similar trends were observed for the other muscle enzymes, as well as in white patients only.
No patients with anti-SRP autoantibodies, which target cytoplasmic autoantigens,33 had elevated antinuclear antibody (ANA) titers. Patients with anti-Mi-2 autoantibodies, which target nuclear autoantigens,33 were more likely to have elevated ANA titers (100%) than patients with anti-synthetase, anti-SRP, or anti-MJ autoantibodies and those who were MSA/MAA negative (0–65%). Patients with anti-p155/140 autoantibodies, another nuclear autoantigen,34 also generally had elevated ANA titers (84% positive), which was significantly more frequent than patients with anti-SRP or anti-MJ autoantibodies and those who were MSA/MAA negative.
Patients with anti-Mi-2 autoantibodies had the highest ANA values (median titer, 1:1280); patients with anti-synthetase or anti-p155/140 autoantibodies had intermediate titers (median, 1:320); and patients with anti-MJ or those who were MSA/MAA negative had the lowest titers (median, 1:80).
Disease Outcomes Among Autoantibody Groups
The most frequent disease course was a chronic disease course (52% overall). All patients in the anti-SRP group had a chronic disease course. Patients with anti-p155/140 autoantibodies were more likely to have a chronic course (65%) than patients who had anti-MJ autoantibodies (46%) or patients without a myositis autoantibody (37%). Myositis autoantibody-negative patients were more likely to have a monocyclic course (39%) compared with patients with anti-p155/140 or anti-SRP autoantibodies (12% and 0%, respectively).
Hospitalization was common in all groups; however, all patients with anti-SRP autoantibodies had a history of hospitalization, whereas approximately 50% of patients with anti-p155/140 autoantibodies and 50% of those who were MSA/MAA negative did (Table 6). Patients with anti-MJ autoantibodies also had a higher frequency of hospitalization (63%) than patients with anti-p155/140 autoantibodies. Patients with anti-SRP autoantibodies had more frequent hospitalizations (median, 2.5 visits) compared with other autoantibody groups.
TABLE 6.
Outcomes of Juvenile Myositis Categorized by Autoantibody
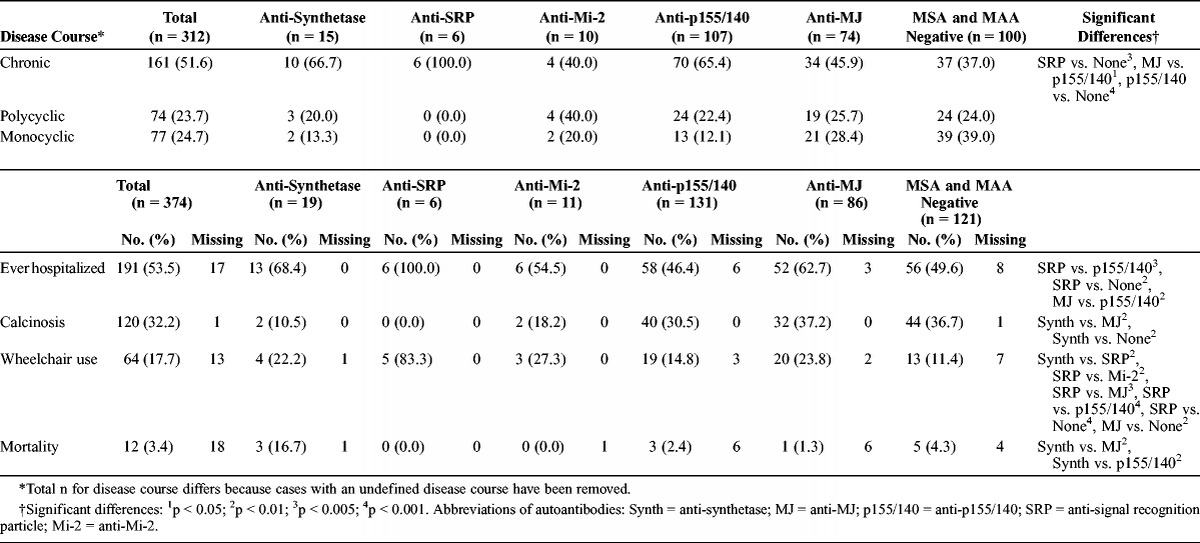
Calcinosis was most common in MSA/MAA-negative patients and in patients with anti-MJ autoantibodies (37% each), and more frequent than in patients with anti-synthetase (10%) or anti-SRP autoantibodies (0%). Wheelchair use was much more common in patients with anti-SRP autoantibodies (83%) than in patients with any other myositis autoantibody (11%–27%). Overall mortality during the follow-up period was 3.4%, but was highest in patients with anti-synthetase autoantibodies (16.7%) compared with patients with anti-p155/140 or anti-MJ autoantibodies (2.4% and 1.3%, respectively), despite a shorter disease duration in the patients with anti-synthetase autoantibodies.
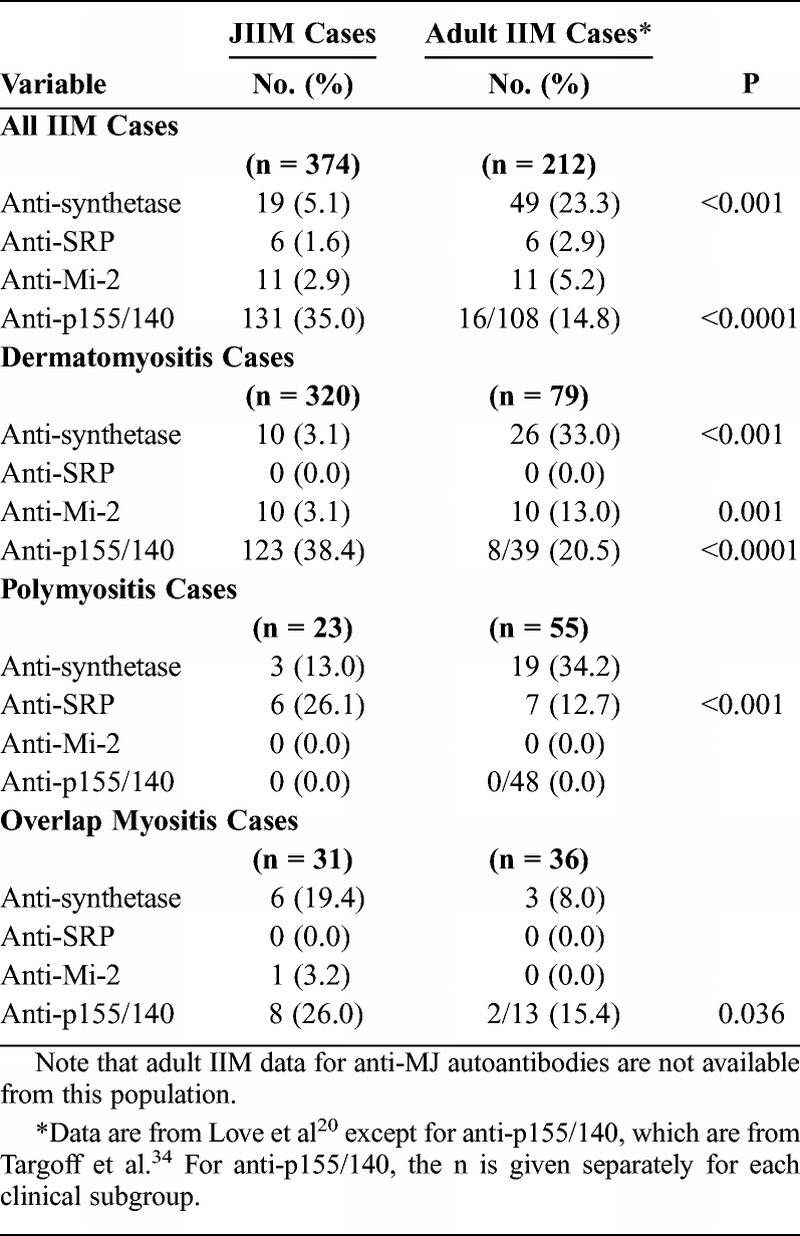
Comparison of Juvenile and Adult-Onset IIM Autoantibody Groups
Adult and juvenile IIM patients had similar distributions of clinical subgroups in the anti-SRP and anti-Mi-2 autoantibody groups (Table 7). All adults and children with anti-SRP autoantibodies had polymyositis (PM), and all adults and children with anti-Mi-2 and anti-p155/140 autoantibodies had DM (or DM with overlap myositis). Adult patients with anti-synthetase autoantibodies were more likely to have PM and less likely to have myositis overlapping with another autoimmune or connective tissue myositis (CTM) compared with JIIM patients with these autoantibodies. By clinical subgroup, a larger proportion of patients with adult DM had anti-synthetase (33%) and anti-Mi-2 (13%) autoantibodies compared with JDM patients (3% each). Children with JDM were more likely to have anti-p155/140 autoantibodies (38%) compared with adults with DM (20%). For patients with PM, adults and children differed only in the proportion with anti-SRP autoantibodies; JPM patients were more likely than adult PM patients to have anti-SRP autoantibodies (26% vs. 13%, respectively). Juvenile CTM patients were more likely to have anti-p155/140 autoantibodies than adult CTM patients (26% vs. 15%), with no other differences in autoantibody distribution among adult and juvenile CTM patients.
TABLE 7.
Comparison of Myositis Autoantibody Distribution in Juvenile vs. Adult Idiopathic Inflammatory Myopathy Patients
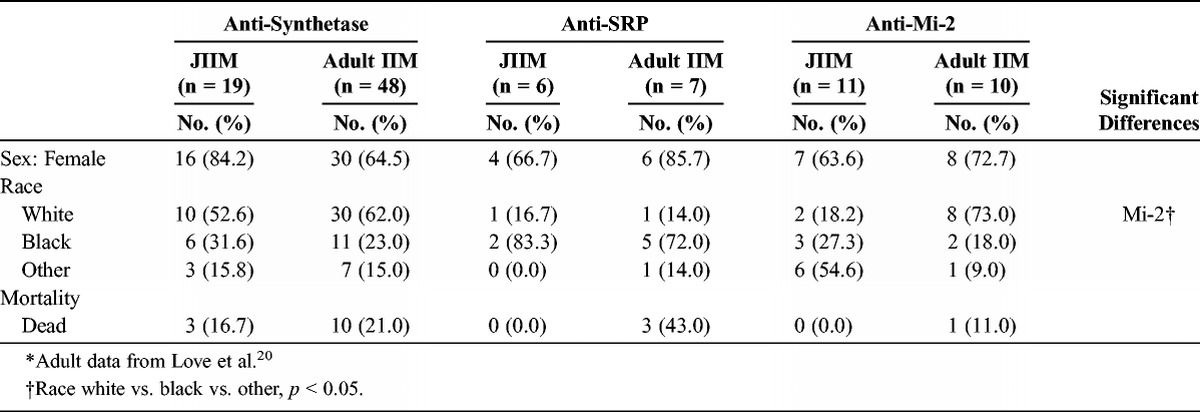
The female predilection was similar in the juvenile and adult IIM autoantibody groups (Table 8). The racial distributions differed for adults and children with anti-Mi-2 autoantibodies. Approximately 55% of anti-Mi-2-positive JIIM patients were characterized as “other race,” which was primarily Hispanic, compared with 9% of adult IIM patients. Although 43% of adults with anti-SRP autoantibodies died, no children with these autoantibodies died.
TABLE 8.
Demographic Features of Patients With Juvenile vs. Adult Idiopathic Inflammatory Myopathies Categorized by Myositis Autoantibodies*
With regard to clinical signs and symptoms of the autoantibody subgroups in juvenile compared with adult IIM, distal weakness, falling, and muscle atrophy were more common in juvenile patients with anti-synthetase autoantibodies (28%–39%) than their adult counterparts (4%) (Table 9). Interstitial lung disease, dyspnea on exertion, Raynaud phenomenon, mechanic’s hands, palpitations, and carpal tunnel syndrome were more common in adult patients with anti-synthetase autoantibodies (33%–94%) compared with JIIM patients (11%–63%). Arthritis and fevers did not differ between children and adults with anti-synthetase autoantibodies. Falling episodes and muscle atrophy (83% and 100%, respectively) were more common in children with anti-SRP autoantibodies compared with adults (33% and 14%, respectively). However, myalgia and palpitations were more common in adults with anti-SRP autoantibodies (100%) compared with children (0–33%).
TABLE 9.
Symptoms and Signs for Patients With Juvenile vs. Adult Idiopathic Inflammatory Myopathies Categorized by Myositis Autoantibodies
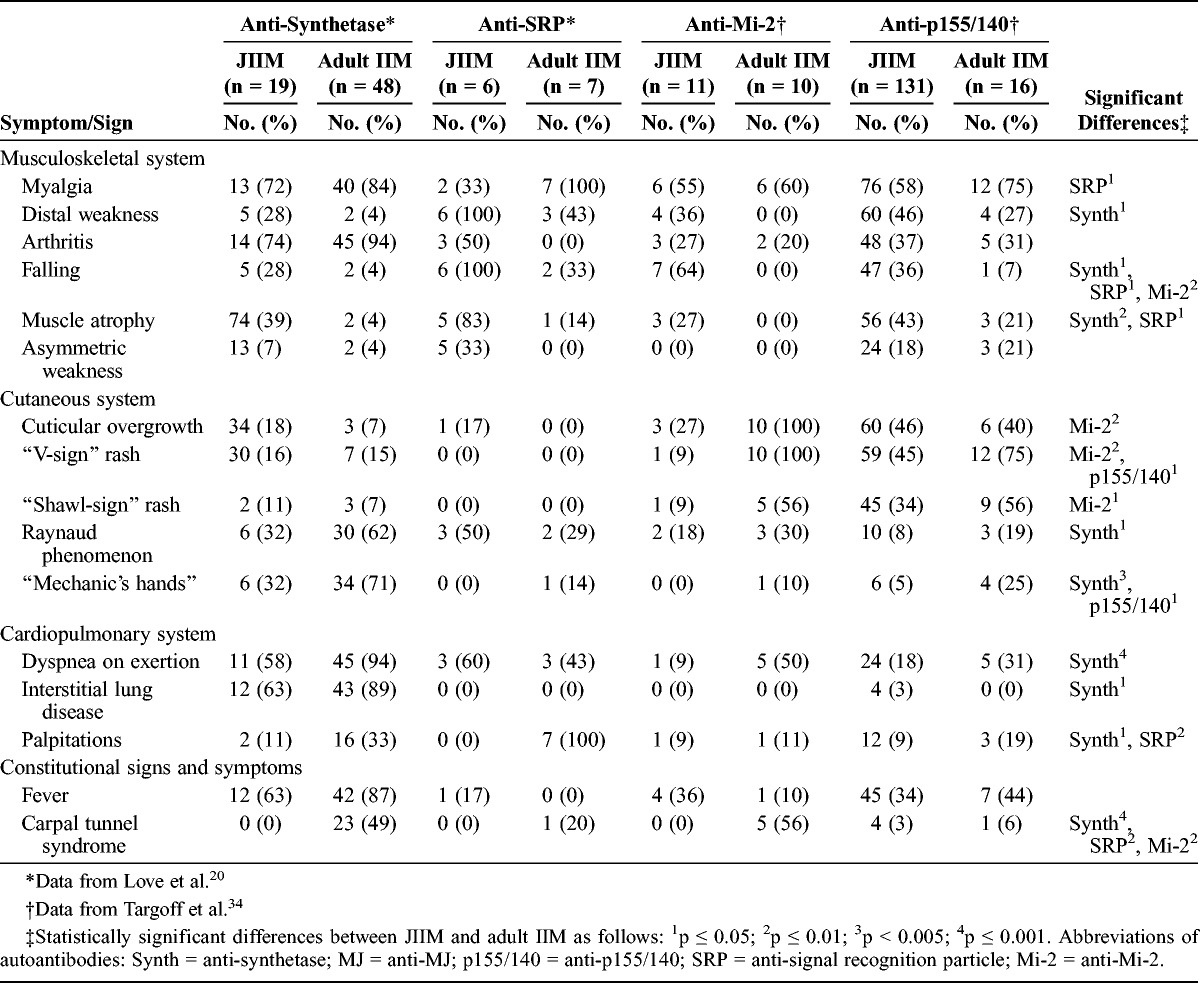
Falling was more common in children (64%) than adults (0%) with anti-Mi-2 autoantibodies. Cuticular overgrowth, V-sign and shawl-sign rashes, and carpal tunnel syndrome (56%–100%) were more prevalent in adult patients with anti-Mi-2 autoantibodies compared with juvenile cases (0–27%). Myalgia and arthritis had similar prevalence in adults and children with anti-Mi-2 autoantibodies. Adults and children with anti-p155/140 autoantibodies shared similar features, except that adults were more likely to have V-sign rash (75%) and mechanic’s hands (25%) compared with children with these autoantibodies (45% and 5%, respectively).
Multivariable Analysis for Anti-p155/140 Autoantibodies
The most important predictors of anti-synthetase compared with anti-p155/140 autoantibodies were CK level (MDA, 80.3), the presence of interstitial lung disease (MDA, 68.8), malar rash (MDA, 65.9), clinical subgroup (MDA, 47.2), age at onset (MDA, 41.7), Gottron papules (MDA, 37.2), photosensitivity (MDA, 32.0), shawl-sign rash (MDA, 25.4), and mechanic’s hands (MDA, 20.4) in a random forest model, with an out-of-bag error rate of 10.7%. Using the top variables from the random forest analysis, interstitial lung disease and malar rash were the only variables that were statistically significant in backward stepwise logistic regression analysis, which was confirmed by exact logistic regression (Table 10). The c statistic measure (0.95) indicated a very good fit for discriminating between these 2 groups. Clinical subgroup was not significant when forced in the model.
TABLE 10.
Final Multivariable Exact Logistic Regression Models for Anti-p155/140 Autoantibodies vs. Classic Myositis-Specific Autoantibodies
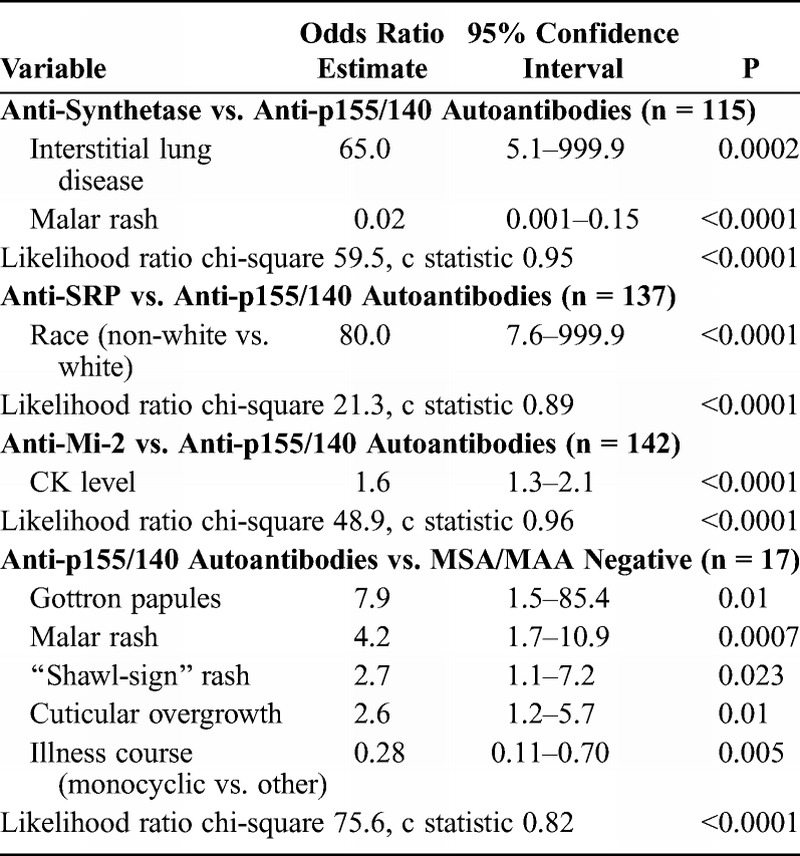
Backward stepwise logistic regression analysis of anti-SRP versus anti-p155/140 autoantibodies yielded race as the only risk factor distinguishing these 2 autoantibody groups. Exact logistic regression (see Table 10) confirmed that being non-white was significant. The c statistic was 0.89, indicating a good fit for discriminating between the 2 groups. Clinical subgroup was not significant when forced in the model. Backward stepwise logistic regression analysis of anti-Mi-2 versus anti-p155/140 autoantibodies yielded only CK level as a risk factor. The c statistic (0.96) indicated a very good fit for discriminating between these 2 autoantibody groups. Clinical subgroup remained insignificant when forced into the model.
In a random forest model, the most important predictors of anti-p155/140 autoantibodies compared with MSA/MAA negative was presence of malar rash (MDA, 99.9), illness course (MDA, 69.2), periungual capillary changes (MDA, 56.0), cuticular overgrowth (MDA, 51.1), shawl-sign rash (MDA, 46.3), and V-sign rash (MDA, 39.6), with an out-of-bag error rate of 26.4%. Using the top variables from the random forest analysis, we found that Gottron papules, malar rash, shawl-sign rash, and cuticular overgrowth were the significant variables in backward stepwise logistic regression, which was confirmed by exact logistic regression. In addition, the anti-p155/140 autoantibody group had a 0.28-fold lower risk of having a monocyclic course compared with MSA/MAA-negative patients. The c statistic (0.82) indicated a good fit for discriminating between these 2 groups. Clinical subgroup was not a significant risk factor when forced into the model.
Multivariable Analysis for Anti-MJ Autoantibodies
In a random forest model, the most important predictors of anti-synthetase compared with anti-MJ autoantibodies were interstitial lung disease (MDA, 97.0), age at onset (MDA, 54.6), mechanic’s hands (MDA, 42.0), and malar rash (MDA, 22.0), with an out-of-bag error rate of 14.1%. Using the top variables from the random forest analysis in backward stepwise logistic regression, interstitial lung disease and mechanic’s hands were the only significant variables, which was confirmed in a final exact logistic regression model (Table 11). The c statistic (0.91) indicated a very good fit for discriminating between these 2 groups. Clinical subgroup was not a significant risk factor when forced into the model.
TABLE 11.
Final Multivariable Exact Logistic Regression Models for Anti-MJ Autoantibodies vs. Other Myositis-Specific Autoantibodies
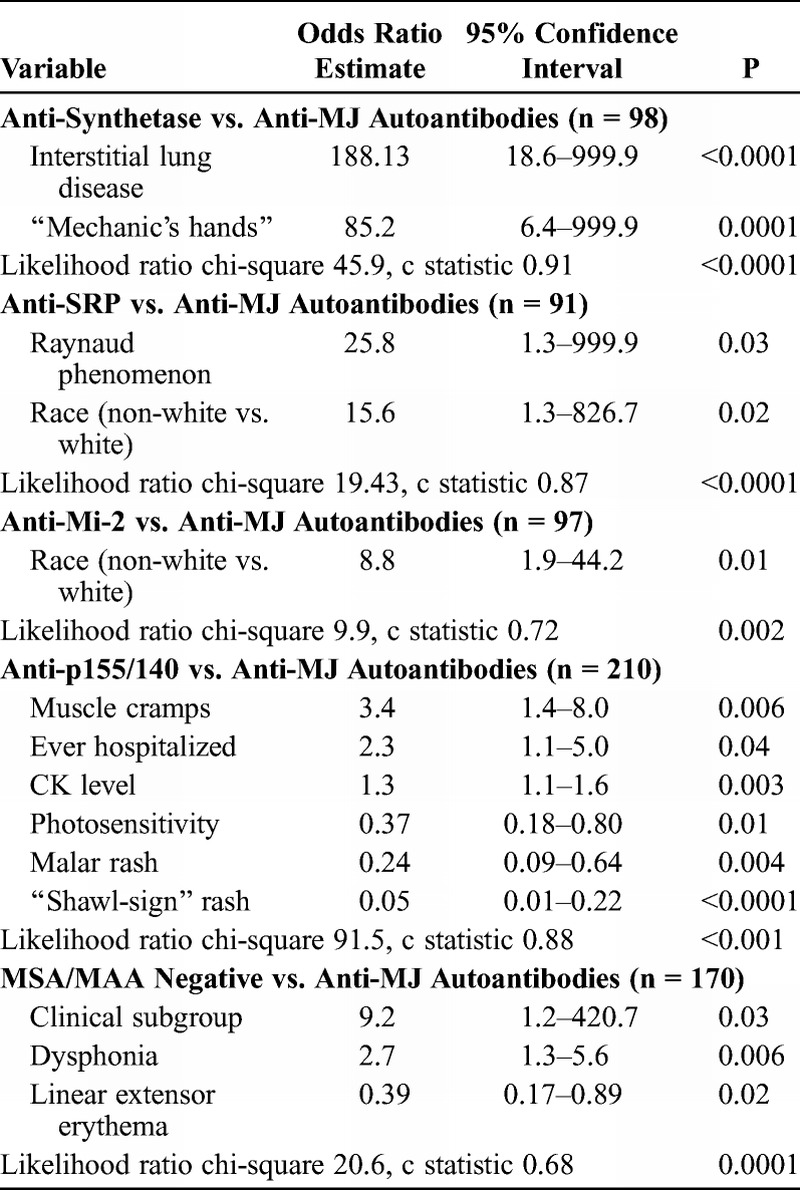
For anti-SRP versus anti-MJ autoantibodies, backward stepwise binary regression analysis with confirmation by exact logistic regression yielded race and Raynaud phenomenon as the only significant risk factors. Patients with anti-SRP autoantibodies had 15.6-fold higher odds of being non-white than patients with anti-MJ autoantibodies. The c statistic (0.87) indicated a good fit for discriminating between the 2 groups, and the likelihood ratio for having anti-SRP over anti-MJ autoantibodies was p < 0.0001, which means that the model differentiated between the 2 groups. Clinical subgroup was not a significant predictor when forced into the model.
Backward stepwise binary regression analysis yielded race as the only significant risk factor for distinguishing anti-Mi-2 from anti-MJ autoantibodies. Exact logistic regression (see Table 11) confirmed that being non-white was significant when comparing patients with anti-Mi-2 versus anti-MJ autoantibodies. The c statistic (0.72) indicated a fair fit for discriminating between the 2 groups. The likelihood ratio for having anti-MJ over anti-Mi-2 autoantibodies means that the model differentiated between the 2 groups (p = 0.002). Clinical subgroup was not a significant predictor when forced into the model.
In a random forest model, the most important predictors of anti-MJ compared with anti-p155/140 autoantibodies were shawl-sign rash (MDA, 98.6), CK level (MDA, 73.6), malar rash (MDA, 46.4), linear extensor erythema (MDA, 28.7), V-sign rash (MDA, 26.8), photosensitivity, (MDA, 25.3) muscle cramps (MDA, 18.2), Gottron papules (MDA, 16.5), illness course (MDA, 14.2), gastrointestinal ulcer or bleeding (MDA, 13.2), and ever hospitalized (MDA, 12.2), with an out-of-bag error rate of 21.9%. Using the top variables from the random forest analysis in backward stepwise logistic regression, followed by exact logistic regression, muscle cramps, ever hospitalized, CK level, photosensitivity, malar rash, and shawl-sign rash were all significant, with the latter 3 more frequent in the anti-p155/140 autoantibody group. The c statistic (0.88) indicated a good fit for discriminating between these 2 autoantibody groups. Clinical subgroup was not a significant predictor when forced into the model.
The most important predictors of anti-MJ autoantibodies compared with the MSA/MAA-negative group were CK level (MDA, 99.6), periungual capillary changes (MDA, 58.4), dysphonia (MDA, 38.9), contractures (MDA, 36.3), linear extensor erythema (MDA, 33.7), clinical subgroup (MDA, 32.9), fever (MDA, 26.5), and wheelchair use (MDA, 24.8) in a random forest model, with an out-of-bag error rate of 35.5%. Using the top variables from the random forest analysis in logistic regression, the final model included clinical subgroup, dysphonia, and linear extensor erythema as significant predictors. The c statistic was 0.68, which indicated a poor fit for discriminating between these 2 subgroups.
Multivariable Analyses of Classic MSA Subgroups
JIIM patients with anti-synthetase autoantibodies were compared to patients with anti-SRP and anti-Mi-2 autoantibodies by using logistic regression and were compared with the MSA/MAA-negative subgroup by using random forest analysis followed by logistic regression.
Backward stepwise binary regression analysis yielded arthralgia as the only risk factor for JIIM patients in the anti-synthetase subgroup compared to JIIM patients with anti-SRP autoantibodies and for anti-synthetase versus anti-Mi-2 autoantibodies, which was confirmed by exact logistic regression (Table 12). The c statistic (0.84) indicated a good fit for the first model in discriminating between anti-synthetase and anti-SRP autoantibodies. For anti-synthetase versus anti-Mi-2 autoantibodies, the c statistic was 0.74, which indicated a fair fit for discriminating between these 2 groups, although the likelihood ratio for being anti-synthetase positive over anti-Mi-2 positive means that the model differentiates between the 2 groups (p = 0.007). Finally, clinical subgroup was included to test for confounding in both models, but this variable was insignificant and therefore omitted from the final models.
TABLE 12.
Final Multivariable Exact Logistic Regression Models for Classic Myositis-Specific Autoantibodies
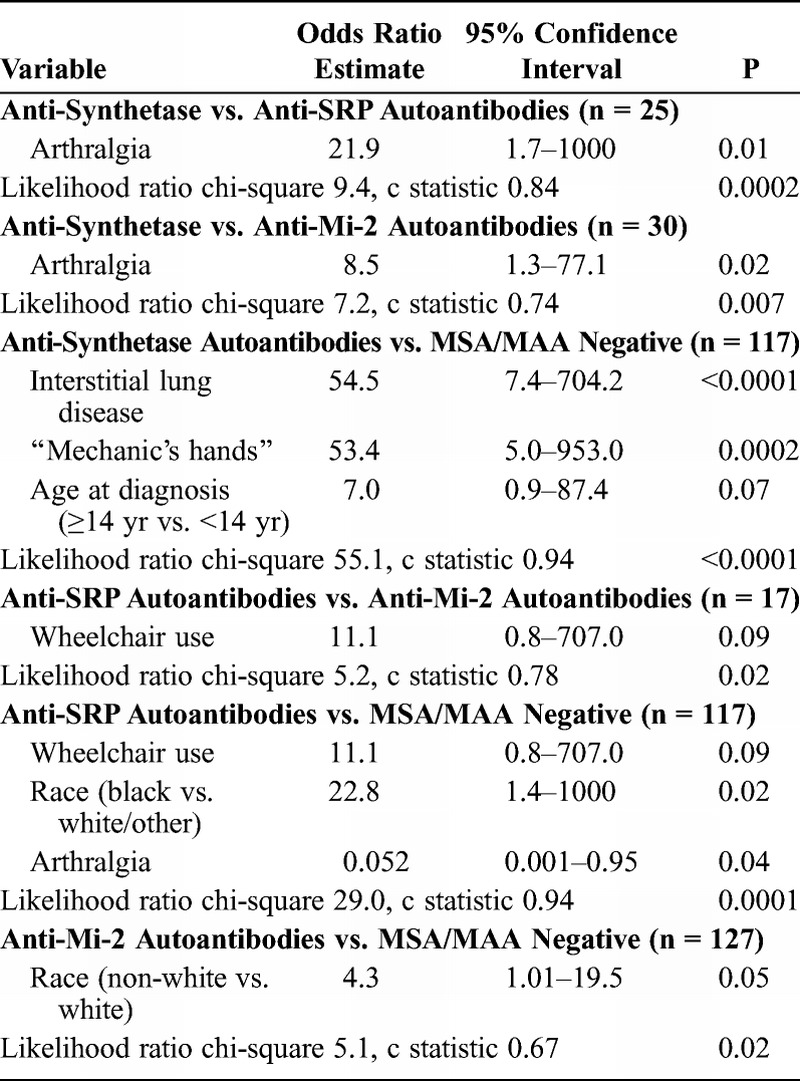
In a random forest classification model, the most important predictors of anti-synthetase autoantibody-positive patients compared to patients who were MSA/MAA negative were the presence of interstitial lung disease (MDA, 90.1), age at diagnosis (MDA, 58.8), age at onset (MDA, 57.6), CK level (MDA, 49.7), and mechanic’s hands (MDA, 43.3), with an out-of-bag error rate of 22.0%. In the final logistic regression model (see Table 12), interstitial lung disease had an odds ratio of 54.5, and mechanic’s hands had an odds ratio of 53.4 for the anti-synthetase autoantibody versus the MSA/MAA-negative group. Patients at least 14 years of age had 7-fold higher odds of having anti-synthetase autoantibodies compared to MSA/MAA-negative patients. The c statistic (0.94) indicated an excellent fit for discriminating between these 2 groups. Clinical subgroup (JDM vs. JPM) was not significant when entered into the model.
Backward stepwise regression analysis yielded wheelchair use as the only risk factor for the anti-SRP autoantibody group compared to the anti-Mi-2 group, which was confirmed by exact logistic regression (see Table 12). The c statistic was 0.78, which indicated a fair fit for discriminating between these 2 groups, but the likelihood ratio was significant (p = 0.02), meaning that the model differentiates between the 2 groups. Clinical subgroup was not significant when forced in the model. Backward stepwise binary regression analysis, with confirmation by exact logistic regression, revealed that wheelchair use, Black race, and arthralgia were significant predictors in distinguishing patients with anti-SRP autoantibodies from MSA/MAA-negative patients (see Table 12). The c statistic (0.94) indicated an excellent fit for discriminating between these groups. Clinical subgroup was not significant when forced in the model.
In comparing patients with anti-Mi-2 autoantibodies and MSA/MAA-negative patients by using backward stepwise logistic regression, race (non-white) was the only potentially significant variable, which was confirmed by exact logistic regression (see Table 12). The c statistic was 0.67, which indicated a poor fit for discriminating between these 2 groups, but a significant likelihood (p = 0.02) suggested that the model differentiates between the 2 groups. Clinical subgroup was not significant when forced in the model.
DISCUSSION
The current study complements our previous report on clinical phenotypes29 and, to our knowledge, is the first study to investigate the association of clinical features with 6 MSAs in a large group of JIIM patients with a large number of demographic, clinical, laboratory, and outcome features. The results of this study suggest that each MSA defines a clinically distinct phenotype and may serve as a predictor of clinical complications and prognosis. The analyses demonstrate that, as is the case for adult myositis, childhood myositis is a heterogeneous group of illnesses with different clinical and demographic characteristics, laboratory features, and outcomes.
Our findings show that MSAs can be an important method for classifying children with myositis. This study dispels suggestions that children with JIIM are a homogeneous group with a low prevalence of autoantibodies.5,9 Anti-p155/140 and anti-MJ autoantibodies were the most prevalent MSA in this population and represent the 2 major serologic subsets of juvenile myositis. We found a higher prevalence of anti-p155/140 autoantibodies (35%) than previously reported by others (21%–23%),8,13,34 although our population is more than 3-fold greater in size. Twenty-three percent of the patients in our database had anti-MJ autoantibodies, which was similar to the frequency reported in the United Kingdom (23%)12 and in an Argentinian JDM population (25%).8 The prevalence of classic MSAs, including anti-synthetase, anti-SRP, and anti-Mi-2 autoantibodies, is consistent with other reports of less than 10% of JDM patients with positive assays.38,39
Adult patients and juvenile patients have different distributions of certain myositis autoantibodies, suggesting that different genetic risk factors or environmental exposures are important in the pathogenesis of these diseases. Adult IIM studies reported that 20%–30% of patients have anti-synthetase autoantibodies.20,37 In contrast, only 5% of our JIIM patients had anti-synthetase autoantibodies. The lower prevalence of anti-synthetase autoantibodies in children with myositis compared to adults suggests that the frequency of possible environmental risk factors for these autoantibodies, such as smoking or certain occupational exposures that are known to differ with age, may play a role in the development of this phenotype.4,19 Anti-SRP autoantibodies were slightly less common in children than adults. Only 1.6% of our juvenile IIM patients had anti-SRP autoantibodies, in contrast to a reported prevalence of 3%–5% in adult-onset IIM patients.35 Anti-Mi-2 autoantibodies represented 3% of our JIIM population, and in previous studies of JIIM, anti-Mi-2 autoantibodies occurred at a frequency of 4%–10%.38,39 This finding is similar to what has been reported in adult IIM (5%–8%).20,38
Specific autoantibodies were highly associated with certain clinical subgroups. For example, anti-SRP autoantibody-positive patients all had JPM. Patients with anti-Mi-2, anti-MJ, or anti-p155/140 autoantibodies displayed classic DM signs and symptoms and almost exclusively had JDM or overlap myositis associated with JDM. Patients with anti-synthetase autoantibodies had significant associations with overlap myositis and JPM. We were able to stratify by clinical subgroup and test for confounding of the clinical subgroup in defining differences between the autoantibody subgroups. However, in most comparisons, subgroup was unrelated to the autoantibody phenotype. Clinical subgroup was important when comparing MSA/MAA-negative and anti-MJ-positive patients and was a potential confounder in that analysis. We were unable to test for clinical subgroup in the comparison of patients with anti-synthetase versus anti-SRP autoantibodies, as the latter all had JPM. Future studies that focus on the association of clinical subgroup and autoantibody group would help to improve the classification of JIIM patients.
Past studies suggest that the classic MSAs are generally mutually exclusive, with only 1 MSA present in a given patient, and that certain autoantibodies are more consistently associated with specific individual clinical profiles.20,37 However, in our population, several patients had coexistence of anti-Mi-2 with 1 of the newer DM-associated MSAs: 6 patients had both anti-Mi-2 and anti-p155/140 autoantibodies, and 1 patient had anti-Mi-2 and anti-MJ autoantibodies. Muro et al23 reported several patients with both anti-TIF1α and anti-Mi-2 autoantibodies, and Fujimoto et al11 reported the coexistence of anti-TIF1α and TIF1γ autoantibodies. The reason for this association is not known, although it is interesting in view of the clinical association with DM. Generally the anti-p155/140 or anti-MJ autoantibodies in sera containing anti-Mi-2 were detected only by immunoprecipitation blotting assay, not by routine immunoprecipitation; this is possibly due to interference by the presence of anti-Mi-2 autoantibodies, which is a complex autoantigen. The more sensitive immunoprecipitation blotting assay may also have detected lower titers of anti-p155/140 or anti-MJ than could be detected by routine immunoprecipitation.36
Characteristics of Distinct JIIM Clinical Subgroups Based on Myositis Autoantibodies and Comparison to Adults With the Same Autoantibody Specificities
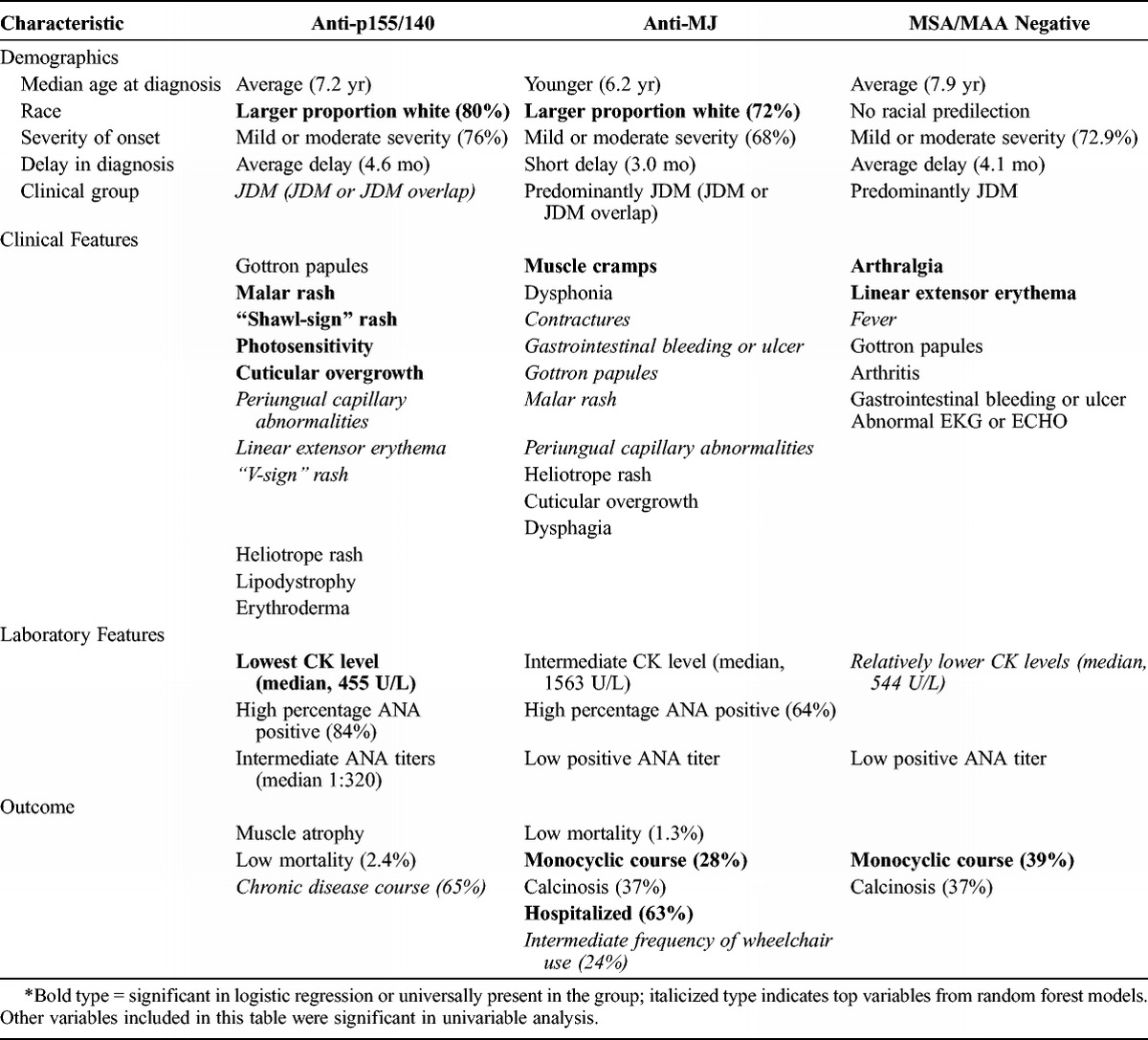
Based on these data, we discovered a number of distinct features associated with the classic MSAs and the 2 recently identified MSAs, anti-p155/140 and anti-MJ autoantibodies. The most essential findings are reported in Tables 13 and 14, with the level of evidence indicated as well in these tables.
TABLE 13.
Characteristics of Juvenile Myositis Classic Autoantibody Phenotypes*
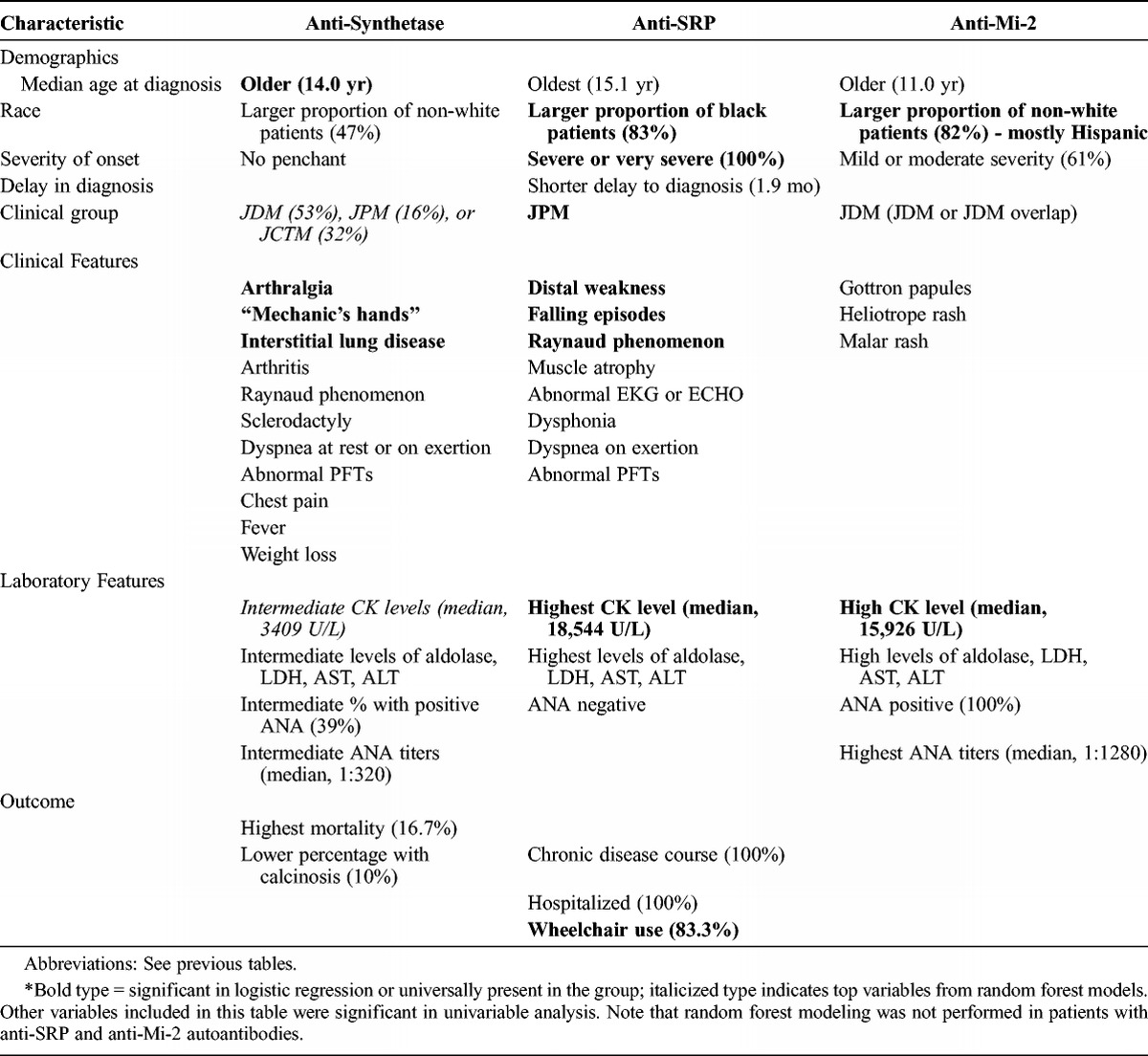
TABLE 14.
Characteristics of Recently Defined Autoantibody Phenotypes in Juvenile Myositis
Anti-synthetase autoantibody-positive patients accounted for 5% of all patients in this large juvenile myositis population; 12 patients were anti-Jo-1 positive and 7 others were positive for the less common anti-synthetase autoantibodies (including anti-alanyl- and glycyl-tRNA synthetase). Important characteristics of patients with anti-synthetase autoantibodies included having an older age at diagnosis and being non-white. Anti-synthetase autoantibodies are highly associated with interstitial lung disease.20,22 In this study, over 60% of patients with anti-synthetase autoantibodies had interstitial lung disease, which is comparable in frequency to some reports in adults but lower than others.7,19,20,38 Dyspnea on exertion, abnormal pulmonary function tests, and dyspnea at rest were also major characteristics of anti-synthetase-positive patients, as in adult patients.38 Other classic features of the anti-synthetase syndrome in adults, such as arthralgia and mechanic’s hands, were also strongly associated with this subgroup of children. Fevers, arthritis, Raynaud phenomenon, and sclerodactyly were also common in the children with anti-synthetase autoantibodies, and they had intermediate to high CK levels, as is the case in adult IIM patients with these autoantibodies.15,17 Anti-synthetase autoantibody-positive patients were more likely to be in the JCTM clinical subgroup than patients with the other MSAs. This might explain why patients with anti-synthetase autoantibodies were more likely to have Raynaud phenomenon, sclerodactyly, interstitial lung disease, arthralgia, and weight loss, which are clinical features we found to be frequent in patients with JCTM.29 However, when we tested for confounding of clinical subgroup in distinguishing anti-synthetase autoantibodies from other MSAs, clinical subgroup was generally not a cofactor in these models, suggesting that these are features of the anti-synthetase autoantibodies themselves. Patients with anti-synthetase autoantibodies tended to have a chronic continuous disease course, similar to adults.20,38 Children with anti-synthetase autoantibodies compared to the other MSAs had higher mortality (16.7% over a median of 2.7 yr), which was similar to the adult population (about 25% over 5 yr) and primarily due to interstitial lung disease.20 One limitation of the current study is that we combined Jo-1 and non-Jo-1 anti-synthetase patients, whose features can differ, particularly in the frequency of interstitial lung disease and severity of muscle weakness.12 The shorter duration of follow-up in the anti-synthetase autoantibody subgroup could also have resulted in symptoms and outcomes that were not as fully developed in this group of patients.
Anti-SRP autoantibody-positive patients accounted for only 1.6% of JIIM patients. Despite the low prevalence, however, patients with anti-SRP autoantibodies were clinically distinct. Anti-SRP autoantibody-positive patients all had JPM, and as in adult patients, all had severe PM.16,20,38 Anti-SRP autoantibody-positive patients were older at diagnosis (median age, 15 yr), and were mostly black females, consistent with previous studies.28,32 Anti-SRP patients, in this JIIM population, had a higher frequency of distal weakness, muscle atrophy, falling episodes, and very high CK levels, suggestive of more severe weakness, consistent with previous reports.20,28,31,35 As in some reports of adults and other children, our patients with anti-SRP autoantibodies also had a high frequency of cardiac abnormalities on electrocardiogram or echocardiogram,20,28,35 but unlike the adult patients, children with anti-SRP did not have frequent palpitations.20,28,35 Similar to Rouster-Stevens et al,28 we also observed frequent Raynaud phenomenon, dysphonia, and dyspnea on exertion. The median CK level for anti-SRP autoantibody-positive patients was very high, similar to other reports.20,31,35 The high CK levels in this subgroup may reflect disruption of the myofiber membrane related to a severe immune-mediated necrotizing myopathy, characterized by myofiber necrosis and little to no muscle inflammation.35 All of our JIIM patients with anti-SRP autoantibodies were hospitalized, most used a wheelchair, and they had a chronic continuous disease course. However, unlike adult patients, none of the juvenile patients died during the follow-up period, which may be the result of better diagnosis and treatment options or a greater capacity for muscle regeneration, including cardiac muscle.
The key associations with anti-Mi-2 autoantibodies, which were present in only 2.9% of JIIM patients, were highest ANA titers and being non-white. Approximately 45% of the patients with anti-Mi-2 autoantibodies were Hispanic. This was consistent with a study by Shamim et al,30 who reported a higher prevalence of anti-Mi-2 autoantibodies in Meso-Americans compared to whites. Patients with anti-Mi-2 autoantibodies had classic DM, since all patients were classified as either JDM or JDM with overlap, confirming that anti-Mi-2 autoantibodies were specific to JDM in JIIM patients.14,20,39 The predilection for JDM explains the higher prevalence of Gottron papules, heliotrope rash, and malar rash in anti-Mi-2 autoantibody-positive patients. Patients with anti-Mi-2 autoantibodies had a high frequency of ANAs, which were also in high titer. Children with anti-Mi-2 autoantibodies often followed a polycyclic illness course. Clinical characteristics of patients with anti-Mi-2 autoantibodies in the current study were generally consistent with those reported in previous studies, in that anti-Mi-2 is associated with typical cutaneous lesions and mild-to-moderate muscle involvement and very low mortality.14,18,20 Different from findings in adults, we did not find the Mi-2 group to be associated with V-sign or shawl-sign rashes or cuticular overgrowth.20 One reason for differences in the features of anti-Mi-2 autoantibodies between children and adults might relate to the fact that some of the adult patients with anti-Mi-2 autoantibodies had cancer-associated DM, whereas the children with this autoantibody did not.20
A recently identified autoantibody, anti-p155/140, was present in 35% of our population as the most common autoantibody identified in JIIM patients, and was associated with JDM or JDM seen with JCTM. The clinical features associated with anti-p155/140 autoantibodies differed from those associated with anti-synthetase and anti-SRP autoantibodies. Patients with anti-p155/140 autoantibodies were mostly white and had mild or moderate disease at illness onset, and they most frequently had a chronic disease course. Similar to anti-MJ, anti-p155/140 autoantibody-positive patients displayed classic JDM features, such as Gottron papules, heliotrope and malar rashes, and periungual capillary abnormalities. In addition, these patients developed frequent photosensitivity, linear extensor erythema, cuticular overgrowth, V-sign and shawl-sign rashes, lipodystrophy, and erythroderma, which are all cutaneous features of DM.6 Patients who were anti-p155/140 autoantibody positive had the lowest CK levels compared to the other autoantibody groups, possibly explaining the lower prevalence of severe muscular manifestations. The commonly associated rashes might be related to their associations with DM, rather than being distinct to p155/140 autoantibodies.34 However, when we tested for confounding of clinical subgroup in our logistic regression models, we did not find clinical subgroup to be a confounder, suggesting that these are indeed part of the features of the autoantibody subgroup itself. Compared to patients studied by Gunawardena et al,13 our patients were slightly older at diagnosis (median age, 7.2 yr compared to 6.0 yr, respectively). Similar to other studies,11,13 the presence of Gottron papules was a significant clinical feature. However, our data did not show that edema or ulceration were significant features of the anti-p155/140 autoantibody subgroup.11,13 Children with anti-p155/140 were less likely to report V-sign rash compared to adults, but still had a higher frequency of this rash than the other autoantibody subgroups. Our study provides additional data on the phenotype of the p155/140 subgroup, including demographic and clinical features and outcomes. One limitation is that we did not distinguish between TIF1γ and TIF1α autoantibodies, which may coexist and have slightly different clinical features.11
Patients with anti-MJ autoantibodies accounted for 23% of the total population, as a second major serologic subset of JIIM, comparable to that of JDM populations in the United Kingdom and South America.8,12 This autoantibody group was most commonly found in JDM patients and JCTM associated with JDM, but was also seen in 3 JPM patients and 1 JCTM patient with associated JPM. Other investigators have also observed that anti-MJ autoantibodies are strongly associated with JDM, although not exclusively.3,8 Patients with anti-MJ autoantibodies in our population had the youngest age at onset and age at diagnosis compared to other classic MSA groups, and most were white. Important clinical features characteristic of the anti-MJ autoantibody-positive patients included muscle cramps, dysphonia, and calcinosis, although the frequency of calcinosis was lower than that previously reported.12 These patients also had more frequent gastrointestinal bleeding and ulcers, dysphagia, and classic JDM rashes, including Gottron papules, heliotrope and malar rashes, and periungual capillary changes. Gunawardena et al12 noted an absence of truncal rashes with anti-MJ autoantibodies, which we did not see in our population. The results of this study were consistent with those in the Argentinean JIIM population, in which there was a greater frequency of muscle cramps and falling episodes in children with anti-MJ autoantibodies, as well as wheelchair use.8 Espada and colleagues8 found that in juvenile patients, anti-MJ autoantibodies were associated with joint contractures, muscle atrophy, and significant compromise of functional status, the first 2 of which were frequent in our population but did not differ from other MSA subgroups. Hospitalization was common among anti-MJ autoantibody-positive patients, even though severity of onset and mortality were low. The present analysis adds to the literature, solidifying that this autoantibody should be classified under the rubric of an MSA and is among the most frequent MSA subgroups in children with myositis.
Although this group was used as a reference for comparison, patients who were MSA/MAA negative displayed unique characteristics compared to those with a particular MSA. These patients predominantly had JDM and were more likely to have a monocyclic illness course. Clinical features that were prevalent in this group included arthralgia and linear extensor erythema as the most distinct, but also frequent arthritis, Gottron papules, gastrointestinal bleeding or ulceration, abnormal electrocardiogram or echocardiogram findings suggesting cardiac involvement, and fever. Although there was no clinical feature that was seen solely in this subgroup, some of these features are characteristic of more severe illness. When comparing this subgroup to the adult MSA/MAA-negative group, we saw little similarity, most probably because the Love et al study20 occurred before the discovery of the newer MSAs. Novel MSAs are still being discovered, and some of our MSA/MAA-negative patients may have autoantibodies of as-yet-undefined specificities.1,22
Limitations
Although the current study is one of the largest registry studies of juvenile myositis, the number of patients with certain MSA is relatively low, which limits the power to test for interactions formally. In fact, in multivariable logistic regression analyses, only a few variables were significant, most likely because they are limited by small sample sizes. Our solution to small sample size was to use exact logistic regression; however, even with the exact regression, in some cases there is suggestion of very wide confidence intervals and possible model instability.
Methods of autoantibody testing differ between laboratories, which may in part explain some of the differences in our results compared to other studies. The methods used in the current study, immunoprecipitation, and for anti-p155/140 and anti-MJ autoantibodies, immunoprecipitation and immunoblotting, are gold standards and repeated to be certain of the results and their reproducibility whenever needed. Immunoblotting of immunoprecipitates was used to confirm that all immunoprecipitation-positive sera were reacting with the same antigen. However, a reverse immunoprecipitation blotting strategy was necessary because two-thirds of sera did not react by immunoblotting directly, suggesting exclusive reaction with conformational epitopes.36 In contrast, some studies for anti-p155/140 and anti-MJ autoantibodies used immunoprecipitation followed by immunodepletion, which is also a relatively specific method.13 Further clarification of these immunoreactivities will be possible when more direct assays have been developed. Additional limitations of this work have been discussed in the previous report by Shah et al.29
CONCLUSIONS
We conducted the current study to develop classification methods, using demographic, clinical, and laboratory features, to better identify stable and mutually exclusive phenotypes of JIIM. The findings show that clinically distinct phenotypes have unique characteristics and that testing for MSAs is important for predicting clinical course, treatment, and prognosis. Based on the findings of this study and our prior report,29 we propose a novel classification system for juvenile myositis patients that includes both clinical and autoantibody phenotypes (Table 15). In this new classification system, JPM and JCTM are unique subgroups in the JIIM family. We also recommend including anti-p155/140 and anti-MJ autoantibodies as part of the MSAs. We recommend testing for MSAs and MAAs in JIIM patients to confirm the diagnosis of JDM, JPM, or JCTM when necessary and to enhance prediction of the clinical features and possible course of the disease.
TABLE 15.
Proposed New Classification of Juvenile Idiopathic Inflammatory Myopathies

ACKNOWLEDGMENTS
The authors thank Drs. Karyl Barron and Michael Ward for valuable comments after their critical reading of the manuscript, and Dr. James Malley for helpful guidance on random forest analysis. We thank Mona Shah’s PhD thesis committee of the George Washington University Department of Epidemiology and Biostatistics for their valuable feedback on this work, including committee members Drs. Heather Young and Yinglei Lai and examiners Drs. Mark Gourley and Margaret Ulfers.
Abbreviations
- ANA
antinuclear antibody
- CK
creatine kinase
- CTM
myositis overlapping with another autoimmune or connective tissue disease
- DM
dermatomyositis
- IIM
idiopathic inflammatory myopathies
- JCTM
juvenile myositis overlapping with another autoimmune or connective tissue disease
- JDM
juvenile dermatomyositis
- JIIM
juvenile idiopathic inflammatory myopathies
- JPM
juvenile polymyositis
- JRA
juvenile rheumatoid arthritis
- MAA
myositis-associated autoantibody
- MDA
mean decrease in accuracy
- MSA
myositis-specific autoantibody
- PM
polymyositis
- PM-Scl
polymyositis-scleroderma
- SLE
systemic lupus erythematosus
- SRP
signal recognition particle
APPENDIX
Childhood Myositis Heterogeneity Collaborative Study Group
Members of the Childhood Myositis Heterogeneity Collaborative Study Group who contributed to this study:
Leslie S. Abramson, Barbara Adams, Daniel A. Albert, Kathy Amoroso, Bita Arabshahi, Eugene R. Arthur, Balu H. Athreya, Alan N. Baer, Imelda M. Balboni, Susan Hyatt Ballinger, C. April Bingham, William P. Blocker, John F. Bohnsack, Gilles Boire, Michael S. Borzy, Gary R. Botstein, Suzanne Bowyer†, Jon M. Burnham, Ruy Carrasco, Victoria W. Cartwright, Gail D. Cawkwell, Chun Peng T. Chao, Randy Q. Cron, Marietta M. DeGuzman, B. Anne Eberhart, Barbara S. Edelheit, John F. Eggert, Andrew H. Eichenfield, Melissa E. Elder, Janet E. Ellsworth, Terri H. Finkel, Irene Flatau, Robert C. Fuhlbrigge, Christos A. Gabriel, Vernon F. Garwood, Abraham Gedalia, Natalie L. Gehringer, Stephen W. George, Harry L. Gewanter, Ellen A. Goldmuntz, Donald P. Goldsmith, Phillip Gorden, Gary V. Gordon, Alexia C. Gospodinoff, Beth S. Gottlieb, Thomas A. Griffin, Brandt P. Groh, Hillary M. Haftel, Melissa Hawkins-Holt, Michael Henrickson, Gloria C. Higgins, George Ho Jr., Mark F. Hoeltzel, J Roger Hollister, Russell J. Hopp, Norman T. Ilowite, Lisa Imundo, Jerry C. Jacobs†, Laura James-Newton, Anna Jansen, James Jarvis, Rita S. Jerath, Courtney R. Johnson, Olcay Y. Jones, Lawrence K. Jung, Thomas V. Kantor, Ildy M. Katona, James D. Katz, Yukiko Kimura, Daniel J. Kingsbury, Steven J. Klein, C. Michael Knee, W. Patrick Knibbe, David K. Kurahara, Bianca A. Lang, Andrew Lasky, Alexander R. Lawton, Julia A. Lee, Johanan Levine, Carol B. Lindsley, Katherine L. Madson, Harold G. Marks, Paul L. McCarthy, John J. Miller III, Stephen R. Mitchell, Hamid Jack Moallem, Chihiro Morishima, Frederick T. Murphy, Terrance O’Hanlon, Kiem G. Oen, Judyann C. Olson, Elif A. Oral, Barbara E. Ostrov, Lauren M. Pachman, Ramesh Pappu, Murray H. Passo, Maria D. Perez, Donald A. Person, Karin S. Peterson, Paul H. Plotz, Marilynn G. Punaro, C. Egla Rabinovich, Charles D. Radis, Linda I. Ray, Ann M. Reed, Robert M. Rennebohm, Peter D. Reuman, Rafael F. Rivas-Chacon, Deborah Rothman, Kenneth N. Schikler, Donald W. Scott, Bracha Shaham, Robert M. Sheets, David D. Sherry, Edward M. Sills, Sara H. Sinal, Abigail Smukler, Sangeeta D. Sule, Robert P. Sundel, Ilona S. Szer, Simeon I. Taylor, Elizabeth S. Taylor-Albert, Richard K. Vehe, Scott A. Vogelgesang, Larry B. Vogler, Carol A. Wallace, Jennifer C. Wargula, Patience H. White, M. Jack Wilkenfeld, Lan Wu, Christianne M. Yung, Lawrence S. Zemel.
†Deceased.
*These authors contributed equally.
†Contributing members listed in the Appendix.
This work was supported in part by the Intramural Research Programs of NIEHS, NIH (project number ES101074), NIAMS, NIH, and CBER, Food and Drug Administration. Gulnara Mamyrova was supported by the Cure JM Foundation and The Myositis Association. Ira Targoff is a consultant to the Oklahoma Medical Research Foundation Clinical Immunology Laboratory.
The authors have no conflicts of interest to disclose.
The contributions of Drs. Rider, Shah, Mamyrova, and Miller are in the public domain.
REFERENCES
- 1. Betteridge ZE, Gunawardena H, McHugh NJ. Novel autoantibodies and clinical phenotypes in adult and juvenile myositis. Arthritis Res Ther. 2011; 13: 209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bohan A, Peter JB. Polymyositis and dermatomyositis. Parts 1 and 2. N Engl J Med. 1975; 292: 344– 347, 3403-3407. [DOI] [PubMed] [Google Scholar]
- 3. Ceribelli A, Fredi M, Taraborelli M, et al. Anti-MJ/NXP-2 autoantibody specificity in a cohort of adult Italian patients with polymyositis/dermatomyositis. Arthritis Res Ther. 2012; 14: R97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chinoy H, Adimulam S, Marriage F, et al. Interaction of HLA-DRB1*03 and smoking for the development of anti-Jo-1 antibodies in adult idiopathic inflammatory myopathies: a European-wide case study. Ann Rheum Dis. 2012; 71: 961– 965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Compeyrot-Lacassagne S, Feldman BM. Inflammatory myopathies in children. Rheum Dis Clin North Am. 2007; 33: 525– 553, vii. [DOI] [PubMed] [Google Scholar]
- 6. Dugan EM, Huber AM, Miller FW, et al. Review of the classification and assessment of the cutaneous manifestations of the idiopathic inflammatory myopathies. Dermatol Online J. 2009; 15: 2. [PubMed] [Google Scholar]
- 7. Dugar M, Cox S, Limaye V, et al. Clinical heterogeneity and prognostic features of South Australian patients with anti-synthetase autoantibodies. Intern Med J. 2011; 41: 674– 679. [DOI] [PubMed] [Google Scholar]
- 8. Espada G, Maldonado Cocco JA, Fertig N, et al. Clinical and serologic characterization of an Argentine pediatric myositis cohort: identification of a novel autoantibody (anti-MJ) to a 142-kDa protein. J Rheumatol. 2009; 36: 2547– 2551. [DOI] [PubMed] [Google Scholar]
- 9. Feldman BM, Reichlin M, Laxer RM, et al. Clinical significance of specific autoantibodies in juvenile dermatomyositis. J Rheumatol. 1996; 23: 1794– 1797. [PubMed] [Google Scholar]
- 10. Feldman BM, Rider LG, Reed AM, et al. Juvenile dermatomyositis and other idiopathic inflammatory myopathies of childhood. Lancet. 2008; 371: 2201– 2212. [DOI] [PubMed] [Google Scholar]
- 11. Fujimoto M, Hamaguchi Y, Kaji K, et al. Myositis-specific anti-155/140 autoantibodies target transcription intermediary factor 1 family proteins. Arthritis Rheum. 2012; 64: 513– 522. [DOI] [PubMed] [Google Scholar]
- 12. Gunawardena H, Wedderburn LR, Chinoy H, et al. Autoantibodies to a 140-kd protein in juvenile dermatomyositis are associated with calcinosis. Arthritis Rheum. 2009; 60: 1807– 1814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gunawardena H, Wedderburn LR, North J, et al. Clinical associations of autoantibodies to a p155/140 kDa doublet protein in juvenile dermatomyositis. Rheumatology (Oxford). 2008; 47: 324– 328. [DOI] [PubMed] [Google Scholar]
- 14. Hamaguchi Y, Kuwana M, Hoshino K, et al. Clinical correlations with dermatomyositis-specific autoantibodies in adult Japanese patients with dermatomyositis: a multicenter cross-sectional study. Arch Dermatol. 2011; 147: 391– 398. [DOI] [PubMed] [Google Scholar]
- 15. Hirakata M, Suwa A, Takada T, et al. Clinical and immunogenetic features of patients with autoantibodies to asparaginyl-transfer RNA synthetase. Arthritis Rheum. 2007; 56: 1295– 1303. [DOI] [PubMed] [Google Scholar]
- 16. Kao AH, Lacomis D, Lucas M, et al. Anti-signal recognition particle autoantibody in patients with and patients without idiopathic inflammatory myopathy. Arthritis Rheum. 2004; 50: 209– 215. [DOI] [PubMed] [Google Scholar]
- 17. Katzap E, Barilla-LaBarca ML, Marder G. Antisynthetase syndrome. Curr Rheumatol Rep. 2011; 13: 175– 181. [DOI] [PubMed] [Google Scholar]
- 18. Komura K, Fujimoto M, Matsushita T, et al. Prevalence and clinical characteristics of anti-Mi-2 antibodies in Japanese patients with dermatomyositis. J Dermatol Sci. 2005; 40: 215– 217. [DOI] [PubMed] [Google Scholar]
- 19. Labirua-Iturburu A, Selva-O’Callaghan A, Vincze M, et al. Anti-PL-7 (anti-threonyl-tRNA synthetase) antisynthetase syndrome: clinical manifestations in a series of patients from a European multicenter study (EUMYONET) and review of the literature. Medicine (Baltimore). 2012; 91: 206– 211. [DOI] [PubMed] [Google Scholar]
- 20. Love LA, Leff RL, Fraser DD, et al. A new approach to the classification of idiopathic inflammatory myopathy: myositis-specific autoantibodies define useful homogeneous patient groups. Medicine (Baltimore). 1991; 70: 360– 374. [DOI] [PubMed] [Google Scholar]
- 21. Malley JD, Malley KG, Pajevic S. Statistical Learning for Biomedical Data. Cambridge: Cambridge University Press; 2011. [Google Scholar]
- 22. Mammen AL. Autoimmune myopathies: autoantibodies, phenotypes and pathogenesis. Nat Rev Neurol. 2011; 7: 343– 354. [DOI] [PubMed] [Google Scholar]
- 23. Muro Y, Sugiura K, Hoshino K, et al. Disappearance of anti-MDA-5 autoantibodies in clinically amyopathic DM/interstitial lung disease during disease remission. Rheumatology (Oxford). 2012; 51: 800– 804. [DOI] [PubMed] [Google Scholar]
- 24. Rider LG, Lindsley C, Cassidy J. Juvenile dermatomyositis. In: Cassidy J, Petty R, Laxer R, Lindsley C, eds. Textbook of Pediatric Rheumatology. New York: Saunders Elsevier; 2011: 375– 413. [Google Scholar]
- 25. Rider LG, Miller FW. Deciphering the clinical presentations, pathogenesis, and treatment of the idiopathic inflammatory myopathies. JAMA. 2011; 305: 183– 190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rider LG, Miller FW, Targoff IN, et al. A broadened spectrum of juvenile myositis. Myositis-specific autoantibodies in children. Arthritis Rheum. 1994; 37: 1534– 1538. [DOI] [PubMed] [Google Scholar]
- 27. Robinson AB, Reed AM. Clinical features, pathogenesis and treatment of juvenile and adult dermatomyositis. Nat Rev Rheumatol. 2011; 7: 664– 675. [DOI] [PubMed] [Google Scholar]
- 28. Rouster-Stevens KA, Pachman LM. Autoantibody to signal recognition particle in African American girls with juvenile polymyositis. J Rheumatol. 2008; 35: 927– 929. [PubMed] [Google Scholar]
- 29. Shah M, Mamyrova G, Targoff IN, et al. ; with the Childhood Myositis Heterogeneity Collaborative Study Group. The clinical phenotypes of the juvenile idiopathic inflammatory myopathies. Medicine (Baltimore). 2013; 92: 25– 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Shamim EA, Rider LG, Pandey JP, et al. Differences in idiopathic inflammatory myopathy phenotypes and genotypes between Mesoamerican Mestizos and North American Caucasians: ethnogeographic influences in the genetics and clinical expression of myositis. Arthritis Rheum. 2002; 46: 1885– 1893. [DOI] [PubMed] [Google Scholar]
- 31. Suzuki S, Hayashi YK, Kuwana M, et al. Myopathy associated with antibodies to signal recognition particle: disease progression and neurological outcome. Arch Neurol. 2012; 69: 728– 732. [DOI] [PubMed] [Google Scholar]
- 32. Suzuki S, Ohta M, Shimizu Y, et al. Anti-signal recognition particle myopathy in the first decade of life. Pediatr Neurol. 2011; 45: 114– 116. [DOI] [PubMed] [Google Scholar]
- 33. Targoff IN. Laboratory testing in the diagnosis and management of idiopathic inflammatory myopathies. Rheum Dis Clin North Am. 2002; 28: 859– 890, viii. [DOI] [PubMed] [Google Scholar]
- 34. Targoff IN, Mamyrova G, Trieu EP, et al. A novel autoantibody to a 155-kd protein is associated with dermatomyositis. Arthritis Rheum. 2006; 54: 3682– 3689. [DOI] [PubMed] [Google Scholar]
- 35. Targoff IN, Nilasena DS, Trieu EP, et al. Clinical features and immunologic testing of patients with anti-Mi-2 antibodies. Arthritis Rheum. 1990; 33: S72. [Google Scholar]
- 36. Trieu EP, Gross JK, Targoff IN. Immunoprecipitation-western blot for proteins of low abundance. Methods Mol Biol. 2009; 536: 259– 275. [DOI] [PubMed] [Google Scholar]
- 37. Troyanov Y, Targoff IN, Tremblay JL, et al. Novel classification of idiopathic inflammatory myopathies based on overlap syndrome features and autoantibodies: analysis of 100 French Canadian patients. Medicine (Baltimore). 2005; 84: 231– 249. [DOI] [PubMed] [Google Scholar]
- 38. Vancsa A, Gergely L, Ponyi A, et al. Myositis-specific and myositis-associated antibodies in overlap myositis in comparison to primary dermatopolymyositis: relevance for clinical classification: retrospective study of 169 patients. Joint Bone Spine. 2010; 77: 125– 130. [DOI] [PubMed] [Google Scholar]
- 39. Wedderburn LR, McHugh NJ, Chinoy H, et al. HLA class II haplotype and autoantibody associations in children with juvenile dermatomyositis and juvenile dermatomyositis-scleroderma overlap. Rheumatology (Oxford). 2007; 46: 1786– 1791. [DOI] [PubMed] [Google Scholar]