

Abstract
Pluronic F-68, an 80% hydrophilic member of the Pluronic family of polyethylene-polypropylene-polyethylene tri-block copolymers, protects non-neuronal cells from traumatic injuries and rescues hippocampal neurons from excitotoxic and oxidative insults. F-68 interacts directly with lipid membranes and restores membrane function after direct membrane damage. Here, we demonstrate the efficacy of Pluronic F-68 in rescuing rat hippocampal neurons from apoptosis after oxygen–glucose deprivation (OGD). OGD progressively decreased neuronal survival over 48 h in a severity-dependent manner, the majority of cell death occurring after 12 h after OGD. Administration of F-68 for 48 h after OGD rescued neurons from death in a dose-dependent manner. At its optimal concentration (30 μm), F-68 rescued all neurons that would have died after the first hour after OGD. This level of rescue persisted when F-68 administration was delayed 12 h after OGD. F-68 did not alter electrophysiological parameters controlling excitability, NMDA receptor-activated currents, or NMDA-induced increases in cytosolic calcium concentrations. However, F-68 treatment prevented phosphatidylserine externalization, caspase activation, loss of mitochondrial membrane potential, and BAX translocation to mitochondria, indicating that F-68 alters apoptotic mechanisms early in the intrinsic pathway of apoptosis. The profound neuronal rescue provided by F-68 after OGD and the high level of efficacy with delayed administration indicate that Pluronic copolymers may provide a novel, membrane-targeted approach to rescuing neurons after brain ischemia. The ability of membrane-active agents to block apoptosis suggests that membranes or their lipid components play prominent roles in injury-induced apoptosis.
Introduction
Despite detailed understanding of mechanisms mediating neuronal death after focal and global brain hypoxia–ischemia (HI), clinical treatments to reduce HI-induced brain injury remain limited to thrombolysis (Albers et al., 2011) and brain hypothermia (Hypothermia after Arrest Study Group, 2002; Jacobs et al., 2007). In addition to the ionic, oxidative, metabolic, gene regulatory, and inflammatory mechanisms that contribute to neuronal death after HI (Zhu et al., 2004; Allen et al., 2005; Wang et al., 2008; Tu et al., 2010; Adelson et al., 2012; Niatsetskaya et al., 2012), increasing evidence indicates that damage to neuronal membranes and changes in membrane function play important roles. HI decreases membrane integrity by damaging phospholipids (Shanta et al., 2012) and alters lipid metabolism, producing apoptosis-inducing lipids (Yu et al., 2000; Soeda et al., 2004). Thus, in mitochondria, production of sphingomyelin metabolites in the outer membrane plays key roles in outer membrane permeabilization during apoptosis (Kanno and Nishizaki, 2011; Chipuk et al., 2012). Similarly, peroxidation of cardiolipin in the inner mitochondrial membrane (Kagan et al., 2005) and its subsequent redistribution to the outer membrane are required for mitochondrial release of proapoptotic factors (Ji et al., 2012). In the plasma membrane, HI-induced blebs (Tanaka et al., 1999) undergo phosphatidylserine externalization and exhibit increased permeability to normally impermeant solutes (Kelly et al., 2009), decreasing membrane integrity and leading to necrosis (Marks et al., 2000). Thus, the multiple mechanisms by which HI-induced membrane damage mediates cell death suggest that repairing membrane damage may be a useful approach to decreasing neuronal death after HI.
Membrane damage has been repaired after mechanical injury to neurons, alveolae, and myocytes using synthetic surfactants (Lee et al., 1992; Serbest et al., 2006; Plataki et al., 2011). These molecules, termed Pluronics, are amphiphilic tri-block copolymers of poly[ethylene oxide] (PEO) and poly[propylene oxide] (PPO) in a PEOm–PPOn–PEOm configuration, in which m and n denote the number of monomers in a block. The ratio of the number of hydrophilic PEO monomers to the number of lipophilic PPO monomers determines the hydrophilic/lipophilic balance (HLB) of the copolymer. The HLB determines how the copolymer interacts with membranes (Wang et al., 2012). Pluronics having lower HLBs (e.g., P85, HLB = 0.50) cross membranes and can transport drugs or DNA across plasma membranes (Batrakova et al., 2003; Yang et al., 2008). Pluronics with higher HLBs (e.g., F-68, HLB = 0.80) insert into lipid bilayers (Firestone et al., 2003) and can restore the integrity of damaged membranes (Lee et al., 1992; Marks et al., 2001).
Administration of Pluronic F-68 (F-68) (PEO76–PPO29–PEO76) improves the function of multiple cell types having damaged or inherently leaky membranes (Serbest et al., 2006; Yasuda et al., 2005; Kilinc et al., 2009; Townsend et al., 2010; Plataki et al., 2011). F-68 also profoundly rescues cultured hippocampal neurons after excitotoxicity and oxidative stress (Marks et al., 2001), central mechanisms of HI-induced neurodegeneration. Here, we describe the efficacy of F-68 in rescuing cultured hippocampal neurons from death after oxygen–glucose deprivation (OGD), a widely used in vitro model of HI brain injury, and determined mechanisms of its action.
Materials and Methods
Materials.
Polymer F-68 was from BASF. DRAQ5 and Hoechst stains were obtained from Axxora. Red SR FLICA Caspase3/7 assay kits were from Immunochemistry Technologies. Anti-cytochrome c (clone 6H2.B4) was from BD Biosciences Pharmingen, anti-BAX (clone Δ21) was from Santa Cruz Biotechnology, and anti-GAPDH was from Abcam.
Cell culture media and supplements, vital dyes, Annexin V fluorescent conjugates, anti-cytochrome oxidase subunit IV (clone 20E8C12), and fluorescence-tagged secondary antibodies were from Invitrogen. All other chemicals were from Sigma.
Hippocampal neuronal cultures.
Hippocampal neurons were prepared as described previously (Plant et al., 2011) from embryonic day 18 Sprague Dawley rat fetuses of either sex, plated onto 15 mm coverslips, and maintained at 37°C in a humidified incubator in which a 5% CO2, 21% O2 environment was maintained. For Western blot studies, 60 mm dishes containing 1 × 106 neurons were used. Neurons were studied between 11 and 15 d in vitro.
OGD.
OGD was induced in a hypoxia workstation (Coy Laboratory Products), in which a humidified, 37°C environment of 1% O2 and 5% CO2, balance N2, was maintained. OGD was induced by immersing neurons on coverslips into Petri dishes containing a glucose-free, bicarbonate-buffered saline composed of (in mm) 95 NaCl, 5.3 KCl, 1.3 NaH2PO4, 1.3 MgSO4, 24 NaHCO3, 25 sucrose, and 2.4 CaCl2 in which the O2 and CO2 tensions and temperature had been equilibrated with those in the hypoxia workstation for 18 h before study. To ensure that control neurons not exposed to OGD were treated identically in all other ways, neurons were immersed in Petri dishes containing the same saline, with equimolar replacement of glucose for sucrose, that had been placed 18 h previously in the 21% O2, 5% CO2 incubator. After OGD or control saline exposure, coverslips were placed back into Petri dishes containing the culture media from which they had been removed and placed back into the incubator.
F-68 administration.
Final concentrations of F-68 were administered by diluting a stock solution of F-68 (10 mm in water) in culture media. Cultures subjected to OGD but not receiving F-68 were provided with the same amount of culture media alone. Cells were incubated in F-68 or media control immediately after or at various times after OGD.
High-content imaging.
Multichannel fluorescence images of neurons stained with various indicators (see Results) were obtained using coverslips with neurons placed in a 24-well plate containing a HEPES-buffered saline composed of (in mm) 110 NaCl, 5.3 KCl, 1.3 NaH2PO4, 2.4 CaCl2, 1 MgCl2, 20 HEPES, 25 glucose, and 1 succinate, and a computer-controlled, high-content image acquisition system (Image Xpress Micro; Molecular Devices), equipped with a plate holder, 300 W xenon illumination, microscope objectives, a cooled CCD camera, an automated xyz stage, autofocus, and a five-position filter cube changer containing matched bandpass emission and excitation filters and dichroic mirrors optimized separately for DAPI, calcein, tetramethylrhodamine methyl ester (TMRM), and Alexa Fluor 647. To ensure unbiased selection of fields on coverslips, 42 equally distributed fields were obtained to sample from the entire 15 mm coverslip (Fig. 1B). Single-wavelength images of labeled neurons were thresholded to allow cell-by-cell segmentation according to the structure identified by the dye, e.g., nucleus from DRAQ5, cytosol from calcein) using MetaXpress software (Molecular Devices). Cells were then analyzed according to user-set criteria to provide assignment of each cell to a category (e.g., live/dead). For other studies, mean intensities with segmentation-defined regions within each cell were measured.
Figure 1.
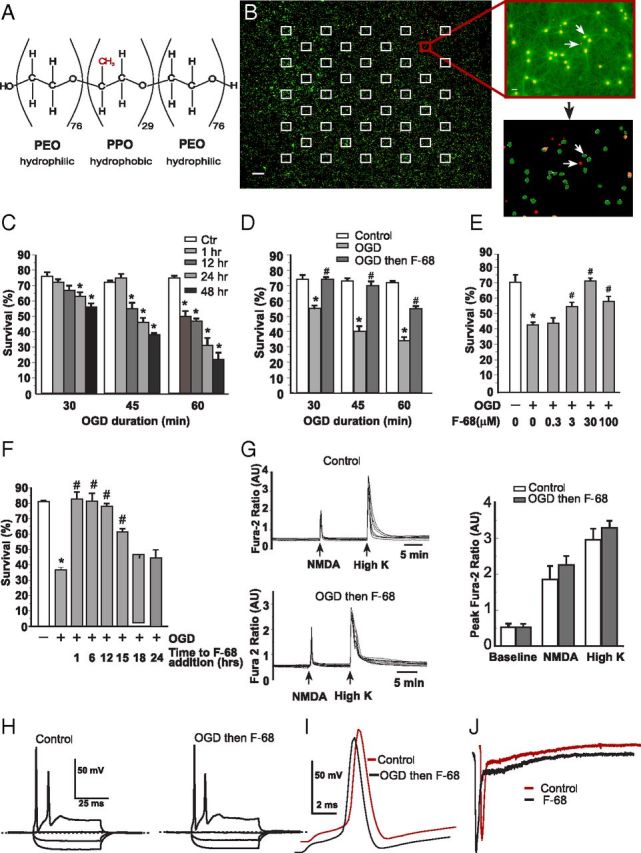
F-68 rescues hippocampal neurons from OGD insult. A, Structure of the tri-block copolymer F-68. B, Left, Pseudocolored photomontage of contiguous 20× fields obtained by high-content imaging of cultured hippocampal neurons over an entire 15-mm-diameter coverslip. Images are overlays of calcein (green) and DRAQ5 (red) images. Squares indicate the location of the 42 fields on the coverslip used for automated cell counting. Scale bar, 500 μm. Top right, Overlaid calcein and DRAQ5 images from the indicated site in the montage. Scale bar, 10 μm. Bottom right, Result of automated image segmentation performed on the image above. Arrows show identified dead (red) and living (green) cells. C–F, Each experimental unit (n) consists of six coverslips per condition, with high-throughput imaging counting ∼1300 cells per coverslip. * indicates significantly different from control (Ctrl); # indicates significantly different from OGD. C, Changes in mean ± SD neuronal survival over time after 30, 45, and 60 min of OGD. n = 3 per condition. D, Changes in mean ± SD percentage neuronal survival induced by F-68 treatment as a function of OGD duration, measured 48 h after OGD (n = 3 per condition per OGD duration). E, Concentration dependence of F-68-induced rescue of neurons after 45 min OGD, measured 48 h later (n = 3 per condition). F, Changes in mean ± SD percentage neuronal survival with increasing delay in F-68 addition after OGD, measured 48 h after OGD (n = 3 per condition). Neuronal rescue from OGD persists when F-68 addition is delayed as much as 12–15 h after OGD. G, Raw, time-dependent [Ca2+]i changes in response to NMDA or potassium-induced membrane depolarization in control neurons (top left) and in neurons 5 d after rescue from 45 min OGD with F-68 (30 μm; bottom right). Right, Mean ± SD peak fura-2 ratios obtained in each condition (n = 3, each study of at least 5 neurons). H, Voltage responses to current injection are similar between control neurons and neurons, 5 d after rescue from OGD-induced death with F-68 (30 μm). I, Action potential morphology is identical between control (orange) neurons and neurons 5 d after rescue from OGD with F-68 (30 μm; black). Traces are offset for clarity. J, Representative NMDA-activated currents in hippocampal neurons studied before (orange) and after 3 min exposure to F-68 (30 μm; black). Mean ± SD current densities were −24.3 ± 2 pA/pF before and −21.4 ± 2 pA/pF after exposure of nine cells to F-68 (30 μm). Mean ± SD times to recovery to half-maximal amplitude were 246 ± 9.5 and 246 ± 8 ms, respectively.
Assessment of neuronal survival.
Cells were incubated in calcein-AM to label living cells and DRAQ5 to identify all nuclei. To restrict our analysis to cells that were alive at the beginning of the experiment, coverslips containing neurons were incubated in membrane-impermeant DNase (900 U/ml) for 1 h to remove exposed nuclei from the coverslip. Images were obtained using high-throughput imaging of 20× fields as described above. Cells positive for calcein and DRAQ5 were classified as living, whereas DRAQ5-positive nuclei without surrounding calcein fluorescence were classified as dead.
Electrophysiology.
Whole-cell patch clamp was performed as described previously (Plant et al., 2011) using an Axopatch 200B amplifier and pClamp software (Molecular Devices) at filter and sampling frequencies of 5 and 25 kHz, respectively, for voltage-clamp experiments and 1 and 10 kHz, respectively, for current-clamp recording. Current-clamp recordings were performed in a bath solution containing the following (in mm): 1 CaCl2, 1 MgCl2, 4 KCl, 140 NaCl, 5 glucose, and 10 HEPES. The pH was adjusted to 7.4 with NaOH. Electrodes were fabricated from borosilicate glass (Clark), were coated with Sigmacote (Sigma) before use, and had a resistance of 4–5 MΩ when filled with a solution containing the following (in mm): 136 KCl, 1 MgCl2, 2 K2ATP, 5 EGTA, and 10 HEPES, adjusted to a pH of 7.2 with KOH. Action potentials were studied in current-clamp mode. Neurons were pulsed with current from −0.2 to 0.5 nA in 0.05 nA intervals every 5 s. The mean voltage (measured at a stable point and not during an action potential or afterhyperpolarization) was plotted against current, and the somatic input resistance was calculated by fitting each set of data with a straight line according to the equation f(x) = mx + b, where m is the gradient, or resistance, in megaohms. All electrophysiology parameters were determined in pClamp and Excel software and are quoted as mean values ± SEM.
NMDA-activated currents were studied in voltage-clamp mode by application of 10 μm NMDA to cells held at −50 mV. Currents were studied in a bath solution containing (in mm) 150 NaCl, 3 KCl, 1 CaCl2, and 10 HEPES adjusted to pH 7.4 with NaOH and a pipette solution containing (in mm) 80 CsF, 60 CsCl, 10 EGTA, 1 MgCl2, and 10 HEPES adjusted to a pH of 7.4 with CsOH. NMDA and F-68 were applied with a computer-controlled MPS-2 multichannel perfusion system (WPI), and experiments were performed at 32°C using a feedback-controlled heated perfusion system (Warner Instruments)
Assessment of apoptotic death.
To restrict our analysis to cells that were alive at the beginning of the experiment, coverslips containing neurons were incubated in membrane-impermeant DNase (900 U/ml) for 1 h to remove exposed nuclei from the coverslip. Apoptotic neuronal death was assayed on neurons fixed with 4% paraformaldehyde, using terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL) staining, according to the protocol of the manufacturer (Roche). All nuclei were identified with Hoechst labeling. Images were obtained using fluorophore-appropriate filters and mirror via high-throughput imaging of 20× fields as described above. Cells positive for TUNEL and Hoechst were counted as apoptotic, whereas Hoechst-positive nuclei without TUNEL staining were counted as non-apoptotic.
Assessment of Annexin V binding.
Living neurons were incubated in Alexa Fluor 647-labeled Annexin V (5 μg/ml) and calcein-AM. Images were acquired with a 63× oil-immersion objective using a laser-scanning confocal microscope (Leica) using the high-content imaging Matrix system. Sequential images of Alexa Fluor 647 and calcein were obtained using laser lines at 630 and 488 nm. Studies were restricted to living neurons defined by calcein labeling. Cells with Annexin V plasma membrane staining were counted and reported as percentage living cells.
Assessment of caspase activity.
To restrict our analysis to cells that were alive at the beginning of the experiment, coverslips containing neurons were incubated in membrane-impermeant DNase (900 U/ml) for 1 h to remove exposed nuclei from the coverslip. The presence of activated caspases was assayed on a cell-by-cell basis using Red SR FLICA Caspase3/7 Assay Kit (Immunochemistry Technologies) according to the protocol of the manufacturer. Cells on coverslips were incubated in cell-permeant SR-DEVD-FMK (sulforhodamine-labeled benzyloxycarbonyl-Asp-Glu-Val-fluoromethyl ketone), which irreversibly binds to activated caspases. Cells were thoroughly washed to remove unbound SR-DEVD-FMK and were stained with calcein-AM (to stain live cells) and Hoechst (to stain all cells). Separate fluorophore-specific images of each 20× field were obtained.
Measurement of mitochondrial membrane potential.
Mitochondrial membrane potential (ΔΨm) was measured using the cationic fluorophore TMRM in unquenched mode as described previously (Marks et al., 2005). TMRM is accumulated in the cell and mitochondria as a function of potential. To allow electrochemical equilibration of TMRM across the plasma and inner mitochondrial membranes, coverslips that contained cells that were placed into wells of a 24-well plate were incubated in TMRM-containing (10 nm) HEPES-buffered saline for 1 h at 37°C, after which calcein-AM and Hoechst were added. During high-throughput imaging, cells were maintained at 37°C by means of a thermostatically controlled plate heater. Imaging of eight 20× fields per coverslip of TMRM, calcein, and Hoechst fluorescence were obtained using fluorophore-specific filter cubes. After acquisition of all coverslips in the plate, ΔΨm was dissipated with carbonyl cyanide-4-(trifluoromethoxy) phenyl-hydrazone (FCCP; 1 μm), and the identical fields were reimaged. Using image segmentation, the soma of each cell was identified with calcein fluorescence and the nucleus of each cell with Hoechst. Mean TMRM intensity in the extranuclear region of each cell was obtained.
Calcium imaging.
Changes in free cytosolic calcium levels ([Ca2+]i) was measured using fura-2 (Invitrogen) as described previously. Neurons were loaded with 1 μm fura-2 AM diluted in culture medium for 30 min at 37°C. After loading, cultures were washed with bicarbonate-buffered saline for 30 min to ensure complete hydrolysis of the AM ester from the dye. Fura-2 dye was sequentially excited at 340 and 380 nm every 20 s by narrow bandpass filters, and the emitted light was imaged in a band centered around 535 nm. Calcium responses of individual neurons to NMDA (300 μm) and KCl (60 mm) were quantified as mean ratio (340 nm/380 nm) on a cell-by-cell basis.
Immunocytochemistry.
For cytochrome c immunocytochemistry, neurons were incubated in 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (40 mg/ml; Tymianski et al., 1997) for 90 min to preserve mitochondrial morphology, followed by paraformaldehyde (4%) for 20 min. For BAX studies, neurons were fixed in paraformaldehyde only. Cells were permeabilized with Triton X-100 (0.2%) and nonspecific binding antibody binding blocked with serum of the species in which the secondary antibodies were raised. Cells were incubated with anti-cytochrome c (5 μg/ml) or anti-BAX (4 μg/ml) for 1 h at room temperature. Cytochrome c immunoreactivity was detected with Alexa Fluor 488-labeled goat anti-mouse IgG. BAX immunoreactivity was detected with Alexa Fluor 594-labeled goat anti-rabbit IgG. Nuclei were counterstained with DRAQ5, and coverslips were mounted in SlowFade.
Quantification of cytochrome c release.
Images were acquired with a laser-scanning confocal microscope (SP5; Leica) via a 100×, 1.45 numerical aperture (NA) oil-immersion objective, with identical illumination acquisition settings between treatment groups. Studies of cytochrome c release at 12 h were done on a different SP5 with identical acquisition settings among the three groups. Three confocal images (500 μm thick) that were contiguous in the z-axis of each neuronal field were acquired so that the volume encompassed the nucleus. Using NIH ImageJ, a region of interest corresponding to the nuclear area was obtained by thresholding the DRAQ5 image, and the mean intensity with that region in the corresponding cytochrome c image was obtained. Mean intensities for each cell were summed across the three images.
Assessment of mitochondrial colocalization of BAX.
Neurons were infected with an adenovirus harboring the sequence of a GFP targeted to the mitochondrial matrix. Neurons were studied 24 h later. Cells were then processed for Alexa Fluor 594-identified BAX immunoreactivity. Images were acquired with a 100×, 1.45 NA oil-immersion objective on a laser-scanning confocal microscope with identical illumination and acquisition settings across fields and conditions. Sequential images of GFP-labeled mitochondria and Alexa Fluor 594-labeled BAX immunoreactivity were obtained and deconvolved to reduce out-of-focus light (Huygens; Scientific Volume Image). Colocalization of BAX with mitochondria was assessed using NIH ImageJ to produce Manders coefficients of colocalization (Manders et al., 1993).
Subcellular fractionation.
Subcellular fractionation was performed according to published methods (Poppe et al., 2001). Sixty-millimeter Petri dishes, each containing 1 × 106 cells, were scraped into 100 μl of isolation buffer containing 50 mm KCl, 70 mm sucrose, 2 mm K2HPO4, 20 mm HEPES, and 1 mm EGTA with the protease inhibitors 1 mm PMSF, 1 μg/ml antipain, 10 μg/ml aprotinin, and 1 μg/ml leupeptin, pH 7.2, and maintained at 4°C for the remainder of the fractionation process. The contents of four dishes per condition (∼4 × 106 cells) were collected into a precooled steel chamber and subjected to nitrogen cavitation (1500 psi for 30 min). The resultant cell lysate was centrifuged at 1060 × g for 10 min. The supernatant was collected and reserved. The pellet was resuspended in isolation buffer and centrifuged again at 1060 × g for 5 min. The two supernatants were pooled and centrifuged at 14,600 × g for 10 min. This supernatant was saved as the cytosolic fraction. The pellet was resuspended and centrifuged three times at 14,600 × g for 10 min to obtain a heavy membrane fraction enriched with mitochondria. Protein was isolated in 50 μl RIPA buffer (Sigma) containing protease inhibitors at the same concentration as the isolation buffer. The protein concentration was measured using the BCA protein assay kit (Pierce) with BSA as the standard.
Western blot analyses.
Equal amounts of protein (20 μg) for each sample were separated using the Any Kd Mini-Protean TGX System (Bio-Rad) at 100 V. Proteins were transferred to nitrocellulose membranes, blocked with 3% BSA in Tris-buffered saline with 0.5% Tween 20, and incubated overnight in anti-BAX (2 mg/ml in blocking buffer). The secondary antibody (0.01 μg/ml) was incubated at room temperature for 1 h before standard enhanced chemiluminescence detection. Blots were stripped with NaOH and reprobed with either anti-cytochrome oxidase subunit IV (2 μg/ml) or anti-GAPDH antibody (2 μg/ml). Protein band intensities were measured as optical density.
Statistical analyses.
For normally distributed data, statistical analysis was performed by one-way ANOVA, followed by Tukey's test for testing of individual means. Values of p < 0.05 were accepted as significant. For analysis of TMRM intensity distributions, the Kolmogorov–Smirnov test was used to obtain D statistics (Lehman and D'Abrera, 2006). To analyze the effects of treatment and time on the total number of neurons counted per coverslip, two-way ANOVA was used, followed by Tukey's test. Statistical tests were performed using GraphPad Prism version 6.01 for Windows (GraphPad Software).
Results
F-68 rescues hippocampal neurons from OGD
F-68 (molecular weight, 8600 Da) is made up of two side chains of 76 PEO units each and a central PPO chain of 29 units (Fig. 1A). Because F-68 rescues neurons from mechanisms underlying HI brain injury, we sought to understand whether F-68 rescues neurons from HI injury itself. Therefore, we used OGD in cultured hippocampal neurons, a well-accepted in vitro model of hypoxic–ischemic brain injury. First, we defined the time course of OGD-induced neuronal death and assessed how the severity of OGD affected neuronal survival. Neurons were exposed to 30, 45, or 60 min OGD or to otherwise-identical, control solutions containing glucose at ambient oxygen tension. After OGD, coverslips were returned to the normoxic incubator in their original, glucose-containing media, and unbiased counts of living and dead neurons were made with high-content imaging at various times after OGD, counting ∼1300 total neurons per coverslip (n = 6 coverslips per condition; Fig. 1B). Forty-eight hours after exposure to control solutions, mean ± SEM neuronal survival was 75 ± 3.4%. OGD significantly decreased neuronal survival at 48 h in a severity-dependent manner, such that 30, 45, and 60 min OGD resulted in ∼55.3 ± 2.6, 40 ± 2.3, and 22.5 ± 3.3% survival, respectively (Fig. 1C). With increasing OGD severity, the rapidity of neuronal death also progressively increased. After 30 min OGD, neuronal survival did not significantly decrease until 24 h after insult. After 45 min OGD, neuronal survival was significantly decreased by 12 h after insult and subsequently decreased more rapidly over time. Notably, after 60 min OGD, neuronal survival was already decreased to 50.6 ± 2.8% by 1 h after insult, followed by a progressive decline over 48 h (Fig. 1C). This early death was not observed after shorter OGD exposures.
Next, we assessed how effectively F-68 treatment rescued neurons from OGD-induced death, by adding F-68 (30 μm) to the media of half of the culture dishes after OGD. Thus, neurons were not exposed to F-68 until 15 min after OGD. Neurons remained in F-68 until assessments of survival at 48 h. After 30 and 45 min OGD, F-68 treatment restored neuronal survival to levels observed after exposure to control buffer, completely preventing all OGD-induced neuronal death (Fig. 1D). After 60 min OGD, F-68 increased survival to 54.8 ± 2.4%, the similar degree of survival seen at 1 h after OGD alone. Thus, F-68 treatment provided profound neuronal rescue after 30 and 45 min OGD and rescued half of the neurons that would have died after 60 min OGD.
The efficacy of F-68-induced neuronal rescue after OGD depended on F-68 concentration: after 45 min OGD, survival progressively increased with F-68 concentration, with maximum survival observed at 30 μm (Fig. 1E). To determine whether F-68 was effective in rescuing neurons from OGD when treatment was delayed after insult, F-68 was added to the culture medium at 1, 6, 12, 15, and 18 h after 45 min OGD. Application of F-68 6 h after insult resulted in complete neuronal rescue (Fig. 1F). Treatment begun at 12 h after OGD was only marginally less effective, whereas progressively later treatment produced less effective rescue. F-68 addition 18 h after OGD provided no neuronal rescue.
Our measure of living neurons was the presence of calcein fluorescence within the soma, which, although commonly used, indicates that cells have intact plasma membranes and functional intracellular esterases. Accordingly, we assessed whether neurons rescued from OGD by F-68 exhibited appropriate changes in intracellular Ca2+ levels induced by membrane depolarization and glutamate receptor activation using time-lapse imaging (Fig 1G, top, bottom). At baseline, mean resting fura-2 ratios did not differ between control neurons and those subjected to OGD and subsequently rescued by F-68 (n = 15–20 neurons on each of 4 coverslips; Fig. 1G, right). Brief (20 s) applications of either NMDA (100 μm) or KCl (60 mm) increased peak fura-2 ratios similarly in control and rescued groups (Fig 1G). Removal of each stimulus was followed by a similar, rapid return of Ca2+ to baseline levels. Accordingly, neurons rescued from OGD by F-68 have normally functioning neuron-specific responses and intact calcium homeostasis mechanisms.
We next asked whether neurons rescued from OGD by F-68 treatment demonstrated altered electrophysiological properties compared with control cells. Excitability was not different between control cells and those subjected to 45 min OGD, followed by F-68 (30 μm) treatment for 48 h. Neurons in both groups (n = 8 per group) had mean resting membrane potentials of −65 ± 1 mV. In addition, none of the key determinants of excitability—cellular capacitance, input resistance, or peak firing frequency—differed significantly between groups (Table 1). In contrast, electrophysiological measurements were not possible from cells exposed to OGD in the absence of F-68, reflecting reduced viability and plasma membrane integrity (data not shown). Action potentials were evoked by current injections of equivalent magnitude and were not different between groups (Fig. 1H,I; Table 1), indicating that ion channel function and cellular energetics were intact after neuronal rescue by F-68 after OGD.
Table 1.
Comparison of biophysical parameters between control neurons and neurons rescued by F-68 from OGD-induced death
| Unit | Control | OGD + F-68 | |
|---|---|---|---|
| Whole-cell parameters | |||
| Resting potential | mV | −65 ± 1.2 | −65 ± 0.7 |
| Capacitance | pF | 20 ± 2.4 | 21 ± 2.4 |
| Input resistance | MΩ | 87 ± 9 | 88 ± 7 |
| Peak firing frequency | Hz | 32.5 ± 4 | 30 ± 3.3 |
| Analysis of first action potential | |||
| Current injection | pA | 70 | 70 |
| Firing threshold | mV | −48 ± 9 | −48 ± 8 |
| Peak deflection | mV | 40 ± 2 | 40 ± 1.7 |
| After hyperpolarization | mV | −69 ± 3.7 | −67 ± 2.4 |
| Half total height | mV | 55 ± 2.6 | 54 ± 1.7 |
| Duration at half height | ms | 1.4 ± 0.04 | 1.4 ± 0.05 |
Values are means ± SEM; n = 8 neurons per condition.
NMDA receptor-mediated excitotoxicity is an important mechanism of neuronal death during and after OGD. Accordingly, we assessed whether F-68 altered NDMA receptor function. Acute exposure to F-68 did not alter NMDA-activated currents in hippocampal neurons: neither mean NMDA current densities (−24.3 ± 1.9 pA/pF for baseline; −21.4 ± 2 pA/pF for F-68) nor the time to half-recovery (246 ± 9.5 ms for baseline; 246 ± 8 ms for F-68) was altered after 3 min perfusion of 30 μm NMDA (n = 9 paired measurements; Fig. 1J). These data indicate that F-68 does not rescue neurons from OGD-induced death through alterations of NMDA receptor function.
F-68 blocks OGD-induced apoptosis
To understand the mechanisms by which F-68 rescues neurons from OGD-induced death, we first characterized the type of death after OGD. With the exception of the sharp decrease in neuronal survival 1 h after 60 min OGD, OGD-induced neuronal death was, in general, progressive over 48 h. Accordingly, we hypothesized that OGD-induced apoptosis was a primary mechanism of death. To exclude the contribution of early (1 h) death, we used 45 min OGD for these and all subsequent studies. Pan-caspase inhibition with Z-VAD-FMK (benzyloxycarbonyl-Val-Ala-Asp-(O-Me)-fluoromethylketone) (50 μm) during and after OGD completely blocked neuronal death (Fig. 2A), confirming that OGD-induced death in this model was by apoptosis.
Figure 2.
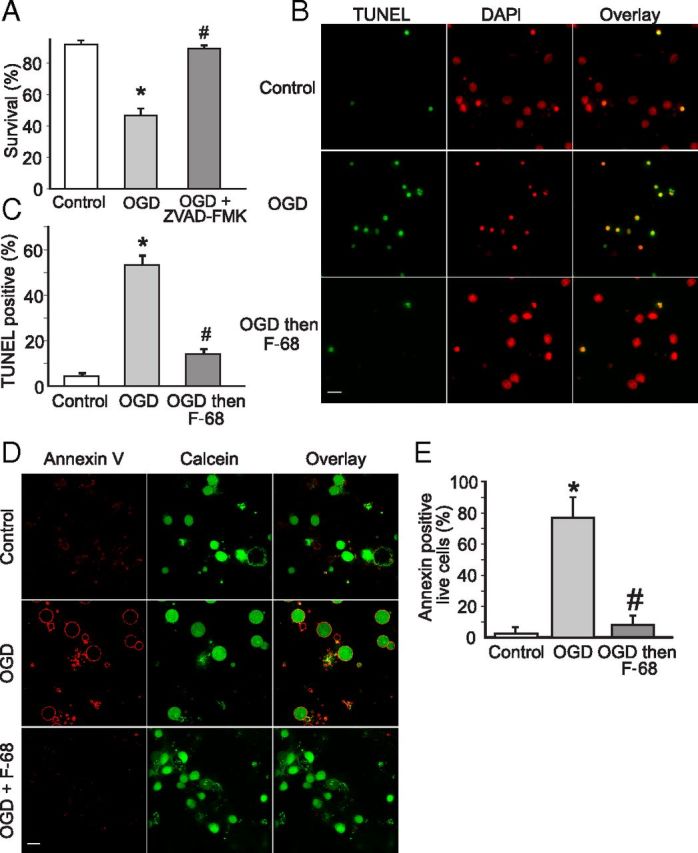
F-68 prevents OGD-induced apoptosis. A, C, Each experimental unit (n) consists of six coverslips per condition, with high-throughput imaging counting ∼1300 cells per coverslip. * indicates significantly different from control; # indicates significantly different from OGD. A, Mean ± SD percentage neuronal survival, 48 h after OGD (45 min) or control, the former in the presence and absence of Z-VAD-FMK (50 μm; n = 3 per condition). B, Photomontage of representative micrographs of neurons labeled with TUNEL (green) for apoptotic neurons and DAPI (red) for all cells. Images obtained 48 h after 45 min of exposure to control or OGD buffers with and without immediate treatment thereafter with F-68 (30 μm). Scale bar, 10 μm. C, Mean ± SD percentage of TUNEL-positive neurons, 48 h after exposure to control buffer, or 45 min OGD with and without immediate treatment thereafter with F-68 (30 μm; n = 3). D, Photomontage of Annexin V labeling (red) of viable neurons (green), obtained 6 h after 45 min exposure to control buffer or OGD buffers with and without immediate treatment thereafter with F-68 (30 μm). Scale bar, 10 μm. E, Mean ± SD percentage of Annexin V-positive neurons by condition (n = 5; for each study, 12 randomly selected fields of 5–10 neurons each, counted on each of 2 coverslips per condition).
To assess the extent to which F-68 treatment decreased apoptosis, we first identified neurons that were dead or dying of apoptosis using TUNEL and counted apoptotic and total cells using high-content imaging (n = 6 coverslips per condition, ∼1500 cells per coverslip). Forty-eight hours after OGD, the mean ± SEM number of TUNEL-positive neurons was markedly increased over control (OGD, 53.15 ± 4%; control, 4.5 ± 1.2%; p < 0.0001; Fig. 2B,C), completely accounting for the OGD-induced decreases in neuronal survival we observed. In contrast, F-68 treatment after OGD almost completely prevented the OGD-induced increase in TUNEL-positive neurons (OGD plus F-68, 14 ± 2.13%; p < 0.0001; Fig. 2B,C). To obtain additional evidence that OGD-induced apoptosis was inhibited by F-68, we assessed the extent of phosphatidylserine externalization on the plasma membrane, an early sign of apoptosis, using confocal microscopy to count fluorescently labeled Annexin V on the surface of calcein-loaded neurons. In control neurons, 6 h after 45 min exposure to control buffer, Annexin V labeling was minimal (1.44 ± 0.33%). In contrast, 6 h after 45 min OGD plasma membrane Annexin V binding was markedly and significantly increased (OGD, 96.81 ± 3%; p = 0.0009; see Fig. 6A,B). Finally, neurons treated with F-68 after OGD demonstrated significantly lower Annexin V staining (9.5 ± 1.7%; p = 0.0014). Together, these data indicate that F-68 treatment markedly decreases OGD-induced apoptosis.
Figure 6.
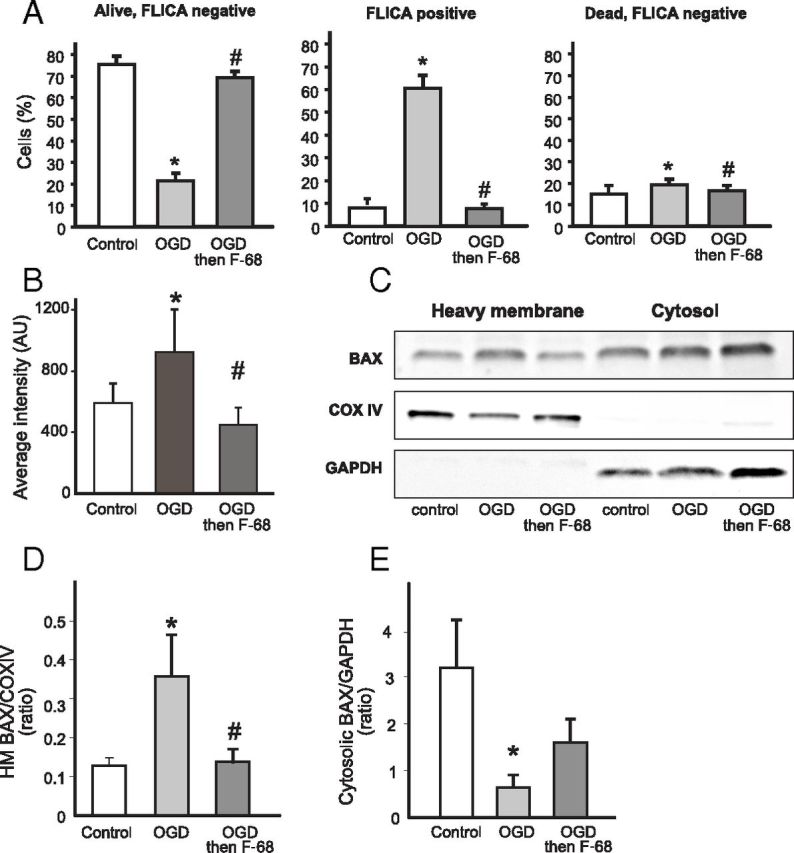
F-68-induced inhibition of apoptosis persists when F-68 treatment is delayed for 12 h after OGD. F-68 (30 μm) or media control were administered 12 h after OGD (45 min), and measurements were made 12 h after F-68 administration. A, Mean ± SD percentage of live/dead and FLICA-positive/negative neurons. Each experimental unit (n) consists of six coverslips per condition, with high-throughput imaging counting ∼1300 cells per coverslip (n = 3). B, Mean ± SD intensity of cytochrome c immunoreactivity over the nuclear volume; n = 6 coverslips per condition. C, Representative Western blots of BAX immunoreactivity in heavy membrane and cytosolic fractions of neurons treated with F-68. Blots were stripped and blotted with the mitochondrial marker COX IV and the cytosolic marker GAPDH to identify fractions and normalize intensities for protein loading. D, E, Quantification of BAX immunoreactivity in heavy membrane (D) and cytosolic (E) fractions. Data are expressed as means ± SD of ratios of BAX/COX IV or BAX/GAPDH (n = 3 experiments, each experiment using 4 × 106 cells per condition). * indicates significantly different from control; # indicates significantly different from OGD.
F-68 prevents OGD-induced caspase activation
Because F-68 blocked OGD-induced apoptosis, we assessed the extent to which it altered caspase activation. Using high-content imaging, we identified total and living cells using Hoechst and calcein. To identify neurons with activated caspases, we simultaneously used a red fluorescent probe that specifically binds to activated caspases (FLICA; ImmunoChemistry Technologies). Using these three markers, we classified cells as living or dead, and within each group, as FLICA-positive or -negative (Fig. 3A).
Figure 3.
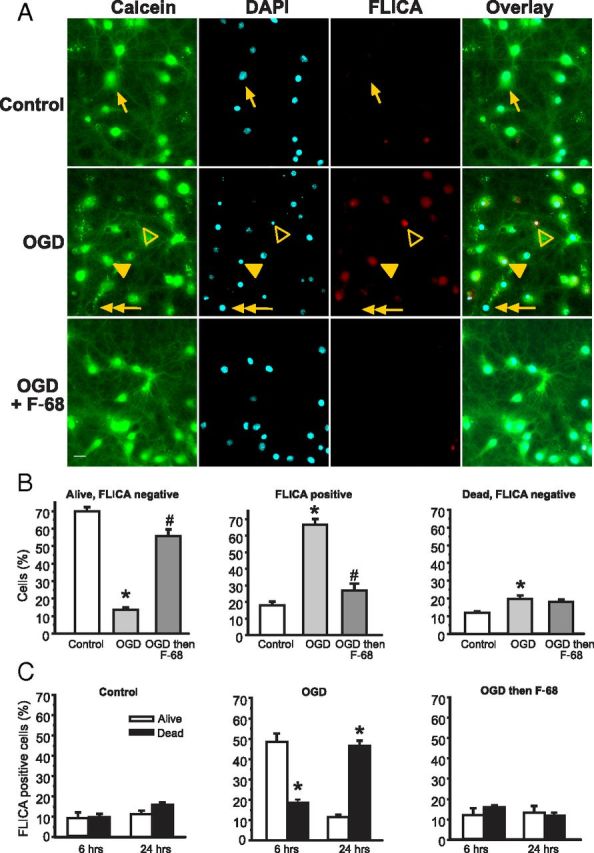
F-68 prevents OGD-induced caspase activation. A, Using calcein (green) to label living neurons, DAPI (cyan) to label all neurons, and FLICA (red) to label caspase-positive neurons allows identification of neurons as living/dead and caspase activation as positive/negative. Images obtained 6 h after 45 min exposure to control buffer or OGD, with and without immediate treatment thereafter with F-68 (30 μm). Scale bar, 10 μm. Arrow, Living neuron, FLICA negative; filled arrowhead, living neuron, FLICA positive; open arrowhead, dead neuron, FLICA positive; double arrowhead, dead neuron, FLICA negative. B, C, Each experimental unit (n) consists of six coverslips per condition, with high-throughput imaging counting ∼1300 cells per coverslip. * indicates significantly different from control; # indicates significantly different from OGD. B, Mean ± SD percentage of total cells at 6 h after OGD or control (n = 3). C, Mean ± SD percentage of FLICA-positive cells over time after OGD or control (n as in B). * indicates significantly different from the paired bar.
Six hours after OGD, the number of FLICA-negative living cells was markedly decreased compared with control (p < 0.0001; Fig. 3B, left). In contrast, in F-68-treated cultures, the percentage of these FLICA-negative, living neurons was greatly increased and not significantly different from control cultures (p < 0.0001; Fig. 3B, left). In like manner, the percentage of all FLICA-positive cells (living and dead) was significantly increased after OGD compared with control, and F-68 treatment markedly decreased the percentage of these FLICA-positive neurons (Fig. 3B, middle). Finally, the numbers of FLICA-negative, dead cells were low in all groups, with statistically significant increases in the OGD group compared with control but not between F-68 and control (Fig. 3B, right). Thus, F-68 treatment prevented the OGD-induced increase in FLICA cells after OGD and preserved the number of living cells without caspase activation.
We next asked how F-68 treatment alters the progression of OGD-induced apoptosis over time, by focusing on living and dead FLICA-positive cells at 6 and 24 h after insult. In cells exposed to normoxic control buffer, we observed no significant differences in the percentages of living or dead FLICA-positive cells at 6 and 24 h after exposure (Fig. 3C, left). After OGD, however, living FLICA-positive cells significantly outnumbered dead FLICA-positive cells at 6 h, by a ratio of 2.6:1. Significant cell death occurred between 6 and 24 h after OGD, because dead FLICA-positive cells significantly outnumbered living cells at 24 h, by a ratio of 4:1 (p < 0.0001; Fig. 3C, middle). Thus, after OGD, neurons destined to die entered the apoptosis cascade by 6 h, with the majority of death occurring by 24 h. In marked contrast, the percentages of FLICA-positive cells in F-68-treated neurons after OGD were not significantly different from control and did not change over time (Fig. 3C, right). The lack of FLICA positivity in F-68-treated neurons suggests that F-68 acts upstream of OGD-induced caspase activation.
F-68 treatment inhibits OGD-induced cytochrome c release from mitochondria
Because OGD-induced caspase activation can occur via the intrinsic, mitochondrial apoptotic pathway, with release of apoptogenic proteins and subsequent activation of caspases (Zimmermann et al., 2000), we assessed whether F-68 alters OGD-induced mitochondrial release of cytochrome c, an important mitochondrial mediator of apoptosis. We used confocal imaging of neurons in which the nucleus was identified with DRAQ5 and cytochrome c identified with immunofluorescence. Neurons exposed to control buffer showed distinct, punctuate cytochrome c staining typical of healthy mitochondria (Fig. 4A). Three hours after OGD, the majority of neurons demonstrated diffuse cytochrome c staining throughout the soma. Staurosporine-treated neurons that were used as positive control for cytochrome c release also demonstrated diffuse cytochrome c staining. In contrast, treatment of neurons with F-68 after OGD showed punctuate staining in most cells, similar to the distribution in control cells.
Figure 4.
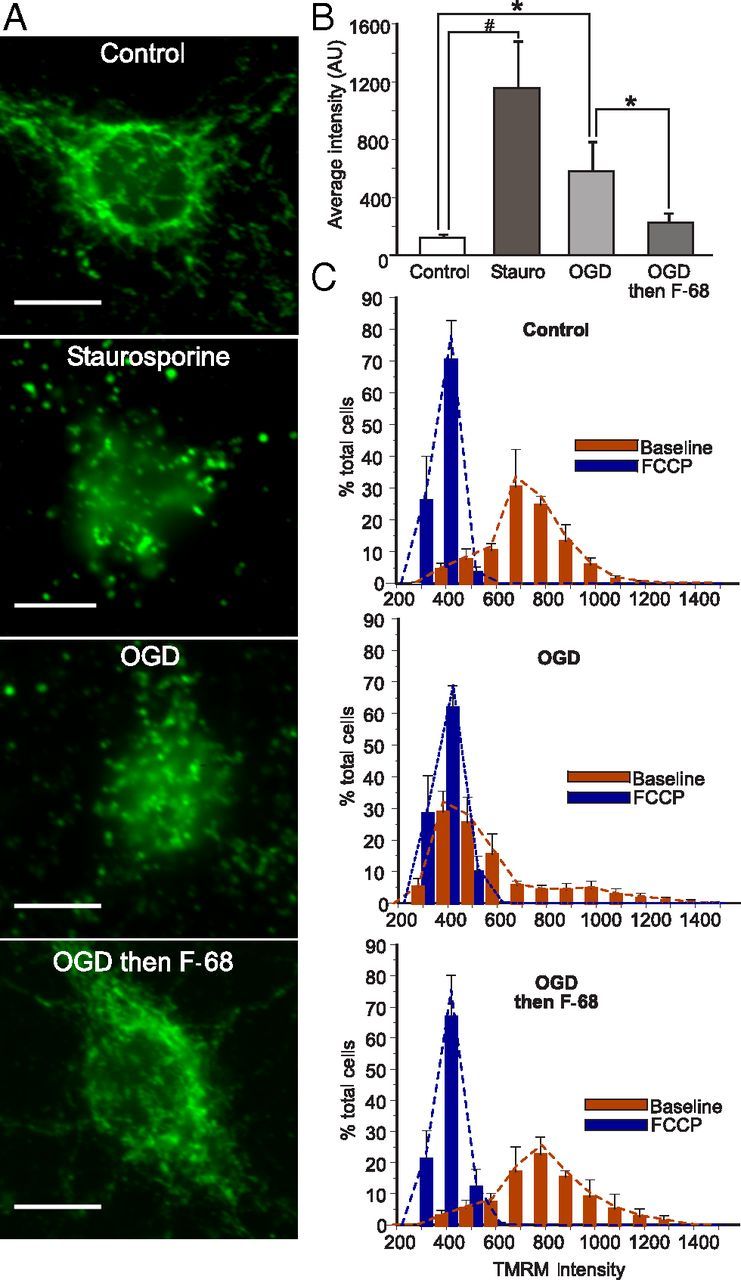
F-68 prevents OGD-induced release of mitochondrial cytochrome c and dissipation of ΔΨm. A, Cellular distribution of cytochrome c immunoreactivity 6 h after staurosporine (1 nm) or 45 min exposure to OGD with or without F-68 or control buffer. Scale bar, 10 μm. B, Mean ± SD intensity of cytochrome c immunoreactivity over the nuclear volume 6 h after OGD; n = 6 coverslips per condition. #p < 0.0001; *p < 0.01. C, Histogram of individual cell TMRM intensities (red bars) 6 h after OGD or control and in the same cells after FCCP to dissipate ΔΨm (blue bars; measured by high-throughput imaging; n = 6 coverslips per condition).
To quantify mitochondrial cytochrome c release on a cell-by-cell basis, we used the confocal images to measure the intensity of fluorescent cytochrome c immunoreactivity throughout the nuclear volume. This volume contains few mitochondria, so that immunofluorescence from free cytochrome c can be attributed to cytochrome c release. However, the diffusion limit of proteins set by the nuclear pore (60 kDa; Wang and Brattain, 2007) allows cytosolic cytochrome c (12 kDa) to equilibrate within the nuclear volume. We first confirmed the validity of the assay using staurosporine. Six hours after staurosporine treatment, cytochrome c immunoreactivity over the nuclear volume was markedly and significantly increased over untreated cells (p < 0.001; Fig. 4B). In OGD-exposed cells, cytochrome c intensity within the nuclear volume was also significantly increased compared with control cells, although not as greatly increased (p = 0.0042; Fig. 4B). Notably, in F-68-treated cells, nuclear volume cytochrome c intensity was significantly decreased from OGD alone and not significantly different from that of control cells (Fig. 4B). Thus, these data suggest that F-68 prevents the release of cytochrome c from the mitochondria.
F-68 treatment inhibits OGD-induced loss of ΔΨm
Cytochrome c release typically precedes dissipation of ΔΨm during apoptosis (Heiskanen et al., 1999; Goldstein et al., 2000). Accordingly, we assessed the extent to which ΔΨm dissipation had occurred by 6 h after OGD. We used high-throughput image acquisition of cells loaded with Hoechst, calcein, and TMRM, a cationic indicator that accumulates within mitochondria as a function of ΔΨm (n = 6 coverslips per condition, ∼150 neurons measured per coverslip). At the end of each experiment, ΔΨm was dissipated with FCCP, and ΔΨm of the same cells was remeasured. We constructed frequency histograms of mean cellular TMRM intensities in each condition (Fig. 4C).
In control cells at baseline, the TMRM intensity histogram was skewed rightward, demonstrating a range of membrane potentials. FCCP treatment produced a much more narrowly distributed, symmetric histogram that was shifted leftward toward zero, demonstrating quite uniform dissipation of ΔΨm across cells (Fig. 4C). In contrast, OGD treatment markedly dissipated ΔΨm in the majority of cells: the histogram peak overlapped the peak of the histogram obtained after treatment of these cells with FCCP, demonstrating ΔΨm dissipation in a large proportion of cells. The distribution of TMRM intensities in control cells differed significantly from that of cells subjected to OGD (D = 0.6621, p < 0.0001). Importantly, in cells treated with F-68 (30 μm) after OGD, TMRM intensities had a similar distribution to that seen in control cells and was significantly different from the distribution seen in cells subjected to OGD alone (D = 0.5666, p < 0.0001). In fact, the peak of the distribution was higher in F-68-treated cells compared with control (D = 0.1737, p < 0.01). Thus, F-68 treatment prevented or alleviated the OGD-induced ΔΨm dissipation.
F-68 inhibits BAX translocation from the cytoplasm to the mitochondria
Mitochondrial cytochrome c release depends on permeabilization of the outer mitochondrial membrane (OMM), which is ultimately induced by formation of pore-forming homo-oligomers of the effector proteins BAX or BAK in the OMM. In the case of BAX, homo-oligomer formation is preceded by translocation of cytosolic BAX to the OMM. Because F-68 treatment prevented OGD-induced loss of mitochondrial cytochrome c, we assessed whether F-68 treatment alters BAX translocation from cytosol to the mitochondria. First, neurons expressing adenovirally transduced mitochondrially targeted GFP (mito-GFP) were subjected to OGD, and images of Alexa Fluor 594-reported BAX immunofluorescence and mito-GFP were obtained with confocal microscopy. To quantify colocalization on a cell-by-cell basis (n = 7–10 cells per condition), unbiased intensity correlation analysis was performed, reported as the Manders coefficient (Manders et al., 1993). In control neurons, mito-GFP exhibited interconnected worm-like structures consistent with healthy mitochondria, and BAX immunoreactivity was primarily cytosolic (Fig. 5A, top, and inset). In contrast, OGD-treated cells exhibited rounded, coalesced mitochondria, and BAX immunoreactivity was similarly coalesced into large puncta, overlapping with mito-GFP (Fig. 5A, middle and inset). Mean Manders colocalization coefficient was markedly and significantly increased in OGD-treated neurons compared with control (p < 0.01; Fig. 5B). In neurons treated with F-68 (30 μm) after OGD, mitochondrial morphology and the distribution of BAX immunoreactivity were indistinguishable from that seen in control neurons (Fig. 5A, bottom and inset). In these cells, mean Manders coefficient was significantly decreased from that in neurons exposed to OGD and not significantly different from control neurons.
Figure 5.
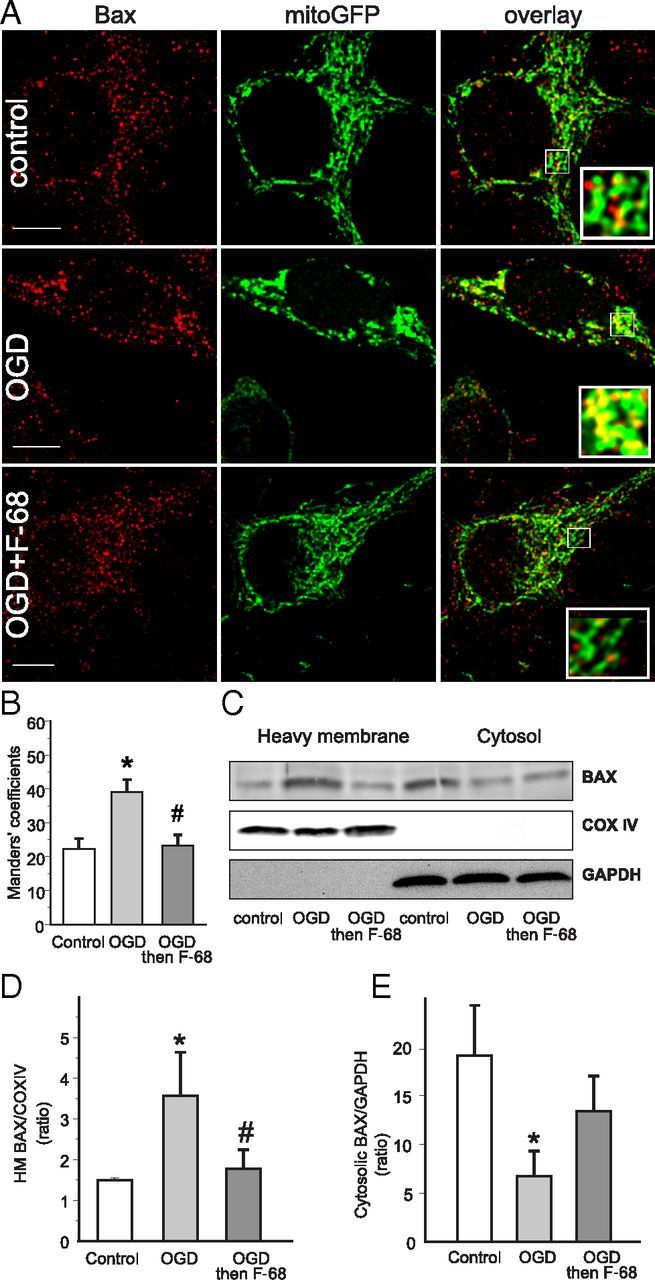
F-68 prevents OGD-induced BAX translocation from cytosol to mitochondria. A, Confocal images of hippocampal neurons expressing mitochondrially targeted GFP (green) and stained for BAX immunoreactivity (red). Scale bar, 10 μm. Note the greater overlap of BAX immunoreactivity with mitochondria after OGD but not OGD followed by F-68. B, Mean ± SD Manders coefficients, unbiased intensity correlation analyses, between BAX and mito-GFP immunoreactivity, demonstrating increased colocalization of BAX with mitochondria after OGD but not after OGD followed by F-68 (n = 7 per group). C, Representative Western blots of BAX immunoreactivity in heavy membrane and cytosolic fractions of neurons subjected to control or OGD 6 h previously. Blots were stripped and blotted with the mitochondrial marker COX IV and cytosolic marker GAPDH to identify fractions and normalize intensities for protein loading. D, E, Quantification of BAX immunoreactivity in heavy membrane (D) and cytosolic (E) fractions. Data are expressed as means ± SD of ratios of BAX/COX IV or BAX/GAPDH (n = 3 experiments, each experiment using 4 × 106 cells per condition). * indicates significantly different from control; # indicates significantly different from OGD.
To confirm these imaging results, we assessed BAX translocation in large populations of neurons using Western blot analysis. BAX levels in cytosolic [GAPDH-positive, cytochrome c oxidase subunit 4 (COX IV-negative)] and heavy membrane (GAPDH-negative, COX IV-positive) fractions of neuronal lysates obtained 6 h after OGD were analyzed (Fig. 5C). After OGD, BAX levels were significantly increased in the heavy membrane fraction and significantly decreased in the cytosolic fraction (p = 0.0220, p = 0.0192; Fig. 5D,E). In neurons treated with F-68 after OGD, BAX levels in the heavy membrane fraction were not significantly different from control levels. Similarly, BAX levels in the cytosolic fraction were not significantly different from control levels. Therefore, these data suggest that F-68 treatment prevents OGD-induced BAX translocation from the cytosol to the OMM.
Inhibition of apoptosis persists when F-68 treatment is delayed 12 h after OGD
When administered 12 or 15 h after OGD, F-68 provided equivalent neuronal rescue to that seen with F-68 administration immediately after OGD (Fig. 1F). However, 6 h after OGD, neurons not exposed to F-68 demonstrated BAX translocation to the mitochondria, release of mitochondrial cytochrome c, and caspase activation. Accordingly, we determined whether F-68 administered 12 h after OGD altered apoptosis, measuring caspase activation, cytochrome c release, and BAX translocation in these neurons. Neurons were exposed to control buffer or OGD and treated with F-68 (30 μm) or media 12 h later. Indices of apoptosis were determined 12 h after treatment, 24 h after OGD.
We first determined whether delayed F-68 administration altered caspase activation, using high-content imaging and triple labeling neurons with FLICA, Hoechst, and calcein to classify and count neurons as dead or alive and either FLICA positive or negative. In control cultures, living, FLICA-negative neurons constituted 74.2 ± 1.1% (mean ± SEM) of neurons. In cultures subjected to OGD alone, the number of these FLICA-negative, living cells was markedly decreased compared with control cultures (p < 0.0001; Fig. 6A, left). In contrast, in cultures treated with F-68 12 h after OGD, the percentage of FLICA-negative, living neurons was not significantly different from control cultures, demonstrating equivalent neuronal rescue when F-68 treatment is delayed by 12 h (p < 0.0001; Fig. 6A, left). In like manner, the percentage of all FLICA-positive cells (living and dead) was significantly increased after OGD compared with control. F-68 treatment provided 12 h after OGD decreased the percentage of these FLICA-positive neurons to control values (Fig. 6A, middle). Finally, the numbers of FLICA-negative, dead cells were low in all groups, with statistically significant increases in the OGD group compared with control and a decrease in the F-68-treated group compared with control (Fig. 6A, right).
Because the total number of cells on coverslips could have been systematically decreased by either the treatment condition (with or without OGD) or by the time after OGD when the cells were counted (6 vs 24 h), we performed a two-way ANOVA of total cells counted on each coverslip, using treatment (control, OGD, OGD + F-68) and time (6 h after OGD, 24 h after OGD) as factors. There were no significant main effects of treatment (F(2,90) = 1.71, p = 0.18) or time (F(1,90) = 0.123, p = 0.73), neither were there any important interactions between treatment and time. Thus, there were no significant differences in total cells between groups to contribute to the differences in cell survival or FLICA positivity that we observed. Together, these data demonstrate that F-68 treatment at 12 h after OGD results in the absence of an increase in caspase activation in surviving neurons 24 h after OGD.
Next, mitochondrial cytochrome c release was quantified 12 h after F-68 treatment on a cell-by-cell basis. In OGD-exposed cells, mean cytochrome c intensity within the nuclear volume was significantly increased compared with control cells (p = 0.0005; Fig. 6B). Notably, in F-68-treated cells, mean cytochrome c intensity over the nuclear volume was significantly decreased from that after OGD and not significantly different from that of control cells (Fig. 6B). Thus, delayed F-68 treatment at 12 h after OGD decreased mitochondrial cytochrome c release at 24 h after OGD.
Finally, the effect of delayed F-68 treatment on BAX translocation was assessed, using Western blots of BAX in heavy membrane and cytosolic fractions of cell lysates. Twelve hours after OGD or control buffer exposure, cultures were treated with F-68 or control buffer. Lysates were harvested 12 h later and fractionated into cytosolic and heavy membrane fractions. In the heavy membrane fraction, normalized BAX levels were increased after OGD compared with control lysates (p = 0.021; Fig. 6D) and decreased in the cytosolic fraction (p = 0.001; Fig. 6E), demonstrating translocation of BAX from cytosol to the mitochondria. Notably, after delayed F-68 treatment, the OGD-induced BAX increase in the heavy membrane fraction was significantly decreased compared with untreated OGD (p = 0.043; Fig. 6D), demonstrating an F-68-mediated decrease in BAX translocation. Therefore, these data indicate that F-68 treatment delayed until 12 h after OGD decreases OGD-induced BAX translocation to the mitochondria seen at 12 h after treatment.
Discussion
In this study, we show that the PEO–PPO–PEO copolymer F-68 provides profound rescue of hippocampal neurons after OGD. In fact, F-68 rescued all neurons that did not die with the first hour after OGD. These data build on our previous findings that F-68 rescues neurons from a variety of excitotoxic and oxidative insults, mechanisms that underlie death from OGD and brain HI in vivo (Marks et al., 2001). The similarity of the [Ca2+]i imaging responses between uninjured and rescued neurons to NMDA receptor activation and membrane depolarization, as well as the identical electrophysiological parameters between these groups, indicate that F-68 treatment restored intact neuronal function after insult. Importantly, this degree of neuronal rescue persisted when F-68 treatment was delayed as late as 12 h after insult. The absence of F-68 effects on NMDA receptor-activated whole-cell inward currents indicate that neuronal rescue is not attributable to F-68-induced NMDA receptor antagonism.
Whether neurons die by apoptosis or necrosis after injury can depend on injury severity (Ankarcrona et al., 1995; Bonfoco et al., 1995). After 60 min OGD, a proportion of neurons died within 1 h of injury. The rapidity of this death, and its appearance only after the most severe injury used, strongly suggest that these neurons died by necrosis. In contrast, the remainder of the death, which began at 6 h and progressed over 48 h, was apoptotic, as demonstrated by TUNEL staining and the sensitivity of this death to pan-caspase inhibition. In fact, the amount of Z-VAD-FMK-sensitive death completely accounted for the number of neurons rescued by F-68 after 45 min of OGD. F-68-induced inhibition of apoptosis has been reported previously in in vitro models of neuronal trauma (Serbest et al., 2005; Kilinc et al., 2007) and chondrocytes (Bajaj et al., 2010).
To understand how F-68 prevents OGD-induced apoptosis, we systematically investigated the points at which F-68 might interfere in the intrinsic apoptotic pathway. F-68 prevented phosphatidylserine externalization, the earliest indicator of apoptosis, and caspase activation. F-68 also prevented OGD-induced ΔΨm dissipation and mitochondrial cytochrome c release, suggesting that F-68 acts upstream of mitochondrial outer membrane permeabilization (MOMP), a central event in apoptosis. Finally, F-68 blocked translocation of cytosolic BAX to mitochondria, an event leading to MOMP in many apoptosis models (for review, see Tait and Green, 2010).
Apoptosis after acute metabolic stress, including OGD, occurs after the activation of multiple processes, including [Ca2+]i deregulation and activation of Ca2+-dependent proteases, ATP consumption, and mitochondrial dysfunction (Budd and Nicholls, 1996; Duchen, 1999; White et al., 2000; Pivovarova and Andrews, 2010). These processes all occur within minutes or several hours of insult. Although F-68 may modulate these early responses, its ability to rescue neurons from apoptosis 12–15 h after OGD strongly suggests that F-68-mediated neuronal rescue does not occur by modulating these processes.
Biophysical approaches have demonstrated that F-68 interacts directly with phospholipid membranes, in supported lipid monolayers (Wu et al., 2004, 2005) and in lipid bilayers (Firestone et al., 2003). Small-angle x-ray scattering (SAXS) has demonstrated that the nature of the interactions of 80% hydrophilic Pluronics with lipid bilayers critically depends on the length of the central PPO chain: copolymers with short PPO chains (e.g., 15 units) exhibit SAXS profiles consistent with the preferential association of the hydrophobic PPO chain with alkyl chains of the membrane phospholipids. With F-68 (29 PPO units), SAXS profiles are consistent with increased anchoring to the membrane, because the increased length of the PPO chain likely increases incorporation into the membrane (Firestone et al., 2003). Using giant unilamellar vesicles, we showed recently that, during osmotic stress, an injury in which lipid packing density is decreased, F-68 acutely adsorbs to the lipid bilayer, retarding the osmotic stress-induced loss of membrane integrity (Wang et al., 2010).
F-68 reverses cell dysfunction arising from plasma membrane defects, including electropermeabilization (Lee et al., 1992; Marks et al., 2001) and genetic defects in myocyte compliance (Yasuda et al., 2005). F-68-mediated abolition of shear stress-induced increases in neuronal plasma membrane permeability (Kilinc et al., 2008) indicates that F-68 interacts with damaged neuronal plasma membranes. Of note, fluorophore-tagged Pluronics applied to cells are rapidly internalized (Sahay et al., 2008), achieving access to intracellular structures.
Six hours after OGD, BAX translocation to the mitochondria was apparent, a key step in MOMP and subsequent release of intra-mitochondrial proapoptotic factors into the cytosol (Putcha et al., 1999; Ghatan et al., 2000; Fan et al., 2012). In contrast, after F-68 treatment, BAX translocation was not seen at 6 h, and neither cytochrome c release nor caspase activation occurred, providing a mechanism by which apoptosis was inhibited. Strikingly, when F-68 treatment was delayed 12 h after OGD, hours after BAX translocation and its downstream effects had occurred, the levels of mitochondrial BAX, cytosolic cytochrome c, and the numbers of FLICA-positive neurons had returned to baseline levels, indicating that F-68 can arrest ongoing OGD-induced apoptosis. Proapoptotic factors are released from individual mitochondria in a one-step process (Goldstein et al., 2005; Bhola et al., 2009). In multiple cell types, this release propagates to adjacent mitochondria in spatially and temporally regulated waves (Bhola et al., 2009; Garcia-Perez et al., 2012), in some studies, in a reactive oxygen species (ROS)-dependent manner (Garcia-Perez et al., 2012). That cytochrome c can be released from at least 15% of the cellular mitochondrial volume without inducing apoptosis (Khodjakov et al., 2004) supports the idea that the number of mitochondria undergoing MOMP by 12 h after OGD has activated insufficient caspases to make apoptosis irreversible. The restoration of mitochondrial BAX levels suggests that F-68 may restore MOMP, as has been observed for plasma membranes after permeabilization (Marks et al., 2001; Plataki et al., 2011; Spurney et al., 2011).The dependence of apoptosis on mitochondrial lipids, including cardiolipin oxidation (Kuwana et al., 2002; Kagan et al., 2005), ceramide, and its metabolites (Siskind et al., 2006; Chipuk et al., 2012), suggest candidate lipid species with which F-68 may interact to restore OMM integrity and prevent progression of apoptosis. The efficacy of endogenous and modified brain lipids, e.g., 16:0 N-acetyl-ethanolamine, in rescuing neurons from focal ischemia (Garg et al., 2010) further support a role for membrane-targeted interventions in rescuing neurons from death. Alternatively, if F-68 acts before BAX translocation to promote neuronal survival, the return of mitochondrial BAX levels to baseline may be attributable to ongoing mitophagy of damaged mitochondria (Zhu et al., 2013).
After mechanical injury, F-68 decreases apoptosis in PC2 cells and chondrocytes, associated with p38 inhibition (Serbest et al., 2005) and activation of Stat-1 (signal transducer and activator of transcription-1) and ATF-2 (activating transcription factor-2) (Bajaj et al., 2010). In both models, specifically inhibiting p38 activation accounts for <50% of the survival benefit afforded by F-68, suggesting that F-68 inhibits apoptosis upstream of p38 activation. In neuronal systems, p38 activation depends on ROS production (Kawasaki et al., 1997; Behrens et al., 1999; Choi et al., 2004). In fact, increased ROS production during and after acute insults are central mechanisms of neuronal death after ischemia (Abramov et al., 2007). For lipid membranes, the primary consequences of injury-induced increases in ROS production are lipid peroxidation and production of 4-hydroxynonenal, a highly reactive lipid electrophile (Liu et al., 2011). These processes can lead to impaired lipid packing density and decreases in membrane integrity. We showed previously that F-68 blocks the ongoing lipid peroxidation in the plasma membrane induced by exogenous Fenton reagents (Marks et al., 2001). Therefore, regardless of the upstream initiating events for ROS production, F-68 may rescue neurons through inhibition of lipid peroxidation, either through acting as a chain-breaking anti-oxidant or as a direct inhibitor of ROS production.
Although the precise membrane mechanisms of neuronal rescue by F-68 remain unclear, this report demonstrates that a synthetic copolymer that interacts with membrane bilayers provides near-complete neuroprotection after OGD and acts by inhibiting apoptosis early in the mitochondrial pathway. The ability of F-68 to rescue injured neurons in vitro suggests that this membrane-targeted approach may also rescue neurons after brain HI in vivo. The efficacy of F-68 when given 12–15 h after OGD positions 80% hydrophilic Pluronics as potential treatments for reperfused stroke, in which patients present hours after the onset of injury. The requirement for F-68 coatings on poly(alkyl cyanoacrylate) nanoparticles to deliver blood–brain barrier-impermeable compounds (Kreuter, 2004; Gelperina et al., 2010) demonstrates that F-68 crosses the blood–brain barrier, removing a potential impediment to its use as an intravenous therapy. In addition, F-68 and related Pluronics have demonstrated acceptable safety profiles in commonly used species (Duvinage et al., 1996; Singh-Joy and McLain, 2008), as well as in humans (Ballas et al., 2004). Finally, the pharmacokinetics of F-68 are well described (Willcox et al., 1978; Jewell et al., 1997; Grindel et al., 2002a,b), allowing protocols for achieving and maintaining desired serum F-68 levels to be developed. Therefore, the ability of F-68 to rescue neurons after delayed administration makes testing its efficacy and the efficacy of related Pluronics in in vivo models of cerebral ischemia important future studies.
Footnotes
This work was funded by National Institutes of Health Grants R01 NS056313 (J.D.M.) and T32 HL 094282 (P.B.S.).
J.D.M. has a financial interest in Maroon Biotech.
References
- Abramov AY, Scorziello A, Duchen MR. Three distinct mechanisms generate oxygen free radicals in neurons and contribute to cell death during anoxia and reoxygenation. J Neurosci. 2007;27:1129–1138. doi: 10.1523/JNEUROSCI.4468-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adelson JD, Barreto GE, Xu L, Kim T, Brott BK, Ouyang YB, Naserke T, Djurisic M, Xiong X, Shatz CJ, Giffard RG. Neuroprotection from stroke in the absence of MHCI or PirB. Neuron. 2012;73:1100–1107. doi: 10.1016/j.neuron.2012.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albers GW, Goldstein LB, Hess DC, Wechsler LR, Furie KL, Gorelick PB, Hurn P, Liebeskind DS, Nogueira RG, Saver JL, STAIR VII Consortium Stroke treatment academic industry roundtable (STAIR) recommendations for maximizing the use of intravenous thrombolytics and expanding treatment options with intra-arterial and neuroprotective therapies. Stroke. 2011;42:2645–2650. doi: 10.1161/STROKEAHA.111.618850. [DOI] [PubMed] [Google Scholar]
- Allen NJ, Káradóttir R, Attwell D. A preferential role for glycolysis in preventing the anoxic depolarization of rat hippocampal area CA1 pyramidal cells. J Neurosci. 2005;25:848–859. doi: 10.1523/JNEUROSCI.4157-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ankarcrona M, Dypbukt JM, Bonfoco E, Zhivotovsky B, Orrenius S, Lipton SA, Nicotera P. Glutamate-induced neuronal death: a succession of necrosis or apoptosis depending on mitochondrial function. Neuron. 1995;15:961–973. doi: 10.1016/0896-6273(95)90186-8. [DOI] [PubMed] [Google Scholar]
- Bajaj S, Shoemaker T, Hakimiyan AA, Rappoport L, Pascual-Garrido C, Oegema TR, Wimmer MA, Chubinskaya S. Protective effect of P188 in the model of acute trauma to human ankle cartilage: the mechanism of action. J Orthop Trauma. 2010;24:571–576. doi: 10.1097/BOT.0b013e3181ec4712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballas SK, Files B, Luchtman-Jones L, Benjamin L, Swerdlow P, Hilliard L, Coates T, Abboud M, Wojtowicz-Praga S, Grindel JM. Safety of purified poloxamer 188 in sickle cell disease: phase I study of a non-ionic surfactant in the management of acute chest syndrome. Hemoglobin. 2004;28:85–102. doi: 10.1081/HEM-120035919. [DOI] [PubMed] [Google Scholar]
- Batrakova EV, Li S, Alakhov VY, Miller DW, Kabanov AV. Optimal structure requirements for pluronic block copolymers in modifying P-glycoprotein drug efflux transporter activity in bovine brain microvessel endothelial cells. J Pharmacol Exp Ther. 2003;304:845–854. doi: 10.1124/jpet.102.043307. [DOI] [PubMed] [Google Scholar]
- Behrens MM, Strasser U, Koh JY, Gwag BJ, Choi DW. Prevention of neuronal apoptosis by phorbol ester-induced activation of protein kinase C: blockade of p38 mitogen-activated protein kinase. Neuroscience. 1999;94:917–927. doi: 10.1016/S0306-4522(99)00212-2. [DOI] [PubMed] [Google Scholar]
- Bhola PD, Mattheyses AL, Simon SM. Spatial and temporal dynamics of mitochondrial membrane permeability waves during apoptosis. Biophys J. 2009;97:2222–2231. doi: 10.1016/j.bpj.2009.07.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonfoco E, Krainc D, Ankarcrona M, Nicotera P, Lipton SA. Apoptosis and necrosis: two distinct events induced, respectively, by mild and intense insults with N-methyl-d-aspartate or nitric oxide/superoxide in cortical cell cultures. Proc Natl Acad Sci U S A. 1995;92:7162–7166. doi: 10.1073/pnas.92.16.7162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budd SL, Nicholls DG. Mitochondria, calcium regulation, and acute glutamate excitotoxicity in cultured cerebellar granule cells. J Neurochem. 1996;67:2282–2291. doi: 10.1046/j.1471-4159.1996.67062282.x. [DOI] [PubMed] [Google Scholar]
- Chipuk JE, McStay GP, Bharti A, Kuwana T, Clarke CJ, Siskind LJ, Obeid LM, Green DR. Sphingolipid metabolism cooperates with BAK and BAX to promote the mitochondrial pathway of apoptosis. Cell. 2012;148:988–1000. doi: 10.1016/j.cell.2012.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi WS, Eom DS, Han BS, Kim WK, Han BH, Choi EJ, Oh TH, Markelonis GJ, Cho JW, Oh YJ. Phosphorylation of p38 MAPK induced by oxidative stress is linked to activation of both caspase-8- and -9-mediated apoptotic pathways in dopaminergic neurons. J Biol Chem. 2004;279:20451–20460. doi: 10.1074/jbc.M311164200. [DOI] [PubMed] [Google Scholar]
- Duchen MR. Contributions of mitochondria to animal physiology: from homeostatic sensor to calcium signalling and cell death. J Physiol. 1999;516:1–17. doi: 10.1111/j.1469-7793.1999.001aa.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duvinage C, Millecamps S, Sagnier A, Guffroy M, Sarsat JP, Belin V. One month intravenous toxicity studies of poloxamer 188 in male Sprague-Dawley rats and in beagle dogs. Toxicol Lett. 1996;88:101–110. [Google Scholar]
- Fan J, Zhang N, Yin G, Zhang Z, Cheng G, Qian W, Long H, Cai W. Edaravone protects cortical neurons from apoptosis by inhibiting the translocation of BAX and increasing the interaction between 14-3-3 and p-BAD. Int J Neurosci. 2012;122:665–674. doi: 10.3109/00207454.2012.707714. [DOI] [PubMed] [Google Scholar]
- Firestone MA, Wolf AC, Seifert S. Small-angle X-ray scattering study of the interaction of poly(ethylene oxide)-b-poly(propylene oxide)-b-poly(ethylene oxide) triblock copolymers with lipid bilayers. Biomacromolecules. 2003;4:1539–1549. doi: 10.1021/bm034134r. [DOI] [PubMed] [Google Scholar]
- Garcia-Perez C, Roy SS, Naghdi S, Lin X, Davies E, Hajnóczky G. Bid-induced mitochondrial membrane permeabilization waves propagated by local reactive oxygen species (ROS) signaling. Proc Natl Acad Sci U S A. 2012;109:4497–4502. doi: 10.1073/pnas.1118244109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg P, Duncan RS, Kaja S, Koulen P. Intracellular mechanisms of N-acylethanolamine-mediated neuroprotection in a rat model of stroke. Neuroscience. 2010;166:252–262. doi: 10.1016/j.neuroscience.2009.11.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelperina S, Maksimenko O, Khalansky A, Vanchugova L, Shipulo E, Abbasova K, Berdiev R, Wohlfart S, Chepurnova N, Kreuter J. Drug delivery to the brain using surfactant-coated poly(lactide-co-glycolide) nanoparticles: Influence of the formulation parameters. Eur J Pharm Biopharm. 2010;74:157–163. doi: 10.1016/j.ejpb.2009.09.003. [DOI] [PubMed] [Google Scholar]
- Ghatan S, Larner S, Kinoshita Y, Hetman M, Patel L, Xia Z, Youle RJ, Morrison RS. p38 MAP kinase mediates bax translocation in nitric oxide-induced apoptosis in neurons. J Cell Biol. 2000;150:335–347. doi: 10.1083/jcb.150.2.335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein JC, Waterhouse NJ, Juin P, Evan GI, Green DR. The coordinate release of cytochrome c during apoptosis is rapid, complete and kinetically invariant. Nat Cell Biol. 2000;2:156–162. doi: 10.1038/35004029. [DOI] [PubMed] [Google Scholar]
- Goldstein JC, Muñoz-Pinedo C, Ricci JE, Adams SR, Kelekar A, Schuler M, Tsien RY, Green DR. Cytochrome c is released in a single step during apoptosis. Cell Death Differ. 2005;12:453–462. doi: 10.1038/sj.cdd.4401596. [DOI] [PubMed] [Google Scholar]
- Grindel JM, Jaworski T, Emanuele RM, Culbreth P. Pharmacokinetics of a novel surface-active agent, purified poloxamer 188, in rat, rabbit, dog and man. Biopharm Drug Dispos. 2002a;23:87–103. doi: 10.1002/bdd.297. [DOI] [PubMed] [Google Scholar]
- Grindel JM, Jaworski T, Piraner O, Emanuele RM, Balasubramanian M. Distribution, metabolism, and excretion of a novel surface-active agent, purified poloxamer 188, in rats, dogs, and humans. J Pharm Sci. 2002b;91:1936–1947. doi: 10.1002/jps.10190. [DOI] [PubMed] [Google Scholar]
- Heiskanen KM, Bhat MB, Wang HW, Ma J, Nieminen AL. Mitochondrial depolarization accompanies cytochrome c release during apoptosis in PC6 cells. J Biol Chem. 1999;274:5654–5658. doi: 10.1074/jbc.274.9.5654. [DOI] [PubMed] [Google Scholar]
- Hypothermia after Arrest Study Group. Mild therapeutic hypothermia to improve the neurologic outcome after cardiac arrest. N Engl J Med. 2002;346:549–556. doi: 10.1056/NEJMoa012689. [DOI] [PubMed] [Google Scholar]
- Jacobs SE, Hunt R, Tarnow-Mordi WO, Inder TE, Davis PG. Cooling for newborns with hypoxic ischaemic encephalopathy. Cochrane Database Syst Rev. 2007;(4):CD003311. doi: 10.1002/14651858.CD003311.pub2. [DOI] [PubMed] [Google Scholar]
- Jewell RC, Khor SP, Kisor DF, LaCroix KA, Wargin WA. Pharmacokinetics of RheothRx injection in healthy male volunteers. J Pharm Sci. 1997;86:808–812. doi: 10.1021/js960491e. [DOI] [PubMed] [Google Scholar]
- Ji J, Kline AE, Amoscato A, Samhan-Arias AK, Sparvero LJ, Tyurin VA, Tyurina YY, Fink B, Manole MD, Puccio AM, Okonkwo DO, Cheng JP, Alexander H, Clark RS, Kochanek PM, Wipf P, Kagan VE, Bayır H. Lipidomics identifies cardiolipin oxidation as a mitochondrial target for redox therapy of brain injury. Nat Neurosci. 2012;15:1407–1413. doi: 10.1038/nn.3195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagan VE, Tyurin VA, Jiang J, Tyurina YY, Ritov VB, Amoscato AA, Osipov AN, Belikova NA, Kapralov AA, Kini V, Vlasova II, Zhao Q, Zou M, Di P, Svistunenko DA, Kurnikov IV, Borisenko GG. Cytochrome c acts as a cardiolipin oxygenase required for release of proapoptotic factors. Nat Chem Biol. 2005;1:223–232. doi: 10.1038/nchembio727. [DOI] [PubMed] [Google Scholar]
- Kanno T, Nishizaki T. Sphingosine induces apoptosis in hippocampal neurons and astrocytes by activating caspase-3/-9 via a mitochondrial pathway linked to SDK/14-3-3 protein/Bax/cytochrome c. J Cell Physiol. 2011;226:2329–2337. doi: 10.1002/jcp.22571. [DOI] [PubMed] [Google Scholar]
- Kawasaki H, Morooka T, Shimohama S, Kimura J, Hirano T, Gotoh Y, Nishida E. Activation and involvement of p38 mitogen-activated protein kinase in glutamate-induced apoptosis in rat cerebellar granule cells. J Biol Chem. 1997;272:18518–18521. doi: 10.1074/jbc.272.30.18518. [DOI] [PubMed] [Google Scholar]
- Kelly CV, Kober MM, Kinnunen P, Reis DA, Orr BG, Banaszak Holl MM. Pulsed-laser creation and characterization of giant plasma membrane vesicles from cells. J Biol Phys. 2009;35:279–295. doi: 10.1007/s10867-009-9167-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khodjakov A, Rieder C, Mannella CA, Kinnally KW. Laser micro-irradiation of mitochondria: is there an amplified mitochondrial death signal in neural cells? Mitochondrion. 2004;3:217–227. doi: 10.1016/j.mito.2003.10.002. [DOI] [PubMed] [Google Scholar]
- Kilinc D, Gallo G, Barbee K. Poloxamer 188 reduces axonal beading following mechanical trauma to cultured neurons. Conf Proc IEEE Eng Med Biol Soc. 2007;2007:5395–5398. doi: 10.1109/IEMBS.2007.4353562. [DOI] [PubMed] [Google Scholar]
- Kilinc D, Gallo G, Barbee KA. Mechanically-induced membrane poration causes axonal beading and localized cytoskeletal damage. Exp Neurol. 2008;212:422–430. doi: 10.1016/j.expneurol.2008.04.025. [DOI] [PubMed] [Google Scholar]
- Kilinc D, Gallo G, Barbee KA. Mechanical membrane injury induces axonal beading through localized activation of calpain. Exp Neurol. 2009;219:553–561. doi: 10.1016/j.expneurol.2009.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreuter J. Influence of the surface properties on nanoparticle-mediated transport of drugs to the brain. J Nanosci Nanotechnol. 2004;4:484–488. doi: 10.1166/jnn.2003.077. [DOI] [PubMed] [Google Scholar]
- Kuwana T, Mackey MR, Perkins G, Ellisman MH, Latterich M, Schneiter R, Green DR, Newmeyer DD. Bid, Bax, and lipids cooperate to form supramolecular openings in the outer mitochondrial membrane. Cell. 2002;111:331–342. doi: 10.1016/S0092-8674(02)01036-X. [DOI] [PubMed] [Google Scholar]
- Lee RC, River LP, Pan FS, Ji L, Wollmann RL. Surfactant-induced sealing of electropermeabilized skeletal muscle membranes in vivo. Proc Natl Acad Sci U S A. 1992;89:4524–4528. doi: 10.1073/pnas.89.10.4524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehman EL, D'Abrera HJM. Nonparametrics: statistical methods based on ranks. Ed 1. New York: Springer; 2006. [Google Scholar]
- Liu W, Porter NA, Schneider C, Brash AR, Yin H. Formation of 4-hydroxynonenal from cardiolipin oxidation: intramolecular peroxyl radical addition and decomposition. Free Radic Biol Med. 2011;50:166–178. doi: 10.1016/j.freeradbiomed.2010.10.709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manders EM, Verbeek FJ, Aten JA. Measurement of co-localization of objects in dual-colour confocal images. J Microsc. 1993;169:375–382. doi: 10.1111/j.1365-2818.1993.tb03313.x. [DOI] [PubMed] [Google Scholar]
- Marks JD, Bindokas VP, Zhang XM. Maturation of vulnerability to excitotoxicity:intracellular mechanisms in cultured postnatal hippocampal neurons. Brain Res Dev Brain Res. 2000;124:101–116. doi: 10.1016/S0165-3806(00)00096-1. [DOI] [PubMed] [Google Scholar]
- Marks JD, Pan CY, Bushell T, Cromie W, Lee RC. Amphiphilic, tri-block copolymers provide potent membrane-targeted neuroprotection. FASEB J. 2001;15:1107–1109. doi: 10.1096/fj.00-0547fje. [DOI] [PubMed] [Google Scholar]
- Marks JD, Boriboun C, Wang J. Mitochondrial nitric oxide mediates decreased vulnerability of hippocampal neurons from immature animals to NMDA. J Neurosci. 2005;25:6561–6575. doi: 10.1523/JNEUROSCI.1450-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niatsetskaya ZV, Sosunov SA, Matsiukevich D, Utkina-Sosunova IV, Ratner VI, Starkov AA, Ten VS. The oxygen free radicals originating from mitochondrial complex I contribute to oxidative brain injury following hypoxia-ischemia in neonatal mice. J Neurosci. 2012;32:3235–3244. doi: 10.1523/JNEUROSCI.6303-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pivovarova NB, Andrews SB. Calcium-dependent mitochondrial function and dysfunction in neurons. FEBS J. 2010;277:3622–3636. doi: 10.1111/j.1742-4658.2010.07754.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plant LD, Dowdell EJ, Dementieva IS, Marks JD, Goldstein SA. SUMO modification of cell surface Kv2.1 potassium channels regulates the activity of rat hippocampal neurons. J Gen Physiol. 2011;137:441–454. doi: 10.1085/jgp.201110604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plataki M, Lee YD, Rasmussen DL, Hubmayr RD. Poloxamer 188 facilitates the repair of alveolus resident cells in ventilator-injured lungs. Am J Respir Crit Care Med. 2011;184:939–947. doi: 10.1164/rccm.201104-0647OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poppe M, Reimertz C, Düssmann H, Krohn AJ, Luetjens CM, Böckelmann D, Nieminen AL, Kögel D, Prehn JH. Dissipation of potassium and proton gradients inhibits mitochondrial hyperpolarization and cytochrome c release during neural apoptosis. J Neurosci. 2001;21:4551–4563. doi: 10.1523/JNEUROSCI.21-13-04551.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putcha GV, Deshmukh M, Johnson EM., Jr BAX translocation is a critical event in neuronal apoptosis: regulation by neuroprotectants, BCL-2, and caspases. J Neurosci. 1999;19:7476–7485. doi: 10.1523/JNEUROSCI.19-17-07476.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahay G, Batrakova EV, Kabanov AV. Different internalization pathways of polymeric micelles and unimers and their effects on vesicular transport. Bioconjug Chem. 2008;19:2023–2029. doi: 10.1021/bc8002315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serbest G, Horwitz J, Barbee K. The effect of poloxamer-188 on neuronal cell recovery from mechanical injury. J Neurotrauma. 2005;22:119–132. doi: 10.1089/neu.2005.22.119. [DOI] [PubMed] [Google Scholar]
- Serbest G, Horwitz J, Jost M, Barbee K. Mechanisms of cell death and neuroprotection by poloxamer 188 after mechanical trauma. FASEB J. 2006;20:308–310. doi: 10.1096/fj.05-4024fje. [DOI] [PubMed] [Google Scholar]
- Shanta SR, Choi CS, Lee JH, Shin CY, Kim YJ, Kim KH, Kim KP. Global changes in phospholipids identified by MALDI MS in rats with focal cerebral ischemia. J Lipid Res. 2012;53:1823–1831. doi: 10.1194/jlr.M022558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh-Joy SD, McLain VC. Safety assessment of poloxamers 101, 105, 108, 122, 123, 124, 181, 182, 183, 184, 185, 188, 212, 215, 217, 231, 234, 235, 237, 238, 282, 284, 288, 331, 333, 334, 335, 338, 401, 402, 403, and 407, poloxamer 105 benzoate, and poloxamer 182 dibenzoate as used in cosmetics. Int J Toxicol. 2008;27(Suppl 2):93–128. doi: 10.1080/10915810802244595. [DOI] [PubMed] [Google Scholar]
- Siskind LJ, Kolesnick RN, Colombini M. Ceramide forms channels in mitochondrial outer membranes at physiologically relevant concentrations. Mitochondrion. 2006;6:118–125. doi: 10.1016/j.mito.2006.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soeda S, Tsuji Y, Ochiai T, Mishima K, Iwasaki K, Fujiwara M, Yokomatsu T, Murano T, Shibuya S, Shimeno H. Inhibition of sphingomyelinase activity helps to prevent neuron death caused by ischemic stress. Neurochem Int. 2004;45:619–626. doi: 10.1016/j.neuint.2004.04.001. [DOI] [PubMed] [Google Scholar]
- Spurney C, Guerron A, Yu Q, Sali A, van der Meulen JH, Hoffman EP, Nagaraju K. Membrane sealant poloxamer P188 protects against isoproterenol induced cardiomyopathy in dystrophin deficient mice. BMC Cardiovasc Disord. 2011;11:20. doi: 10.1186/1471-2261-11-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tait SW, Green DR. Mitochondria and cell death: outer membrane permeabilization and beyond. Nat Rev Mol Cell Biol. 2010;11:621–632. doi: 10.1038/nrm2952. [DOI] [PubMed] [Google Scholar]
- Tanaka E, Yamamoto S, Inokuchi H, Isagai T, Higashi H. Membrane dysfunction induced by in vitro ischemia in rat hippocampal CA1 pyramidal neurons. J Neurophysiol. 1999;81:1872–1880. doi: 10.1152/jn.1999.81.4.1872. [DOI] [PubMed] [Google Scholar]
- Townsend D, Turner I, Yasuda S, Martindale J, Davis J, Shillingford M, Kornegay JN, Metzger JM. Chronic administration of membrane sealant prevents severe cardiac injury and ventricular dilatation in dystrophic dogs. J Clin Invest. 2010;120:1140–1150. doi: 10.1172/JCI41329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu W, Xu X, Peng L, Zhong X, Zhang W, Soundarapandian MM, Balel C, Wang M, Jia N, Zhang W, Lew F, Chan SL, Chen Y, Lu Y. DAPK1 interaction with NMDA receptor NR2B subunits mediates brain damage in stroke. Cell. 2010;140:222–234. doi: 10.1016/j.cell.2009.12.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tymianski M, Bernstein GM, Abdel-Hamid KM, Sattler R, Velumian A, Carlen PL, Razavi H, Jones OT. A novel use for a carbodiimide compound for the fixation of fluorescent and non-fluorescent calcium indicators in situ following physiological experiments. Cell Calcium. 1997;21:175–183. doi: 10.1016/S0143-4160(97)90042-7. [DOI] [PubMed] [Google Scholar]
- Wang HK, Park UJ, Kim SY, Lee JH, Kim SU, Gwag BJ, Lee YB. Free radical production in CA1 neurons induces MIP-1α expression, microglia recruitment, and delayed neuronal death after transient forebrain ischemia. J Neurosci. 2008;28:1721–1727. doi: 10.1523/JNEUROSCI.4973-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JY, Chin J, Marks JD, Lee KY. Effects of PEO-PPO-PEO triblock copolymers on phospholipid membrane integrity under osmotic stress. Langmuir. 2010;26:12953–12961. doi: 10.1021/la101841a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JY, Marks J, Lee KY. Nature of interactions between PEO-PPO-PEO triblock copolymers and lipid membranes: (I) The effect of polymer hydrophobicity on its ability to protect liposomes from peroxidation. Biomacromolecules. 2012;13:2616–2623. doi: 10.1021/bm300847x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R, Brattain MG. The maximal size of protein to diffuse through the nuclear pore is larger than 60kDa. FEBS Lett. 2007;581:3164–3170. doi: 10.1016/j.febslet.2007.05.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White BC, Sullivan JM, DeGracia DJ, O'Neil BJ, Neumar RW, Grossman LI, Rafols JA, Krause GS. Brain ischemia and reperfusion: molecular mechanisms of neuronal injury. J Neurol Sci. 2000;179:1–33. doi: 10.1016/S0022-510X(00)00386-5. [DOI] [PubMed] [Google Scholar]
- Willcox ML, Newman MM, Paton BC. A study of labeled pluronic F-68 after intravenous injection into the dog. J Surg Res. 1978;25:349–356. doi: 10.1016/0022-4804(78)90130-0. [DOI] [PubMed] [Google Scholar]
- Wu G, Majewski J, Ege C, Kjaer K, Weygand MJ, Lee KY. Lipid corralling and poloxamer squeeze-out in membranes. Phys Rev Lett. 2004;93 doi: 10.1103/PhysRevLett.93.028101. 028101. [DOI] [PubMed] [Google Scholar]
- Wu G, Majewski J, Ege C, Kjaer K, Weygand MJ, Lee KY. Interaction between lipid monolayers and poloxamer 188: an x-ray reflectivity and diffraction study. Biophys J. 2005;89:3159–3173. doi: 10.1529/biophysj.104.052290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Sahay G, Sriadibhatla S, Kabanov AV. Amphiphilic block copolymers enhance cellular uptake and nuclear entry of polyplex-delivered DNA. Bioconjug Chem. 2008;19:1987–1994. doi: 10.1021/bc800144a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda S, Townsend D, Michele DE, Favre EG, Day SM, Metzger JM. Dystrophic heart failure blocked by membrane sealant poloxamer. Nature. 2005;436:1025–1029. doi: 10.1038/nature03844. [DOI] [PubMed] [Google Scholar]
- Yu ZF, Nikolova-Karakashian M, Zhou D, Cheng G, Schuchman EH, Mattson MP. Pivotal role for acidic sphingomyelinase in cerebral ischemia-induced ceramide and cytokine production, and neuronal apoptosis. J Mol Neurosci. 2000;15:85–97. doi: 10.1385/JMN:15:2:85. [DOI] [PubMed] [Google Scholar]
- Zhu S, Li M, Figueroa BE, Liu A, Stavrovskaya IG, Pasinelli P, Beal MF, Brown RH, Jr, Kristal BS, Ferrante RJ, Friedlander RM. Prophylactic creatine administration mediates neuroprotection in cerebral ischemia in mice. J Neurosci. 2004;24:5909–5912. doi: 10.1523/JNEUROSCI.1278-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Massen S, Terenzio M, Lang V, Chen-Lindner S, Eils R, Novak I, Dikic I, Hamacher-Brady A, Brady NR. Modulation of serines 17 and 24 in the LC3-interacting region of Bnip3 determines pro-survival mitophagy versus apoptosis. J Biol Chem. 2013;288:1099–1113. doi: 10.1074/jbc.M112.399345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann KC, Waterhouse NJ, Goldstein JC, Schuler M, Green DR. Aspirin induces apoptosis through release of cytochrome c from mitochondria. Neoplasia. 2000;2:505–513. doi: 10.1038/sj.neo.7900120. [DOI] [PMC free article] [PubMed] [Google Scholar]