

Abstract
While sialylation plays important functions in the nervous system, the complexity of glycosylation pathways and limitations of genetic approaches preclude the efficient analysis of these functions in mammalian organisms. Drosophila has recently emerged as a promising model for studying neural sialylation. Drosophila sialyltransferase, DSiaT, was shown to be involved in the regulation of neural transmission. However, the sialylation pathway was not investigated in Drosophila beyond the DSiaT-mediated step. Here we focused on the function of Drosophila cytidine monophosphate-sialic acid synthetase (CSAS), the enzyme providing a sugar donor for DSiaT. Our results revealed that the expression of CSAS is tightly regulated and restricted to the CNS throughout development and in adult flies. We generated CSAS mutants and analyzed their phenotypes using behavioral and physiological approaches. Our experiments demonstrated that mutant phenotypes of CSAS are similar to those of DSiaT, including decreased longevity, temperature-induced paralysis, locomotor abnormalities, and defects of neural transmission at neuromuscular junctions. Genetic interactions between CSAS, DSiaT, and voltage-gated channel genes paralytic and seizure were consistent with the hypothesis that CSAS and DSiaT function within the same pathway regulating neural excitability. Intriguingly, these interactions also suggested that CSAS and DSiaT have some additional, independent functions. Moreover, unlike its mammalian counterparts that work in the nucleus, Drosophila CSAS was found to be a glycoprotein-bearing N-glycans and predominantly localized in vivo to the Golgi compartment. Our work provides the first systematic analysis of in vivo functions of a eukaryotic CSAS gene and sheds light on evolutionary relationships among metazoan CSAS proteins.
Introduction
Sialylation stands out among other types of glycosylation because of the unique properties of sialic acid (Sia), including its negative charge, relatively large molecular size, and typically terminal position in glycan chains, all making Sia a crucial player in many biological processes, including nervous system development and functioning (Schauer and Kamerling, 1997; Varki, 2007). The pivotal role of sialylation in the nervous system was demonstrated in the context of gangliosides that affect nerve regeneration (Vyas et al., 2002), and polysialylation of NCAM that controls axonal fasciculation, neural migration, and synaptic plasticity (Rutishauser, 2008; Muhlenhoff et al., 2009). Sia also has an important function in neural transmission, albeit the mechanism underlying this function is less studied. Enzymatic removal of sialylated glycans suggested that sialylation influences network excitability and modulates voltage-gated Na+ channels in brain neurons (Isaev et al., 2007). Vertebrate voltage-gated Na+ channels, the major determinants of neural excitability, are heavily sialylated, which affects their gating properties in vitro (Recio-Pinto et al., 1990; Bennett, 2002). The role of sialylation in the regulation of these channels has been mostly attributed to a large negative charge due to the channels' prominent polysialylation (Zuber et al., 1992; Montpetit et al., 2009). However, polysialylation and sialylation appear to have different effects on voltage-gated Na+ channels, and the regulation of these channels was shown to be affected by some “functional” Sia residues rather than their total charge (Stocker and Bennett, 2006; Ahrens et al., 2011). At the same time, in vertebrate organisms, it is difficult to dissect these effects, and the function of sialylation in neural transmission has not been directly analyzed in vivo and remains poorly understood.
Drosophila has recently emerged as a promising model for studying animal sialylation. Sialylation was characterized in Drosophila embryos and adult brains by mass spectrometry (Aoki et al., 2007; Koles et al., 2007). It is mediated by a sole sialyltransferase, DSiaT, which is closely related to the ST6Gal family of mammalian enzymes (Koles et al., 2004). Recent experiments revealed that DSiaT has a nervous system-specific function that regulates neural transmission (Repnikova et al., 2010). The evolutionary conservation of the sialyltransferase and its substrate specificity, along with the presence of other vertebrate-type enzymes potentially working in the Drosophila sialylation pathway, suggest that this neural function of sialylation is conserved between flies and mammals (Kim et al., 2002; Viswanathan et al., 2006; Koles et al., 2009). However, the Drosophila sialylation pathway was not investigated in vivo beyond the DSiaT-mediated step, and the genetic mechanisms of the pathway remained largely unknown. In the present study, we focused on the function of Drosophila cytidine monophosphate (CMP)-sialic acid synthetase (CSAS), the enzyme predicted to work immediately upstream of DSiaT and generate a sugar donor, CMP-Sia. Our results provide the first insight into CSAS function in vivo and establish its involvement in the regulation of neural transmission. While being consistent with the hypothesis that Drosophila CSAS works upstream of DSiaT, our data also suggest that these genes have additional, independent functions in the nervous system.
Materials and Methods
Drosophila strains.
Wild-type control w1118 Canton-S was obtained from Josh Dubnau (Dubnau et al., 2001). paraLK5 (null allele), parats1 [hypomorph, conditional temperature-sensitive (TS) allele], paraDp (Dp(1;4)r+f+, a duplication of para on the fourth chromosome), and seits1 (null, TS paralytic allele) were received from Barry Ganetzky (Ganetzky, 1984; Titus et al., 1997). UAS-ManII-eGFP flies were obtained from Bing Ye (Ye et al., 2007). C155-GAL4 (Lin and Goodman, 1994), Mi[ET1]MB04236, SM6a P{w[+mC]=hsILMiT}2.4 (source of Minos transposase; Metaxakis et al., 2005), and Df(3L)XS2182/TM3 [CSAS deficiency (CSASDf)] were from the Bloomington Stock Center (Indiana University). PDI::GFP protein-trap strain was kindly provided by Alain Debec (Bobinnec et al., 2003). DSiaTS23 and DsiaTL22 are null alleles that were previously described (Repnikova et al., 2010). All Drosophila strains were reared in a controlled environment incubator (25°C, 39% humidity, 12 h light/dark cycle) on standard cornmeal-malt-yeast medium.
CSAS mutants.
To obtain a CSAS mutant allele, we searched the collection of Drosophila stocks with randomly integrated Minos transposons (Metaxakis et al., 2005). We identified a transposon insertion, Mi{ET1}MB04236 (designated here as CSASMi), that could potentially disrupt the function of CSAS due to its location within the CSAS transcribed region (see Fig. 4A). CSASMi represented a hypomorphic allele, as it was revealed by our experiments. To generate a null allele of CSAS, the Minos element in CSASMi was mobilized by the Minos transposase using the P{w[+mC]=hsILMiT} construct (Metaxakis et al., 2005). The flies with mobilized transposon were screened for potential deletions generated by the transposon excision within the CSAS locus. In this screen, we isolated a revertant, CSASR3, and a deletion within the CSAS gene, CSAS21. These generated alleles were confirmed by genomic PCR and sequencing. The following primers were used for the genomic PCR analysis of CSAS mutant alleles: CSAS-genome-up, 5′-CTGCCTTATGGAGTGGAGACC-3′; CSAS-genome-down, 5′-AAGCCTAAACTACAAAATCAAACC-3′. CSAS21 includes a 431 bp deletion removing the start codon and two exons encoding the N-terminal part of CSAS required for the enzymatic activity of CMP-Sia synthetases (Münster-Kühnel et al., 2004; Viswanathan et al., 2006; see Fig. 4A,B). Thus, CSAS21 represented a null mutation, which was also confirmed by the fact that it was indistinguishable from the CSASDf allele in all our assays.
Figure 4.
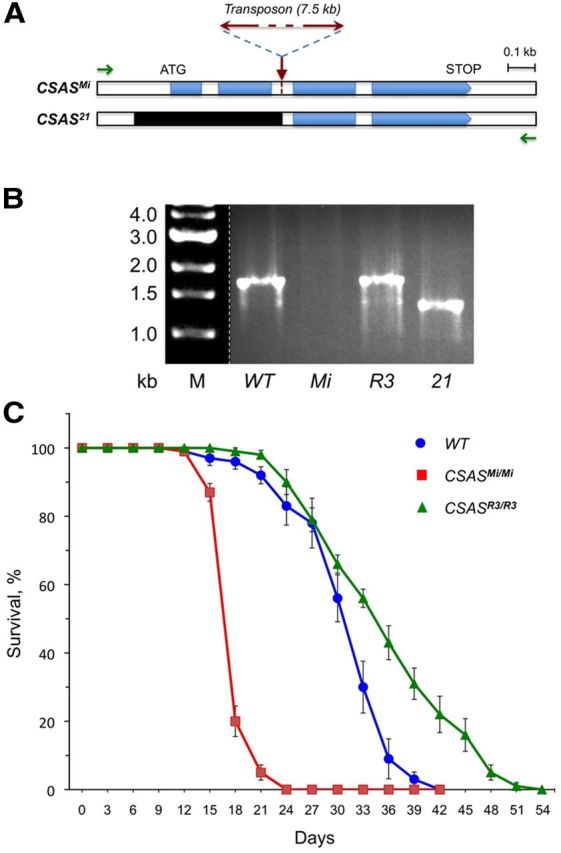
Schematic of CSAS mutants, their molecular analysis, and longevity phenotypes. A, CSAS gene region of CSASMi mutants with indicated position of the Minos transposon insertion. B, Corresponding genomic region of CSAS21 mutants with indicated deletion (black). Green arrows indicate the position of primers that were used for genomic PCR analysis of the mutants. B, Genomic PCR analysis of CSAS genotypes homozygous for wild-type (WT), Mi, R3, and 21 alleles, as indicated. The genomic PCR amplification of the region shown in A generates a 1.7 kb band for wild-type and R3 (revertant) alleles. A ∼1.3 kb PCR band is generated for CSAS21 mutants having a 431 bp deletion, while no PCR amplification occurs for CSASMi mutants due to the large transposon insertion. C, CSAS mutants have significantly decreased longevity. Survival curves for wild type (WT), CSASMi mutants, and CSASR3 revertants. At least 100 flies were analyzed for each genotype. The CSASMi allele was outcrossed 10 times to wild-type flies to minimize potential influence of genetic background. Error bars represent SEM.
CSAS rescue.
CSAS rescue construct was generated using DmCSAS cDNA plasmid (Viswanathan et al., 2006) by standard molecular cloning techniques. To facilitate the detection of CSAS expression, we engineered a tag by adding the 3FLAG-coding sequence (Sigma-Aldrich) fused in-frame after the last codon of the CSAS reading frame. Details on molecular cloning of CSAS-3FLAG are available on request. The CSAS-3FLAG sequence was subcloned into the pUAST vector for in vivo expression (Brand et al., 1994), and several transgenic Drosophila lines were generated by P-element-mediated germline transformation. Due to its leaking expression (which was confirmed by immunostaining with anti-FLAG antibody using several independent chromosomal insertions of UAS-CSAS-3FLAG), this construct could rescue the phenotypes of CSAS mutants without the presence of a GAL4 driver. Thus, UAS-CSAS-3FLAG was used in all rescue experiments without a driver, with the exception of evoked excitatory junction potential (EJP) experiments when C155-GAL4 driver was also present in the rescue genotype.
Behavioral assays.
Behavioral assays were performed essentially as described previously (Repnikova et al., 2010). Briefly, for longevity assays, adult male flies were kept individually and transferred to a new vial with fresh food every 3 d. Dead flies were counted every day. For locomotion and paralysis assays, individual flies were collected on the day of enclosure and aged for 5 d, during which they were transferred once (on day 3) to a fresh-food vial. In the righting assay, each fly was placed in an empty vial and allowed to adapt for 10 min. Thereafter, the vial with a single fly was gently banged five times onto a soft rubber pad. The time it took the fly to right itself after falling on its back was recorded. For each fly, the assay was performed with two trials and 10 min recovery in between. For TS-paralysis assays, individual flies were transferred to empty vials and temperature was shifted to 38°C by submerging vials in a controlled-temperature water bath. We defined paralysis as a condition when a fly lies on the bottom of a vial and is unable to stand and walk for at least 1 min. Paralysis phenotype can be sensitive to genetic background, and it is expressed differently in males and females. Thus, same-sex animals were compared in any particular experiment, as indicated. To decrease the influence of different genetic backgrounds, genotypes compared in experiments were outcrossed to the same reference genetic background (e.g., w1118 Canton S “wild type”) and/or represented siblings produced by the same parents. Although behavioral phenotypes can be quantitatively compared within each experiment, generally they cannot be quantitatively compared across different experiments.
CSAS in situ hybridization.
In situ hybridization was performed on Drosophila embryos and dissected tissues essentially as described previously (Koles et al., 2004) using digoxigenin-labeled RNA probe produced with Drosophila CSAS cDNA as a template. Anti-digoxigenin antibody conjugated with alkaline phosphatase was used for the detection of hybridized probe (Roche Applied Science). Images were obtained with Axioplan 2 Zeiss microscope.
Dissections and immunostaining.
Brains were dissected in ice-cold Ringer's solution, washed, and fixed in fresh fixative solution (4% paraformaldehyde, 50 mm NaCl, 0.1 m Pipes, pH 7.2) for 20 min at room temperature with gentle agitation. Fixed tissues were analyzed by immunostaining and microscopy. Immunostaining was performed using fluorescent secondary antibodies essentially as described earlier (Lyalin et al., 2006). The following primary antibodies and corresponding dilutions were used for immunostaining: rabbit anti-Lava lamp (anti-Lva) 1:2000 (a gift from John Sisson (Sisson et al., 2000); rabbit anti-HA, 1:1,000 (Covance); mouse anti-KDEL, 1:100 (Stressgen); rabbit anti-GFP, 1:800 (Invitrogen); mouse anti-FLAG M2, 1:2000 (Sigma-Aldrich). The following fluorescent secondary antibodies were used (with corresponding dilution): anti-mouse-Cy3 (1:250), anti-rabbit-FITC (1:150), anti-mouse-Cy5 (1:250) (all from donkey, Jackson Laboratories), and goat anti-rabbit Alexa-488 (1:140) (Invitrogen).
Microscopy and image analyses.
Immunofluorescent staining was examined using an Olympus FV1000 confocal microscope with 100× objective and a Zeiss Axioplan 2 with 63× objective and ApoTome module for optical sectioning. Colocalization was analyzed on multichannel confocal images. We used ImageJ software with JACoP module to quantify colocalization and calculate Manders' correlation coefficients (MCCs) that measure co-occurrence of fluorescent signals recorded in the same focal plane in different channels (Bolte and Cordelières, 2006; Dunn et al., 2011).
Electrophysiology.
Current-clamp intracellular recordings were performed from muscle 1 neuromuscular junction (NMJ) of dissected third instar larvae as previously described (Repnikova et al., 2010). Briefly, Drosophila third instar larvae were dissected in ice-cold Ca2+-free HL3 solution and electrophysiological recordings were performed at room temperature. Microelectrodes were filled with 3 m KCl and had an input resistance of 8–16 MΩ. Membrane potentials were amplified by the Axoclamp 200B amplifier, digitized and analyzed on a Dell PC using pClamp10 software (Molecular Devices). Spontaneous miniature EJP (miniEJP) recordings were performed at 1 mm extracellular Ca2+ and analyzed using a semiautomated protocol (Repnikova et al., 2010). Evoked EJP analysis was performed by averaging 10 events for each NMJ recording and considering the average amplitude as one measurement.
Statistical analyses.
Unpaired two-tailed t test was used for comparing two groups of data. For multiple-groups comparison, one-way ANOVA was used as a first step to reveal possible presence of significant differences in mean (Kruskal–Wallis p < 0.0001). As a post hoc test, Wilcoxon each pair nonparametric test was applied to assess differences between the groups.
Results
Drosophila CSAS is expressed primarily in the CNS throughout development
To shed light on the in vivo function of CSAS, we examined the expression of the CSAS gene during different developmental stages using in situ hybridization. No maternal mRNA was detected at early embryonic stages. Initiation of zygotic expression was detected during late embryogenesis, at stage 16 (∼14 h of embryonic development), in the developing CNS. CSAS expression was upregulated in many cells throughout the ventral ganglion and brain hemispheres in the late embryo. This broad expression continues in the brain and the ventral ganglion during all larval stages (Fig. 1A,B). CSAS was also expressed in the adult brain, with more prominent expression being present in the optic lobes (Fig. 1C). Thus, the expression of CSAS is developmentally regulated, dynamic, and appears to be differentially regulated between different cells within the CNS. We did not detect the expression of CSAS outside of the CNS at any examined developmental stages. Thus the function of CSAS appears to be primarily restricted to the nervous system.
Figure 1.

The expression of CSAS in Drosophila brain during different developmental stages. A, CSAS expression in the late embryo at stage 17 is detected in the developing CNS, including the ventral ganglion (arrowheads) and brain hemispheres (arrows). Lateral view, Anterior is to the right. B, Third instar larval brain shows a widespread CSAS expression. Arrowheads, Expression in the lateral regions of the ventral ganglion. Arrows, Expression in brain hemispheres. Ventrolateral view, Anterior is to the right. C, CSAS is expressed broadly in the adult brain, including the optic lobes (arrows). Frontal view, Only one optic lobe is shown.
CSAS is a glycoprotein that localizes in vivo to the secretory compartment
The subcellular localization of mammalian CSAS is strictly regulated, with all CSAS proteins studied so far being localized to the nucleus. In contrast to its vertebrate counterparts, Drosophila CSAS was reported to be present in the Golgi when it was introduced transgenically into mammalian cultured cells. However, the expression of CSAS was not previously examined in Drosophila cells. To reveal the subcellular distribution of CSAS in vivo, we expressed a FLAG-tagged CSAS in the Drosophila CNS using the UAS-GAL4 system. The functionality of the FLAG-tagged CSAS was confirmed by rescue experiments (see Figs. 5, 6A, 7B). We characterized the localization of CSAS by immunofluorescence using double staining for several markers of subcellular compartments (Fig. 2). Colocalization between CSAS and these markers was analyzed on confocal images and quantified using Manders' correlation coefficients (MCCs) that measure co-occurrence of fluorescent signals recorded in the same focal plane in different channels (Dunn et al., 2011). Most prominent colocalization was detected between CSAS and DSiaT (Fig. 2A–C; MCC = 0.73, representing a fraction of CSAS overlapping with sialyltransferase, a marker of trans-Golgi compartment (Stanley et al., 2009; Repnikova et al., 2010)). We also found significant colocalization of CSAS with α-mannosidase-II (ManII; Fig. 2D–F), a marker of medial-Golgi, and Lva (Fig. 2G–I), a marker of cis-Golgi (MCC = 0.46 and 0.43, respectively) (Sisson et al., 2000; Ye et al., 2007; Stanley et al., 2009). Further decreased amount of CSAS was detected in the endoplasmic reticulum (ER) by minimal colocalization with ER markers, KDEL, and protein disulfide-isomerase (MCC = 0.29 and 0.26, respectively; Fig. 2J–O (Munro and Pelham, 1987; Morin et al., 2001; Lyalin et al., 2006)). No colocalization was found with DAPI staining of the nucleus (MCC = 0.0; Fig. 2P–R). Thus, these experiments indicated that CSAS is localized in the secretory compartment of the cell, predominantly in the trans-Golgi. Because secretory pathway proteins are commonly glycosylated, we decided to examine whether the CSAS is modified with N-linked glycans. To this end, FLAG-tagged CSAS was purified from Drosophila heads and then analyzed by Western blotting (Fig. 3). The CSAS protein appeared on Western blots as two closely positioned bands in the range of 35–39 kDa. To test for the possible presence of N-glycans, we analyzed the CSAS protein that was treated with peptide N-glycosidase F (PNGase F) (Tarentino et al., 1990). PNGase F specifically removes N-glycan chains from glycoproteins, and hence the decrease of molecular mass upon PNGase F treatment is indicative of N-glycosylation. Indeed, we found that PNGase F treatment increased the electrophoretic mobility of CSAS, resulting in the collapse of the two CSAS bands into a new, lower-molecular-mass band. We did not detect any nonglycosylated CSAS isoform, which indicated that CSAS is an obligatory secretory pathway glycoprotein bearing N-linked glycans. These data confirmed the results of our immunolocalization experiments.
Figure 5.

CSAS mutations cause TS paralysis. TS-paralysis assays of flies with different combinations of CSAS alleles. CSASMi mutants have a slightly milder paralysis phenotype than CSAS21, which suggests that CSASMi is a strong hypomorphic mutation. At least 20 flies were assayed for each genotype. WT, Wild-type control; CSASMi/Mi, CSASMi homozygous mutant; CSAS21/21, CSAS21 homozygous mutant; CSASMi/21 and CSASMi/Df, heteroallelic CSAS mutants carrying combinations of CSAS mutant alleles; CSASR3/R3, homozygous for a revertant allele; CSAS+ CSASMi/Df, CSASMi/Df mutant rescued by transgenic expression of CSAS-FLAG. Statistical analysis was performed using one-way ANOVA followed by post hoc multiple-comparison Wilcoxon each pair test. *p < 0.05. Error bars represent SEM.
Figure 6.

CSAS mutants have a defect of neural transmission at larval NMJ. A, Evoked EJP amplitudes recorded from abdominal muscle 1 in third instar larvae. EJP is significantly reduced in CSASMi and CSAS21 mutants, compared with wild-type larvae. CSAS+ CSASMi/Mi, EJP defect was rescued in CSASMi homozygous mutants by transgenic expression of CSAS-FLAG. At least 14 larvae were analyzed for each genotype. Statistical analysis was performed by one-way ANOVA followed with multiple-comparison Wilcoxon each pair test. *p < 0.05, ***p < 0.001, statistically significant differences. B, Representative traces of evoked EJPs. C, The evoked EJP defect of CSAS mutants can be rescued by increased Ca2+ concentration. Ten larvae were assayed for each data point. Error bars represent SEM. See Figure 5 for explanation of genotypes.
Figure 7.
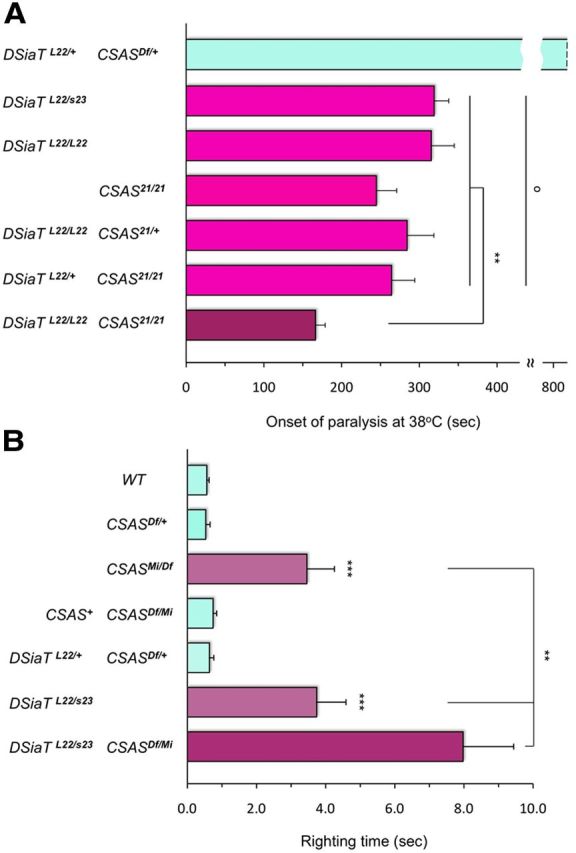
Genetic interactions between null alleles of CSAS and DSiaT analyzed by behavioral assays. A, Interactions between null alleles of CSAS and DSiaT analyzed by TS-paralysis assays. While CSAS and DSiaT single mutants show a quantitatively similar TS-paralysis phenotype, the paralysis of CSAS-DSiaT double mutants is more severe than that of the single mutants. For the genotype combining a homozygous mutation in one gene with a heterozygous mutation of another gene, the phenotypes are not significantly different from each other and from the phenotypes of single homozygous mutants. At least 20 flies (males) were assayed for each genotype.DSiaTL22/+ CSASDf/+, double heterozygote for CSAS and DSiaT combining one copy of null mutation for each gene; DSiaTL22/s23, heteroallelic DSiaT mutant combining two null alleles; DSiaTL22/L22, homozygous null mutant; CSAS21/21, homozygous null mutant; DSiaTL22/L22 CSAS221/+, combination of DSiaTL22 homozygote with CSAS221 heterozygote; DSiaTL22/+ CSAS221/221, combination of DSiaTL22 heterozygote with CSAS221 homozygote; DSiaTL22/L22 CSAS221/221, double homozygous null mutant for DSiaT and CSAS. B, Interactions analyzed by locomotion assays. CSAS and DSiaT homozygous single mutants have prominent locomotion phenotypes that are qualitatively and quantitatively indistinguishable from each other. At the same time, CSAS-DSiaT double mutants have a significantly stronger phenotype than that of single mutants. The locomotion defect of CSAS mutants can be fully rescued by transgenic expression of the CSAS-FLAG construct (CSAS+ CSASMi/Df). See above and Figure 5 for explanation of genotypes. A, B, Statistical analysis was performed using one-way ANOVA followed by post hoc multiple-comparison Wilcoxon each pair test. **p < 0.01, ***p < 0.001, statistically significant differences; o, no significant differences (p > 0.24). Error bars are SEM.
Figure 2.
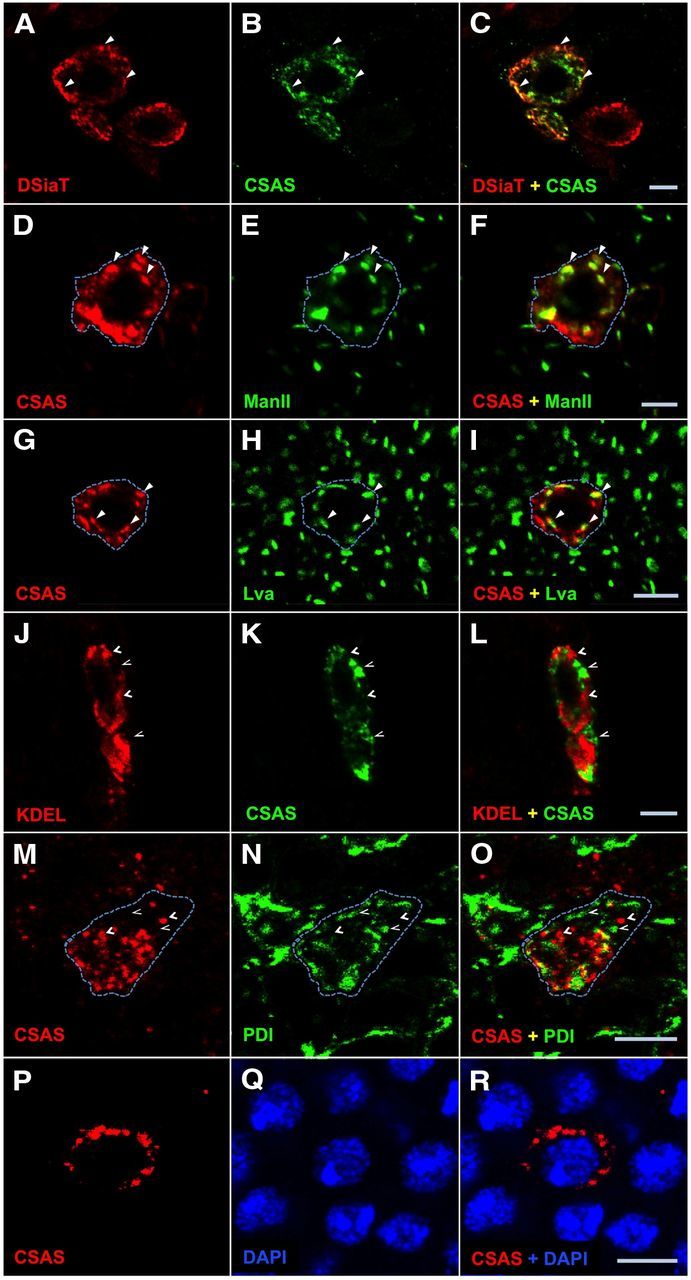
CSAS protein localizes to the secretory pathway compartment in Drosophila neurons. A–C, CSAS is largely colocalized with sialyltransferase (DSiaT) within the cell. A, DSiaT (red). B, CSAS (green). C, Overlay of A and B. D–F, Simultaneous detection of CSAS (D, red) and ManII (E, green) reveals significant colocalization of these proteins. F, Overlay of D and E. G–I, CSAS (G, red) shows substantial colocalization with Lva (H, green). I, Overlay of G and H. J–L, Codetection of KDEL (J, red) and CSAS (K, green) shows a minimal overlap between their localizations. L, Overlay of J and K. M–O, CSAS (J, red) and protein disulfide-isomerase (PDI; K, green) are minimally colocalized within the cell. O is the overlay of M and N. P–R, CSAS (J, red) and DAPI (K, green) have no colocalization within the cell. R is the overlay of P and Q. Solid and open arrowheads indicate examples of colocalization and the absence of colocalization, respectively. Dashed line helps visualize an approximate area occupied by a single cell with CSAS expression. Images represent confocal sections of third instar larval brains. CSAS-FLAG, ManII, and DSiaT proteins were expressed in the brain using C155 neuronal driver. PDI-GFP is a protein-trap that expresses endogeneous PDI tagged with GFP. Scale bars, 5 μm.
Figure 3.

CSAS protein is modified with N-linked glycans when expressed in vivo. Western-blot analysis of CSAS-FLAG expressed in Drosophila heads. Lane 1, Cell lysate from heads. Lane 2, Affinity purified CSAS-FLAG, PNGase F mock-treated control. Lane 3, Affinity purified CSAS-FLAG treated with PNGase F. The decrease of CSAS molecular mass upon PNGase treatment indicates the presence of N-linked glycans. IgHC and IgLC indicate Ig chains leached from FLAG-affinity beads in denaturing conditions. Approximate positions of molecular mass markers are shown on the left.
CSAS mutants have significantly decreased life span
To elucidate the function of CSAS in vivo, we characterized the phenotypes of CSAS mutants. We started with the analysis of the CSASMi allele. CSASMi has a Minos transposon insertion in the second intron of the CSAS gene (Fig. 4A). We outcrossed this allele 10 times to an isogenized wild-type strain (w1118 Canton-S). The outcrossed CSASMi mutants were homozygous viable, without any noticeable morphological defects. However, the flies appeared to be weak and short-lived. We found that the longevity of the mutant flies was decreased by 43% compared with the wild-type control flies with matching genetic background (average lifespan at 29°C of wild-type and CSASMi flies ± SD, respectively: 30.1 ± 5.7 d and 17.2 ± 1.8 d, t test p = 5E-43). Although outcrossing had ensured the minimal influence of genetic background in this experiment, we wanted to confirm that the phenotype was indeed associated with the transposon insertion. By mobilizing the Minos element, we generated a revertant, CSASR3, that lost the transposon, having the fully restored endogenous gene sequence (Fig. 4B; see details in Materials and Methods). The reversion restored the longevity (average lifespan of CSASR3 flies at 29°C ± SD: 35.4 ± 8.1 d), which confirmed that the phenotype was associated with the transposon insertion within the CSAS gene (Fig. 4C).
CSAS mutants are paralyzed at elevated temperature
Mutations in DSiaT were shown to result in TS paralysis (Repnikova et al., 2010). Considering the hypothesis that DSiaT and CSAS function in the same pathway, we tested whether CSAS mutants have similar phenotype. We found that CSASMi flies are also paralyzed at elevated temperature, with the onset of paralysis being within 3–7 min at 38°C (Fig. 5). Both the longevity and the paralysis phenotypes of CSAS mutants were very similar to that of DSiaT-null mutants (Repnikova et al., 2010), which suggested that the CSASMi allele possibly represents a strong hypomorph or a null allele. To be able to use the CSASMi allele in comparative studies, we decided to characterize it in more detail. We included in our analyses other CSAS mutant alleles, i.e., CSASDf, a deficiency removing the whole CSAS locus (McQuilton et al., 2012), and CSAS21, a null mutant that we generated by mobilizing the Minos element in the CSASMi allele (Fig. 4; Materials and Methods). TS-paralysis assays revealed that CSASMi homozygotes had a slightly milder phenotype than CSAS21 homozygous mutants, or mutants with heteroallelic combinations with CSAS21 or the deficiency (Fig. 5). These results indicated that CSASMi indeed represents a strong hypomorphic allele. The CSAS21 allele was indistinguishable from the deficiency allele, which confirmed that CSAS21 is a null mutation. No paralysis was detected in heterozygous mutants, indicating that the CSAS mutations are recessive. The phenotype was restored to wild type in the CSASR3 revertants, while the transgenic expression of FLAG-tagged CSAS fully rescued the phenotype of CSAS mutants (Fig. 5). Together, these results confirmed that the mutant phenotype was associated with the CSAS defect.
CSAS affects neural transmission
The CNS-specific expression and mutant phenotypes of CSAS suggested that CSAS plays an important role in the regulation of neural functions. To investigate this role in more detail, we used electrophysiological assays to characterize neural transmission in CSAS mutants at the level of individual NMJs in third instar larvae. We focused our analyses on the NMJ of abdominal muscle 1. This NMJ was previously found to be affected in DSiaT mutants (Repnikova et al., 2010). No significant difference was detected in the resting membrane potential of muscle cells between mutant and wild-type larvae (CSAS21, −63.8 ± 2.2 mV; CSASMi, −62.6 ± 2.2 mV; wild-type control, −62.6 ± 3.8 mV; errors are SD; sample size for each genotype N > 12), which suggested that muscle physiology is not affected in CSAS mutants. This result is consistent with the fact that neither CSAS nor DSiaT was found to be expressed in muscles by in situ hybridization (Repnikova et al., 2010; this study). Next, using intracellular recordings, we analyzed evoked EJPs and found that EJP amplitude was strongly reduced in CSAS mutants (Fig. 6A,B). EJP amplitude was more affected in CSAS21, compared with CSASMi, which was consistent with the result of paralysis assays indicating that CSASMi represents a hypomorphic allele associated with low residual activity of CSAS. As in the case of behavioral phenotypes, the defect in the EJP of CSAS mutants was fully rescued by CSAS transgenic expression, which confirmed that the EJP phenotype was caused by the compromised function of CSAS. Since the phenotype of decreased EJP amplitude in most cases results from either a defect in synaptic transmission or an abnormal excitability of neurons, we further investigated these two possibilities. To shed more light on synaptic functions, we analyzed the spontaneous NMJ synaptic transmission in the absence of stimulation by examining miniEJPs. No significant difference between mutants and wild-type larvae was found when we analyzed the amplitude and frequency of miniEJP: amplitude was 1.12 ± 0.09 mV and 0.95 ± 0.08 mV, and frequency was 1.35 ± 0.06 Hz and 1.41 ± 0.04 Hz for wild-type larvae and CSASMi mutants, respectively (errors are SEM, 10 larvae were analyzed for each genotype, t test p > 0.16). These results suggested that CSAS mutants have no significant defects in stimulus-independent synaptic vesicle exocytosis, the amount of neurotransmitter per synaptic vesicle, and the postsynaptic response to neurotransmitter (Verstreken and Bellen, 2001; Repnikova et al., 2010). Next, we analyzed the evoked EJP at elevated external Ca2+ concentration, the condition that increases the probability of synaptic vesicle fusion and neurotransmitter release up to a saturated level. This condition is expected to rescue defects associated with decreased probability of release, but would not rescue the phenotypes with diminished number of active zones at the synapse or an impairment of Ca2+-mediated exocytosis (Aravamudan et al., 1999; Stewart et al., 2000; Bao et al., 2005; Long et al., 2008). We found that Ca2+ concentrations of >2 mm could fully rescue the EJP (Fig. 6C), which suggested that the number of active zones and Ca2+-triggered exocytosis are probably not affected in CSAS mutants. All these phenotypes revealed that the effect of CSAS on neural transmission closely resembles that of DSiaT (Repnikova et al., 2010), which is consistent with the hypothesis that CSAS and DSiaT function within the same pathway. Together, our results support the notion that CSAS is involved in the regulation of neural transmission. Since synaptic function is not significantly affected by CSAS, its activity probably affects neuronal excitability.
Relation between CSAS and DSiaT functions at organismal and cellular levels
To reveal the relationship between CSAS and DSiaT functions in more detail, we created and analyzed double mutants with different combinations of CSAS and DSiaT alleles. First, we characterized the phenotype of these mutants at the organismal level. Using TS-paralysis assays and null alleles, we found that homozygous CSAS or DSiaT mutants, as well as the genotypes combining the homozygous mutation of one gene with the heterozygous mutation of another gene (tests for dominant enhancement), all have statistically indistinguishable phenotypes (Fig. 7A). Double heterozygotes were found to be similar to wild-type flies, suggesting that removing one functional copy for each of those genes is not sufficient to compromise the pathway. Unexpectedly, we found that the phenotype of double homozygotes was more severe than the phenotype of single mutants, indicating that CSAS and DSiaT mutations have additive effects on neural functions. This result suggested some independent roles for DSiaT and CSAS in the nervous system. Thus, these genes are involved in more complex interactions than those predicted by a linear genetic pathway.
In addition to temperature-induced paralysis, we also assayed the locomotion of the mutants. We analyzed the ability of flies to right themselves after being momentarily knocked down by a gentle agitation. DSiaT mutants were previously found to have a prominent defect in locomotion, including coordination and righting ability (Repnikova et al., 2010). When we compared CSAS and DSiaT mutants using the righting assay, we found that the phenotypes of DSiaT homozygous null mutants and CSAS mutants combining a deficiency and a strong hypomorphic allele were indistinguishable. At the same time, the phenotype of the double mutants was significantly stronger than that of single mutants (Fig. 7B). This enhancement of the phenotype of DSiaT mutants by CSAS mutations suggested that CSAS exerts some additional, DSiaT-independent effect on neural function. These results were consistent with the data of paralysis assays (Fig. 7A). Additionally, they indicated that neural functions of CSAS mutants are affected not solely at elevated temperature, but also at regular temperature, and thus they are generally compromised.
In addition to behavioral approaches, we also used electrophysiological assays to characterize the phenotype of CSAS-DSiaT double mutants at the cellular level (i.e., at individual NMJs). The evoked EJP at abdominal muscle 1 NMJs was significantly reduced in the double mutants compared with DSiaT single mutants (Fig. 8). This was consistent with the more pronounced behavioral defects of the double mutants as revealed by paralysis and righting assays (Fig. 7). Interestingly, unlike the behavioral phenotypes, the evoked EJP phenotype was more pronounced in CSAS mutants than in DSiaT mutants, while no significant difference was observed between CSAS mutants and CSAS-DSiaT double mutants.
Figure 8.

Interactions between CSAS and DSiaT analyzed by electrophysiological assays. A, The EJP defect of CSAS-DSiaT double mutants is stronger than that of DSiaT single mutants, while the double mutants and CSAS single mutants have similar EJP phenotypes. ** and o indicate statistically significant (p < 0.01) and not significant (p > 0.06) differences, respectively. At least 10 larvae were analyzed for each genotype. Error bars are SEM. B, Representative traces of evoked EJPs. Assays were performed on muscle 1 NMJ of third instar larvae. See Figure 7 for explanation of genotypes.
CSAS shows strong genetic interactions with ion channel genes
DSiaT potentiates neural excitability and strongly interacts with the voltage-gated Na+ channel gene paralytic (para), while the function of the Para channel was found to be affected in DSiaT mutants (Repnikova et al., 2010). Our electrophysiological assays suggested that CSAS is also involved in the regulation of neural excitability. Para is a major player of neural excitability pathway in Drosophila (Loughney et al., 1989). Hence we decided to test whether CSAS and para exhibit genetic interactions. Using two different para mutant alleles, the TS allele parats1 and the null allele paraLk5, we investigated the effect of combining CSAS and para mutations on the TS paralysis. While para heterozygous mutants were not paralytic, the substitution of one normal copy of para in CSAS homozygous mutants with parats1 or paraLk5 resulted in a dramatic enhancement of the paralysis phenotype (Fig. 9A). This synergistic effect of para mutations on the paralysis of CSAS mutants could be fully rescued by adding a copy of para duplication, which confirmed the specificity of interactions. Moreover, the phenotype of CSAS mutants was ameliorated in the presence of three normal copies of the para locus (para+/+/Dp). Thus, the phenotype of CSAS mutants is very sensitive to the dosage of Para.
Figure 9.
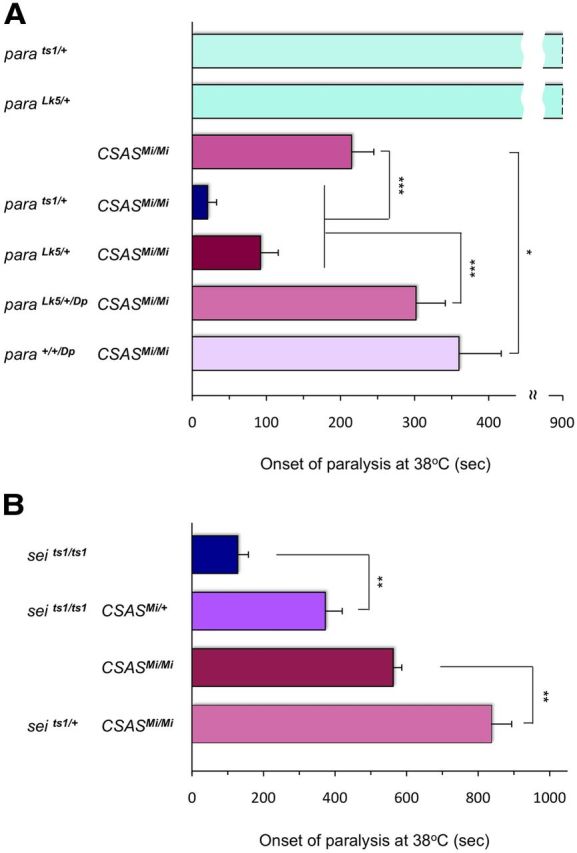
Genetic interactions between CSAS and voltage-gated ion channel genes. A, CSAS and para show strong synergistic interactions. TS-paralysis assays of flies with different combinations of CSAS and para mutant alleles. Whereas heterozygous para mutations do not have TS paralysis, they significantly exacerbate the paralysis defect of CSAS mutants. This synergistic enhancement of the phenotype is rescued by a para genetic duplication (paraDp). The presence of three copies of normal para locus (para+/+/Dp) can ameliorate the phenotype of CSAS mutants even further. parats1/+ and paraLk5/+, heterozygotes for para TS and null alleles, respectively; CSASMi/Mi, CSAS homozygous mutant; parats1/+ CSASMi/Mi and paraLk5/+ CSASMi/Mi, combinations of two different para heterozygous alleles with CSASMi homozygote; paraLk5/+/Dp CSASMi/Mi, combination of para heterozygous null allele, para duplication, and CSASMi homozyous mutant; para+/+/Dp CSASMi/Mi, combination of para homozygous wild-type allele, para duplication, and CSASMi homozyous mutant. At least 20 five-day-old female flies were assayed for each genotype. Statistical analysis was performed using one-way ANOVA followed by multiple-comparison Wilcoxon each pair test. *p < 0.05 and ***p < 0.001, statistically significant differences. B, CSAS and sei exhibit genetic interactions while alleviating each other's paralysis phenotype. TS-paralysis assays of single and double mutants for CSAS and sei. Adding a single mutant allele of CSAS to seits1 homozygotes significantly diminishes their TS-paralysis phenotype. A heterozygous seits1 mutation has a similar effect on CSAS homozygous mutants. At least 20 five-day-old flies were assayed for each genotype. Compared genotypes represent siblings obtained from the same parents. t test, **p < 0.01, statistically significant differences. Error bars are SEM.
Together with voltage-gated Na+ channels, voltage-gated K+ channels also play an essential role in determining neural excitability. However, the activities of the Na+ and K+ channels have opposing effects on the membrane potential, with K+ channels being responsible for membrane repolarization during the declining phase of the action potential (Wu and Ganetzky, 1992; Bean, 2007). We decided to test interactions between CSAS and seizure, the Drosophila erg K+ channel gene that is broadly expressed in the CNS (Titus et al., 1997; Wang et al., 1997). We used the recessive loss-of-function allele seits1 that is associated with hyperactivity and TS-paralysis phenotype (Titus et al., 1997; Wang et al., 1997). Since sei mutations increase membrane excitability, they are expected to counteract the decrease of neural excitability resulting from compromised sialylation. Indeed, we found that a single copy of seits1 could significantly ameliorate the paralysis of CSAS mutants (Fig. 9B). Moreover, adding a heterozygous CSAS mutation to seits1 homozygous mutants resulted in the diminishing of TS-paralysis phenotype. Thus, CSAS and sei show prominent interaction, while the genetic mode of this interaction is opposite to the mode of interaction between CSAS and para.
Discussion
This work offers the first systematic investigation of in vivo functions and mutant phenotypes of a eukaryotic CSAS gene. We found that Drosophila CSAS is expressed specifically in the CNS throughout development (Fig. 1) and its expression is developmentally regulated, starting in the late embryogenesis (stage 16) and continuing broadly within the developing CNS and the adult brain. This pattern suggested that CSAS has a nervous system-specific function, which was confirmed by the phenotype analyses of CSAS mutants generated in our experiments. These analyses revealed that CSAS mutations are associated with prominent neurological abnormalities. At the organismal level, the phenotypes included significantly shortened life span, TS paralysis, and locomotion defects (Figs. 4, 5, 7B). At the cellular level, we found that neural transmission was severely compromised at individual NMJs. Electrophysiological assays demonstrated that CSAS mutants exhibited strongly diminished EJP amplitudes, while no significant defects of muscle physiology or synaptic transmission per se were detected (Fig. 6). All these mutant phenotypes closely resemble those of DSiaT mutants (Repnikova et al., 2010). These results indicate that the function of CSAS impinges on the same aspects of neural transmission that are affected by DSiaT and suggest that this function concentrates mainly on the regulation of neural excitability. Data presented here are consistent with the hypothesis that CSAS collaborates with DSiaT in vivo, while working at the penultimate step of the sialylation pathway.
The notion that neural excitability is compromised in CSAS mutants is further supported by the strong genetic interactions between CSAS and the two voltage-gated channel genes, para and sei, that control membrane excitability in the CNS (Fig. 9). Para is the predominant voltage-gated Na+ channel ubiquitously expressed in the nervous system and responsible for the generation of action potential in most Drosophila neurons (Loughney et al., 1989). Sei encodes the erg voltage-dependent outwardly rectifying K+ channel that is broadly expressed throughout the CNS (Titus et al., 1997; Wang et al., 1997). Mutations in each of these channel genes result in TS paralysis. However, they exert opposite effects on neural excitability: while para mutations decrease excitability of neurons, the loss of sei leads to neural hyperactivity (Wu and Ganetzky, 1992; Titus et al., 1997). Consistent with their distinct effects on excitability, these channel genes have different genetic modes of interaction with CSAS: para shows a strong synergistic interaction with CSAS in producing TS paralysis; sei and CSAS mutations, on the other hand, ameliorate the paralysis phenotype of each other. It is interesting to note that an extra copy of para could partially rescue the CSAS phenotype (Fig. 9A). This suggests that CSAS mutants may have a decreased amount of Para, which could also explain the defect of neural excitability observed in these mutants. In this scenario, the CSAS phenotype is expected to be ameliorated by an extra copy of para, the condition leading to an increased amount of functional Para (Wu and Ganetzky, 1992). This scenario is consistent with the hypothesis that compromised sialylation results in the reduction of cell surface density of cardiac sodium channel in a mouse model of heart failure (Ufret-Vincenty et al., 2001). Similarly, a role of sialylated glycans in promoting cell surface expression of a channel protein was also suggested by cell culture experiments with voltage-gated potassium channels (Watanabe et al., 2007; Noma et al., 2009). However, further experiments are required to test more directly whether sialylation indeed regulates the amount of Para on the cell surface.
To shed light on the regulatory mechanisms of Drosophila sialylation and the relationship between DSiaT and CSAS, we quantitatively compared their phenotypes. We also created genotypes with various combinations of DSiaT and CSAS mutations to be included in this analysis. Using TS-paralysis assays, we found that double heterozygotes did not show mutant phenotypes, indicating that just one normal copy for each of those genes can fully restore the function of sialylation pathway. At the same time, the phenotypes of Diet-null and CSAS-null homozygotes were not significantly different, while combining a homozygous null mutation of one gene with a heterozygous mutation of another gene did not influence the mutant phenotype that was similar for all these genetic combinations (Fig. 7A). This absence of dominant enhancement was consistent with the idea that these two genes participate in the same pathway. However, we found that double homozygotes for null alleles of CSAS and DSiaT had more prominent defects than the corresponding single mutants. These conclusions were confirmed by locomotion assays that additionally demonstrated that CSAS neurological phenotypes are expressed not only at elevated temperatures but also in normal environments (Fig. 7B). The exacerbation of defects in double-null mutants indicated that CSAS and DSiaT have additive effects on neural functions. The roles of CSAS and DSiaT in the biosynthesis of sialylation predict a simple linear pathway where CSAS and DSiaT mediate two consecutive steps (Angata and Varki, 2002). However, our results revealed more complex interactions and suggested that these genes have some independent functions impinging on neural transmission.
We investigated the CSAS–DSiaT relationship using electrophysiological recording from individual NMJs and found that the defect in evoked EJPs was significantly more pronounced in CSAS-DSiaT double mutants than in DSiaT single mutants (Fig. 8). This observation was consistent with more severe behavioral phenotypes of the double mutants, compared with DSiaT mutants (Fig. 7A,B). At the same time, EJP assays revealed that defect in neural transmission was stronger in CSAS mutants than in DSiaT mutants, while the phenotype of CSAS-DSiaT double mutants was not significantly different from that of CSAS mutants (Fig. 8). This contrasts the results of behavioral assays that demonstrated that CSAS and DSiaT mutants had quantitatively similar phenotypes. The EJP phenotypes are consistent with the idea that CSAS functions upstream of DSiaT in the sialylation pathway, indicating that genetically CSAS is epistatic to DSiaT in NMJ neural transmission. However, EJP assays also suggest that CSAS has some additional, DSiaT-independent function. The difference between the results of electrophysiological and behavioral assays probably reflects the fact that CSAS and DSiaT have distinct functional relations at different developmental stages (i.e., larval vs adult) and/or these genes may interact differently while controlling diverse neural functions (e.g., NMJ neural transmission vs CNS neural circuits). Together, our results demonstrate that CSAS and DSiaT have a complex mode of genetic interactions that do not conform with the scenario of a simple linear-pathway relationship between these genes. To the best of our knowledge, this is the first example of such complex functional interactions between CSAS and a sialyltransferase in animals. It is tempting to speculate that these genes may have novel functions that possibly involve some nonenzymatic activities of their protein products. However, further studies will be necessary to elucidate the mechanism underlying these interactions and test whether this mechanism is evolutionarily conserved.
To explore CSAS functions at the cellular level, we characterized the subcellular localization of the CSAS protein in vivo. All reported mammalian CSAS orthologs are nuclear-localized proteins, which is one of the most intriguing and unique features of these enzymes (Kean et al., 2004; Buschiazzo and Alzari, 2008). The nuclear localization was found to be not required for the enzymatic role of these proteins, and thus the function of the nuclear localization remains unknown (Munster et al., 2002). Albeit Drosophila CSAS shows significant sequence conservation when compared with mammalian counterparts, the CSAS protein was detected in the Golgi when expressed transgenically in mammalian cell culture (Viswanathan et al., 2006). However, the expression of the CSAS protein was not previously analyzed in Drosophila cells. Thus, we investigated in detail the localization of Drosophila CSAS in vivo. We used a FLAG-tagged CSAS protein that could fully rescue the phenotypes of CSAS mutants and thus represented a functional CSAS protein. Our immunostaining experiments revealed that CSAS was present in the secretory compartment of the cell, predominantly localizing to the Golgi in the CNS neurons (Fig. 2). This conclusion was confirmed by the analysis of CSAS glycosylation that demonstrated that CSAS is a secretory pathway glycoprotein bearing N-linked carbohydrate modifications (Fig. 3). Interestingly, we found significant colocalization between CSAS and DSiaT, which indicated that CSAS concentrates largely in the trans-Golgi, the locale of the secretory pathway where sialylation occurs (Varki et al., 2009). This suggests a possibility that DSiaT and CSAS may function within the same molecular complex. The difference between subcellular localization of Drosophila CSAS and its mammalian orthologs reflects a remarkable evolutionary event, possibly driven by an increased demand for the amount and variety of sialylated structures in deuterostomes, including mammals, that favored the spatial separation of the last two enzymatic steps of the sialylation pathway. To our knowledge, this difference in the localization between Drosophila and mammalian CMP-Sia synthetases represents so far the only example of such a radical evolutionary change in the subcellular localization demonstrated for any orthologous eukaryotic proteins. It is tempting to speculate that this evolutionary change was associated with the acquisition of additional regulatory mechanisms of sialylation. Further comparative studies of sialylation pathway in different organisms should shed light on this intriguing aspect of sialobiology.
Footnotes
This work was supported by National Institutes of Health Grants GM069952 and NS075534 to V.M.P. We thank Hong Bao and Bing Zhang for their invaluable help with the methodology of NMJ electrophysiology. We are grateful to Barry Ganetzky, Bing Ye, Josh Dubnau, and Alain Debec for fly strains; Michael Betenbaugh and Rita Gerardy-Schahn for CMP-Sia synthetase cDNA; and John Sisson for Lva antibody. We thank Michael Tiemeyer, Naosuke Nakamura, and Dmitry Lyalin for discussions. We appreciate the assistance of Niraj Kc with genetic experiments and the help of Stan Vitha with confocal microscopy. We are thankful to Paul Hardin and Darya Panina for comments on this manuscript. We acknowledge the use of Texas A&M Microscopy and Imaging Center confocal facility, antibodies from Developmental Studies Hybridoma Bank (University of Iowa), and Drosophila strains from the Bloomington Stock Center (Indiana University).
References
- Ahrens J, Foadi N, Eberhardt A, Haeseler G, Dengler R, Leffler A, Mühlenhoff M, Gerardy-Schahn R, Leuwer M. Defective polysialylation and sialylation induce opposite effects on gating of the skeletal Na+ channel NaV1.4 in Chinese hamster ovary cells. Pharmacology. 2011;87:311–317. doi: 10.1159/000327389. [DOI] [PubMed] [Google Scholar]
- Angata T, Varki A. Chemical diversity in the sialic acids and related alpha-keto acids: an evolutionary perspective. Chem Rev. 2002;102:439–469. doi: 10.1021/cr000407m. [DOI] [PubMed] [Google Scholar]
- Aoki K, Perlman M, Lim JM, Cantu R, Wells L, Tiemeyer M. Dynamic developmental elaboration of N-linked glycan complexity in the Drosophila melanogaster embryo. J Biol Chem. 2007;282:9127–9142. doi: 10.1074/jbc.M606711200. [DOI] [PubMed] [Google Scholar]
- Aravamudan B, Fergestad T, Davis WS, Rodesch CK, Broadie K. Drosophila UNC-13 is essential for synaptic transmission. Nat Neurosci. 1999;2:965–971. doi: 10.1038/14764. [DOI] [PubMed] [Google Scholar]
- Bao H, Daniels RW, MacLeod GT, Charlton MP, Atwood HL, Zhang B. AP180 maintains the distribution of synaptic and vesicle proteins in the nerve terminal and indirectly regulates the efficacy of Ca2+-triggered exocytosis. J Neurophysiol. 2005;94:1888–1903. doi: 10.1152/jn.00080.2005. [DOI] [PubMed] [Google Scholar]
- Bean BP. The action potential in mammalian central neurons. Nat Rev Neurosci. 2007;8:451–465. doi: 10.1038/nrn2148. [DOI] [PubMed] [Google Scholar]
- Bennett ES. Isoform-specific effects of sialic acid on voltage-dependent Na+ channel gating: Functional sialic acids are localized to the S5–S6 loop of domain I. J Physiology. 2002;538:675–690. doi: 10.1113/jphysiol.2001.013285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bobinnec Y, Marcaillou C, Morin X, Debec A. Dynamics of the endoplasmic reticulum during early development of Drosophila melanogaster. Cell Motil Cytoskeleton. 2003;54:217–225. doi: 10.1002/cm.10094. [DOI] [PubMed] [Google Scholar]
- Bolte S, Cordelières FP. A guided tour into subcellular colocalization analysis in light microscopy. J Microsc. 2006;224:213–232. doi: 10.1111/j.1365-2818.2006.01706.x. [DOI] [PubMed] [Google Scholar]
- Brand AH, Manoukian AS, Perrimon N. Ectopic expression in Drosophila. Methods Cell Biol. 1994;44:635–654. doi: 10.1016/S0091-679X(08)60936-X. [DOI] [PubMed] [Google Scholar]
- Buschiazzo A, Alzari PM. Structural insights into sialic acid enzymology. Curr Opin Chem Biol. 2008;12:565–572. doi: 10.1016/j.cbpa.2008.06.017. [DOI] [PubMed] [Google Scholar]
- Dubnau J, Grady L, Kitamoto T, Tully T. Disruption of neurotransmission in Drosophila mushroom body blocks retrieval but not acquisition of memory. Nature. 2001;411:476–480. doi: 10.1038/35078077. [DOI] [PubMed] [Google Scholar]
- Dunn KW, Kamocka MM, McDonald JH. A practical guide to evaluating colocalization in biological microscopy. Am J Physiol Cell Physiol. 2011;300:C723–C742. doi: 10.1152/ajpcell.00462.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganetzky B. Genetic studies of membrane excitability in Drosophila: lethal interaction between two temperature-sensitive paralytic mutations. Genetics. 1984;108:897–911. doi: 10.1093/genetics/108.4.897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaev D, Isaeva E, Shatskih T, Zhao Q, Smits NC, Shworak NW, Khazipov R, Holmes GL. Role of extracellular sialic acid in regulation of neuronal and network excitability in the rat hippocampus. J Neurosci. 2007;27:11587–11594. doi: 10.1523/JNEUROSCI.2033-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kean EL, Münster-Kühnel AK, Gerardy-Schahn R. CMP-sialic acid synthetase of the nucleus. Biochim Biophys Acta. 2004;1673:56–65. doi: 10.1016/j.bbagen.2004.04.006. [DOI] [PubMed] [Google Scholar]
- Kim K, Lawrence SM, Park J, Pitts L, Vann WF, Betenbaugh MJ, Palter KB. Expression of a functional Drosophila melanogaster N-acetylneuraminic acid (Neu5Ac) phosphate synthase gene: evidence for endogenous sialic acid biosynthetic ability in insects. Glycobiology. 2002;12:73–83. doi: 10.1093/glycob/12.2.73. [DOI] [PubMed] [Google Scholar]
- Koles K, Irvine KD, Panin VM. Functional characterization of Drosophila sialyltransferase. J Biol Chem. 2004;279:4346–4357. doi: 10.1074/jbc.M309912200. [DOI] [PubMed] [Google Scholar]
- Koles K, Lim JM, Aoki K, Porterfield M, Tiemeyer M, Wells L, Panin V. Identification of N-glycosylated proteins from the central nervous system of Drosophila melanogaster. Glycobiology. 2007;17:1388–1403. doi: 10.1093/glycob/cwm097. [DOI] [PubMed] [Google Scholar]
- Koles K, Repnikova E, Pavlova G, Korochkin LI, Panin VM. Sialylation in protostomes: a perspective from Drosophila genetics and biochemistry. Glycoconj J. 2009;26:313–324. doi: 10.1007/s10719-008-9154-4. [DOI] [PubMed] [Google Scholar]
- Lin DM, Goodman CS. Ectopic and increased expression of Fasciclin II alters motoneuron growth cone guidance. Neuron. 1994;13:507–523. doi: 10.1016/0896-6273(94)90022-1. [DOI] [PubMed] [Google Scholar]
- Long AA, Kim E, Leung HT, Woodruff E, 3rd, An L, Doerge RW, Pak WL, Broadie K. Presynaptic calcium channel localization and calcium-dependent synaptic vesicle exocytosis regulated by the Fuseless protein. J Neurosci. 2008;28:3668–3682. doi: 10.1523/JNEUROSCI.5553-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loughney K, Kreber R, Ganetzky B. Molecular analysis of the para locus, a sodium channel gene in Drosophila. Cell. 1989;58:1143–1154. doi: 10.1016/0092-8674(89)90512-6. [DOI] [PubMed] [Google Scholar]
- Lyalin D, Koles K, Roosendaal SD, Repnikova E, Van Wechel L, Panin VM. The twisted gene encodes Drosophila protein O-mannosyltransferase 2 and genetically interacts with the rotated abdomen gene encoding Drosophila protein O-mannosyltransferase 1. Genetics. 2006;172:343–353. doi: 10.1534/genetics.105.049650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McQuilton P, St Pierre SE, Thurmond J, Thurmond J. FlyBase 101—the basics of navigating FlyBase. Nucleic Acids Res. 2012;40:D706–D714. doi: 10.1093/nar/gkr1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metaxakis A, Oehler S, Klinakis A, Savakis C. Minos as a genetic and genomic tool in Drosophila melanogaster. Genetics. 2005;171:571–581. doi: 10.1534/genetics.105.041848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montpetit ML, Stocker PJ, Schwetz TA, Harper JM, Norring SA, Schaffer L, North SJ, Jang-Lee J, Gilmartin T, Head SR, Haslam SM, Dell A, Marth JD, Bennett ES. Regulated and aberrant glycosylation modulate cardiac electrical signaling. Proc Natl Acad Sci U S A. 2009;106:16517–16522. doi: 10.1073/pnas.0905414106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morin X, Daneman R, Zavortink M, Chia W. A protein trap strategy to detect GFP-tagged proteins expressed from their endogenous loci in Drosophila. Proc Natl Acad Sci U S A. 2001;98:15050–15055. doi: 10.1073/pnas.261408198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mühlenhoff M, Oltmann-Norden I, Weinhold B, Hildebrandt H, Gerardy-Schahn R. Brain development needs sugar: the role of polysialic acid in controlling NCAM functions. Biol Chem. 2009;390:567–574. doi: 10.1515/BC.2009.078. [DOI] [PubMed] [Google Scholar]
- Munro S, Pelham HR. A C-terminal signal prevents secretion of luminal ER proteins. Cell. 1987;48:899–907. doi: 10.1016/0092-8674(87)90086-9. [DOI] [PubMed] [Google Scholar]
- Munster AK, Weinhold B, Gotza B, Muhlenhoff M, Frosch M, Gerardy-Schahn R. Nuclear localization signal of murine CMP-Neu5Ac synthetase includes residues required for both nuclear targeting and enzymatic activity. J Biol Chem. 2002;277:19688–19696. doi: 10.1074/jbc.M201093200. [DOI] [PubMed] [Google Scholar]
- Münster-Kühnel AK, Tiralongo J, Krapp S, Weinhold B, Ritz-Sedlacek V, Jacob U, Gerardy-Schahn R. Structure and function of vertebrate CMP-sialic acid synthetases. Glycobiology. 2004;14:43R–51R. doi: 10.1093/glycob/cwh113. [DOI] [PubMed] [Google Scholar]
- Noma K, Kimura K, Minatohara K, Nakashima H, Nagao Y, Mizoguchi A, Fujiyoshi Y. Triple N-glycosylation in the long S5-P loop regulates the activation and trafficking of the Kv12.2 potassium channel. J Biol Chem. 2009;284:33139–33150. doi: 10.1074/jbc.M109.021519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Recio-Pinto E, Thornhill WB, Duch DS, Levinson SR, Urban BW. Neuraminidase treatment modifies the function of electroplax sodium channels in planar lipid bilayers. Neuron. 1990;5:675–684. doi: 10.1016/0896-6273(90)90221-Z. [DOI] [PubMed] [Google Scholar]
- Repnikova E, Koles K, Nakamura M, Pitts J, Li H, Ambavane A, Zoran MJ, Panin VM. Sialyltransferase regulates nervous system function in Drosophila. J Neurosci. 2010;30:6466–6476. doi: 10.1523/JNEUROSCI.5253-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutishauser U. Polysialic acid in the plasticity of the developing and adult vertebrate nervous system. Nat Rev Neurosci. 2008;9:26–35. doi: 10.1038/nrn2285. [DOI] [PubMed] [Google Scholar]
- Schauer R, Kamerling JP. Chemistry, biochemistry and biology of sialic acids. In: Montreuil J, Vliegenthart JFG, Schachter H, editors. Glycoproteins II. Amsterdam: Elsevier; 1997. pp. 243–402. [Google Scholar]
- Sisson JC, Field C, Ventura R, Royou A, Sullivan W. Lava lamp, a novel peripheral Golgi protein, is required for Drosophila melanogaster cellularization. J Cell Biol. 2000;151:905–918. doi: 10.1083/jcb.151.4.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley P, Schachter H, Taniguchi N. N-glycans. In: Varki A, Cummings RD, Esko JD, Freeze HH, Stanley P, Bertozzi CR, Hart GW, Etzler ME, editors. Essentials of glycobiology. 2nd Edition. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory; 2009. [PubMed] [Google Scholar]
- Stewart BA, Mohtashami M, Trimble WS, Boulianne GL. SNARE proteins contribute to calcium cooperativity of synaptic transmission. Proc Natl Acad Sci U S A. 2000;97:13955–13960. doi: 10.1073/pnas.250491397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stocker PJ, Bennett ES. Differential sialylation modulates voltage-gated Na+ channel gating throughout the developing myocardium. J General Physiology. 2006;127:253–265. doi: 10.1085/jgp.200509423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarentino AL, Quinones G, Trumble A, Changchien LM, Duceman B, Maley F, Plummer TH., Jr Molecular cloning and amino acid sequence of peptide-N4-(N-acetyl-beta-D-glucosaminyl)asparagine amidase from flavobacterium meningosepticum. J Biol Chem. 1990;265:6961–6966. [PubMed] [Google Scholar]
- Titus SA, Warmke JW, Ganetzky B. The Drosophila erg K+ channel polypeptide is encoded by the seizure locus. J Neurosci. 1997;17:875–881. doi: 10.1523/JNEUROSCI.17-03-00875.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ufret-Vincenty CA, Baro DJ, Lederer WJ, Rockman HA, Quinones LE, Santana LF. Role of sodium channel deglycosylation in the genesis of cardiac arrhythmias in heart failure. J Biol Chem. 2001;276:28197–28203. doi: 10.1074/jbc.M102548200. [DOI] [PubMed] [Google Scholar]
- Varki A. Glycan-based interactions involving vertebrate sialic-acid-recognizing proteins. Nature. 2007;446:1023–1029. doi: 10.1038/nature05816. [DOI] [PubMed] [Google Scholar]
- Varki A, Esko DF, Colley KJ. Cellular organization of glycosylation. In: Varki A, Cummings RD, Esko JD, Freeze HH, Stanley P, Bertozzi CR, Hart GW, Etzler ME, editors. Essentials of glycobiology. 2nd edition. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory; 2009. pp. 37–46. [Google Scholar]
- Verstreken P, Bellen HJ. Neuroscience. The meaning of a mini. Science. 2001;293:443–444. doi: 10.1126/science.1063293. [DOI] [PubMed] [Google Scholar]
- Viswanathan K, Tomiya N, Park J, Singh S, Lee YC, Palter K, Betenbaugh MJ. Expression of a functional Drosophila melanogaster CMP-sialic acid synthetase. Differential localization of the Drosophila and human enzymes. J Biol Chem. 2006;281:15929–15940. doi: 10.1074/jbc.M512186200. [DOI] [PubMed] [Google Scholar]
- Vyas AA, Patel HV, Fromholt SE, Heffer-Lauc M, Vyas KA, Dang J, Schachner M, Schnaar RL. Gangliosides are functional nerve cell ligands for myelin-associated glycoprotein (MAG), an inhibitor of nerve regeneration. Proc Natl Acad Sci U S A. 2002;99:8412–8417. doi: 10.1073/pnas.072211699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XJ, Reynolds ER, Déak P, Hall LM. The seizure locus encodes the Drosophila homolog of the HERG potassium channel. J Neurosci. 1997;17:882–890. doi: 10.1523/JNEUROSCI.17-03-00882.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe I, Zhu J, Sutachan JJ, Gottschalk A, Recio-Pinto E, Thornhill WB. The glycosylation state of Kv1.2 potassium channels affects trafficking, gating, and simulated action potentials. Brain Res. 2007;1144:1–18. doi: 10.1016/j.brainres.2007.01.092. [DOI] [PubMed] [Google Scholar]
- Wu CF, Ganetzky B. Neurogenetic studies of ion channels in Drosophila. Ion Channels. 1992;3:261–314. doi: 10.1007/978-1-4615-3328-3_9. [DOI] [PubMed] [Google Scholar]
- Ye B, Zhang Y, Song W, Younger SH, Jan LY, Jan YN. Growing dendrites and axons differ in their reliance on the secretory pathway. Cell. 2007;130:717–729. doi: 10.1016/j.cell.2007.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuber C, Lackie PM, Catterall WA, Roth J. Polysialic acid is associated with sodium channels and the neural cell adhesion molecule N-CAM in adult rat brain. J Biol Chem. 1992;267:9965–9971. [PubMed] [Google Scholar]