

Abstract
Lipid droplets (LDs) are dynamic, cytosolic lipid-storage organelles found in nearly all cell types. Too many or too few LDs during excess or deficient fat storage lead to many different human diseases. Recent insights into LD biology and LD protein functions shed new light on mechanisms underlying those metabolic pathologies. These findings will likely provide opportunities for treatment of diseases associated with too much or too little fat.
Keywords: atherosclerosis, lipid droplet, lipodystrophy, metabolic syndrome, triglyceride storage
Introduction
Balancing fluctuations in availability and requirements of metabolic energy is important for life. With their high energy content, triglycerides (TGs) in cellular lipid droplets (LDs) are the largest energy reservoir in most organisms. Among different cell types, the plasticity of TG storage is impressive. However, saturating or exceeding the fat storage capacity leads to disease in humans. This is most obvious in obesity and linked pathologies. Less apparent is that deficiencies in forming or maintaining fat stores also are detrimental. While obesity and related diseases in most humans have multifactorial etiologies, recent discoveries in LD biology have begun to shed light on mechanisms that contribute to these metabolic diseases. Here, we examine metabolic disease from the LD viewpoint, reviewing the basic mechanisms that contribute to the development of pathologies associated with imbalances of LDs and fat storage (Fig 1).
Figure 1. The balance of fat is important for human health.
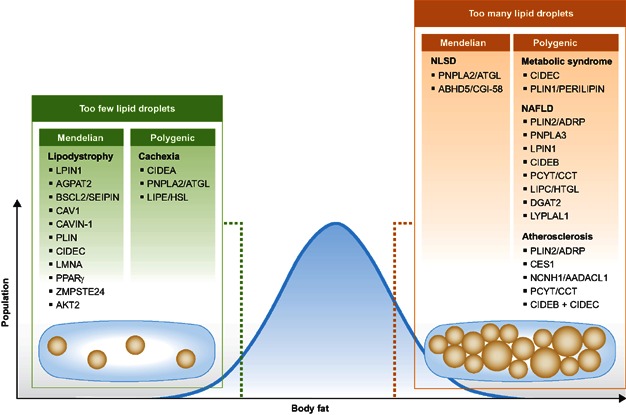
Within the human population, too much or too little body fat results in different monogenic or polygenic pathologies. Human genes that are linked to these extremes of body fat and LD storage are shown and are discussed in the text.
Lipid droplets: how cells store fat
To store excess lipids, such as sterols or fatty acids, cells esterify these lipids, forming neutral lipids, and package them into cytosolic LDs. In humans, adipocytes in white and brown adipose tissue are specialized for storing lipids in LDs. However, other cells also store lipids in LDs, including hepatocytes, enterocytes, macrophages and adrenocortical cells. Fat can also be prominent in tissues of blood formation or maturation, such as the bone marrow or thymus.
LDs have a nonpolar, neutral lipid core. In white adipocytes or macrophages, the core is mainly TG or sterol esters, respectively. In liver stellate cells, retinol esters are prominent. The LD core is surrounded by a phospholipid monolayer. In mammalian cells, the monolayer is mostly phosphatidylcholine with lesser amounts of phosphatidylethanolamine, phosphatidylinositol, lyso-phosphatidylcholine, and lyso-phosphatidylethanolamine (Liu et al, 2007). Phosphatidylcholine in particular is crucial as a surfactant to prevent LD coalescence, which might contribute to diseases of lipid storage excess. Depletion of proteins that change the phospholipid content dramatically changes LD morphology (Fei et al, 2011; Goodman et al, 2007; Guo et al, 2008; Krahmer et al, 2011).
Some proteins bind LD surfaces and regulate LD size and number. These include the PAT proteins [e.g., perilipin1, perilipin2/adipophilin (ADRP), perilipin3/Tip47, perilipin4/S3-12], CIDE (Cell Death Inducing DNA Fragmentation Factor) proteins, and several lipases. However, the LD proteome is much more complex: hundreds of proteins have been found in purified LDs fractions of different cell types, although many of these proteins may be contaminants from biochemical purifications (Martin et al, 2009). Recent quantitative proteomics approaches provide a means to distinguish bona fide LD proteins from contaminants with high confidence. In Drosophila cells, such an approach yielded ∼100 LD-enriched proteins (Krahmer et al, 2013).
LDs are dynamic organelles, constantly forming, growing or shrinking. They are mostly formed in the endoplasmic reticulum (ER), where enzymes catalyzing neutral lipid synthesis (e.g., acyl-CoA cholesterol acyltransferases (ACATs) for sterol esters and acyl CoA:diacylglycerol acyltransferases (DGATs) for TGs) predominantly reside (Farese et al, 2000, 2008). In fatty acid excess conditions, LDs rapidly increase their volumes, as seen for example in cell culture or murine small intestine. In part, this represents expansion of individual LDs by relocalization of TG synthesis to LD surfaces (Farese et al, 2009; Stone et al, 2009; Wilfling et al, 2013; Xu et al, 2012). During LD growth, the synthesis of neutral lipids and phospholipids is coordinated: as LD volume increases, surfaces expand, and phospholipids are needed to shield the neutral lipid core, reduce surface tension, and prevent LD coalescence (Krahmer et al, 2011). Thus, changes in LD phospholipid composition are important in determining their morphology and may play a role in diseases with altered lipid storage.
The main lipid storage tissue is the adipose tissue. In contrast to other cell types that form multiple small LDs with diameters of 100 nm to 5 µm, white adipocytes mainly form one giant, unilocular LD as large as 100 µm. These unilocular LDs likely provide the most efficient packaging of energy per volume. Several proteins such as the CIDE proteins were found to promote the formation of unilocular LDs.
LD degradation and mobilization of stored lipids are highly regulated processes (see Ahmadian et al, 2009; Lass et al, 2011). When fatty acids are required for energy generation or membrane lipid synthesis, lipids from LDs are used. In adipose tissue, catecholamines induce signalling pathways that activate adipose triglyceride lipase (ATGL) and hormone sensitive lipase (HSL), the lipases that act in concert on the LDs to degrade TGs. TGs may also be mobilized by lipases in autophagosomes (Czaja et al, 2009). Changes in lipolysis and lipid mobilization contribute to some lipid storage diseases.
LDs also function in multiple cellular processes, such as protein storage and degradation, viral replication and regulation of activities of associated enzymes (see Murphy, 2012; Thiele & Spandl, 2008; Walther & Farese, 2012).
Glossary
ACAT 1
Acyl-CoA:cholesterol acyltransferase 1 is one of two ER proteins that catalyze the formation of cholesterol esters by using cholesterol and fatty acyl CoA as substrates.
Adiponutrin/PNPLA3
Patatin-like phospholipase domain-containing protein 3 has TG lipase and acylglycerol O-acyltransferase activities.
AGPAT2
1-Acylglycerol-3-phosphate O-acyltransferase 2 catalyzes TG synthesis by using 1-acylglycerol-3 phosphate and fatty acyl CoA as substrates.
AKT2
AKT2, also called protein kinase B (PKB), is a serine/threonine protein kinase involved in insulin signalling.
ATGL
Adipose triglyceride lipase is a LD protein catalyzing the initial step in TG hydrolysis, the hydrolysis of TG into diacylglycerol.
AZGP1
Zinc-alpha-2-glycoprotein stimulates lipid degradation in adipocytes and causes the extensive fat losses associated with some advanced cancers. May bind polyunsaturated fatty acids.
Caveolin 1
Acts as a scaffolding protein within the specialized plasma membrane domains caveolae, and is involved in signalling.
Cavin
Structural protein of caveolae.
CIDE proteins
Cell Death Inducing DNA Fragmentation Factor proteins, are a family of LD proteins including three members CIDEA, CIDEB, CIDEC/FSP27. They were initially found to be involved in cell death. CIDEC, and possibly other CIDE proteins, promote LD growth by mediating directional TG transfer from smaller to larger LDs at LD contact sites.
CGI-58
Comparative gene identification-58, a coactivator of ATGL, which also interacts with perilipin1 and perlipin2/ADRP.
CTα
Also called CCT. CTP:phosphocholine cytidylyltransferase is the rate-limiting enzyme for phosphatidylcholine synthesis and catalyzes the reaction: CTP + phosphocholine → diphosphate + CDP-choline.
DGAT1
Acyl-CoA:diacylglycerol acyltransferase, one of two ER proteins catalyzing TG synthesis by using diacylglycerol and fatty acyl CoA as substrates.
DGAT2
Acyl-CoA:diacylglycerol acyltransferases, LD and ER protein catalyzing TG synthesis by using diacylglycerol and fatty acyl CoA as substrates.
HSL
Hormone sensitive lipase, a LD protein primarily catalyzing the second step in TG hydrolysis, the conversion of diacylglycerol into monoacylglycerol.
LaminA
Nuclear protein forming nuclear lamina to provide a framework for the nuclear envelope.
Lipin1
A phosphatidate phosphatase which catalyzes the conversion of phosphatidic acid to diacylglycerol for TG synthesis and localizes to the ER, nucleus and LDs.
PAT proteins
Family of LD proteins including Perilipin 1, Perilipin2/adipophilin (ADRP), Perilipin3/TIP47, Perilipin4/S3-12, Perilipin 5 (OXPAT). They interact with lipases and regulate their access to LDs. Moreover they are suggested to be involved in LD biogenesis and LD growth.
Seipin/BSCL2
Berardinelli–Seip congenital lipodystrophy type 2 protein localizes to ER–LD junctions and regulates neutral lipid storage. Its molecular function is unknown.
TGH
An ER luminal TG hydrolase.
ZMPSTE24
Zinc metallopeptidase STE24 homologue that activates laminA/C by proteolytic cleavage.
Pathologies associated with too few lipid droplets
Genetic defects in lipid storage: lipodystrophies
Human lipodystrophies are the clinical manifestations of total (congenital generalized lipodystrophy, CGL) or partial (familial partial lipodystrophy, FPL) loss of body fat (see Garg & Agarwal, 2009; Huang-Doran et al, 2010; Vigouroux et al, 2011). Lipodystrophies cause severe changes of whole-body energy metabolism and are commonly associated with insulin resistance, hepatic steatosis, hypertension and other metabolic dysfunctions. Inability to store TGs in white adipose tissue gives rise to lipid storage in other tissues and tissue lipotoxicity. Lack of white adipose tissue leads to leptin deficiency and associated metabolic defects, such as insulin resistance. Many defects of severe lipodystrophies can be corrected by leptin supplementation (Oral et al, 2002; Shimomura et al, 1999).
Numerous gene defects cause CGL or FPL (reviewed in Garg, 2011). Here we review lipodystrophies with established connections to LD biology. Many of the lipodystrophy genes encode TG synthesis or storage proteins, and some act at the LD surface and are involved in LD formation and regulation. Still others regulate adipogenesis, thereby affecting LDs indirectly.
Two CGL loci encode proteins that regulate de novo TG synthesis: 1-acylglycerol-3-phosphate O-acyltransferase 2 (AGPAT2) and lipin1 (LPIN1). AGPAT2 catalyzes the formation of phosphatidic acid from lyso-phosphatidic acid and fatty acyl-CoA. Lipin1, a phosphatidic acid phosphatase, removes the phosphate group from phosphatidic acid to form diacylglycerol, the direct precursor of TG. AGPAT2 mutations account for ∼50% of CGL (Van Maldergem et al, 2002). So far, none of the other AGPAT isoforms appears to be implicated in CGL. This suggests a specialized role for AGPAT2, at least in adipocytes. In contrast to AGPAT2, LPIN1 mutations have been found to be associated with lipodystrophy only in mouse models; there are no known human LPIN1 mutations causing lipodystrophy.
How AGPAT2 and LPIN1 mutations cause lipodystrophy is uncertain. Mutation of either enzyme causes reduced TG levels as well as accumulation of lipid synthesis intermediates, and phospholipids in tissues. For example, both Lipin1 and Agpat2 knockout mice have increased phosphatidic acid and lyso-phosphatidic acid levels in the adipose tissue (Gale et al, 2006; Peterfy et al, 2001). One model posits that lipid synthesis products, such as phosphatidic acid or lyso-phosphatidic acid, accumulate and inhibit adipocyte differentiation, perhaps by influencing PPARγ signalling (Gale et al, 2006; Yang et al, 2011).
Mutations of Berardinelli–Seip congenital lipodystrophy 2 gene (BSCL2/SEIPIN) also lead to severe CGL. BSCL2 mutations in lipodystrophy patients result mostly in null alleles (Agarwal et al, 2004). BSCL2 mutations exhibit more severe lipodystrophy and metabolic alterations than AGPAT2 mutations (Van Maldergem et al, 2002).
How BSCL2 deficiency causes lipodystrophy remains unclear. The BSCL2 protein seipin is an ER protein, embedded in the membrane by a hairpin-type hydrophobic sequence. BSCL2 is expressed in adipocytes and up-regulated during adipocyte differentiation in 3T3L1 cells (Chen et al, 2009). BSCL2 knockdown inhibits adipocyte differentiation and suppresses PPARγ expression in this cell line (Chen et al, 2009). Moreover, it causes strong alterations in LD size, distribution, and number (usually fewer LDs) in multiple cell types and organisms, leading to markedly altered LD morphology (Fei et al, 2008; Szymanski et al, 2007; Tian et al, 2011). The yeast seipin orthologue forms oligomers and localizes near LDs (Binns et al, 2010; Lundin et al, 2006; Szymanski et al, 2007), suggesting a role in LD formation. However, seipin might also function in lipid biosynthesis pathways: its knockout alters cell phospholipids in yeast and Drosophila (Fei et al, 2011; Tian et al, 2011) and fatty acid composition in lymphoblastoid cell-lines, the latter from reduced desaturase activity (Boutet et al, 2009). In yeast and Drosophila, seipin knockout leads to accumulation of phosphatidic acid, which might induce LD fusion and alter LD morphology (Fei et al, 2011). Phosphatidic acid excess may also influence signalling pathways for adipocyte differentiation. In Drosophila, BSCL2/seipin depletion leads to loss of fat body lipids and to ectopic TG accumulation (Tian et al, 2011).
Deficiency of the membrane protein caveolin 1 also causes lipodystrophy. A homozygous nonsense mutation with complete loss of CAVEOLIN1 (CAV1) expression causes CGL, and heterozygous mutations were found in patients with atypical partial lipodystrophy (Cao et al, 2008; Kim et al, 2008). Caveolin 1 is an essential organizer of caveolae, specialized cholesterol-rich microdomains in the plasma membrane that form invaginations. Caveolin 1 also localizes to LDs (Ostermeyer et al, 2001) and is highly expressed in adipocytes (Lisanti & Razani, 2001). A related protein, cavin 1, another component of caveolae (Hill et al, 2008), was found to be mutated in a patient with lipodystrophy (Matsuo et al, 2010; Shastry et al, 2010). Cavins interact with caveolin, and loss of cavin 1 leads to loss of caveolae and caveolin 1 mislocalization (Liu et al, 2008). Since mutations in CAV1 and cavin 1 profoundly affect whole-body TG storage, caveolae may be important in lipid storage (Pilch & Liu, 2011). Caveolae are thought to function in the cellular response to mechanical stress, endocytosis, transcytosis, fatty acid uptake, LD formation, and lipid trafficking (Parton & Bastiani, 2010). The relationship between caveolae and LDs is still unclear.
Mutations in two LD proteins, perilipin1 and CIDEC, have been reported to cause FPL. A homozygous CIDEC mutation in a FPL patient led to a truncated protein that does not target LDs (Rubio-Cabezas et al, 2009) and cannot induce formation of unilocular LDs in adipocytes. Instead multiple small LDs form, similar to the murine knockout phenotype (Nishino et al, 2008). Two frame-shift mutations in the C-terminus of perilipin1 are associated with FPL and insulin resistance. Unlike wild-type perilipin1, the mutated perlipin1 fails to bind to and inhibit comparative gene identification-58 (CGI-58), leading to constitutive ATGL activation by CGI-58, resulting in increased basal lipolysis (Gandotra et al, 2011). While those mutations account for FPL in very few individuals, most FPL cases are caused by mutation of lamin A (LMNA). As a component of the nuclear lamina, LMNA is important for maintaining the nuclear envelope functions, loss of which leads to premature cell death and loss of adipocytes (Garg, 2004). This might also happen in FPL patients carrying mutations of ZMPSTE24, encoding a metalloprotease required for processing lamin A (Agarwal et al, 2003). In several other FPL patients, mutations in the genes for transcription factor PPARγ and AKT2, a factor involved in insulin signalling, were reported (George et al, 2004; Savage et al, 2003). Those mutations lead to defective adipocyte differentiation and thereby disturb TG storage. It is unclear why certain mutations cause FPL rather than CGL. Perhaps FPL is caused by deficient TG storage in existing adipocytes, and CGL is due to failure to form adipocytes.
In contrast to genetic inherited forms of lipodystrophies, acquired forms due to autoimmune diseases or drug treatment are much more common (see Garg, 2004). Protease inhibitors used to treat HIV infection are the most frequent cause of acquired lipodystrophy. The mechanism of the pathogenesis is not well understood. HIV protease inhibitors change the expression and localization of transcription factors, such as PPARγ or SREBP, which mediate adipocyte differentiation (Caron et al, 2001). Moreover, they increase basal and stimulated lipolysis in adipocytes, as shown in 3T3-L1 cells. This effect is mediated by a decrease in perilipin levels on the LDs by increased lysosomal perilipin degradation (Adler-Wailes et al, 2005).
Rapid loss of triglyceride stores: cancer cachexia
Cachexia, a complex metabolic syndrome, is common in cancer patients, particularly gastrointestinal, prostate and lung cancer (Tisdale, 2005). Unlike lipodystrophy, which features chronic deficiency of adipose tissue, cachexia is an acute wasting disease. Lipid metabolism is fundamentally changed, leading to dramatically reduced body weight, caused early by a loss of adipose tissue and later by atrophy of skeletal muscle (Bing & Trayhurn, 2009). Those changes are associated with poor response to chemotherapy and high mortality: 15–20% of cancer deaths are caused by cachexia (Tisdale, 2002). Increased lipolysis is a key factor in cachexia, and cachexia patients show elevated blood glycerol and fatty acids (Shaw & Wolfe, 1987).
The role of LDs in cachexia is beginning to be unravelled. Recent advances reveal that ATGL, and not HSL as previously thought, mediates increased lipolysis in cachexia (Das et al, 2011). Atgl knockout mice are completely protected from adipose tissue wasting and had no increased lipolysis after inducing cachexia, despite high levels of lipid-mobilizing factors, such as zinc-alpha-2-glycoprotein 1 (AZGP1), tumour necrosis factor α (TNF-α), or interleukin-1 (Das et al, 2011). Thus, inflammatory and lipolytic mediators that activate ATGL, potentially secreted by the tumour, might cause uncontrolled loss of adipose tissue in cachexia. Intriguingly, skeletal muscle loss was also absent in Atgl knockout mice.
Pathologies associated with too many lipid droplets
An increasingly sedentary lifestyle and unhealthy eating habits have made obesity and its associated pathologies a dramatic global health issue. These diseases are characterized by over-accumulation of TGs and LDs. Additionally, some genetic diseases are associated with over-accumulation of LDs. Understanding these rare conditions might provide therapeutic insights for their cure and uncover new obesity treatments. Here, we review molecular insights provided by these more rare conditions.
Monogenetic diseases of triglyceride and lipid droplet accumulation
Neutral lipid storage disease (NLSD) is a rare, autosomal recessive disorder characterized by substantial systemic TG accumulation (Schweiger et al, 2009). Mutations in genes encoding adipose triglyceride lipase (ATGL) and its cofactor CGI-58 cause NLSD. Although these proteins collaborate during lipolysis, mutations in each cause different symptoms. Mutations in CGI-58 are associated with ichthyosis, a permeability barrier defect of the skin; ATGL mutations cause severe cardiomyopathy and systemic lipid accumulation in humans and mice (Lefevre et al, 2001; Schweiger et al, 2009). The differences suggest that CGI-58 has other, ATGL-independent functions.
New insights into the molecular mechanism of cardiomyopathy in ATGL-deficient NLSD reveal a role for LDs in cellular signalling (Haemmerle et al, 2011). Hydrolysis of TGs from LDs by ATGL releases activators of the PPARα/peroxisome proliferator-activated receptor-γ coactivator 1 (PGC1) complex in cardiac myocytes. Activity of the complex increases with fatty acid supplies in the cell and regulates mitochondrial oxidative capacity. Cardiac ATGL deficiency decreases PGC-1 expression, leading to mitochondrial dysfunction and lipid accumulation in the heart, thus causing heart failure. Treatment of Atgl KO mice with pharmaceutical PPARα agonists reverses mitochondrial dysfunction and restores heart function. Which LD proteins are involved in regulating lipolysis for PPARα signalling and which specific products of the ATGL reaction mediate PPARα-PGC1 activation remain unknown. Nonetheless, these findings suggest PPARα agonists will be useful in treating patients with NLSD cardiomyopathy.
Lipid droplets in obesity and associated diseases
Adipose tissue is critical in regulating lipid and glucose metabolism. It buffers lipid excess by sequestering fatty acids into TGs, thereby protecting the body from lipotoxicity, and releases fatty acids and glycerol for peripheral tissues during starvation. Adipose tissue also exerts an important endocrine function by secreting factors, such as leptin and adiponectin that regulate insulin sensitivity or inflammation (Capeau et al, 2011; Friedman, 2009). Obesity can lead to altered systemic metabolism, including the metabolic syndrome, which increases the risk of type II diabetes, steatohepatitis and coronary heart disease. With adipocyte hypertrophy, insufficient amounts of adipokines are secreted to maintain insulin sensitivity (Hotamisligil, 2003), and pro-inflammatory cytokines, such as monocyte chemoattractant protein-1 (MCP-1) and TNF-α, are secreted that induce macrophage infiltration and inflammation (Hotamisligil et al, 1993). TNF-α increases lipolysis and down-regulates proteins that promote TG storage and protect LDs from lipolysis (Guilherme et al, 2008). TNF-α reduces cellular perilipin levels, likely leading to increased basal lipolysis and free fatty acid levels in the blood (Souza et al, 1998a). These insulin resistance-promoting effects of TNF-α can be antagonized by anti-diabetic agents, such as thiazolidinediones (Souza et al, 1998b).
Normally, the adipose tissue sequesters free fatty acids and other lipids in form of inert TGs. However, under certain conditions, such as the metabolic syndrome, the neutral lipid storage capacity of the adipose tissue is exceeded. Free fatty acids flow from adipose to peripheral tissues, and ectopic lipids accumulate in skeletal muscle, heart or liver (van Herpen & Schrauwen-Hinderling, 2008). Excess bioactive lipids, such as diacylglycerol, free fatty acids, and related derivatives, may cause lipotoxicity, with lipids interfering with signalling pathways and promoting insulin resistance in skeletal muscle and hepatic tissue (Schaffer, 2003; Virtue & Vidal-Puig, 2010). Usually, there is also accompanying leptin resistance, which promotes insulin resistance (El-Haschimi et al, 2000).
Although multiple genetic and environmental factors contribute to the development of the metabolic syndrome, at the cellular level TG accumulation equates with massive over-accumulation of LDs in adipose and other tissues. LD-associated proteins in adipose tissue can in turn influence the pathology of excess TG storage in some individuals. Proteins, such as FSP27/CIDEC and perilipin1, are crucial for unilocular LDs in adipose tissue. For example, FSP27/CIDEC influences the development of the metabolic syndrome by regulating TG storage in adipocytes. FSP27/CIDEC polymorphisms influence obesity risk (Dahlman et al, 2005; Zhang et al, 2008, 2011). Studies examining expression of CIDE proteins and perilipin as a function of insulin sensitivity found that the levels of mRNAs that encode these LD-associated proteins correlate inversely with insulin sensitivity subjects similar body mass index (Guilherme et al, 2008). This indicates that high levels of expression of these TG storage-promoting proteins might help to sequester lipids in the adipose tissue and to protect against insulin resistance. However, paradoxically, FSP27/CIDEC knockout mice exhibit increased energy expenditure and are protected from diet-induced obesity and insulin resistance (Nishino et al, 2008). All three members of the CIDE family (CIDEA, CIDEB, FSP27/CIDEC) localize to LDs via their C-terminal CIDE-domain, as shown in different cell lines (Gong et al, 2009). In adipocytes, FSP27/CIDEC is important in forming unilocular LDs. Depleting FSP27/CIDEC prevents their formation, and overexpressing it in other cell types induces larger and fewer LDs (Gong et al, 2011; Jambunathan et al, 2011; Nian et al, 2010; Nishino et al, 2008; Puri et al, 2007). FSP27/CIDEC localizes mainly to contact sites between LDs, where it seems to facilitate lipid transfer from smaller to larger LDs in 3T3L1 cells (Gong et al, 2011).
Perilipin1 also regulates TG storage in white adipocytes (Greenberg et al, 2011). Perilipin1 knockout mice are lean and protected from diet-induced obesity (Martinez-Botas et al, 2000; Tansey et al, 2001). Perilipin1 localizes to LD surfaces and is an important regulator of lipolysis, protecting LDs from basal lipolysis (Zhai et al, 2010). However, when lipolysis is stimulated, perilipin1 regulates access of lipases to LDs (Sztalyrd et al, 2003).
In humans, expression of PERILIPIN and FSP27/CIDEC in adipose tissue inversely correlates with insulin resistance (Puri et al, 2008), and insulin-sensitive obese individuals have higher levels of perilipin1 and CIDEC in adipose tissue than insulin-resistant subjects of the same weight, suggesting that higher expression of these proteins promotes TG storage in adipose tissue and protects from lipotoxicity. These findings in humans contrast with findings in mice, in which the lack of perilipin1 or FSP27 leads to increased insulin sensitivity (Martinez-Botas et al, 2000; Nishino et al, 2008; Tansey et al, 2001). Such findings highlight the difficulties in extrapolating results from experiments in mice to human pathologies.
Lipid droplets in hepatic steatosis and liver disease
The metabolic syndrome is often accompanied by nonalcoholic fatty liver disease (NAFLD), the accumulation of TG-containing LDs in hepatocytes. Several studies show that during steatogenenesis, the expression pattern of several LD-associated PAT proteins changes in a PPARγ-dependent manner (Inoue et al, 2005; Matsusue et al, 2008; Schadinger et al, 2005). Notably, perilipin1, normally only in adipose tissue, is expressed in human hepatocytes of fatty liver (Fujii et al, 2008; Straub et al, 2008). Also ADRP levels are up-regulated in steatosis in humans and in mice (Motomura et al, 2006). ADRP-deficient mice are resistant to diet-induced fatty liver, implicating ADRP in hepatic lipid accumulation (Chang et al, 2006).
A polymorphism of another LD protein PNPLA3/adiponutrin was linked to increased risk for NAFLD development (Romeo et al, 2010). The molecular mechanism underlying PNPLA3/adiponutrin function in hepatic lipid metabolism is controversial. A recent study reported LPA acyltransferase activity for PNPLA3/adiponutrin (Kumari et al, 2012) in contrast to other studies detecting TG hydrolase activity (Jenkins et al, 2004). The mutation associated with increased risk for NAFLD was characterized as gain of function mutation leading to TG accumulation and thereby explaining the increase in NAFLD disposition (Kumari et al, 2012; Li et al, 2012).
Genetic variants in genes encoding two proteins with TG lipase activity, hepatic lipase (LIPC/HTGL) (Yamada et al, 2011) and lysophospholipase-like1 (LYPLAL1) (Speliotes et al, 2011), and the DGAT2 enzyme involved in TG synthesis (Kantartzis et al, 2009) have also been associated with the risk of developing hepatic steatosis. Interestingly the reported genetic variants were only associated with increased risk for hepatic steatosis but not insulin resistance (see Farese et al, 2012), consistent with the paradigm that sequestration of lipids into liver TG might protect from free fatty acid-induced lipotoxicity that promotes insulin resistance.
The liver secretes TG-rich very low-density lipoproteins (VLDL) for distributing lipids throughout the body. Blocking VLDL secretion leads to NAFLD but not always insulin resistance (Sun & Lazar, 2013). Tissue-specific ablation of hepatic CTP:phosphocholine cytidylyltransferase α (CTα), the rate-limiting step in phosphatidylcholine synthesis, blocks VLDL secretion and leads to hepatic steatosis in mice (Jacobs et al, 2004). CTα targeting to LDs, required for LD expansion (Krahmer et al, 2011), suggests that loss of this capability contributes to the phenotype. However, how cytosolic LDs are coordinated with VLDL secretion is not understood. This complex process might be regulated by the coordinated action of multiple proteins. Liver TG hydrolase (TGH) might play an important role in mobilizing TG from LD for VLDL secretion as ectopic TGH expression leads to increased VLDL secretion and a reduction of cellular TG pools (Lehner & Vance, 1999). However, as TGH is an ER enzyme with its active site localizing to the ER lumen (Lehner et al, 1999), it remains enigmatic how TGH accesses TGs in the LD core. The expression and compartmentalization of lipin may also be important for the regulation of VLDL formation. TGs synthesized by expression of lipin-1 were reported to be mainly channelled into VLDL in McA-RH7777 cells (Khalil et al, 2009). CIDEB is localized to ER and LDs, interacts with apoB, and promotes TG secretion in VLDL in murine hepatocytes (Ye et al, 2009). CIDEB, by interacting with LDs and the ER at the cytosolic face, might channel TGs stored in cytosolic LDs into VLDL for secretion.
Hepatitis C virus (HCV) infection also strongly increases the risk for hepatic steatosis by inducing metabolic changes in infected hepatocytes. The HCV core protein is targeted to LDs by a DGAT1-dependent mechanism (Herker et al, 2010), and LDs are important for the assembly of infectious viral particles (Miyanari et al, 2007). LD targeting of HCV core induces clustering and cellular redistribution of LDs in cultured cells (Boulant et al, 2008). The expression of a genetic variant of core is sufficient to induce steatosis in transgenic mice (Moriya et al, 1997). Although numerous mechanisms might contribute to steatosis caused by HCV core (Herker & Ott, 2011), at least in part HCV induces LD accumulation by inhibiting lipolysis and stabilizing LDs (Harris et al, 2011). This induction of steatosis also apparently depends on DGAT1.
Lipid droplets in cardiovascular disease
Atherosclerosis is a leading cause of death in industrial countries. Accumulation of cholesterol esters in arteries is tightly linked to atherosclerosis and can lead to myocardial infarction, stroke, or sudden cardiac death. Cholesterol esters in arteries are stored mainly in LDs of macrophage foam cells, named for their appearance by microscopy Atherogenic apoB-containing lipoproteins (low density lipoprotein (LDL), chylomicron and VLDL remnants), containing large amounts of cholesterol, accumulate in the subendothelial space and are taken up by macrophages (Moore & Tabas, 2011). In macrophages, internalized lipoproteins are hydrolyzed, and the free cholesterol is re-esterified by ACAT enzymes, in particular ACAT1 in macrophages, to cholesterol esters for storage in LDs. The specific protein composition of cholesterol ester droplets is unknown, and besides ACAT1, specific factors for forming and regulating cholesterol esters LDs are unknown. Recently, specific PAT proteins were shown preferentially on TG- or cholesterol ester-containing LDs in different cell lines (Hsieh et al, 2012), indicating that LD protein composition might vary depending on the neutral lipid content of the LD.
Macrophages that accumulate large amounts of cholesterol esters become foam cells. Cholesterol esterification in foam cells seems to be protective, since free cholesterol is toxic to cells and pro-inflammatory (Kellner-Weibel et al, 1998). Cholesterol in LDs continuously undergoes a cycle of cholesterol ester hydrolysis and re-esterification (Brown et al, 1980). Free cholesterol effluxes from macrophages to extracellular acceptors, such as nascent high-density lipoprotein (HDL) and apolipoprotein-AI, which mediate reverse cholesterol transport from peripheral tissues to liver and are inversely correlated with atherosclerosis (Khera et al, 2011). Thus, cholesterol ester formation and storage in macrophages likely provide buffering capacity until cholesterol can be removed from the arterial wall by cholesterol efflux and reverse cholesterol transport (Moore & Tabas, 2011). Consistent with this notion, ACAT1 knockout in macrophages of hyperlipidemic mice leads to a pro-inflammatory state in skin and, if anything, increased atherosclerosis (Accad et al, 2000; Fazio et al, 2001; Su et al, 2005). If cholesterol accumulation overwhelms removal mechanisms, inflammation and pathology progress, similar to that of a wound, leading to plaque formation, rupture and thrombosis (Bornfeldt & Tabas, 2011).
Several LD proteins (e.g., ADRP, CIDEB and CIDEC) are up-regulated in foam cells and may promote storage of cholesterol esters in LDs, thereby protecting from cellular toxicity from free cholesterol (Yuan et al, 2012). However, peritoneal macrophages from Adrp knockout mice accumulate fewer LDs upon cholesterol loading, and in atherosclerosis-prone apoE knockout mice, ADRP depletion decreases the number of LDs in foam cells, yet protects mice from atherosclerosis (Paul et al, 2008).
For cholesterol efflux, the rate-limiting step is cholesterol ester hydrolysis from LDs. Different lipases may act on the LD surface to mobilize cholesterol (Hua et al, 2009). HSL was thought to provide most of cholesterol ester hydrolase activity, but deleting HSL does not reduce cholesterol ester hydrolysis (Osuga et al, 2000). Other candidates include CES1 (carboxylesterase 1), which decreases cholesterol ester storage in human macrophages and promotes cholesterol efflux (Ghosh et al, 2003). Its expression in macrophages reduces atherosclerosis in LDL-receptor deficient mice (Zhao et al, 2007). Arylacetamide deacetylase-like 1 (AADACL1) depletion increases atherosclerosis and cholesterol ester storage in murine macrophages (Sekiya et al, 2009), and its overexpression in THP-1 cells reduces cholesterol ester storage (Igarashi et al, 2010). However, CES1 and AADACL1 localize to the ER with their active sites facing the lumen and not the LD surface (Quiroga & Lehner, 2011), indicating they likely function in cholesterol ester hydrolysis at a different site from LDs. Other lipases might be responsible for cholesterol ester mobilization from macrophage LDs.
Besides cytosolic cholesterol ester hydrolysis on the LD surface, cholesterol can be mobilized from LDs by autophagy, leading to degradation of cholesterol ester by lysosomal acid lipases (Marcel et al, 2011). The released free cholesterol provides a major source of ABC-AI-dependent cholesterol efflux to apoAI and a minor proportion of HDL-mediated efflux. Macrophages from autophagy-deficient autophagy protein-5 (Atg5) knockout mice show a reduced cholesterol efflux (Marcel et al, 2011). In foam cells, autophagy seems to be activated in response to cholesterol loading. These findings suggest that cholesterol ester accumulation in macrophages triggers autophagy and that accelerators of this pathway slow atherosclerosis.
In addition to esterifying excess cholesterol, macrophages up-regulate phosphatidylcholine synthesis to protect against cytotoxicity (Shiratori et al, 1994). Such up-regulation is induced in Drosophila cells and mammalian macrophages, at least in part, by relocalizing the rate-limiting enzyme choline–phosphate cytidylyltransferase to membranes and LDs (Krahmer et al, 2011), thereby increasing enzyme activity. Posttranslational regulation leads to increased phosphatidylcholine synthesis, helping to maintain a constant phosphatidylcholine:free cholesterol ratio in the cell membrane and preventing free cholesterol toxicity (Tabas, 2000). This mechanism might help to adapt cellular phosphatidylcholine synthesis to elevated needs during LD formation. Macrophages from CTα knockout mice die faster under cholesterol loading (Zhang et al, 2000). Macrophage cell death is an important factor in the progression of unstable plaques, and thus, phospholipid synthesis pathways allow intervention to prevent advanced lesions.
Summary and outlook
The balance of fat is important for human health. Too much or too little is linked to common disease pathologies. LDs store fat, but despite their importance for cell and organismal physiology, relatively little is known about many basic processes in LDs in different tissues. Identification of key players in LD biology will uncover molecular processes underlying lipid storage and promote better understanding of LD-linked diseases. Important proteins regulating lipid storage have been recently identified, and a number of genes encoding LD proteins have been linked to metabolic diseases. Some of these molecular insights could lead to therapies. For example, ATGL inhibitors could offer the possibility of treating cachexia, or DGAT1 inhibitors could be tested as a means to prevent hepatitis C infection. The recent upsurge in LD biology research will lead to more knowledge of lipid storage and novel molecular-based approaches for treating diseases due to over- or under-storage of fat.
Pending Issues
What are the physiological functions of different LD populations, e.g. generated by different enzymes and containing different lipids? Do specific factors regulate their formation and consumption?
Why do mutations in some lipodystrophy genes cause congenital generalized lipodystrophy and others familial partial lipodystrophy?
What role does altered phospholipid levels play in the pathogenesis of lipodystrophies in patients with AGPAT2 and BSCL2/SEIPIN mutations? What is the mechanism of action for these proteins?
Which factors related to ATGL-mediated lipolysis trigger cancer cachexia?
What determines the capacity of cells to store neutral lipids? What are the mechanisms of lipotoxicity leading to atherosclerosis, insulin resistance, and other pathologies?
How do FSP27/CIDEC and PAT proteins influence insulin sensitivity? Why are CIDE knockout mice protected from obesity and insulin resistance whereas in humans, expression levels inversely correlate with insulin sensitivity?
How are neutral lipid storage in LDs and neutral lipid secretion via VLDL balanced and regulated? Which proteins regulate those processes and which enzymes mobilize neutral lipids for secretion? Is there the possibility for pharmacological intervention for the treatment of NAFLD and its progression to NASH?
Acknowledgments
We thank Drs. Douglas Mashek and Ivana Semova for comments on the manuscript and Gary Howard for editorial assistance. Research on lipid droplets in the Walther and Farese laboratories is supported by NIH–GMS R01s GM097194 and GM099844, respectively, the Gladstone Institutes (RF), and the Mathers Foundation (TW).
The authors declare that they have no conflict of interest.
References
- Accad M, Smith SJ, Newland DL, Sanan DA, King LE, Jr, Linton MF, Fazio S, Farese RV., Jr Massive xanthomatosis and altered composition of atherosclerotic lesions in hyperlipidemic mice lacking acyl CoA:cholesterol acyltransferase 1. J Clin Invest. 2000;105:711–719. doi: 10.1172/JCI9021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adler-Wailes DC, Liu H, Ahmad F, Feng N, Londos C, Manganiello V, Yanovski JA. Effects of the human immunodeficiency virus-protease inhibitor, ritonavir, on basal and catecholamine-stimulated lipolysis. J Clin Endocrinol Metab. 2005;90:3251–3261. doi: 10.1210/jc.2004-2194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal AK, Fryns JP, Auchus RJ, Garg A. Zinc metalloproteinase, ZMPSTE24, is mutated in mandibuloacral dysplasia. Hum Mol Genet. 2003;12:1995–2001. doi: 10.1093/hmg/ddg213. [DOI] [PubMed] [Google Scholar]
- Agarwal AK, Barnes RI, Garg A. Genetic basis of congenital generalized lipodystrophy. Int J Obes Relat Metab Disord. 2004;28:336–339. doi: 10.1038/sj.ijo.0802487. [DOI] [PubMed] [Google Scholar]
- Ahmadian M, Duncan RE, Sul HS. The skinny on fat: lipolysis and fatty acid utilization in adipocytes. Trends Endocrinol Metab. 2009;20:424–428. doi: 10.1016/j.tem.2009.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bing C, Trayhurn P. New insights into adipose tissue atrophy in cancer cachexia. Proc Nutr Soc. 2009;68:385–392. doi: 10.1017/S0029665109990267. [DOI] [PubMed] [Google Scholar]
- Binns D, Lee S, Hilton CL, Jiang QX, Goodman JM. Seipin is a discrete homooligomer. Biochemistry. 2010;49:10747–10755. doi: 10.1021/bi1013003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bornfeldt KE, Tabas I. Insulin resistance, hyperglycemia, and atherosclerosis. Cell Metab. 2011;14:575–585. doi: 10.1016/j.cmet.2011.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulant S, Douglas MW, Moody L, Budkowska A, Targett-Adams P, McLauchlan J. Hepatitis C virus core protein induces lipid droplet redistribution in a microtubule- and dynein-dependent manner. Traffic. 2008;9:1268–1282. doi: 10.1111/j.1600-0854.2008.00767.x. [DOI] [PubMed] [Google Scholar]
- Boutet E, El Mourabit H, Prot M, Nemani M, Khallouf E, Colard O, Maurice M, Durand-Schneider AM, Chretien Y, Gres S, et al. Seipin deficiency alters fatty acid Delta9 desaturation and lipid droplet formation in Berardinelli-Seip congenital lipodystrophy. Biochimie. 2009;91:796–803. doi: 10.1016/j.biochi.2009.01.011. [DOI] [PubMed] [Google Scholar]
- Brown MS, Ho YK, Goldstein JL. The cholesteryl ester cycle in macrophage foam cells – continual hydrolysis and re-esterification of cytoplasmic cholesteryl esters. J Biol Chem. 1980;255:9344–9352. [PubMed] [Google Scholar]
- Cao H, Alston L, Ruschman J, Hegele RA. Heterozygous CAV1 frameshift mutations (MIM 601047) in patients with atypical partial lipodystrophy and hypertriglyceridemia. Lipids Health Dis. 2008;7:3. doi: 10.1186/1476-511X-7-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capeau J, Vigouroux C, Caron-Debarle M, Le Dour C, Magre J. Molecular mechanisms of human lipodystrophies: from adipocyte lipid droplet to oxidative stress and lipotoxicity. Int J Biochem Cell Biol. 2011;43:862–876. doi: 10.1016/j.biocel.2011.03.002. [DOI] [PubMed] [Google Scholar]
- Caron M, Auclair R, Vigouroux C, Glorian M, Forest C, Capeau J. The HIV protease inhibitor indinavir impairs sterol regulatory element-binding protein-1 intranuclear localization, inhibits preadipocyte differentiation, and induces insulin resistance. Diabetes. 2001;50:1378–1388. doi: 10.2337/diabetes.50.6.1378. [DOI] [PubMed] [Google Scholar]
- Chang BHJ, Li L, Paul A, Taniguchi S, Nannegari V, Heird WC, Chan L. Protection against fatty liver but normal adipogenesis in mice lacking adipose differentiation-related protein. Mol Cell Biol. 2006;26:1063–1076. doi: 10.1128/MCB.26.3.1063-1076.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Yechoor VK, Chang BH, Li MV, March KL, Chan L. The human lipodystrophy gene product Berardinelli-Seip congenital lipodystrophy 2/seipin plays a key role in adipocyte differentiation. Endocrinology. 2009;150:4552–4561. doi: 10.1210/en.2009-0236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czaja MJ, Singh R, Kaushik S, Wang YJ, Xiang YQ, Novak I, Komatsu M, Tanaka K, Cuervo AM. Autophagy regulates lipid metabolism. Nature. 2009;458:1131–1135. doi: 10.1038/nature07976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahlman I, Kaaman M, Jiao H, Kere J, Laakso M, Arner P. The CIDEA gene V115F polymorphism is associated with obesity in Swedish subjects. Diabetes. 2005;54:3032–3034. doi: 10.2337/diabetes.54.10.3032. [DOI] [PubMed] [Google Scholar]
- Das SK, Eder S, Schauer S, Diwoky C, Temmel H, Guertl B, Gorkiewicz G, Tamilarasan KP, Kumari P, Trauner M, et al. Adipose triglyceride lipase contributes to cancer-associated cachexia. Science. 2011;333:233–238. doi: 10.1126/science.1198973. [DOI] [PubMed] [Google Scholar]
- El-Haschimi K, Pierroz DD, Hileman SM, Bjorbaek C, Flier JS. Two defects contribute to hypothalamic leptin resistance in mice with diet-induced obesity. J Clin Invest. 2000;105:1827–1832. doi: 10.1172/JCI9842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farese RV, Buhman KF, Accad M. Mammalian acyl-CoA: cholesterol acyltransferases. Biochim Biophys Acta. 2000;1529:142–154. doi: 10.1016/s1388-1981(00)00144-x. [DOI] [PubMed] [Google Scholar]
- Farese RV, Yen CLE, Stone SJ, Koliwad S, Harris C. DGAT enzymes and triacylglycerol biosynthesis. J Lipid Res. 2008;49:2283–2301. doi: 10.1194/jlr.R800018-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farese RV, Stone SJ, Levin MC, Zhou P, Han JY, Walther TC. The endoplasmic reticulum enzyme DGAT2 is found in mitochondria-associated membranes and has a mitochondrial targeting signal that promotes its association with mitochondria. J Biol Chem. 2009;284:5352–5361. doi: 10.1074/jbc.M805768200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farese RV, Zechner R, Newgard CB, Walther TC. The problem of establishing relationships between hepatic steatosis and hepatic insulin resistance. Cell Metab. 2012;15:570–573. doi: 10.1016/j.cmet.2012.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fazio S, Major AS, Swift LL, Gleaves LA, Accad M, Linton MF, Farese RV., Jr Increased atherosclerosis in LDL receptor-null mice lacking ACAT1 in macrophages. J Clin Invest. 2001;107:163–171. doi: 10.1172/JCI10310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fei W, Shui G, Gaeta B, Du X, Kuerschner L, Li P, Brown AJ, Wenk MR, Parton RG, Yang H. Fld1p, a functional homologue of human seipin, regulates the size of lipid droplets in yeast. J Cell Biol. 2008;180:473–482. doi: 10.1083/jcb.200711136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fei W, Shui G, Zhang Y, Krahmer N, Ferguson C, Kapterian TS, Lin RC, Dawes IW, Brown AJ, Li P, et al. A role for phosphatidic acid in the formation of “supersized” lipid droplets. PLoS Genet. 2011;7:e1002201. doi: 10.1371/journal.pgen.1002201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman JM. Leptin at 14 y of age: an ongoing story. Am J Clin Nutr. 2009;89:973s–979s. doi: 10.3945/ajcn.2008.26788B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii H, Ikura Y, Iezzoni JC, Park S, Itabe H, Kawada N, Ueda M, Caldwell SH. Expression of perilipin and adipophilin in human nonalcoholic fatty liver disease and their relation to oxidative damage. J Hepatol. 2008;48:S349–S349. doi: 10.5551/jat.2055. [DOI] [PubMed] [Google Scholar]
- Gale SE, Frolov A, Han X, Bickel PE, Cao L, Bowcock A, Schaffer JE, Ory DS. A regulatory role for 1-acylglycerol-3-phosphate-O-acyltransferase 2 in adipocyte differentiation. J Biol Chem. 2006;281:11082–11089. doi: 10.1074/jbc.M509612200. [DOI] [PubMed] [Google Scholar]
- Gandotra S, Le Dour C, Bottomley W, Cervera P, Giral P, Reznik Y, Charpentier G, Auclair M, Delepine M, Barroso I, et al. Perilipin deficiency and autosomal dominant partial lipodystrophy. N Engl J Med. 2011;364:740–748. doi: 10.1056/NEJMoa1007487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg A. Medical progress – acquired and inherited lipodystrophies. N Engl J Med. 2004;350:1220–1234. doi: 10.1056/NEJMra025261. [DOI] [PubMed] [Google Scholar]
- Garg A. Clinical review#: lipodystrophies: genetic and acquired body fat disorders. J Clin Endocrinol Metab. 2011;96:3313–3325. doi: 10.1210/jc.2011-1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg A, Agarwal AK. Lipodystrophies: disorders of adipose tissue biology. Biochim Biophys Acta. 2009;1791:507–513. doi: 10.1016/j.bbalip.2008.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George S, Rochford JJ, Wolfrum C, Gray SL, Schinner S, Wilson JC, Soos MA, Murgatroyd PR, Williams RM, Acerini CL, et al. A family with severe insulin resistance and diabetes due to a mutation in AKT2. Science. 2004;304:1325–1328. doi: 10.1126/science.1096706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh S, St Clair RW, Rudel LL. Mobilization of cytoplasmic CE droplets by overexpression of human macrophage cholesteryl ester hydrolase. J Lipid Res. 2003;44:1833–1840. doi: 10.1194/jlr.M300162-JLR200. [DOI] [PubMed] [Google Scholar]
- Gong J, Sun Z, Li P. CIDE proteins and metabolic disorders. Curr Opin Lipidol. 2009;20:121–126. doi: 10.1097/MOL.0b013e328328d0bb. [DOI] [PubMed] [Google Scholar]
- Gong JY, Sun ZQ, Wu LZ, Xu WY, Schieber N, Xu DJ, Shui GH, Yang HY, Parton RG, Li P. Fsp27 promotes lipid droplet growth by lipid exchange and transfer at lipid droplet contact sites. J Cell Biol. 2011;195:953–963. doi: 10.1083/jcb.201104142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman JM, Szymanski KM, Binns D, Bartz R, Grishin NV, Li WP, Agarwal AK, Garg A, Anderson RGW. The lipodystrophy protein seipin is found at endoplasmic reticulum lipid droplet junctions and is important for droplet morphology. Proc Natl Acad Sci USA. 2007;104:20890–20895. doi: 10.1073/pnas.0704154104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg AS, Coleman RA, Kraemer FB, McManaman JL, Obin MS, Puri V, Yan QW, Miyoshi H, Mashek DG. The role of lipid droplets in metabolic disease in rodents and humans. J Clin Invest. 2011;121:2102–2110. doi: 10.1172/JCI46069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guilherme A, Virbasius JV, Puri V, Czech MP. Adipocyte dysfunctions linking obesity to insulin resistance and type 2 diabetes. Nat Rev Mol Cell Biol. 2008;9:367–377. doi: 10.1038/nrm2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y, Walther TC, Rao M, Stuurman N, Goshima G, Terayama K, Wong JS, Vale RD, Walter P, Farese RV. Functional genomic screen reveals genes involved in lipid-droplet formation and utilization. Nature. 2008;453:657–661. doi: 10.1038/nature06928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haemmerle G, Moustafa T, Woelkart G, Buttner S, Schmidt A, van de Weijer T, Hesselink M, Jaeger D, Kienesberger PC, Zierler K, et al. ATGL-mediated fat catabolism regulates cardiac mitochondrial function via PPAR-alpha and PGC-1. Nat Med. 2011;17:1076–1085. doi: 10.1038/nm.2439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris C, Herker E, Farese RV, Jr, Ott M. Hepatitis C virus core protein decreases lipid droplet turnover: a mechanism for core-induced steatosis. J Biol Chem. 2011;286:42615–42625. doi: 10.1074/jbc.M111.285148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herker E, Ott M. Unique ties between hepatitis C virus replication and intracellular lipids. Trends Endocrinol Metab. 2011;22:241–248. doi: 10.1016/j.tem.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herker E, Harris C, Hernandez C, Carpentier A, Kaehlcke K, Rosenberg AR, Farese RV, Jr, Ott M. Efficient hepatitis C virus particle formation requires diacylglycerol acyltransferase-1. Nat Med. 2010;16:1295–1298. doi: 10.1038/nm.2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill MM, Bastiani M, Luetterforst R, Kirkham M, Kirkham A, Nixon SJ, Walser P, Abankwa D, Oorschot VM, Martin S, et al. PTRF-Cavin, a conserved cytoplasmic protein required for caveola formation and function. Cell. 2008;132:113–124. doi: 10.1016/j.cell.2007.11.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotamisligil GS. Inflammatory pathways and insulin action. Int J Obes Relat Metab Disord. 2003;27(Suppl. 3):S53–S55. doi: 10.1038/sj.ijo.0802502. [DOI] [PubMed] [Google Scholar]
- Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science. 1993;259:87–91. doi: 10.1126/science.7678183. [DOI] [PubMed] [Google Scholar]
- Hsieh K, Lee YK, Londos C, Raaka BM, Dalen KT, Kimmel AR. Perilipin family members preferentially sequester to either triacylglycerol-specific or cholesteryl-ester-specific intracellular lipid storage droplets. J Cell Sci. 2012;125:4067–4076. doi: 10.1242/jcs.104943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua Z, Song S, Luo C, Wang J, Wang Y. [Effect of angelica polysaccharides co-erythropoietin on JAK2/STAT5 signal transduction pathway in hematopoietic stem/progenitor cells] Zhongguo Zhong Yao Za Zhi. 2009;34:3268–3271. [PubMed] [Google Scholar]
- Huang-Doran I, Sleigh A, Rochford JJ, O'Rahilly S, Savage DB. Lipodystrophy: metabolic insights from a rare disorder. J Endocrinol. 2010;207:245–255. doi: 10.1677/JOE-10-0272. [DOI] [PubMed] [Google Scholar]
- Igarashi M, Osuga J, Uozaki H, Sekiya M, Nagashima S, Takahashi M, Takase S, Takanashi M, Li Y, Ohta K, et al. The critical role of neutral cholesterol ester hydrolase 1 in cholesterol removal from human macrophages. Circ Res. 2010;107:1387–1395. doi: 10.1161/CIRCRESAHA.110.226613. [DOI] [PubMed] [Google Scholar]
- Inoue M, Ohtake T, Motomura W, Takahashi N, Hosoki Y, Miyoshi S, Suzuki Y, Saito H, Kohgo Y, Okumura T. Increased expression of PPARgamma in high fat diet-induced liver steatosis in mice. Biochem Biophys Res Commun. 2005;336:215–222. doi: 10.1016/j.bbrc.2005.08.070. [DOI] [PubMed] [Google Scholar]
- Jacobs RL, Devlin C, Tabas I, Vance DE. Targeted deletion of hepatic CTP:phosphocholine cytidylyltransferase alpha in mice decreases plasma high density and very low density lipoproteins. J Biol Chem. 2004;279:47402–47410. doi: 10.1074/jbc.M404027200. [DOI] [PubMed] [Google Scholar]
- Jambunathan S, Yin J, Khan W, Tamori Y, Puri V. FSP27 promotes lipid droplet clustering and then fusion to regulate triglyceride accumulation. PLoS One. 2011;6:e28614. doi: 10.1371/journal.pone.0028614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins CM, Mancuso DJ, Yan W, Sims HF, Gibson B, Gross RW. Identification, cloning, expression, and purification of three novel human calcium-independent phospholipase A2 family members possessing triacylglycerol lipase and acylglycerol transacylase activities. J Biol Chem. 2004;279:48968–48975. doi: 10.1074/jbc.M407841200. [DOI] [PubMed] [Google Scholar]
- Kantartzis K, Machicao F, Machann J, Schick F, Fritsche A, Haring HU, Stefan N. The DGAT2 gene is a candidate for the dissociation between fatty liver and insulin resistance in humans. Clin Sci. 2009;116:531–537. doi: 10.1042/CS20080306. [DOI] [PubMed] [Google Scholar]
- Kellner-Weibel G, Jerome WG, Small DM, Warner GJ, Stoltenborg JK, Kearney MA, Corjay MH, Phillips MC, Rothblat GH. Effects of intracellular free cholesterol accumulation on macrophage viability: a model for foam cell death. Arterioscler Thromb Vasc Biol. 1998;18:423–431. doi: 10.1161/01.atv.18.3.423. [DOI] [PubMed] [Google Scholar]
- Khalil MB, Sundaram M, Zhang HY, Links PH, Raven JF, Manmontri B, Sariahmetoglu M, Tran K, Reue K, Brindley DN, et al. The level and compartmentalization of phosphatidate phosphatase-1 (lipin-1) control the assembly and secretion of hepatic VLDL. J Lipid Res. 2009;50:47–58. doi: 10.1194/jlr.M800204-JLR200. [DOI] [PubMed] [Google Scholar]
- Khera AV, Cuchel M, de la Llera-Moya M, Rodrigues A, Burke MF, Jafri K, French BC, Phillips JA, Mucksavage ML, Wilensky RL, et al. Cholesterol efflux capacity, high-density lipoprotein function, and atherosclerosis. N Engl J Med. 2011;364:127–135. doi: 10.1056/NEJMoa1001689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim CA, Delepine M, Boutet E, El Mourabit H, Le Lay S, Meier M, Nemani M, Bridel E, Leite CC, Bertola DR, et al. Association of a homozygous nonsense caveolin-1 mutation with Berardinelli-Seip congenital lipodystrophy. J Clin Endocrinol Metab. 2008;93:1129–1134. doi: 10.1210/jc.2007-1328. [DOI] [PubMed] [Google Scholar]
- Krahmer N, Guo Y, Wilfling F, Hilger M, Lingrell S, Heger K, Newman HW, Schmidt-Supprian M, Vance DE, Mann M, et al. Phosphatidylcholine synthesis for lipid droplet expansion is mediated by localized activation of CTP:phosphocholine cytidylyltransferase. Cell Metab. 2011;14:504–515. doi: 10.1016/j.cmet.2011.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krahmer N, Hilger M, Kory N, Wilfling F, Stoehr G, Mann M, Farese RV, Walther TC. Protein correlation profiles identify lipid droplet proteins with high confidence. Mol Cell Proteomics. 2013;12:1115–1126. doi: 10.1074/mcp.M112.020230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumari M, Schoiswohl G, Chitraju C, Paar M, Cornaciu I, Rangrez AY, Wongsiriroj N, Nagy HM, Ivanova PT, Scott SA, et al. Adiponutrin functions as a nutritionally regulated lysophosphatidic acid acyltransferase. Cell Metab. 2012;15:691–702. doi: 10.1016/j.cmet.2012.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lass A, Zimmermann R, Oberer M, Zechner R. Lipolysis – a highly regulated multi-enzyme complex mediates the catabolism of cellular fat stores. Prog Lipid Res. 2011;50:14–27. doi: 10.1016/j.plipres.2010.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefevre C, Jobard F, Caux F, Bouadjar B, Karaduman A, Heilig R, Lakhdar H, Wollenberg A, Verret JL, Weissenbach J, et al. Mutations in CGI-58, the gene encoding a new protein of the esterase/lipase/thioesterase subfamily, in Chanarin-Dorfman syndrome. Am J Hum Genet. 2001;69:1002–1012. doi: 10.1086/324121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehner R, Vance DE. Cloning and expression of a cDNA encoding a hepatic microsomal lipase that mobilizes stored triacylglycerol. Biochem J. 1999;343:1–10. [PMC free article] [PubMed] [Google Scholar]
- Lehner R, Cui Z, Vance DE. Subcellullar localization, developmental expression and characterization of a liver triacylglycerol hydrolase. Biochem J. 1999;338:761–768. [PMC free article] [PubMed] [Google Scholar]
- Li JZ, Huang Y, Karaman R, Ivanova PT, Brown HA, Roddy T, Castro-Perez J, Cohen JC, Hobbs HH. Chronic overexpression of PNPLA3I148M in mouse liver causes hepatic steatosis. J Clin Invest. 2012;122:4130–4144. doi: 10.1172/JCI65179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisanti MP, Razani B. Caveolin-deficient mice: insights into caveolar function and human disease. J Clin Invest. 2001;108:1553–1561. doi: 10.1172/JCI14611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu PS, Bartz R, Li WH, Venables B, Zehmer JK, Roth MR, Welti R, Anderson RGW, Chapman KD. Lipidomics reveals that adiposomes store ether lipids and mediate phospholipid traffic. J Lipid Res. 2007;48:837–847. doi: 10.1194/jlr.M600413-JLR200. [DOI] [PubMed] [Google Scholar]
- Liu L, Brown D, McKee M, Lebrasseur NK, Yang D, Albrecht KH, Ravid K, Pilch PF. Deletion of Cavin/PTRF causes global loss of caveolae, dyslipidemia, and glucose intolerance. Cell Metab. 2008;8:310–317. doi: 10.1016/j.cmet.2008.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundin C, Nordstrom R, Wagner K, Windpassinger C, Andersson H, von Heijne G, Nilsson I. Membrane topology of the human seipin protein. FEBS Lett. 2006;580:2281–2284. doi: 10.1016/j.febslet.2006.03.040. [DOI] [PubMed] [Google Scholar]
- Marcel YL, Ouimet M, Franklin V, Mak E, Liao XH, Tabas I. Autophagy regulates cholesterol efflux from macrophage foam cells via lysosomal acid lipase. Cell Metab. 2011;13:655–667. doi: 10.1016/j.cmet.2011.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin S, Murphy S, Parton RG. Lipid droplet-organelle interactions; sharing the fats. Biochim Biophys Acta. 2009;1791:441–447. doi: 10.1016/j.bbalip.2008.07.004. [DOI] [PubMed] [Google Scholar]
- Martinez-Botas J, Anderson JB, Tessier D, Lapillonne A, Chang BHJ, Quast MJ, Gorenstein D, Chen KH, Chan L. Absence of perilipin results in leanness and reverses obesity in Lepr db db mice. Nat Genet. 2000;26:474–479. doi: 10.1038/82630. [DOI] [PubMed] [Google Scholar]
- Matsuo M, Dwianingsih EK, Takeshima Y, Itoh K, Yamauchi Y, Awano H, Malueka RG, Nishida A, Ota M, Yagi M. A Japanese child with asymptomatic elevation of serum creatine kinase shows PTRF-CAVIN mutation matching with congenital generalized lipodystrophy type 4. Mol Genet Metab. 2010;101:233–237. doi: 10.1016/j.ymgme.2010.06.016. [DOI] [PubMed] [Google Scholar]
- Matsusue K, Kusakabe T, Noguchi T, Takiguchi S, Suzuki T, Yamano S, Gonzalez FJ. Hepatic steatosis in leptin-deficient mice is promoted by the PPAR gamma target gene Fsp27. Cell Metab. 2008;7:302–311. doi: 10.1016/j.cmet.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyanari Y, Atsuzawa K, Usuda N, Watashi K, Hishiki T, Zayas M, Bartenschlager R, Wakita T, Hijikata M, Shimotohno K. The lipid droplet is an important organelle for hepatitis C virus production. Nat Cell Biol. 2007;9:1089–1097. doi: 10.1038/ncb1631. [DOI] [PubMed] [Google Scholar]
- Moore KJ, Tabas I. Macrophages in the pathogenesis of atherosclerosis. Cell. 2011;145:341–355. doi: 10.1016/j.cell.2011.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriya K, Yotsuyanagi H, Shintani Y, Fujie H, Ishibashi K, Matsuura Y, Miyamura T, Koike K. Hepatitis C virus core protein induces hepatic steatosis in transgenic mice. J Gen Virol. 1997;78:1527–1531. doi: 10.1099/0022-1317-78-7-1527. [DOI] [PubMed] [Google Scholar]
- Motomura W, Inoue M, Ohtake T, Takahashi N, Nagamine M, Tanno S, Kohgo Y, Okumura T. Up-regulation of ADRP in fatty liver in human and liver steatosis in mice fed with high fat diet. Biochem Biophys Res Commun. 2006;340:1111–1118. doi: 10.1016/j.bbrc.2005.12.121. [DOI] [PubMed] [Google Scholar]
- Murphy DJ. The dynamic roles of intracellular lipid droplets: from archaea to mammals. Protoplasma. 2012;249:541–585. doi: 10.1007/s00709-011-0329-7. [DOI] [PubMed] [Google Scholar]
- Nian ZQ, Sun ZQ, Yu LX, Toh SY, Sang JL, Li P. Fat-specific protein 27 undergoes ubiquitin-dependent degradation regulated by triacylglycerol synthesis and lipid droplet formation. J Biol Chem. 2010;285:9604–9615. doi: 10.1074/jbc.M109.043786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishino N, Tamori Y, Tateya S, Kawaguchi T, Shibakusa T, Mizunoya W, Inoue K, Kitazawa R, Kitazawa S, Matsuki Y, et al. FSP27 contributes to efficient energy storage in murine white adipocytes by promoting the formation of unilocular lipid droplets. J Clin Invest. 2008;118:2808–2821. doi: 10.1172/JCI34090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oral EA, Simha V, Ruiz E, Andewelt A, Premkumar A, Snell P, Wagner AJ, DePaoli AM, Reitman ML, Taylor SI, et al. Leptin-replacement therapy for lipodystrophy. N Engl J Med. 2002;346:570–578. doi: 10.1056/NEJMoa012437. [DOI] [PubMed] [Google Scholar]
- Ostermeyer AG, Paci JM, Zeng Y, Lublin DM, Munro S, Brown DA. Accumulation of caveolin in the endoplasmic reticulum redirects the protein to lipid storage droplets. J Cell Biol. 2001;152:1071–1078. doi: 10.1083/jcb.152.5.1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osuga J, Ishibashi S, Oka T, Yagyu H, Tozawa R, Fujimoto A, Shionoiri F, Yahagi N, Kraemer FB, Tsutsumi O, et al. Targeted disruption of hormone-sensitive lipase results in male sterility and adipocyte hypertrophy, but not in obesity. Proc Natl Acad Sci USA. 2000;97:787–792. doi: 10.1073/pnas.97.2.787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parton RG, Bastiani M. Caveolae at a glance. J Cell Sci. 2010;123:3831–3836. doi: 10.1242/jcs.070102. [DOI] [PubMed] [Google Scholar]
- Paul A, Chang BHJ, Li L, Yechoor VK, Chan L. Deficiency of adipose differentiation-related protein impairs foam cell formation and protects against atherosclerosis. Circ Res. 2008;102:1492–1501. doi: 10.1161/CIRCRESAHA.107.168070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterfy M, Phan J, Xu P, Reue K. Lipodystrophy in the fld mouse results from mutation of a new gene encoding a nuclear protein, lipin. Nat Genet. 2001;27:121–124. doi: 10.1038/83685. [DOI] [PubMed] [Google Scholar]
- Pilch PF, Liu L. Fat caves: caveolae, lipid trafficking and lipid metabolism in adipocytes. Trends Endocrinol Metab. 2011;22:318–324. doi: 10.1016/j.tem.2011.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puri V, Konda S, Ranjit S, Aouadi M, Chawla A, Chouinard M, Chakladar A, Czech MP. Fat-specific protein 27, a novel lipid droplet protein that enhances triglyceride storage. J Biol Chem. 2007;282:34213–34218. doi: 10.1074/jbc.M707404200. [DOI] [PubMed] [Google Scholar]
- Puri V, Ranjit S, Konda S, Nicoloro SM, Straubhaar J, Chawla A, Chouinard M, Lin C, Burkart A, Corvera S, et al. Cidea is associated with lipid droplets and insulin sensitivity in humans. Proc Natl Acad Sci USA. 2008;105:7833–7838. doi: 10.1073/pnas.0802063105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quiroga AD, Lehner R. Role of endoplasmic reticulum neutral lipid hydrolases. Trends Endocrinol Metab. 2011;22:218–225. doi: 10.1016/j.tem.2011.03.003. [DOI] [PubMed] [Google Scholar]
- Romeo S, Sentinelli F, Dash S, Yeo GSH, Savage DB, Leonetti F, Capoccia D, Incani M, Maglio C, Iacovino M, et al. Morbid obesity exposes the association between PNPLA3 I148M (rs738409) and indices of hepatic injury in individuals of European descent. Int J Obesity. 2010;34:190–194. doi: 10.1038/ijo.2009.216. [DOI] [PubMed] [Google Scholar]
- Rubio-Cabezas O, Puri V, Murano I, Saudek V, Semple RK, Dash S, Hyden CS, Bottomley W, Vigouroux C, Magre J, et al. Partial lipodystrophy and insulin resistant diabetes in a patient with a homozygous nonsense mutation in CIDEC. EMBO Mol Med. 2009;1:280–287. doi: 10.1002/emmm.200900037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savage DB, Tan GD, Acerini CL, Jebb SA, Agostini M, Gurnell M, Williams RL, Umpleby AM, Thomas EL, Bell JD, et al. Human metabolic syndrome resulting from dominant-negative mutations in the nuclear receptor peroxisome proliferator-activated receptor-gamma. Diabetes. 2003;52:910–917. doi: 10.2337/diabetes.52.4.910. [DOI] [PubMed] [Google Scholar]
- Schadinger SE, Bucher NL, Schreiber BM, Farmer SR. PPARgamma2 regulates lipogenesis and lipid accumulation in steatotic hepatocytes. Am J Physiol Endocrinol Metab. 2005;288:E1195–E1205. doi: 10.1152/ajpendo.00513.2004. [DOI] [PubMed] [Google Scholar]
- Schaffer JE. Lipotoxicity: when tissues overeat. Curr Opin Lipidol. 2003;14:281–287. doi: 10.1097/00041433-200306000-00008. [DOI] [PubMed] [Google Scholar]
- Schweiger M, Lass A, Zimmermann R, Eichmann TO, Zechner R. Neutral lipid storage disease: genetic disorders caused by mutations in adipose triglyceride lipase/PNPLA2 or CGI-58/ABHD5. Am J Physiol Endocrinol Metab. 2009;297:E289–E296. doi: 10.1152/ajpendo.00099.2009. [DOI] [PubMed] [Google Scholar]
- Sekiya M, Osuga J, Nagashima S, Ohshiro T, Igarashi M, Okazaki H, Takahashi M, Tazoe F, Wada T, Ohta K, et al. Ablation of neutral cholesterol ester hydrolase 1 accelerates atherosclerosis. Cell Metab. 2009;10:219–228. doi: 10.1016/j.cmet.2009.08.004. [DOI] [PubMed] [Google Scholar]
- Shastry S, Delgado MR, Dirik E, Turkmen M, Agarwal AK, Garg A. Congenital generalized lipodystrophy, type 4 (CGL4) associated with myopathy due to novel PTRF mutations. Am J Med Genet A. 2010;152A:2245–2253. doi: 10.1002/ajmg.a.33578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw JH, Wolfe RR. Fatty acid and glycerol kinetics in septic patients and in patients with gastrointestinal cancer. The response to glucose infusion and parenteral feeding. Ann Surg. 1987;205:368–376. doi: 10.1097/00000658-198704000-00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimomura I, Hammer RE, Ikemoto S, Brown MS, Goldstein JL. Leptin reverses insulin resistance and diabetes mellitus in mice with congenital lipodystrophy. Nature. 1999;401:73–76. doi: 10.1038/43448. [DOI] [PubMed] [Google Scholar]
- Shiratori Y, Okwu AK, Tabas I. Free cholesterol loading of macrophages stimulates phosphatidylcholine biosynthesis and up-regulation of CTP: phosphocholine cytidylyltransferase. J Biol Chem. 1994;269:11337–11348. [PubMed] [Google Scholar]
- Souza SC, de Vargas LM, Yamamoto MT, Lien P, Franciosa MD, Moss LG, Greenberg AS. Overexpression of perilipin A and B blocks the ability of tumor necrosis factor alpha to increase lipolysis in 3T3-L1 adipocytes. J Biol Chem. 1998a;273:24665–24669. doi: 10.1074/jbc.273.38.24665. [DOI] [PubMed] [Google Scholar]
- Souza SC, Yamamoto MT, Franciosa MD, Lien P, Greenberg AS. BRL 49653 blocks the lipolytic actions of tumor necrosis factor-alpha – a potential new insulin-sensitizing mechanism for thiazolidinediones. Diabetes. 1998b;47:691–695. doi: 10.2337/diabetes.47.4.691. [DOI] [PubMed] [Google Scholar]
- Speliotes EK, Yerges-Armstrong LM, Wu J, Hernaez R, Kim LJ, Palmer CD, Gudnason V, Eiriksdottir G, Garcia ME, Launer LJ, et al. Genome-wide association analysis identifies variants associated with nonalcoholic fatty liver disease that have distinct effects on metabolic traits. PLoS Genet. 2011;7:e1001324. doi: 10.1371/journal.pgen.1001324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone SJ, Levin MC, Zhou P, Han JY, Walther TC, Farese RV. The endoplasmic reticulum enzyme DGAT2 is found in mitochondria-associated membranes and has a mitochondrial targeting signal that promotes its association with mitochondria. J Biol Chem. 2009;284:5352–5361. doi: 10.1074/jbc.M805768200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straub BK, Stoeffel P, Heid H, Zimbelmann R, Schirmacher P. Differential pattern of lipid droplet-associated proteins and de novo perilipin expression in hepatocyte steatogenesis. Hepatology. 2008;47:1936–1946. doi: 10.1002/hep.22268. [DOI] [PubMed] [Google Scholar]
- Su YR, Dove DE, Major AS, Hasty AH, Boone B, Linton MF, Fazio S. Reduced ABCA1-mediated cholesterol efflux and accelerated atherosclerosis in apolipoprotein E-deficient mice lacking macrophage-derived ACAT1. Circulation. 2005;111:2373–2381. doi: 10.1161/01.CIR.0000164236.19860.13. [DOI] [PubMed] [Google Scholar]
- Sun Z, Lazar MA. Dissociating fatty liver and diabetes. Trends Endocrinol Metab. 2013;24:4–12. doi: 10.1016/j.tem.2012.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sztalyrd C, Xu GH, Dorward H, Tansey JT, Contreras JA, Kimmel AR, Londos C. Perilipin A is essential for the translocation of hormone-sensitive lipase during lipolytic activation (vol 161, pg 1093, 2003) J Cell Biol. 2003;162:353–353. doi: 10.1083/jcb.200210169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szymanski KM, Binns D, Bartz R, Grishin NV, Li WP, Agarwal AK, Garg A, Anderson RG, Goodman JM. The lipodystrophy protein seipin is found at endoplasmic reticulum lipid droplet junctions and is important for droplet morphology. Proc Natl Acad Sci USA. 2007;104:20890–20895. doi: 10.1073/pnas.0704154104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabas I. Cholesterol and phospholipid metabolism in macrophages. Biochim Biophys Acta. 2000;1529:164–174. doi: 10.1016/s1388-1981(00)00146-3. [DOI] [PubMed] [Google Scholar]
- Tansey JT, Sztalryd C, Gruia-Gray J, Roush DL, Zee JV, Gavrilova O, Reitman ML, Deng CX, Li C, Kimmel AR, et al. Perilipin ablation results in a lean mouse with aberrant adipocyte lipolysis, enhanced leptin production, and resistance to diet-induced obesity. Proc Natl Acad Sci USA. 2001;98:6494–6499. doi: 10.1073/pnas.101042998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiele C, Spandl J. Cell biology of lipid droplets. Curr Opin Cell Biol. 2008;20:378–385. doi: 10.1016/j.ceb.2008.05.009. [DOI] [PubMed] [Google Scholar]
- Tian Y, Bi J, Shui G, Liu Z, Xiang Y, Liu Y, Wenk MR, Yang H, Huang X. Tissue-autonomous function of Drosophila seipin in preventing ectopic lipid droplet formation. PLoS Genet. 2011;7:e1001364. doi: 10.1371/journal.pgen.1001364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tisdale MJ. Cachexia in cancer patients. Nat Rev Cancer. 2002;2:862–871. doi: 10.1038/nrc927. [DOI] [PubMed] [Google Scholar]
- Tisdale MJ. Molecular pathways leading to cancer cachexia. Physiology (Bethesda) 2005;20:340–348. doi: 10.1152/physiol.00019.2005. [DOI] [PubMed] [Google Scholar]
- van Herpen NA, Schrauwen-Hinderling VB. Lipid accumulation in non-adipose tissue and lipotoxicity. Physiol Behav. 2008;94:231–241. doi: 10.1016/j.physbeh.2007.11.049. [DOI] [PubMed] [Google Scholar]
- Van Maldergem L, Magre J, Khallouf TE, Gedde-Dahl T, Jr, Delepine M, Trygstad O, Seemanova E, Stephenson T, Albott CS, Bonnici F, et al. Genotype-phenotype relationships in Berardinelli-Seip congenital lipodystrophy. J Med Genet. 2002;39:722–733. doi: 10.1136/jmg.39.10.722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigouroux C, Caron-Debarle M, Le Dour C, Magre J, Capeau J. Molecular mechanisms of human lipodystrophies: from adipocyte lipid droplet to oxidative stress and lipotoxicity. Int J Biochem Cell Biol. 2011;43:862–876. doi: 10.1016/j.biocel.2011.03.002. [DOI] [PubMed] [Google Scholar]
- Virtue S, Vidal-Puig A. Adipose tissue expandability, lipotoxicity and the Metabolic Syndrome – an allostatic perspective. Biochim Biophys Acta. 2010;1801:338–349. doi: 10.1016/j.bbalip.2009.12.006. [DOI] [PubMed] [Google Scholar]
- Walther TC, Farese RV., Jr Lipid droplets and cellular lipid metabolism. Annu Rev Biochem. 2012;81:687–714. doi: 10.1146/annurev-biochem-061009-102430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilfling F, Wang H, Haas JT, Krahmer N, Gould TJ, Uchida A, Cheng JX, Graham M, Christiano R, Frohlich F, et al. Triacylglycerol synthesis enzymes mediate lipid droplet growth by relocalizing from the ER to lipid droplets. Dev Cell. 2013;24:384–399. doi: 10.1016/j.devcel.2013.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu NY, Zhang SBO, Cole RA, McKinney SA, Guo FL, Haas JT, Bobba S, Farese RV, Mak HY. The FATP1-DGAT2 complex facilitates lipid droplet expansion at the ER-lipid droplet interface. J Cell Biol. 2012;198:895–911. doi: 10.1083/jcb.201201139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada M, Wolfe D, Han G, French SW, Ross MG, Desai M. Early onset of fatty liver in growth-restricted rat fetuses and newborns. Congenit Anom. 2011;51:167–173. doi: 10.1111/j.1741-4520.2011.00336.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang HY, Fei WH, Du XM. Seipin, adipogenesis and lipid droplets. Trends Endocrinol Metab. 2011;22:204–210. doi: 10.1016/j.tem.2011.02.004. [DOI] [PubMed] [Google Scholar]
- Ye J, Li JZ, Liu Y, Li X, Yang T, Ma X, Li Q, Yao Z, Li P. Cideb, an ER- and lipid droplet-associated protein, mediates VLDL lipidation and maturation by interacting with apolipoprotein B. Cell Metab. 2009;9:177–190. doi: 10.1016/j.cmet.2008.12.013. [DOI] [PubMed] [Google Scholar]
- Yuan Y, Li P, Ye J. Lipid homeostasis and the formation of macrophage-derived foam cells in atherosclerosis. Protein Cell. 2012;3:173–181. doi: 10.1007/s13238-012-2025-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhai W, Xu C, Ling Y, Liu S, Deng J, Qi Y, Londos C, Xu G. Increased lipolysis in adipose tissues is associated with elevation of systemic free fatty acids and insulin resistance in perilipin null mice. Horm Metab Res. 2010;42:247–253. doi: 10.1055/s-0029-1243599. [DOI] [PubMed] [Google Scholar]
- Zhang D, Tang W, Yao PM, Yang C, Xie B, Jackowski S, Tabas I. Macrophages deficient in CTP:phosphocholine cytidylyltransferase-alpha are viable under normal culture conditions but are highly susceptible to free cholesterol-induced death. Molecular genetic evidence that the induction of phosphatidylcholine biosynthesis in free cholesterol-loaded macrophages is an adaptive response. J Biol Chem. 2000;275:35368–35376. doi: 10.1074/jbc.M007099200. [DOI] [PubMed] [Google Scholar]
- Zhang L, Miyaki K, Nakayama T, Muramatsu M. Cell death-inducing DNA fragmentation factor alpha-like effector A (CIDEA) gene V115F (G→T) polymorphism is associated with phenotypes of metabolic syndrome in Japanese men. Metabolism. 2008;57:502–505. doi: 10.1016/j.metabol.2007.11.011. [DOI] [PubMed] [Google Scholar]
- Zhang L, Dai Y, Bian L, Wang W, Muramatsu M, Hua Q. Association of the cell death-inducing DNA fragmentation factor alpha-like effector A (CIDEA) gene V115F (G/T) polymorphism with phenotypes of metabolic syndrome in a Chinese population. Diabetes Res Clin Pract. 2011;91:233–238. doi: 10.1016/j.diabres.2010.10.016. [DOI] [PubMed] [Google Scholar]
- Zhao B, Song JM, Chow WN, Clair RWS, Rudel LL, Ghosh S. Macrophage-specific transgenic expression of cholesteryl ester hydrolase significantly reduces atherosclerosis and lesion necrosis in Ldlr(−)/(−) mice. J Clin Invest. 2007;117:2983–2992. doi: 10.1172/JCI30485. [DOI] [PMC free article] [PubMed] [Google Scholar]