

Abstract
Entry of vitamin C or ascorbate into most tissues requires its movement across the endothelial cell barrier of vessels. If trans-cellular ascorbate movement occurs, then it should be evident as ascorbate efflux from endothelial cells. Cultured EA.926 endothelial cells that had been loaded to about 3.5 mM intracellular ascorbate lost 70–80% of ascorbate to the medium over several hours at 37 °C via a non-saturable process that was insensitive to anion transport inhibitors and thiol reagents. Oxidation of this extracellular ascorbate by ascorbate oxidase or ferricyanide enhanced apparent ascorbate efflux, suggesting that efflux of the vitamin was countered in part by its re-uptake on ascorbate transporters. Although basal ascorbate efflux was not calcium-dependent, increased entry of calcium into the cells enhanced ascorbate release. These results support the hypothesis that ascorbate efflux reflects trans-endothelial cell ascorbate movement out of the blood vessel.
Keywords: ascorbate transport, ascorbate efflux, sulfinpyrazone, trans-cellular transport, ascorbate recycling, endothelial cells
Introduction
Endothelial cells lining blood vessel walls are constantly exposed to ascorbic acid concentrations in the plasma of 30–60 μM in normal individuals [1]. These concentrations tend to decline with age [2–4], but in humans remain a function of dietary intake in both men and women [2]. Although in vivo measurements of intracellular ascorbate concentrations in endothelial cells are not feasible, exposure of cells in culture to such ascorbate concentrations results in intracellular concentrations in the low millimolar range [5, 6]. This gradient across the plasma membrane is likely generated by the SVCT2, a specific ascorbate transporter that has mediates sodium- and energy-dependent ascorbate uptake in other cells [7]. The two-electron oxidized form of ascorbate, termed dehydroascorbate, can also enter cells on ubiquitous glucose transporters [8]. Once inside cells, DHA is rapidly reduced back to ascorbate, resulting in a net accumulation of the vitamin in the cells [9–11]. Of the two mechanisms, uptake of ascorbate on its specific transporter probably contributes much more to the trans-plasma membrane ascorbate gradient than does uptake and reduction of DHA, since concentrations of the latter are usually very low in plasma [12], since DHA competes with 5 mM D-glucose for glucose transporters, and since human erythrocytes, which lack ascorbate transporters [13], have the same intracellular ascorbate concentration as in plasma [1, 11]. Ascorbate appears to be trapped in most cells because of its negative charge at physiologic pH, although ascorbate efflux has been described in hepatocytes [14], in brain cells under glutamate-induced stress [15], and in endothelial cells [14, 16]. Loss of ascorbate from cells has also been documented following its oxidation as efflux of DHA on glucose transporters [17]. One might expect this to be a significant route in cells under oxidant stress, such as in cells cultured at ambient oxygen concentrations. However, there have been no studies comparing rates of ascorbate efflux as ascorbate versus DHA. This could be an important issue, since ascorbate leaving cells as DHA might not readily re-enter cells due to competition from glucose or to opening of its unstable lactone ring and thus permanent loss from the system.
The question of ascorbate efflux is especially relevant for endothelial cells, which in many capillary beds form tight junctions and thus must somehow move ascorbate out of the vessel lumen and into the extracellular space of tissues. In this work we studied ascorbate efflux and re-uptake by cultured human-derived endothelial cells (EA.hy926). These cells retain endothelial cell features, including cobblestone appearance with formation of capillary-like tubes in culture [18], expression of factor VIII antigen [19], oxidative modification of human LDL [20], and calcium-dependent endothelial nitric oxide synthase activation [20, 21]. We found that loss of the vitamin from EA.hy926 cells occurs largely as ascorbate and not as DHA under standard culture conditions in oxygenated buffer, that a significant fraction of ascorbate efflux is counteracted by its re-uptake on the ascorbate transporter, and that ascorbate efflux is enhanced by increases in intracellular calcium.
Materials and methods
Materials
Sigma/Aldrich Chemical Co. (St. Louis, MO) supplied the A23187, dehydroascorbic acid (DHA), 4,4′-diisothiocyanatostilbene-2,2′-disulfonic acid (DIDS), diethylenetriamine-pentaacetic acid, ethylenediamine-tetraacetic acid (EDTA), N-2-hydroxyethylpiperazine N’-2-ethanesulfonic acid (Hepes), A23187, phenylarsine oxide, and sulfinpyrazone. Nimodipine and mibefradil were a gift from Dr. Katherine Murray. Perkin-Elmer Life and Analytical Sciences, Inc. (Boston, MA) supplied the L-[1-14C]ascorbic acid, which was dissolved in deionized water containing 0.1 mM acetic acid and stored in multiple aliquots at -20 °C until use.
Methods
Cell Culture
EA.hy926 cells were a generous gift from Dr. Dr. Cora Edgell (University of North Carolina, Chapel Hill, NC, USA). The cells were cultured in Dulbecco’s minimal essential medium and 10% (v/v) heat-inactivated fetal bovine serum, which contained 20 mM D-glucose and HAT media supplement (Sigma/Aldrich Chemical Co., St. Louis, MO). Cells were cultured to near confluence at 37 ºC in humidified air containing 5% CO2. Just before an experiment, cells were rinsed 3 times in 2 ml of Krebs-Ringer Hepes (KRH) buffer at 37 °C. KRH buffer consisted of 20 mM Hepes, 128 mM NaCl, 5.2 mM KCl, 1 mM NaH2PO4, 1.4 mM MgSO4, and 1.4 mM CaCl2, pH 7.4. In some experiments, calcium was omitted from the KRH.
Assay of radioactive ascorbate efflux
EA.hy926 cells in 12-well plates were rinsed twice in 2 ml of KRH to remove culture medium, and then incubated for 1 h at 37 °C in KRH that contained 5 mM D-glucose, 0.5 mM GSH, and 0.05 μCi of L-[1-14C]ascorbic acid. The total ascorbate concentration was 0.3 μM, unless otherwise stated. To start the efflux assay, the supernatant was aspirated and the cells were rinsed three times in 2 ml of KRH at 37 °C and the cells were treated with 1 ml of fresh KRH that contained 20 mM D-glucose and additives as noted. After incubation for the times indicated, an aliquot of the supernatant was removed for radioactive counting, and the cells were rinsed twice in ice-cold KRH. The cell monolayer was then treated with 1 ml of 0.05 N NaOH, the cells were scraped from the plate, and the extract was added to 5 ml of Ecolume liquid scintillation fluid (ICN, Costa Mesa, CA) with mixing. The radioactivity of duplicate samples was measured in a Packard CA-2200 liquid scintillation counter.
Assay of ascorbate and GSH
After incubations as noted in 6-well plates, the medium was aspirated and the cells were gently rinsed twice with 2 ml of ice-cold KRH. The last rinse was removed and the cell monolayer was treated with 0.1 ml of 25% metaphosphoric acid (w/v) for several minutes, followed by 0.35 ml of a buffer containing 0.1M Na2HPO4 and 0.05 mM EDTA, pH 8.0. The remaining cell material was scraped from the plate, and the lysate was removed and centrifuged at 3 °C for 1 min at 13,000 x g. Intracellular GSH was measured by the method of Hissin and Hilf [22]. Assay of ascorbic acid was performed in duplicate by high performance liquid chromatography as previously described [23]. In some experiments, ascorbate was also measured in 0.1 ml of the incubation medium by adding 0.1 ml of 25% metaphosphoric acid (w/v), mixing, neutralizing with 0.35 ml of the above phosphate/EDTA buffer, and centrifuging to remove any precipitated solids before assay of ascorbate. Intracellular concentrations of ascorbate were calculated based on the intracellular distribution space of 3-O-methylglucose in EA.hy926 cells. We previously determined this value for endothelial cells in culture to be 3.6 ± 1.2 μl/mg protein [21] and used this in the calculations.
Data Analysis
Results are shown as mean + standard error. Statistical comparisons were made using SigmaStat 2.0 software (Jandel Scientific, San Rafael, CA). Differences between treatments were assessed by two-way analysis of variance with post-hoc testing using Dunnett’s test.
Results
Ascorbate efflux from endothelial cells
To assess the efflux of ascorbate from endothelial cells, confluent EA.hy926 cells were loaded by a short incubation with 0.5 mM DHA to intracellular ascorbate concentrations of about 3.5 mM (Fig. 1). DHA is very unstable in physiologic buffers, with a half-life of about 6 min [24, 25]. However, it is rapidly taken into cells on glucose transporters, this DHA is rapidly reduced to ascorbate. As long as D-glucose is present this reduction is not toxic to the cells [26]. After several rinses to remove any extracellular ascorbate, the cells were incubated in KRH containing 5 mM D-glucose as an energy source for various times up to 4 h. As shown in Fig. 1A, intracellular ascorbate decreased fairly rapidly to about 20% of starting levels at 2 h, and then stabilized at about 0.6 mM out to 4 h (circles). As expected, extracellular ascorbate was very low at the beginning of the incubation, but increased over the 4 h incubation, although more slowly after 2 h to reach about 6 μM (Fig. 1B, circles). The total ascorbate in the cells and buffer decreased to less than 50% of the initial value over the first two hours of incubation, but then stabilized when most (> 70%) of the ascorbate remaining in the 1 ml system was extracellular (Fig. 1C, circles). Although most of the ascorbate was outside the cells, it should be noted that there was still a 67-fold concentration gradient of ascorbate across the plasma membrane.
Fig. 1.
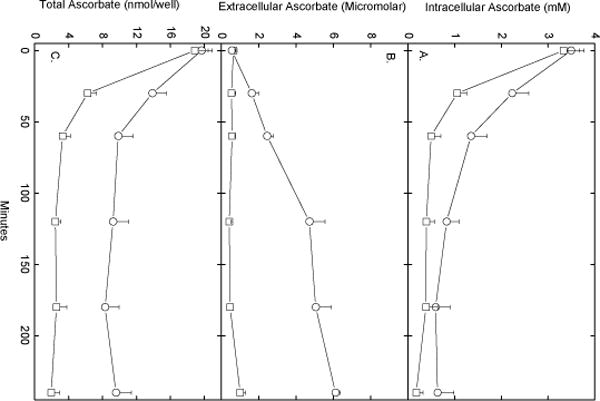
Ascorbate efflux from EA.hy926 cells. Confluent cells were incubated at 37 °C in KRH that contained 5 mM D-glucose and 500 μM DHA. After 30 min, the medium was aspirated can and the cells were rinsed twice in 2 ml of KRH and incubated at 37 °C in fresh KRH that contained 5 mM D-glucose without (circles) or with (squares) 0.5 units/ml ascorbate oxidase. At the times indicated, aliquots of the medium were removed for assay of ascorbate, and the cells were rinsed 3 times in KRH before they were taken for assay of intracellular ascorbate. Panel A shows intracellular ascorbate, Panel B shows medium concentrations of ascorbate, and Panel C shows total ascorbate in each well. Results are from 5 experiments.
Ascorbate re-uptake during its efflux
To assess the extent to which re-uptake of ascorbate that had escaped the cells contributed to maintaining intracellular ascorbate, ascorbate oxidase was added to incubations of ascorbate-loaded EA.hy926 cells. Ascorbate oxidase will not appreciably penetrate the cells and will destroy extracellular ascorbate, thus preventing its re-uptake on the ascorbate transporter. As shown by the squares in Fig. 1A, ascorbate oxidase caused a greater decrease in intracellular ascorbate over the first hour of incubation compared to cells in the same experiment not incubated with the enzyme. Ascorbate oxidase destroyed most of the extracellular ascorbate (Fig. 1B), and decreased the total amount of ascorbate in the system by more than 50% at each time point after the first (Fig. 1C). Similar results were observed when ferricyanide was added to oxidize extracellular ascorbate during 2 h of incubation (results not shown). At least during short-term incubations, ferricyanide, like ascorbate oxidase, does not appreciably enter cells [27] and thus will selectively oxidize extracellular ascorbate. Together, the ascorbate oxidase and ferricyanide results suggest that there is a substantial ascorbate efflux from ascorbate-loaded EA.hy926 cells, and this is partially offset by reuptake of the vitamin.
Evaluation of the mechanism of ascorbate efflux
Efflux of ascorbate was also evident when EA.hy926 cells were loaded for 1 h with 0.3 μM radioactive ascorbate (Fig. 2, circles), although the relative intracellular depletion (Fig. 2, squares) was not as severe as observed with higher initial intracellular ascorbate concentrations (e.g., Fig. 1). Whereas the total amount of intra- and extracellular radioactivity in each well were equal after 90 min of efflux, even with some trapping of extracellular ascorbate by the cells, the intracellular radioactive ascorbate concentration was over 100-fold that outside the cells.
Fig. 2.
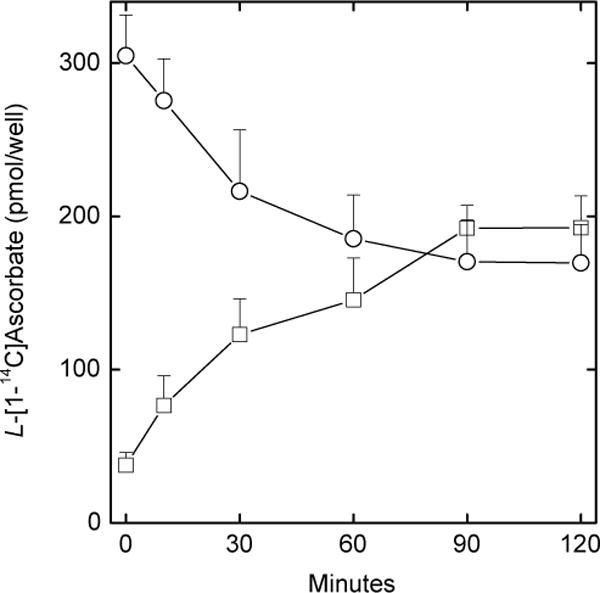
Efflux of radioactive ascorbate from EA.hy926 cells. Ascorbate loss from cells (circles) and appearance in the extracellular buffer (squares) was measured at the times indicated in confluent cells as described under Methods. Results are shown from 3 experiments.
Accurate measurement of rates of ascorbate efflux in EA.hy926 cells is complicated by its re-uptake on the ascorbate transporter. This can be prevented by oxidizing extracellular ascorbate with ascorbate oxidase and by assessing efflux using radiolabeled ascorbate. The extracellular accumulation of radiolabel (even though its chemical form is L-[1-14C]DHA and its breakdown products) reflects efflux of ascorbate. To prevent uptake of radiolabeled DHA on glucose transporters, the medium glucose concentration was increased to 20 mM. Using this approach, we evaluated the kinetics of radiolabeled ascorbate efflux by increasing intracellular ascorbate and following efflux of radiolabeled ascorbate. Since ascorbate efflux in the presence of ascorbate oxidase was rapid for 30 min (Fig. 1B), this time was used for the efflux studies. Cells were simultaneously loaded for 1 h with tracer amounts of radiolabeled ascorbate and with increasing amounts of dehydroascorbate. The latter will be converted to unlabeled ascorbate inside the cells, which will then compete for efflux with the radiolabeled ascorbate if there is a saturable efflux mechanism. It was determined in separate studies not shown that cells loaded with increasing amounts of DHA and the same concentration of radioactive ascorbate in fact contained the same amount of radiolabeled ascorbate independent of the loading DHA concentration. Whereas DHA loading progressively increased intracellular ascorbate concentrations (Fig. 3, X-axis values), there was no effect on the efflux of radiolabeled ascorbate from the cells at 60 min (Fig. 3, Y-axis values). This shows that efflux is not due to membrane damage or cell lysis from DHA loading or reduction and that it is independent of the intracellular ascorbate concentration.
Fig. 3.
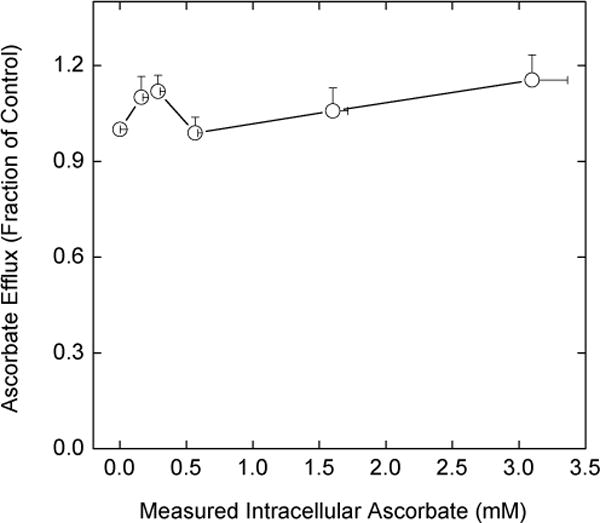
Failure of intracellular ascorbate to affect ascorbate efflux. Rinsed cells were incubated at 37 °C in KRH that contained 5 mM D-glucose, 0.05 μCi of [1-14C]ascorbate, and one of the following DHA concentrations: 0, 0.05, 0.1, 0.1, 0.5, or 1 mM. After 1 h, the cells were rinsed with warm KRH and incubated at 37 °C in KRH that contained 20 mM D-glucose and 0.5 unit/ml ascorbate oxidase. Efflux was allowed to continue for 30 min in this buffer before sampling of the extracellular radioactivity. The latter was expressed as a fraction of the radioactivity outside cells that were treated only with radiolabeled ascorbate, but not DHA. In separate experiments under identical conditions, cells were loaded with only with DHA as noted above for 1 h, rinsed in KRH, and taken for determination of intracellular ascorbate. The results from 7 experiments involving radioactive ascorbate and 4 experiments involving DHA loading only are plotted as a function of the measured intracellular ascorbate concentrations.
To further examine the mechanism of ascorbate efflux from EA.hy926 cells, the effects of adding various agents during the efflux phase was studied. Under the conditions of Fig. 1, the following agents at the concentrations noted had no effect on ascorbate efflux over 60 min: DIDS (100 μM), phenylarsine oxide (100 μM), p-chloromercuribenzene sulfonic acid (200 μM), L-cysteine (5 mM), oxidized glutathione (5 mM), and ATP (0.3 mM). However, GSH at 5 mM and the calcium ionophore A23187 at 10 μM markedly enhanced efflux. Neither agent affected intracellular GSH, showing that the results were not due to cell lysis. The effect of GSH was evident by 30 min of efflux, but was maximal at 1 h (results not shown). The concentration-dependence of the effect of GSH at 1 h is shown in Fig. 4. Whereas GSH concentrations of 1 mM or less had no effect on ascorbate efflux, GSH at 2.5 mM and higher caused nearly complete loss of ascorbate from the cells (Fig. 4A) and a tripling of the ascorbate concentration in the incubation medium (Fig. 4B). The efflux of radiolabeled ascorbate from cells into the incubation medium showed similar concentration dependence when carried out under the conditions of Fig. 2 at 2 h (results not shown). In the experiments shown in Fig. 4C, the efflux of ascorbate was accompanied by a decrease in the total ascorbate in the system at 2.5 and 5 mM GSH, which could indicate an oxidative loss of ascorbate due to GSH. However, the GSH-induced efflux of ascorbate was not affected by the presence of up to 1 mM diethylenetriamine-pentaacetic acid, 0.01 mM EDTA, or 100 U of catalase added to the cells just before the efflux phase (results not shown). On the other hand, removal of calcium from the efflux buffer completely prevented both the effect of GSH and A23187 to increase ascorbate efflux (Fig. 5). Removal of calcium from the incubation medium did not affect either intracellular (Fig. 5A, left-hand pair of bars) or extracellular (Fig. 5B, left-hand pair of bars) ascorbate. A23187 treatment decreased intracellular ascorbate (Fig. 5A) and caused a corresponding increase in extracellular ascorbate (Fig. 5B). Calcium-free buffer prevented both the decrease in intracellular ascorbate due to A23187 (Fig. 5A) and the increase in the incubation buffer (Fig. 5B). As expected from the results of Fig. 4, GSH at 5 mM caused a marked depletion of intracellular ascorbate (Fig. 5A) and a corresponding increase in extracellular ascorbate (Fig. 5B). As with A23187, the effect of GSH was completely prevented by removal of calcium from the incubation buffer. The GSH effect to increase ascorbate efflux was not prevented by the selective calcium channel blockers nimodipine and mibefradil, both at 40 μM (results not shown).
Fig. 4.
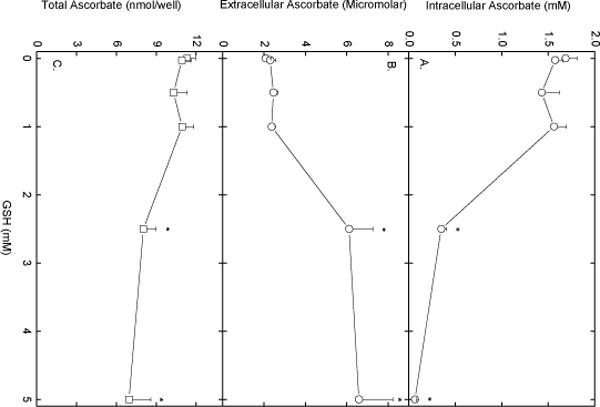
Concentration-dependent acceleration of ascorbate efflux by GSH. EA.hy926 cells were loaded with ascorbate as described in the Fig. 1 legend, rinsed twice in KRH, and incubated in fresh KRH that contained 5 mM D-glucose and the indicated concentration of GSH. After 60 min, the buffer was sampled for extracellular ascorbate, the cells were rinsed 3 times in KRH and taken for assay of intracellular ascorbate. Panel A shows intracellular ascorbate, Panel B shows extracellular ascorbate, and Panel C shows total ascorbate in the system from 5 experiments. An asterisk (*) indicates p < 0.05 compared to the cells not exposed to GSH.
Fig. 5.
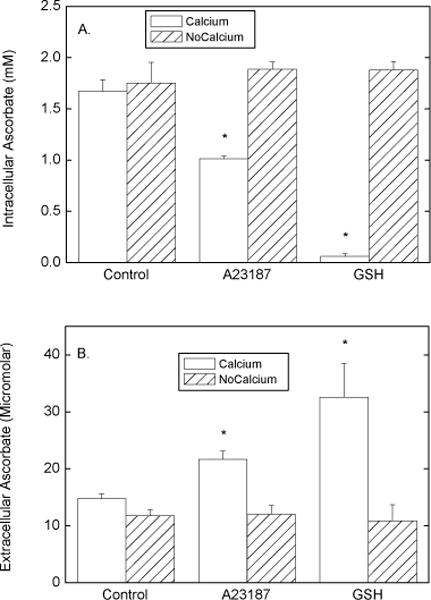
Calcium effects on ascorbate efflux in EA.hy926 cells. Rinsed cells were loaded with ascorbate during 30 min of incubation with 0.5 mM DHA as described in the legend to Fig. 1. The cells were rinsed 3 times with either KRH or where noted with KRH that did not contain calcium and incubated at 37 °C in the appropriate KRH that contained 5 mM D-glucose with other additions as noted: 10 μM A23187, or 5 mM GSH. After 1 h, the medium was sampled for ascorbate (Panel B), the cells were rinsed 3 times in KRH, and the cells were taken for assay of ascorbate (Panel A). Results are shown from 4 experiments, with an asterisk (*) indicating p < 0.05 compared to the adjacent bar and to the respective control sample.
Mechanism of ascorbate re-uptake by EA.hy926 endothelial cells
We next investigated the mechanism of the apparent ascorbate re-uptake, since this could occur as ascorbate on the ascorbate transporter, or as DHA on the glucose transporter, with subsequent reduction and trapping within the cells. In results not shown, in cells loaded with 0.5 mM DHA as described for the studies shown in Fig. 1, no effects of 30 mM D-glucose, 40 mM 3-O-methylglucose, or 25 μM cytochalasin B were observed on either intra- or extracellular ascorbate concentrations at 1 h. This suggests that re-uptake of DHA on the glucose transporter is not involved in the net ascorbate uptake. On the other hand, unlabeled extracellular ascorbate accelerated the loss of radioactive ascorbate from EA.hy926 cells. When cells were first loaded with tracer (< 1 μM) amounts of radioactive ascorbate, rinsed to remove extracellular ascorbate, and then incubated for 60 min with increasing concentrations of unlabeled ascorbate, net efflux of the tracer was enhanced. This is depicted in Fig. 6 as decreases in intracellular radiolabeled ascorbate (circles) and as increases in extracellular radiolabel (squares) with increasing initial extracellular concentrations of unlabeled ascorbate. These results fit with the notion that the ascorbate transporter was involved in re-uptake of ascorbate by the cells.
Fig. 6.
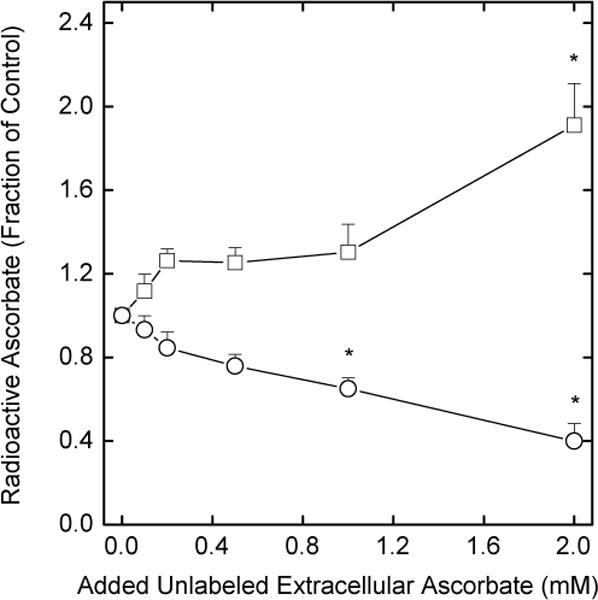
Acceleration of radiolabeled ascorbate efflux by extracellular ascorbate. Rinsed cells were incubated at 37 °C in the presence of 5 mM D-glucose and 0.05 μCi of L-[1-14C]ascorbate. After 1 h, the cells were rinsed 3 times in KRH and incubated at 37 °C in KRH that contained 5 mM D-glucose and the indicated concentration of unlabeled ascorbate. Changes in intracellular (circles) and extracellular (squares) radiolabel were assessed after 30 min of incubation and expressed as a fraction of the value in a sample loaded with tracer ascorbate only. Results are shown from 7 experiments with an asterisk (*) indicating p < 0.05 compared to cells not treated with unlabeled ascorbate.
Additional evidence that the ascorbate transporter mediates re-uptake of ascorbate was also sought using sulfinpyrazone, a known inhibitor of ascorbate transport in endothelial cells [28]. As shown in Fig. 7, inclusion of 1 mM sulfinpyrazone during the efflux phase of the experiment significantly decreased intracellular ascorbate (Fig. 7A, squares) compared to untreated control cells (Fig. 7A, circles). It also tended to increase extracellular ascorbate, although this difference was not significant in these experiments (Fig. 7B). Further, when cells were loaded for 60 min with radioactive ascorbate (as in the experiments shown in Fig. 2), then allowed to efflux for another 60 min with or without 1 mM sulfinpyrazone, extracellular radioactivity was increased by 52% in cells treated with sulfinpyrazone compared to control (p < 0.02, data not shown). Sulfinpyrazone had no effect on intracellular GSH content, showing that it did not cause cell lysis.
Fig. 7.
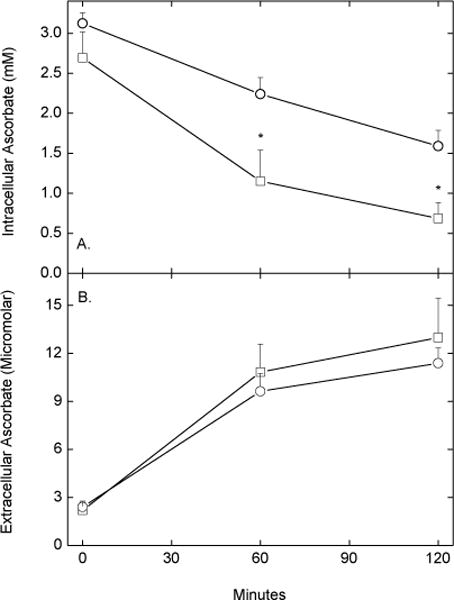
Acceleration of apparent ascorbate efflux by sulfinpyrazone. Rinsed, confluent EA.hy926 cells were incubated for 30 min at 37 °C with 0.5 mM DHA in KRH containing 5 mM D-glucose. The cells were then rinsed twice in KRH, followed by incubation at 37 °C in KRH that contained 5 mM D-glucose in the absence (circles) or presence (squares) of 1 mM sulfinpyrazone. At the times indicated, cells (Panel A) and medium aliquots (Panel B) were taken for assay of ascorbate. Results are shown from 5 experiments with an asterisk (*) indicating p < 0.05 compared to treatment without sulfinpyrazone.
We next assessed whether efflux and re-uptake of ascorbate is significant in cells cultured overnight in complete culture medium. Cells were first loaded with increasing concentrations of ascorbate for 1 h, rinsed several times to remove extracellular ascorbate, and cultured 16–18 h in fresh complete medium not containing ascorbate. As shown in Fig. 8A, intracellular ascorbate concentrations increased with increasing amounts of ascorbate added, but were halved by addition of ascorbate oxidase during culture. The measured extracellular concentration of ascorbate was below 1 μM, even in the absence of ascorbate oxidase (Fig. 8B, circles). In the presence of the oxidase, extracellular ascorbate was decreased further, but not to zero. Comparison of the extra- and intracellular ascorbate concentrations again showed a very steep gradient of over 1000-fold. Thus, efflux and re-uptake of ascorbate during longer term culture in a bicarbonate-based buffer system is again a significant mechanism for maintaining intracellular ascorbate.
Fig. 8.
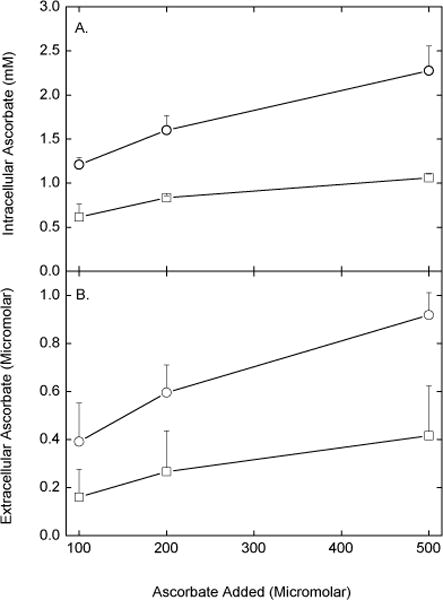
Efflux and re-uptake of ascorbate by EA.hy926 cells in overnight culture. Cells in culture medium were treated for 6 h at 37 °C with the indicated concentration of ascorbate. The medium was removed and the cells were rinsed once in KRH, followed by addition of new complete culture medium in the absence (circles) or presence (squares) of 1 unit/ml ascorbate oxidase. After 16–18 h in culture, aliquots of the incubation medium were taken for assay of ascorbate (Panel B), the cells were rinsed 3 times in KRH, and taken for assay of ascorbate (Panel A). Results are shown for 3 experiments. The curve for ascorbate oxidase was significantly lower than the curve in the absence of the oxidase.
Discussion
Although EA.hy926 endothelial cells concentrate ascorbate against a gradient, when loaded with relatively high intracellular ascorbate concentrations, they show substantial rates of ascorbate efflux. Over several hours of incubation in physiologic buffer, ascorbate concentrations inside of cells loaded to 3 - 4 mM ascorbate decrease to steady-state concentrations of about 0.6 mM. This plateau reflects a balance between ascorbate efflux and its re-uptake. In cells cultured for 16–18 h in medium supplemented at the beginning of the incubation with ascorbate, extracellular ascorbate was very low. This was probably due to its destruction in oxygenated culture medium containing trace amounts of iron. However, the intracellular ascorbate concentration was 1.2 – 2.8 mM, depending on the initial amount of ascorbate loaded into the cell. Thus, in both short and long term incubations, there was a steep ascorbate concentration gradient across the plasma membrane (70-fold and greater), which is likely due to an energy-dependent ascorbate transporter such as the SVCT2 [7]. If endothelial cells in vivo could maintain even a 40-fold concentration gradient from plasma into the cells, at a normal ascorbate concentration of 40 μM, intracellular ascorbate concentrations would be on the order of 1.6 mM.
Ascorbate is a hydrophilic molecule and also carries a negative charge at physiologic pH. It diffuses slowly out of a simple cell such as an erythrocyte, which loses 10–20% of ascorbate over 40 min of incubation, depending on the initial intracellular concentration [14, 17, 29]. Efflux is more rapid from endothelial cells than from erythrocytes [14]. We show here that endothelial cells containing about 3 mM ascorbate lose 50–60% of this ascorbate over an hour of incubation in physiologic buffer. Efflux of ascorbate at lower intracellular concentrations is relatively less rapid. This efflux could represent vectorial transport of ascorbate, which in vivo would be efflux from the basal side of the cells that permits movement of ascorbate from the vessel lumen to the interstitium. A robust system has been described for intestinal Caco-2 cells involving polarized ascorbate transport out of the intestinal lumen and across epithelial cells that is initiated by ascorbate uptake on the SVCT1 in the brush-border membrane side of the cell [30]. Whether such a system exists for endothelial cells remains to be determined. It is possible that the SVCT2 is involved in such vectorial transport of ascorbate across capillary walls. Support for this notion comes from observations that ascorbate does not rapidly cross the blood-brain barrier in vivo[31, 32] and that, in contrast to endothelial cells from other vessel types, the SVCT2 is not present in vivo in the endothelial cells that form the blood-brain barrier [33, 34]. Nonetheless, such an effect will need to be determined experimentally.
One of the major findings of this work is that efflux of ascorbate out of endothelial cells is countered by its re-uptake. Thus, removal of extracellular ascorbate by destruction by ascorbate oxidase doubled apparent rates of ascorbate loss from endothelial cells and halved the intracellular concentration of ascorbate that the cells could maintain, either after several hours in physiologic buffer or after 18 h of culture. Similar results were obtained with ferricyanide in short term incubations. Both ferricyanide and ascorbate oxidase cause a one-electron oxidation of ascorbate, resulting in the ascorbate radical. Two molecules of the ascorbate radical dismutate to form one molecule each of ascorbate and DHA [35]. Any extracellular DHA would rapidly re-enter the cells on the glucose transporter followed by intracellular reduction to ascorbate. If DHA uptake and reduction accounted for the ascorbate re-uptake, then treatment with either agent would have decreased net ascorbate efflux, not increased it, as was observed (e.g., Fig. 1). Further, inhibition of glucose transporters with D-glucose, 3-O-methylglucose, and cytochalasin B failed to affect rates of net ascorbate efflux. On the other hand, ascorbate loss was enhanced by sulfinpyrazone, as expected if it inhibited ascorbate re-uptake on the ascorbate transporter. It must be noted, however, that sulfinpyrazone has been shown to inhibit a component of DHA uptake in cerebral astrocytes that is insensitive to D-glucose [36] and we have also observed this effect in EA.hy926 cells (May, J.M., unpublished results). Since the findings noted above suggest that ascorbate re-uptake occurs as ascorbate and not as DHA, and especially since cytochalasin B is known to inhibit > 90% of such D-glucose-insensitive DHA uptake and reduction [37], we conclude that the effect of sulfinpyrazone was most likely on the ascorbate transporter. Thus, re-uptake of ascorbate on its transporter rather than uptake of DHA on the glucose transporter is the mechanism of ascorbate recovery by the cells.
If the efflux of ascorbate in EA.hy926 cells reflects vectorial transport of ascorbate from the luminal plasma membrane to that on the basal side of the cell, the then maintaining high intracellular ascorbate concentrations via uptake from the blood is important for facilitating this process. It is also likely to be important for ascorbate functions within endothelial cells, such as serving as an antioxidant and as a co-factor for the prolyl and lysyl hydroxylases that regulate HIF-1α levels and that hydroxylate collagen [38]. Regarding the latter, we have previously shown that optimal rates of type IV collagen synthesis in EA.hy926 cells required intracellular ascorbate concentrations of about 2 mM [6]. Whether or not the SVCT2 participates in trans-cellular ascorbate movement, its major effect is to maintain intracellular ascorbate concentrations.
The mechanism of ascorbate efflux from endothelial cells is non-saturable with respect to the intracellular ascorbate concentration, suggesting simple diffusion or movement across a channel. In cultured HepG2 liver cells, which have higher rates of efflux on a per cell basis than do endothelial cells, efflux was saturable, temperature-dependent, and inhibited by phloretin and DIDS [14]. This suggests a specific transporter involved in efflux from these cells. Such a transporter could serve to export newly synthesized ascorbate from hepatocytes. Although HepG2 cells are derived from a human hepatoma and thus do not make ascorbate, they could have retained this mechanism of ascorbate export. Upston and colleagues [14] showed that substantial ascorbate also was lost from perfused rat and guinea pig livers. Rats synthesize ascorbate in hepatocytes [39], while guinea pigs, like humans, lack gulonolactone oxidase, the last step in the biosynthesis of ascorbate. Still, the presence of ascorbate efflux in hepatocytes from all three species suggests the presence of a specific transporter in the liver. Although non-saturable efflux observed in endothelial cells in the present work could still reflect gradient-dependent movement of ascorbate across a membrane channel, our finding that sulfinpyrazone and other agents known to inhibit anion channels did not decrease ascorbate efflux suggests either simple diffusion or a channel insensitive to these agents.
The constitutive or basal efflux of ascorbate from endothelial cells can be enhanced by calcium influx. Davis and colleagues [16] previously demonstrated calcium-inducible efflux of ascorbate from pig coronary artery endothelial cells that was not present in pig smooth muscle arterial cells. Ascorbate efflux was increased both by A23187 and by ATP, the latter acting through P2Y2-like receptors. Ascorbate efflux stimulated by ATP did not occur on the ascorbate transporter, but was inhibited by anion channel blockers (including sulfinpyrazone) and by omitting extracellular calcium or chelating it intracellularly [16]. We also found that A23187 increased ascorbate efflux in cells incubated in calcium-containing medium, although we did not see an increase in ascorbate efflux due to ATP in EA.hy926 cells. This could indicate lack of functional cell surface nucleotide receptors on EA.hy926 cells. Omission of the calcium from the cell incubation buffer prevented A23187-induced ascorbate efflux, but it did not affect basal efflux. The latter suggests that non-stimulated ascorbate efflux seen both in this and in the study by Davis, et al. [16] is not calcium-dependent. We found that GSH concentrations of 2.5 mM and higher also stimulated ascorbate efflux, an effect that was again completely prevented by removal of calcium in the medium. Supraphysiologic extracellular GSH concentrations have been noted to cause oxidant stress by redox cycling [40]. The thiol group on GSH is not a good donor of electrons, but if GSH is converted to cysteinyl-glycine by γ-glutamyltransferase (E.C. 2.3.2.2), the thiol becomes much more reactive and can initiate redox cycling with ferric iron [40]. Although oxidant species generated in this reaction might cause opening of calcium channels, the effect of GSH was not mimicked by the more reactive L-cysteine or by direct treatment with H2O2. It was also not blocked by catalase or extracellular iron chelators, which is further evidence against a redox cycling medium. Although the mechanism of the GSH effect remains to be determined, the observation does confirm the findings of Davis, et al. [16] that influx of calcium causes efflux of ascorbate from endothelial cells. As also noted by Davis, et al. [16], endothelial cell release of ascorbate could be beneficial in atherosclerosis and other inflammatory vascular conditions in which nitric oxide release and function is impaired, since ascorbate could preserve nitric oxide by scavenging extracellular superoxide.
In conclusion, in this work we found that ascorbate-loaded EA.hy926 endothelial cells lost significant amounts of ascorbate through a non-saturable mechanism that was partially counteracted by re-uptake of the vitamin on the ascorbate transporter. In addition, increased flux of calcium into the cells due to the calcium ionophore A23187 or to GSH markedly augmented rates of ascorbate loss from the cell, although probably by a different mechanism. It is possible that ascorbate efflux relates to trans-cellular movement of ascorbate across the endothelium from the vessel lumen into the interstitium, but it is clear that the primary role for the SVCT2 is to retain ascorbate in the cells.
Acknowledgments
This work was supported by NIH grant DK 50435 and by the Vanderbilt Diabetes Research and Training Center (DK 20593).
Abbreviations
- DHA
dehydroascorbic acid
- DIDS
4,4′-diisothiocyanatostilbene-2,2′-disulfonic acid
- EDTA
ethylenediamine-tetraacetic acid
- GSH
reduced glutathione
- Hepes
N-2-hydroxyethylpiperazine-NN-2-ethanesulfonic acid
- KRH
Krebs-Ringer Hepes
References
- 1.Evans RM, Currie L, Campbell A. The distribution of ascorbic acid between various cellular components of blood, in normal individuals, and its relation to the plasma concentration. Br J Nutr. 1982;47:473–482. doi: 10.1079/bjn19820059. [DOI] [PubMed] [Google Scholar]
- 2.Brubacher D, Moser U, Jordan P. Vitamin C concentrations in plasma as a function of intake: a meta-analysis. Int J Vitam Nutr Res. 2000;70:226–237. doi: 10.1024/0300-9831.70.5.226. [DOI] [PubMed] [Google Scholar]
- 3.Leibovitz BE, Siegel BV. Aspects of free radical reactions in biological systems: aging. J Gerontol. 1980;35:45–56. doi: 10.1093/geronj/35.1.45. [DOI] [PubMed] [Google Scholar]
- 4.Sasaki R, Kurokawa T, Tero-Kubota S. Ascorbate radical and ascorbic acid level in human serum and age. J Gerontol. 1983;38:26–30. doi: 10.1093/geronj/38.1.26. [DOI] [PubMed] [Google Scholar]
- 5.Martin A, Frei B. Both intracellular and extracellular vitamin C inhibit atherogenic modification of LDL by human vascular endothelial cells. Arterioscler Thromb Vasc Biol. 1997;17:1583–1590. doi: 10.1161/01.atv.17.8.1583. [DOI] [PubMed] [Google Scholar]
- 6.May JM, Qu ZC. Transport and intracellular accumulation of vitamin C in endothelial cells: relevance to collagen synthesis. Arch Biochem Biophys. 2005;434:178–186. doi: 10.1016/j.abb.2004.10.023. [DOI] [PubMed] [Google Scholar]
- 7.Tsukaguchi H, Tokui T, Mackenzie B, et al. A family of mammalian Na+-dependent L-ascorbic acid transporters. Nature. 1999;399:70–75. doi: 10.1038/19986. [DOI] [PubMed] [Google Scholar]
- 8.Vera JC, Rivas CI, Fischbarg J, et al. Mammalian facilitative hexose transporters mediate the transport of dehydroascorbic acid. Nature. 1993;364:79–82. doi: 10.1038/364079a0. [DOI] [PubMed] [Google Scholar]
- 9.Christine L, Thomson G, Iggo B, et al. The reduction of dehydroascorbic acid by human erythrocytes. Clin Chim Acta. 1956;1:557–569. doi: 10.1016/0009-8981(56)90043-2. [DOI] [PubMed] [Google Scholar]
- 10.Hughes RE. Reduction of dehydroascorbic acid by animal tissues. Nature. 1964;203:1068–1069. doi: 10.1038/2031068a0. [DOI] [PubMed] [Google Scholar]
- 11.Mendiratta S, Qu Z-C, May JM. Erythrocyte ascorbate recycling: Antioxidant effects in blood. Free Radic Biol Med. 1998;24:789–797. doi: 10.1016/s0891-5849(97)00351-1. [DOI] [PubMed] [Google Scholar]
- 12.Dhariwal KR, Hartzell WO, Levine M. Ascorbic acid and dehydroascorbic acid measurements in human plasma and serum. Am J Clin Nutr. 1991;54:712–716. doi: 10.1093/ajcn/54.4.712. [DOI] [PubMed] [Google Scholar]
- 13.May JM, Qu ZC, Qiao H, et al. Maturational loss of the vitamin C transporter in erythrocytes. Biochem Biophys Res Commun. 2007;360:295–298. doi: 10.1016/j.bbrc.2007.06.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Upston JM, Karjalainen A, Bygrave FL, et al. Efflux of hepatic ascorbate: a potential contributor to the maintenance of plasma vitamin C. Biochem J. 1999;342:49–56. [PMC free article] [PubMed] [Google Scholar]
- 15.Miele M, Boutelle MG, Fillenz M. The physiologically induced release of ascorbate in rat brain is dependent on impulse traffic, calcium influx and glutamate uptake. Neuroscience. 1994;62:87–91. doi: 10.1016/0306-4522(94)90316-6. [DOI] [PubMed] [Google Scholar]
- 16.Davis KA, Samson SE, Best K, et al. Ca(2+)-mediated ascorbate release from coronary artery endothelial cells. Br J Pharmacol. 2006;147:131–139. doi: 10.1038/sj.bjp.0706492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.May JM, Qu Z-C, Whitesell RR. Ascorbic acid recycling enhances the antioxidant reserve of human erythrocytes. Biochemistry. 1995;34:12721–12728. doi: 10.1021/bi00039a031. [DOI] [PubMed] [Google Scholar]
- 18.Bauer J, Margolis M, Schreiner C, et al. In vitro model of angiogenesis using a human endothelium-derived permanent cell line: contributions of induced gene expression, G-proteins, and integrins. J Cell Physiol. 1992;153:437–449. doi: 10.1002/jcp.1041530302. [DOI] [PubMed] [Google Scholar]
- 19.Edgell CJ, McDonald CC, Graham JB. Permanent cell line expressing human factor VIII-related antigen established by hybridization. Proc Natl Acad Sci USA. 1983;80:3734–3737. doi: 10.1073/pnas.80.12.3734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pech-Amsellem MA, Myara I, Pico I, et al. Oxidative modifications of low-density lipoproteins (LDL) by the human endothelial cell line EA.hy 926. Experientia. 1996;52:234–238. doi: 10.1007/BF01920713. [DOI] [PubMed] [Google Scholar]
- 21.Jones W, Li X, Perriott LM, et al. Uptake, recycling, and antioxidant functions of α-lipoic acid in endothelial cells. Free Radic Biol Med. 2002;33:83–93. doi: 10.1016/s0891-5849(02)00862-6. [DOI] [PubMed] [Google Scholar]
- 22.Hissin PJ, Hilf R. A fluorometric method for determination of oxidized and reduced glutathione in tissues. Anal Biochem. 1976;74:214–226. doi: 10.1016/0003-2697(76)90326-2. [DOI] [PubMed] [Google Scholar]
- 23.May JM, Qu Z-C, Mendiratta S. Protection and recycling of α-tocopherol in human erythrocytes by intracellular ascorbic acid. Arch Biochem Biophys. 1998;349:281–289. doi: 10.1006/abbi.1997.0473. [DOI] [PubMed] [Google Scholar]
- 24.Deutsch JC, Santhosh-Kumar CR. Dehydroascorbic acid undergoes hydrolysis on solubilization which can be reversed with mercaptoethanol. J Chromatogr A. 1996;724:271–278. [Google Scholar]
- 25.Koshiishi I, Mamura Y, Liu J, et al. Degradation of dehydroascorbate to 2,3-diketogulonate in blood circulation. Biochim Biophys Acta. 1998;1425:209–214. doi: 10.1016/s0304-4165(98)00073-7. [DOI] [PubMed] [Google Scholar]
- 26.May JM, Qu Z-C, Li X. Ascorbic acid blunts oxidant stress due to menadione in endothelial cells. Arch Biochem Biophys. 2003;411:136–144. doi: 10.1016/s0003-9861(02)00715-4. [DOI] [PubMed] [Google Scholar]
- 27.Székely M, Mányai S, Straub FB. Über den Mechanismus der osmotischen Hämolyse. Acta Physiol Acad Sci Hung. 1952;3:571–583. [PubMed] [Google Scholar]
- 28.Best KA, Holmes ME, Samson SE, et al. Ascorbate uptake in pig coronary artery endothelial cells. Mol Cell Biochem. 2005;271:43–49. doi: 10.1007/s11010-005-3442-0. [DOI] [PubMed] [Google Scholar]
- 29.Hornig D, Weber F, Wiss O. Uptake and release of [I-14C]ascorbic acid and [I- 14C]dehydroascorbic acid by erythrocytes of guinea pigs. Clin Chim Acta. 1971;31:25–35. doi: 10.1016/0009-8981(71)90358-5. [DOI] [PubMed] [Google Scholar]
- 30.Maulen NP, Henriquez EA, Kempe S, et al. Upregulation and polarized expression of the sodium-ascorbic acid transporter SVCT1 in post-confluent differentiated CaCo-2 cells. J Biol Chem. 2003;278:9035–9041. doi: 10.1074/jbc.M205119200. [DOI] [PubMed] [Google Scholar]
- 31.Agus DB, Gambhir SS, Pardridge WM, et al. Vitamin C crosses the blood-brain barrier in the oxidized form through the glucose transporters. J Clin Invest. 1997;100:2842–2848. doi: 10.1172/JCI119832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huang J, Agus DB, Winfree CJ, et al. Dehydroascorbic acid, a blood-brain barrier transportable form of vitamin C, mediates potent cerebroprotection in experimental stroke. Proc Natl Acad Sci USA. 2001;98:11720–11724. doi: 10.1073/pnas.171325998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.García ML, Salazar K, Millán C, et al. Sodium vitamin C cotransporter SVCT2 is expressed in hypothalamic glial cells. Glia. 2005;50:32–47. doi: 10.1002/glia.20133. [DOI] [PubMed] [Google Scholar]
- 34.Qiao H, May JM. Development of ascorbate transport in brain capillary endothelial cells in culture. Brain Res. 2008;1208:79–86. doi: 10.1016/j.brainres.2008.02.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bielski BH, Allen AO, Schwarz HA. Mechanism of disproportionation of ascorbate radicals. J Am Chem Soc. 1981;103:3516–3518. [Google Scholar]
- 36.Daskalopoulos R, Korcok J, Tao L, et al. Accumulation of intracellular ascorbate from dehydroascorbic acid by astrocytes is decreased after oxidative stress and restored by propofol. Glia. 2002;39:124–132. doi: 10.1002/glia.10099. [DOI] [PubMed] [Google Scholar]
- 37.Siushansian R, Tao L, Dixon SJ, et al. Cerebral astrocytes transport ascorbic acid and dehydroascorbic acid through distinct mechanisms regulated by cyclic AMP. J Neurochem. 1997;68:2378–2385. doi: 10.1046/j.1471-4159.1997.68062378.x. [DOI] [PubMed] [Google Scholar]
- 38.Arrigoni O, De Tullio MC. Ascorbic acid: much more than just an antioxidant. Biochim Biophys Acta Gen Subj. 2002;1569:1–9. doi: 10.1016/s0304-4165(01)00235-5. [DOI] [PubMed] [Google Scholar]
- 39.Chatterjee IB, Chatterjee GC, Ghosh NC, et al. Biological synthesis of L-ascorbic acid in animal tissues: conversion of L-gulonolactone into L-ascorbic acid. Biochem J. 1960;74:193–203. doi: 10.1042/bj0740193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Paolicchi A, Dominici S, Pieri L, et al. Glutathione catabolism as a signaling mechanism. Biochem Pharmacol. 2002;64:1027–1035. doi: 10.1016/s0006-2952(02)01173-5. [DOI] [PubMed] [Google Scholar]