

Abstract
Advances in high-throughput sequencing have enabled technologies that probe the adaptive immune system with unprecedented depth. We have developed a multiplex PCR method to sequence tens of millions of T cell receptors (TCRs) from a single sample in a few days. A method is presented to test the precision, accuracy, and sensitivity of this assay. T cell clones, each with one fixed productive TCR rearrangement, are doped into complex blood cell samples. TCRs from a total of eleven samples are sequenced, with the doped T cell clones ranging from 10% of the total sample to 0.001% (one cell in 100,000). The assay is able to detect even the rarest clones. The precision of the assay is demonstrated across five orders of magnitude. The accuracy for each clone is within an overall factor of three across the 100,000 fold dynamic range. Additionally, the assay is shown to be highly repeatable.
Keywords: TCR, repertoire, MRD
1. Introduction
Diversity of the T cell receptor (TCR) repertoire allows the human cellular adaptive immune system to protect the body against a vast of array of potential pathogens. The TCR is a heterodimeric receptor, comprised of an α and a β chain, that recognizes antigen presented by MHC molecules. These chains are somatically rearranged from individual gene segments to create millions of different surface receptors, with the majority of T cells expressing a single productively rearranged TCR α and β chain allele. Until recently, the immense size of the TCR repertoire has prevented measurement of the global state, or profile, of the T cell clones constituting the cellular adaptive immune system.
Utilizing the power of high-throughput sequencing, we have developed a method to sequence millions of TCRβ chains simultaneously (Robins et al.; Robins et al., 2009). A few other groups have employed different methods to sequence both TCR and BCR sequences at high-throughput (Boyd et al.; Wang et al.; Warren et al.; Boyd et al., 2009; Freeman et al., 2009; Weinstein et al., 2009). The strategy we employ to accomplish this sequencing at such high volume is based on a multiplex PCR method. The nucleotide sequence encoding the TCRβ chain is determined by somatic rearrangement of V, D, and J segments along with a set of non-templated insertions and deletions at the junctions between the V and D, and the D and J segments. We have designed a pool of primers to all V and J pairs such that they specifically amplify the complete VDJ junction region, known as the CDR3 region, as well as a portion of the J and V segments that is sufficient for identification.
This methodology maximizes throughput for immune profiling because only the minimal region containing the antigen-specific nucleotide information for each TCRβ is amplified and sequenced. Each TCRβ sequence is identified with one short read (60 nucleotides) compared with other methodologies that sequence longer reads from each TCRβ chain, which require either lower throughput/longer read technology, or fractionation of each template and higher coverage (Wang et al.; Freeman et al., 2009). In addition, our assay applies to both genomic DNA as well as cDNA. Methods using a single pair of primers are not appropriate for genomic DNA (with presently available sequencing capabilities) due to the size of the intronic sequence between the J segments and the downstream constant region. Using genomic DNA as template, our method has the potential to accurately capture the frequency of individual TCR clonotypes in biologic samples.
In this manuscript we present a direct test of the precision, accuracy, and sensitivity of our methodology to identify TCRβ chains sequenced from genomic DNA. Two independent laboratories challenged our technology with doped samples. These labs each started with complex samples of blood cells and spiked in a total of seven different T cell clones of known TCRβ CDR3 sequence at a range of 10 to 100,000 cells in a million. We performed deep sequencing of the TCRβ locus for each sample, comparing the sequencing results from each complex mix with the independently attained sequences for each spike-in clone. The full TCRβ nucleotide sequence is a nearly unique tag that allows us to identify and track individual T cell clones. For antigen experienced T cells, each unique beta chain is expected to pair with only one alpha chain (Arstila et al., 1999). The results show that our TCRβ technology is sufficiently sensitive to reliably detect clones at a level of one cell in 100,000. The precision is within 16% and the overall accuracy has a standard deviation of less than a factor of three for random clonotypes across a 100,000 fold dynamic range.
In addition, a set of experiments are reported that test the repeatability of the assay. A large sample of PBMCs was split into parallel arms prior to the three main steps in the sequencing process; DNA extraction, PCR amplification, and sequencing. Using the TCRβ sequences to tag each clone, the repertoires from each step are compared. The assay is reproducible.
2. Materials & Methods
2.1. Samples
Samples were obtained from two independent sources. Three mixes of CD4+ T cell clones were prepared using previously described T cell clones specific for GAD65 in the context of DRB1*0404 (Reijonen et al., 2002). The five CD4+ T cells clones CD4+ 372, CD4+ 339, CD4+ 303, CD4+ 310, and CD4+ 340 have unique TCRβ CDR3 region sequences (Supplementary Table 1A). The background cells for these doped samples were CD4+ CD45RA+ naïve T cells sorted from fresh PBMC from a control donor obtained with informed consent. The clones were sorted as CD3+ CD4+ T cells and added to a background of one million naïve CD4+ T cells in the numbers indicated in Supplementary Table 1A using flow cell counts as a measure of cell number. The doped samples were pelleted and DNA prepared using the DNeasy kit (Qiagen Inc., Valencia CA). DNA was also prepared from separate samples of purified clone cells to validate the TCRβ CDR3 sequence.
Two CD4+ T cell clones each displaying a unique TCRbeta CDR3 sequence and recognizing the tumor-associated NY-ESO-1 antigen were generated as previously described (Hunder et al., 2008) (Supplementary Table 1B). These clones were doped into 2 million autologous peripheral blood mononuclear cells at counts of 20, 200, 2 × 10, 2 × 10 clones to yield titrations of 1:100,000, 1:10,000, 1:1,000 and 1:100, respectively.
2.2. Sequencing of TCRB CDR3 regions
TCRβ CDR3 regions were amplified and sequenced using protocols described by Robins et al. (Robins et al., 2009). Briefly, a multi-plexed PCR method was employed to amplify all possible rearranged genomic TCRβ sequences using 45 forward primers, each specific to a functional TCR Vβ segment, and 13 reverse primers, each specific to a TCR Jβ segment. For clones CD8+ Clone1, CD8+ Clone2, CD4+ 339, CD4+ 303, CD4+ 310, and CD4+ 340, the same primer set was used. For CD4+ 372, the 13 J primers were moved downstream 10 nucleotides. Reads of length 60bp were obtained using the Illumina GAII System. Raw GA sequence data were preprocessed to remove errors in the primary sequence of each read, and to compress the data. A nearest neighbor algorithm was used to collapse the data into unique sequences by merging closely related sequences, to remove both PCR and sequencing errors. The TCR CDR3 region was defined according to the IMGT collaboration (reference), beginning with the second conserved cysteine encoded by the 3′ portion of the Vβgene segment and ending with the conserved phenylalanine encoded by the 5′ portion of the Jβ gene segment. The number of nucleotides between these codons determines the length and therefore the frame of the CDR3 region.
2.3. Identification of spiked-in clones
TCR amino acid sequences for each spiked-in clone were either obtained from the supplier of the clone, or confirmed by direct sequencing using the Adaptive TCR Immunoseq assay. The copy count of sequences in each mix matching at the amino acid level was obtained from the processed sequence data. These counts were used to calculate the frequency of each clone in each mix. Doping levels were used to estimate the expected frequencies of each clone for each mix.
2.4. Characterization of TCRB Jβ deletion length
In order to establish a distribution of the observed number of TCRB J deletions, TCR sequencing results from 14 samples, consisting of CD8+ naïve and memory populations from seven individuals (Robins et al. 2010), were combined and the length of the J deletion for each distinct TCR sequence was determined. A total of 2,632,129 TCR CDR3 sequences were analyzed with J deletions ranging in length from 0 to 15 nucleotides, with a median deletion length of 3 nucleotides.
3. Results
3.1. Precise and sensitive detection of T cell clones spiked into complex cell mixtures
Two separate sets of doped samples were generated to test the accuracy and sensitivity of our TCRβ sequencing technology. The first experiment consisted of three background samples and five CD4+ T cell clones that recognize GAD65 in the context of DRB1*0404 (ref Reijonen). The background samples were fresh CD4+ CD45RA+ naïve T cells sorted from a DRB1*0404 control donor, with a million T cells in each of the three samples. The five clones were spiked into the three samples at different levels, with one sample (Mix 2) consisting of each clone at equal concentration and the other two samples (Mixes 1 and 3) consisting of clones present in a dynamic range of 10 to 100,000 clone cells in a million naïve CD4+ T cells (Supplementary Table 1A). The second experiment consisted of eight samples, each of which had a background of two million PBMCs. A single CD8+ T cell clone was spiked into each sample at 10 to 10,000 clone cells per million PBMC (Supplementary Table 1B).
Genomic DNA was prepared from the doped samples and a TCRβ chain CDR3 library was generated by PCR using one or two million genomes of DNA. We amplified a 200 bp fragment encompassing the CDR3 regions of TCRβ chains using 45 V primers and 13 J primers which anneal with similar binding characteristics to all 54 Vs and 13 Js in the TCRβ chain locus (Supplementary Figure 1A) (Robins et al., 2009). The PCR library was then sequenced on an Illumina system, starting from the J side, generating 60 bp reads. The sequencing primers capture all TCRβ chains with J deletions less than 14 nucleotides in length. This strategy is designed to capture over 99% of rearranged TCRβ’s in humans. The distribution of J deletions compiled from millions of TCRβ chains sequenced from human blood is presented in Supplementary Figure 1B.
For the doped samples in Supplementary Table 1A, we sequenced each of the three samples particularly deep, with an average of 28.4 million TCRβ sequence reads per sample (Supplementary Table 1C). Four of the five clones were detected in each of the three samples at levels comparable to the expected frequency (Supplementary Table 1D, Figure 1). The fifth clone was not detected in any of the three samples, so we hypothesized that this clone must have an anomalously long J deletion. To test this hypothesis, we designed a new set of J primers located 20 bases further away from the beginning of each of the J segments. Although the PCR and sequencing protocols have not been optimized for this set of primers, we were readily able to detect the missing clone. It did indeed have a 16 nucleotide deletion (Supplementary Figure 1B).
The second set of eight doped samples consisted of four samples for each of the two spiked-in CD8+ T cell clones. For these samples, we sequenced between three and five million reads per sample (Supplementary Table 1E). The appropriate spiked-in clone was detected in all eight samples. Since the background of this second set of samples was PBMCs, we determined the percentage of these cells which are T cells by sorting on the surface marker CD3 (Supplementary Figure 1C). Using these numbers, we calculated the fraction of T cells represented by each spiked in clone. These results are compared to the expected doped quantities in Figure 1 and Supplementary Table 1.
Figure 1. Expected versus observed frequencies for 6 spiked-in clones in multiple mixes.
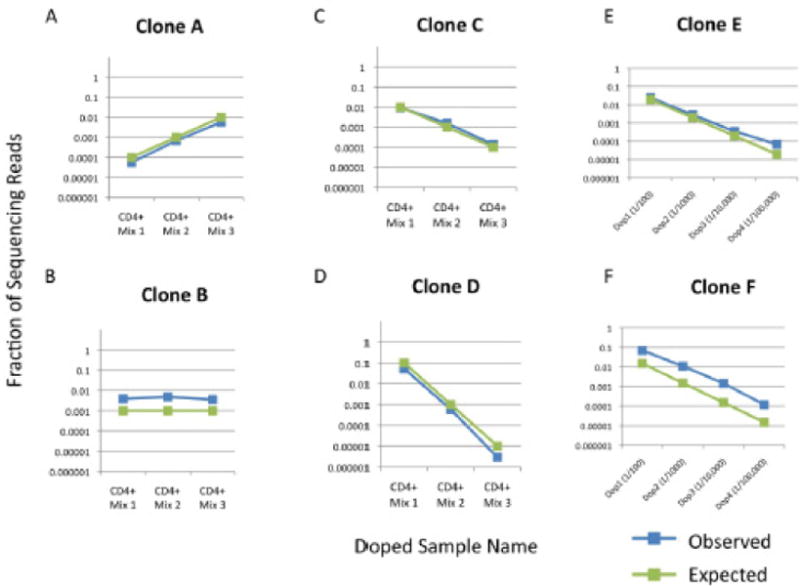
Four CD4+ clones (A, B, C, D) and two CD8+ clones (E & F) and were spiked into either complex PBMC or sorted CD4+ T cell populations at concentrations spanning five orders of magnitude. Results show concordance between expected and observed clone frequencies.
Considering both of the experiments together, three different clones were spiked in at one part in 100,000. These were all detected. For the CD4+ spike-in experiment, we found 10 clone cells in a background of one million and in the CD8+ spike in experiment, we found both sets of 20 clone cells in backgrounds of two million. These results illustrate the ability of the method to track low frequency clones.
The precision of the assay is assessed by comparison of the measured clone values across the different doped levels. The variation of the ratios of the measured clone values compared to the expected clone value was computed for all clone sequences and values, excluding the 1/100,000 levels where we expect significant statistical fluctuations. The standard deviation of the percent difference from the expected ratios is 16%, so the assay is very precise.
The accuracy of the assay was also assessed. The six doped clones accounted for five of the 54 V segments and four of the 13 J segments. The methodology tracked each of these clones with standard deviation 196%. The spiked in samples were created by independent laboratories, so the particular VJ pairs can be considered random tests of our technology. Since there are a total of 13*54 = 702 possible VJ combinations, testing all combinations is not feasible in one set of experiments. However, we expect to measure over half the VJ pairs within a factor of two.
The two clones, B and F in Figure 1, were over amplified by a consistent fraction for all six spiked in quantities. Both of these clones have the same J gene, Jβ2–7, and therefore are amplified by the same J primer. A feasible solution to this issue is scaling back the quantity of this particular primer in our pool to bring these values back to the true underlying values. This exemplifies both the difficulty and, yet, feasibility of getting accurate amplification from multiplex PCR assays. The difficulty is that it requires a series of iterations to fine tune the individual primers. However, these iterations are possible because amplification bias from any particular J (or V) gene primer affects the full set of opposing V (or J) primers nearly equivalently. In the case of sample B and F, Jβ2–7 is over amplified, but the amount of over amplification is similar for the six spike in values despite clones B and F utilizing two different V genes, Vβ6.7 and Vβ7.2 respectively.
3.2 Assessment of reproducibility through repetition experiments
We have assessed the sensitivity, precision, and accuracy of the assay. To show that it is also repeatable, we carried out an additional set of experiments outlined in the schematic in Figure 2A. Beginning with a blood draw, the sample preparation process involves two steps: DNA extraction and PCR amplification. We designed an experiment that splits our samples at each step, completes the preparation and sequencing, then compares the results. The total amount of DNA added to the PCR reaction at each step was 3μg, which is equivalent to approximately one million haploid genomes. The initial blood sample contained tens of millions of T cells. The comparisons of the repeats at the different steps in the sample preparation are presented in Figure 2. We generated 9,817,529 reads on average for each TCRβ molecule in our starting template. As expected, the results from the same PCR library (Figure 2B) have little variation across the full dynamic range in the log-log plot. A total of 97.5% of all sequencing reads obtained for SeqA1a were derived from a total of 150,612 distinct TCR sequences that were common to both replicates, while 97.4% of all reads obtained for SeqA1b were derived from common sequences. The comparisons between split samples from both DNA extraction (Figure 2C) and cells (Figure 2D) produce comparable results. For DNA extraction replicates, 54.9% of all reads obtained from sample A1a were from 25,673 distinct TCR sequences shared with sample A2. For sample A2, 55.9% of all reads obtained were derived from shared sequences. For replicates where the blood draw was split in half and extracted, 55% and 54.5% of all reads obtained for samples A1a and B1 were derived from shared TCR sequences, respectively. Since the blood draw is a small subset of the total cells in the blood, we expect variation due to finite sampling. However, for greatly expanded clones, we expect the ratio in both samples to converge.
Figure 2. Experimental design and results of repeatability experiment.

A) Schematic of experimental design. B) Sequence copy counts of TCRB clones obtained by sequencing the same PCR library on two different lanes of a flow cell. Each point represents a unique clone in this log-log scatter plot. The points in red are found in both repetitions, while those in blue where only found in the one lane. The total number of overlapping clones plotted on this graph is 122,000, with 20,000 clones sequenced that were unique to each lane. C) Sequence copy counts obtained by sequencing two separate PCR reactions performed using template from the same DNA extraction. D) Sequence copy counts obtained by sequencing two different DNA extractions from the same blood draw.
The similar results from splitting the sample after the blood draw and after the DNA extraction is expected. We began with a large number of T cells from the blood draw. So, the total amount of extracted DNA from each sample was much larger than the 3μg we amplified. For both cases, we sampled a small subset of the total pool of T cells or DNA, so the overlap in both cases is expected to be driven by the number of template genomes amplified, which is the same in both cases. The overlap of ~55% is determined by the distribution of T cells clones in the peripheral blood. We do not know the form of the full distribution of clonotypes in blood. However, the total diversity of both naïve and memory T cells have been estimated, with 2–4 million unique naïve T cells and 100K-400K memory T cells (Arstila et al., 1999; Robins et al., 2009). Roughly, we expect to find a majority of the memory T cells overlapping in our experiment and a small fraction of naives as we use 3μg of DNA which corresponds to 1 million haploid genomes or DNA from 500,000 cells. So, a rough estimate of the expected overlap between repeated sampling of 500,000 T cells from blood should approximate the ratio of memory to naïve T cells, which is ~60% in our donor.
4. Discussion
The sensitivity of this adaptive immune receptor sequencing method has multiple near term clinical applications. This assay’s ability to repeatedly measure T cell clones at a level of one cell in a 100,000 has the potential to outperform the present technology for monitoring minimal residual (MRD) in T cell leukemia and lymphoma patients (and the strategy can be readily extended to IgH receptors in B cells to monitor B cell leukemia and lymphoma). There are currently two competing assays for monitoring MRD; one utilizes a flow cell procedure and the other involves designing specific primers for an individual patient’s tumor clone (Ladetto et al., 2000; Rawstron et al., 2007; Sayala et al., 2007). The commonly utilized flow sort technology is sensitive to a detection level of approximately one cell in 10,000, while the specific primer design technique is labor intensive and very costly. At this point, our high-throughput assay is at least ten times more sensitive than the flow strategy, yet can be performed at a competitive cost. In fact, the results presented were not designed to test the true depth of our sensitivity. We are currently able to sequence 70 million TCR sequences per flow cell lane using the Illumina HiSeq machine. With 16 lanes available, we are able to sequence over a billion TCR sequences in a few days on a single machine. We use redundancy for error correction and require a five-fold coverage, which means we are presently able to sequence every rearranged T cell in over 100 million T cells on each run of a single machine. So, our true potential sensitivity is greater than one T cell in 100 million. However, this sequencing depth would add expense. The presentation in this manuscript is designed to be readily transportable to the new faster, cheaper, and lower throughput sequencing machines that would be clinically viable. For example, the Illumina MiSeq is reported to deliver nearly 5 million reads of length 60 nucleotides in less than eight hours.
Further, this procedure can be used to track with high precision the population of transferred T cells in patients receiving adoptive cellular therapy. The use of ex vivo expanded tumor-infiltrating T lymphocytes (TIL) in combination with a pre-infusion conditioning regimen is effective treatment for patients with advanced refractory melanoma (Weber et al.); efficacy is directly related to the capacity of transferred cells to persist long term (Robbins et al., 2004). An accurate measure of the in vivo persistence of a polyclonal T cell product such as a TIL infusion requires that every individual and unique TCR be sequenced and quantified, a feat possible only with the massively parallel sequencing capabilities of this assay. As these and other forms of adoptive cellular therapy become increasingly accepted treatment modalities, a routine means to reliably monitor transferred T cells will be essential (Jorritsma et al.; Yee; Rosenberg and Dudley, 2009).
Supplementary Material
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Arstila TP, Casrouge A, Baron V, Even J, Kanellopoulos J, Kourilsky P. A direct estimate of the human alphabeta T cell receptor diversity. Science. 1999;286:958–61. doi: 10.1126/science.286.5441.958. [DOI] [PubMed] [Google Scholar]
- Boyd SD, Gaeta BA, Jackson KJ, Fire AZ, Marshall EL, Merker JD, Maniar JM, Zhang LN, Sahaf B, Jones CD, Simen BB, Hanczaruk B, Nguyen KD, Nadeau KC, Egholm M, Miklos DB, Zehnder JL, Collins AM. Individual variation in the germline Ig gene repertoire inferred from variable region gene rearrangements. J Immunol. 184:6986–92. doi: 10.4049/jimmunol.1000445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyd SD, Marshall EL, Merker JD, Maniar JM, Zhang LN, Sahaf B, Jones CD, Simen BB, Hanczaruk B, Nguyen KD, Nadeau KC, Egholm M, Miklos DB, Zehnder JL, Fire AZ. Measurement and clinical monitoring of human lymphocyte clonality by massively parallel VDJ pyrosequencing. Sci Transl Med. 2009;1:12ra23. doi: 10.1126/scitranslmed.3000540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman JD, Warren RL, Webb JR, Nelson BH, Holt RA. Profiling the T-cell receptor beta-chain repertoire by massively parallel sequencing. Genome Res. 2009;19:1817–24. doi: 10.1101/gr.092924.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunder NN, Wallen H, Cao J, Hendricks DW, Reilly JZ, Rodmyre R, Jungbluth A, Gnjatic S, Thompson JA, Yee C. Treatment of metastatic melanoma with autologous CD4+ T cells against NY-ESO-1. N Engl J Med. 2008;358:2698–703. doi: 10.1056/NEJMoa0800251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorritsma A, Schotte R, Coccoris M, de Witte MA, Schumacher TN. Prospects and Limitations of T Cell Receptor Gene Therapy. Curr Gene Ther. doi: 10.2174/156652311796150390. [DOI] [PubMed] [Google Scholar]
- Ladetto M, Donovan JW, Harig S, Trojan A, Poor C, Schlossnan R, Anderson KC, Gribben JG. Real-Time polymerase chain reaction of immunoglobulin rearrangements for quantitative evaluation of minimal residual disease in multiple myeloma. Biol Blood Marrow Transplant. 2000;6:241–53. doi: 10.1016/s1083-8791(00)70006-1. [DOI] [PubMed] [Google Scholar]
- Rawstron AC, Villamor N, Ritgen M, Bottcher S, Ghia P, Zehnder JL, Lozanski G, Colomer D, Moreno C, Geuna M, Evans PA, Natkunam Y, Coutre SE, Avery ED, Rassenti LZ, Kipps TJ, Caligaris-Cappio F, Kneba M, Byrd JC, Hallek MJ, Montserrat E, Hillmen P. International standardized approach for flow cytometric residual disease monitoring in chronic lymphocytic leukaemia. Leukemia. 2007;21:956–64. doi: 10.1038/sj.leu.2404584. [DOI] [PubMed] [Google Scholar]
- Reijonen H, Novak EJ, Kochik S, Heninger A, Liu AW, Kwok WW, Nepom GT. Detection of GAD65-specific T-cells by major histocompatibility complex class II tetramers in type 1 diabetic patients and at-risk subjects. Diabetes. 2002;51:1375–82. doi: 10.2337/diabetes.51.5.1375. [DOI] [PubMed] [Google Scholar]
- Robbins PF, Dudley ME, Wunderlich J, El-Gamil M, Li YF, Zhou J, Huang J, Powell DJ Jr, Rosenberg SA. Cutting edge: persistence of transferred lymphocyte clonotypes correlates with cancer regression in patients receiving cell transfer therapy. J Immunol. 2004;173:7125–30. doi: 10.4049/jimmunol.173.12.7125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robins HS, Campregher PV, Srivastava SK, Wacher A, Turtle CJ, Kahsai O, Riddell SR, Warren EH, Carlson CS. Comprehensive assessment of T-cell receptor beta-chain diversity in alphabeta T cells. Blood. 2009;114:4099–107. doi: 10.1182/blood-2009-04-217604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robins HS, Srivastava SK, Campregher PV, Turtle CJ, Andriesen J, Riddell SR, Carlson CS, Warren EH. Overlap and effective size of the human CD8+ T cell receptor repertoire. Sci Transl Med. 2:47ra64. doi: 10.1126/scitranslmed.3001442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg SA, Dudley ME. Adoptive cell therapy for the treatment of patients with metastatic melanoma. Curr Opin Immunol. 2009;21:233–40. doi: 10.1016/j.coi.2009.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayala HA, Rawstron AC, Hillmen P. Minimal residual disease assessment in chronic lymphocytic leukaemia. Best Pract Res Clin Haematol. 2007;20:499–512. doi: 10.1016/j.beha.2007.03.004. [DOI] [PubMed] [Google Scholar]
- Wang C, Sanders CM, Yang Q, Schroeder HW, Jr, Wang E, Babrzadeh F, Gharizadeh B, Myers RM, Hudson JR, Jr, Davis RW, Han J. High throughput sequencing reveals a complex pattern of dynamic interrelationships among human T cell subsets. Proc Natl Acad Sci USA. 107:1518–23. doi: 10.1073/pnas.0913939107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren RL, Freeman JD, Zeng T, Choe G, Munro S, Moore R, Webb JR, Holt RA. Exhaustive T-cell repertoire sequencing of human peripheral blood samples reveals signatures of antigen selection and a directly measured repertoire size of at least 1 million clonotypes. Genome Res. doi: 10.1101/gr.115428.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber J, Atkins M, Hwu P, Radvanyi L, Sznol M, Yee C. White Paper on Adoptive Cell Therapy for Cancer with Tumor-Infiltrating Lymphocytes: A Report of the CTEP Subcommittee on Adoptive Cell Therapy. Clin Cancer Res. 17:1664–1673. doi: 10.1158/1078-0432.CCR-10-2272. [DOI] [PubMed] [Google Scholar]
- Weinstein JA, Jiang N, White RA, 3rd, Fisher DS, Quake SR. High-throughput sequencing of the zebrafish antibody repertoire. Science. 2009;324:807–10. doi: 10.1126/science.1170020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yee C. Adoptive therapy using antigen-specific T-cell clones. Cancer J. 16:367–73. doi: 10.1097/PPO.0b013e3181eacba8. [DOI] [PubMed] [Google Scholar]
- Yousfi Monod M, Giudicelli V, Chaume D, Lefranc MP. IMGT/JunctionAnalysis: the first tool for the analysis of the immunoglobulin and T cell receptor complex V-J and V-D-J JUNCTIONs. Bioinfomatics. 2004;20(suppl1):i379–i385. doi: 10.1093/bioinformatics/bth945. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.