

Abstract
We have previously demonstrated that Sox17 regulates cell cycle exit and differentiation in oligodendrocyte progenitor cells. Here we investigated its function in white matter (WM) development and adult injury with a newly generated transgenic mouse overexpressing Sox17 in the oligodendrocyte lineage under the CNPase promoter. Sox17 overexpression in CNP-Sox17 mice sequentially promoted postnatal oligodendrogenesis, increasing NG2 progenitor cells from postnatal day (P) 15, then O4+ and CC1+ cells at P30 and P120, respectively. Total Olig2+ oligodendrocyte lineage cells first decreased between P8 and P22 through Sox17-mediated increase in apoptotic cell death, and thereafter significantly exceeded WT levels from P30 when cell death had ceased. CNP-Sox17 mice showed increased Gli2 protein levels and Gli2+ cells in WM, indicating that Sox17 promotes the generation of oligodendrocyte lineage cells through Hedgehog signaling. Sox17 overexpression prevented cell loss after lysolecithin-induced demyelination by increasing Olig2+ and CC1+ cells in response to injury. Furthermore, Sox17 overexpression abolished the injury-induced increase in TCF7L2/TCF4+ cells, and protected oligodendrocytes from apoptosis by preventing decreases in Gli2 and Bcl-2 expression that were observed in WT lesions. Our study thus reveals a biphasic effect of Sox17 overexpression on cell survival and oligodendrocyte formation in the developing WM, and that its potentiation of oligodendrocyte survival in the adult confers resistance to injury and myelin loss. This study demonstrates that overexpression of this transcription factor might be a viable protective strategy to mitigate the consequences of demyelination in the adult WM.
Introduction
Oligodendrogenesis from oligodendrocyte (OL) progenitor cells (OPCs) to mature, myelinating OLs is spatially and temporally regulated by transcription factors under the control of multiple signaling pathways, including canonical Wnt, Sonic hedgehog, Notch, bone and morphogenetic proteins (Nicolay et al., 2007; Fancy et al., 2009). Members of the SRY-box (Sox) transcription factors have emerged as crucial regulators of OL development and regeneration. Sox transcription factors that contain a conserved high mobility domain that binds the DNA minor groove (Gubbay et al., 1990), are essential for the differentiation and maturation of OLs in the developing nervous system (Chew and Gallo, 2009; Stolt and Wegner, 2010). Sox9 has an early function in maintaining the OPC population (Stolt et al., 2003), while Sox10 is essential for terminal differentiation and myelin gene expression (Stolt et al., 2002). Inhibitory Sox factors 4, 5, and 6 are also critical for timing OL specification and terminal differentiation (Potzner et al., 2007).
Sox17 was found in the postnatal mouse white matter (WM) to be developmentally associated with the expression of multiple myelin genes, and its pattern of expression in vivo supports a role in proliferative arrest (Sohn et al., 2006). In cultured OPCs, Sox17 was shown to perform the dual functions of promoting OPC cycle exit and maturation to OLs (Sohn et al., 2006; Chew et al., 2011). Sox17 downregulation by siRNA increases OPC proliferation and attenuates differentiation. In addition, Sox17 knockdown upregulates β-catenin and its targets cyclin D1 and Axin2. Conversely, Sox17 overexpression in vitro (1) increases OPC cell cycle exit, (2) decreases cyclinD1 levels, and the levels and activity of b-catenin, (3) promotes degradation of b-catenin, (4) relieves Wnt repression of myelin protein levels, and (5) enhances myelin promoter activity (Sohn et al., 2006; Chew et al., 2011). These findings identify Sox17 as a Wnt/β-catenin antagonist in the lineage and suggest that ectopic Sox17 expression in vivo may promote OL formation through Wnt modulation.
To study the function of Sox17 in OLs in vivo, we generated a transgenic mouse strain in which a full-length mouse Sox17 cDNA fragment is expressed under the control of the CNP gene promoter. The CNP (2',3'-cyclic nucleotide 3'- phosphodiesterase) promoter has been shown to provide robust OL lineage-specific expression in the WM (Yuan et al., 2002). We wanted to determine whether Sox17 overexpression would lead to increased development of OLs. Since demyelination upregulates Wnt signaling (Fancy et al., 2009), we also wanted to determine whether Sox17 overexpression could block Wnt signaling and alter the course of demyelination in the adult WM. Our present analysis constitutes the first in vivo study of Sox17 function in WM. Sox17 overexpression increased WM levels of the Hedgehog mediator Gli2, regulated β-catenin-expressing cells and development of the OL lineage in biphasic fashion, and ultimately produced supranormal numbers of OL cells. As lysolecithin-induced demyelination injury failed to increase cell death, or affect MBP levels, Gli2 and the antiapoptotic protein Bcl-2 in the adult CNP-Sox17 mouse, we propose that Sox17 potentiates Hedgehog signaling in its attenuation of WM damage.
Materials and Methods
Plasmid construct and generation of transgenic mice.
The plasmid for generating transgenic mice was constructed as follows: (1) the CNP promoter plasmid CNP4.2 (Gravel et al., 1998) was modified by introducing restriction enzyme AgeI site at HindIII site to obtain CNP3.9 vector; (2) a full length of IRES-ZsGreen1 with added AgeI site at 5′ and XhoI site at 3′ was amplified by PCR from pIRES-ZsGreen1 plasmid (Cat#632478, Clontech) and subsequently inserted into CNP3.9 to obtain pCNP-IRES-ZsGreen1; and (3) the full-length mouse Sox17 cDNA fragment (Kanai et al., 1996; Chew et al., 2011) with added AgeI sites at both 5′ and 3′ ends was amplified by PCR and inserted into AgeI site of pCNP-IRES-ZsGreen1 to obtain pCNP-Sox17-IRES-ZsGreen1 (Fig. 1). A 6.79 kb XbaI/XhoI fragment of pCNP-Sox17-IRES-ZsGreen1 was excised, purified, and sent to the University of Michigan Transgenic Animal Model Core for microinjection into (C57BL/6XSJL) F1 eggs. Founders were bred with C57BL/6 male and female mice from the Jackson Laboratory. All mouse lines for the studies were maintained as hemizygotes and had previously been bred with C57BL/6 for at least 10 generations. To identify positive transgenic animals, a pair of primers based on ZsGreen1 coding sequence and another pair of primers based on Sox17 coding sequence were used for tail biopsies. ZsGreen1 primers were as follows: forward: ZsGr405F, 5′_-CCCCGTGATGAAGAAGATGA-_3′; reverse: ZsGr614R, 5′_-GTCAGCTTGTGCTGGATGAA-_3′. Sox17 primers were as follows: forward: Sox277F, 5_-CTGCACAACGCAGAGCTAAGC-_3′; reverse: Sox448R, 5′_-CGCTTCATGCGCTTCACCTGC-_3′. The Sox17 cDNA lacks a 431 bp intron between exons 4 and 5 (Ensembl: ENSMUST00000027035). The cycling conditions were as follows: 95°C for 5 min followed by 35 cycles of 95°C for 60 s, 60°C for 60 s, 72°C for 60 s, and a final extension step of 72°C for 5 min. PCR amplification of the transgene thus yields a 171 bp fragment in founders whereas a 602 bp fragment is amplified in WT mice. With this genotyping approach, we identified a total of five transgenic mouse founders that were positive for both ZsGreen1 and recombinant Sox17, and that showed germline transmission of the transgene. The copy number of transgene in transgenic mouse was identified by real-time PCR (Joshi et al., 2008). The primers used for copy number determination were ZsGR405F and ZsGr614R as described for genotyping. Twenty-five microliters of PCR mixtures contain 1 ng of genomic DNA, 1 μl of 10 μm primer mixture, and 12.5 μl of SYBR GreenER qPCR SuperMix Universal (11762-100, Invitrogen). PCR was performed on an ABI Prism 7900 HT Sequence Detection System. The cycling conditions were as follows: 50°C for 2 min, 95°C for 10 min followed by 40 cycles of 95°C for 30 s, 60°C for 45 s, and 72°C for 30 s.
Figure 1.

Generation of CNPase promoter-driven Sox17 transgenic mice leads to elevated levels of Sox17 in postnatal WM tissue. A, Diagram showing transgenic construct containing full-length mouse Sox17 cDNA fragment between CNPase promoter and IRES-EGFP. B, Immunohistochemical analysis of total Sox17-expressing cells in the CNP-Sox17 P30 subcortical WM. Confocal microscope images showing colocalization of ZsGreen fluorescence with Sox17 in WM, indicating expression of the transgene. C, Confocal images showing colocalization of ZsGreen fluorescence with NG2 (red), O4 (red), and CC1 (red) in subcortical WM at P30. Scale bar, 20 μm. D, Western blot showing total Sox17 protein levels in corpus callosum (CC) in the CNP-Sox17 transgenic (Trans) compared with WT mice. E, Quantitation of Western blots including those in D. Densitometric values of Sox17 protein normalized against actin are plotted as a function of postnatal age. Values represent the mean fold change of Sox17 protein abundance, relative to actin, in transgenic (Trans) over WT. Increased levels of Sox17 are observed in the CNP-Sox17 (Trans) at P15 and P60. Values shown are mean and SEM obtained from 1–2 mice from each of four independent litters. **p < 0.01 versus WT, unpaired Student's t test.
Western blots.
WM tissue was microdissected from brains of WT and CNP-Sox17 transgenic mouse brain at postnatal day (P) 8, P22, P30, P60, and P120. Fresh coronal sections 200–300 μm thick containing the anterior WM regions of corpus callosum, cingulum, and external capsule were prepared on a tissue chopper. Under a dissecting scope, the WM tissue was separated from gray matter regions and striatum from these slices with fine forceps while submerged in HBSS. WM tissue from three such sections per brain were homogenized in 100–200 μl of RIPA lysis buffer (10 mm HEPES, pH 7.9, 1 mm EDTA, pH 8.0, 150 mm NaCl, 1 mm DTT, 1 mm PMSF, 0.5 mm sodium vanadate, 1% NP-40) on ice and then centrifuged at 12,000 rpm at 4°C for 15 min. Each lysate was mixed with equal volume of 2× loading buffer (Sigma-Aldrich) and boiled for 5 min. Ten-to-twenty micrograms of protein samples were separated on 4–12% NuPage Bis-Tris precast gel (Invitrogen) and transferred to PVDF membranes (Millipore). The membranes were blocked in Odyssey blocking buffer (part#: 927-40000, Odyssey Infrared Image System, LI-COR) and then incubated with diluted primary antibodies overnight at 4°C. This was followed by washing three times with PBS containing 0.05% Tween 20. The membranes were incubated with LI-COR Odyssey secondary antibodies in LI-COR Odyssey blocking buffer for 1 h at room temperature with agitation. After washing three times with PBS containing 0.05% Tween 20, the membranes were analyzed with Odyssey Infrared Image System (LI-COR).
Tissue section preparation and immunohistochemistry.
Mouse brains were fixed with 4% paraformaldehyde by transcardial perfusion as previously described (Yuan et al., 2002). Tissue sections of brains were prepared on a sliding Microtome 860 (American Optical). Sections were cut (40 μm) and stored in 1× PBS, 0.05% sodium azide solution. For immunohistochemistry, floating tissue sections were blocked at room temperature for at least 1 h in blocking solution (1% BSA, 0.3% Triton X-100, and 20% normal goat or horse serum in 1× PBS) and then incubated with primary antibodies at 4°C overnight. After three washes with carrier solution (1% BSA, 0.3% Triton X-100, 1% normal serum in 1× PBS) at room temperature, secondary antibodies were added and the sections incubated for another 1 h, followed by three washes of 1× PBS per 10 min. Epi-fluorescence images were obtained on Olympus BX61TRF and confocal images were obtained on a Zeiss LSM510 single photon laser confocal microscope. Images were analyzed with ImageJ software, and means of cell counts obtained from a total of 5–8 animals from each mouse strain or group, comprising pairs of transgenic offspring and littermate negative controls from at least 3–4 independent litters.
Antibodies.
The primary and secondary antibodies used in our studies are listed in Tables 1 and 2.
Table 1.
Primary antibodies used in this study
| Primary antibody | Catalog No. | Clone | Isotype | Host | Dilution for immunohistology | Dilution for Western blot | Manufacturer |
|---|---|---|---|---|---|---|---|
| Active-β-catenin | 05-665 | Ig | Mouse | 1:1000 | Millipore | ||
| MBP | SMI-99 | IgG2b | Mouse | 1:1000 | 1:2000 | Covance | |
| Gli2 | ab7195 | IgG | Rabbit | 1:500 | 1:200 | Abcam | |
| CNPase | SMI91R | IgG1 | Mouse | 1:1000 | Covance | ||
| MAG | sc-15324 | H-300 | Rabbit | 1:500 | Santa Cruz Biotechnology | ||
| Myelin oligodendrocyte glycoprotein | ab32760 | Ig3G | Rabbit | 1:1000 | Abcam | ||
| Human biotinylated SOX17 | BAF1924 | Asp177-Val414 | IgG | Goat | 1:100 | 1:50 | R&D Systems |
| NG2 | Ab5320 | Ig | Rabbit | 1;100 | Millipore | ||
| O4 | MAB1326 | O4 | IgM | Mouse | 1:200 | R&D Systems | |
| Cleaved caspase-3 | 9664S | IgG | Rabbit | 1:50 | Cell Signaling Technology | ||
| Olig2 | AB9610 | Ig | Rabbit | 1:500 | Millipore | ||
| Adenomatous polyposis coli | OP80 | CC1 | IgG2b | Mouse | 1:500 | Calbiochem | |
| Human biotinylated Olig2 for dual label | BAF2418 | Met1-Lys323 | IgG | Goat | 1:400 | R&D Systems | |
| Ki67 | M7240 | MIB-1 | IgG1κ | Mouse | 1:100 | Dako | |
| Tcf4 | Sc-8632 | C-19 | IgG | Goat | 1:500 | Santa Cruz Biotechnology | |
| Bcl-2 | Ab793 | Rabbit | 1:500 | Abcam | |||
| Olig2 for dual label | MABN50 | 211F1.1 | IgG2ak | Mouse | 1:100 | Millipore | |
| NG2 for dual label | MAB5384 | 132.39 | IgG1 | Mouse | 1:100 | Millipore |
Table 2.
Secondary antibodies used in this study
| Secondary antibody | Catalog No. | Isotype | Host | Anti- | Dilution for immunohistochemistry | Dilution for Western blot | Company |
|---|---|---|---|---|---|---|---|
| IRDye 800CW | 926-32210 | IgG (heavy and light chains) | Goat | Mouse | 1:2000 | Odyssey (LI-COR) | |
| IRDye 800CW | 926-32213 | IgG (heavy and light chains) | Donkey | Rabbit | 1:2000 | Odyssey (LI-COR) | |
| IRDye 680LT | 926-68020 | IgG (heavy and light chains) | Goat | Mouse | 1:2000 | Odyssey (LI-COR) | |
| IRDye 680LT | 926-68023 | IgG (heavy and light chains) | Donkey | Rabbit | 1:2000 | Odyssey (LI-COR) | |
| IRDye 680LT | 926-68031 | Conjugate streptavidin | Biotinylated antibody | 1:1000 | Odyssey (LI-COR) | ||
| Rhodamine | 111-295-144 | IgG (heavy and light chains) | Goat | Rabbit | 1:250 | Jackson ImmunoResearch Laboratories | |
| Cy5 | 111-605- Immuno144 | IgG (heavy and light chains) | Goat | Rabbit | 1:200 | Jackson ImmunoResearch Laboratories | |
| Alexa Fluor 647 | A31573 | IgG (heavy and light chains) | Donkey | Rabbit | 1:300 | Invitrogen | |
| Alexa Fluor 647 | A21237 | IgG (heavy and light chains) | Goat | Mouse | 1:300 | Invitrogen | |
| Alexa Fluor 546 | A10036 | IgG (heavy and light chains) | Donkey | Mouse | 1:500 | Invitrogen | |
| Alexa Fluor 546 | A11071 | IgG (heavy and light chains) | Goat | Rabbit | 1:500 | Invitrogen | |
| Alexa Fluor 594 | S32356 | Conjugate streptavidin | Biotinylated antibody | 1:500 | Invitrogen | ||
| Alexa Fluor 647 | S32357 | Conjugate streptavidin | Biotinylated antibody | 1:300 | Invitrogen |
Lysolecithin-induced focal demyelination.
Focal demyelination was performed and analyzed as described previously (Aguirre et al., 2007). Adult mice at P35–P40 were anesthetized with an intraperitoneal injection of a ketamine/xylaxine mixture (10 mg per gram of body weight) and positioned in a stereotaxic frame (Stoelting). Focal lesion was induced by injection of 2 μl of 1% lysolecithin (Cat 440-154, CalBiochem/Millipore) in 0.9% saline by a 5 μl Hamilton microliter syringe. Controls were injected with 2 μl of saline. Lysolecithin or saline was injected unilaterally into the corpus callosum using stereotaxic coordinates of 2.5 mm anterior to the bregma, 1 mm lateral, and 2.5 mm deep from the skull surface. The day of lysolecithin injection was designated as day 0 [0 days postlesion (DPL)]. Mice were killed and processed for immunohistochemical analysis at 3 and 7 DPL after lysolecithin injection.
Results
CNP-Sox17 transgenic mice express increased levels of Sox17 in WM tissue
Previous studies have shown that Sox17 promoted OL lineage progression in cultured OPCs (Sohn et al., 2006). To investigate the function of Sox17 by overexpression in the OL lineage in vivo, we designed a transgene under the control of the CNPase promoter that is shown in Figure 1A. The CNPase promoter has been demonstrated to label OL lineage cells at all developmental stages in transgenic mice (Yuan et al., 2002; Aguirre and Gallo, 2004; Aguirre et al., 2004). A full-length fragment of the mouse Sox17 cDNA was ligated upstream of a PCR fragment of the ZsGreen reporter. The ZsGreen was expressed independently of the Sox17 protein via an internal ribosome entry site (IRES-ZsGreen, Clontech). Five apparently healthy, normally ambulating, and stably transmitting founders were found to be double positive for recombinant Sox17 and ZsGreen, which was further verified by immunofluorescence and Western blotting for Sox17. Each of these was backcrossed with C57BL6 over at least 10 generations. These founders also showed normal life span. Real-time PCR determined that each heterozygote founder contained an average of 3–7 copies of the transgene (data not shown). A unique founder number 653 was selected from among the five lines for all subsequent analysis. A second founder number 642 was randomly selected for confirmation of selected Western blotting and histological results.
Figure 1B shows colocalization of ZsGreen reporter fluorescence and Sox17 immunolabeling in the subcortical WM at P30. We have previously shown that endogenous Sox17 is not OL-specific, and is also expressed in neurons and astrocytes (Sohn et al., 2006). The ZsGreen reporter is also colocalized with NG2 (OPC), O4 (pre-OL), and CC1 (OL) markers at P30 (Fig. 1C), showing the potential for recombinant Sox17 to impact multiple developmental stages in the lineage. To quantify Sox17 protein levels in microdissected WM, Western blotting was performed. These results indicate that, at both P15 and P60, the total levels of Sox17 detected in the CNP-Sox17 mouse were significantly higher than that in WT controls (Fig. 1D,E).
Sox17 overexpression shows a biphasic effect on postnatal OL development
Based on our findings of Sox17 function in OL cell differentiation in vitro (Sohn et al., 2006; Chew et al., 2011), we hypothesized that Sox17 might also promote OL maturation in vivo. Since the mouse CNP promoter is active in OL progenitor cells (OPCs; Yuan et al., 2002), we performed a postnatal time course analysis to determine when and how lineage progression would be modulated by recombinant Sox17. In comparison with WT controls, the subcortical WM in CNP-Sox17 (Trans) first showed a reduction of the NG2+ OPC population at P8, followed by a surprising increase from P15 through P60 (Fig. 2A,B). No change in O4+ cells was observed until P30, indicating that increased maturation to premyelinating OLs only occurred after the first increase in NG2+ cells (Fig. 2C,D). Coincident with the increase in NG2+ cells, the number of mature myelinating OLs labeled with CC1 was reduced from P15 to P30, indicating a relatively immature population. However, this rose to normal levels by P60 (Fig. 3A,B), and then clearly exceeded WT levels at P120. To confirm these changes in the CC1+ population, Western blotting was performed with microdissected WM tissue from the CNP-Sox17 mouse. Consistent with the findings in CC1 cells, the levels of myelin proteins (MBP, MAG, and myelin OL glycoprotein) also showed similar decreases until P30 (Fig. 3C,D), and thereafter were found not to be significantly different from WT at P60 (Fig. 3D) and P120 (data not shown), indicating that OL maturation was delayed relative to the WT. The sequential increases observed beginning at P15 in NG2+, O4+, and CC1+ cells would be consistent with an effect of recombinant Sox17 on promoting the generation of the lineage as a whole, starting with the OPC stage. The normal levels of myelin proteins despite the increased numbers of CC1+ cells at P120 is not understood.
Figure 2.

Developmental analysis of OL lineage cells in the postnatal CNP-Sox17 transgenic mouse. A, Confocal images of NG2+ progenitor cells (red) in corpus callosum of WT and CNP-Sox17 transgenic (Trans) mouse strains. Nuclei are counterstained with DAPI (blue). B, Quantitative analysis of cells positive for NG2+. The density of NG2+ cells is plotted as a function of postnatal age. Trans WM first show decreased density at P8 followed by increases through P60. C, Confocal images of O4+ pre-OL cells (red) in cingulate WM of WT and CNP-Sox17 transgenic (Trans) mouse strains. Nuclei are counterstained with DAPI (blue). D, Quantitative analysis of cells positive for O4+. The density of O4+ cells is plotted as a function of postnatal age. Trans WM shows increases starting at P30. Mean and SEM were obtained with mice from at least three independent litters. *p < 0.05, **p < 0.01 versus WT, unpaired Student's t test.
Figure 3.

Developmental analysis of OL cells and myelin proteins in the postnatal CNP-Sox17 transgenic mouse. A, Confocal images of CC1+ OL cells (red) in corpus callosum of WT and CNP-Sox17 transgenic (Trans) mouse strains. Nuclei are counterstained with DAPI (blue). B, Quantitative analysis of CC1+ cells. The density of CC1+ cells is plotted as a function of postnatal age. Trans WM show decreased density until P60 followed by an increase at P120. Values represent means ± SEM from at least three independent litters. *p < 0.05, **p < 0.01 versus WT, unpaired Student's t test. C, Western blot of microdissected WM corpus callosum samples from postnatal mice between P8 and P60. Each lane contains 5 μg of protein from WM of one animal. D, Quantitative analysis of Western blots of myelin proteins including those shown in C. Densitometric values of myelin proteins are plotted as a function of postnatal age. Values represent the fold change of relative myelin protein abundance (normalized against actin) in transgenic (Trans) over WT. The pattern of expression of MBP, MAG, and myelin OL glycoprotein (MOG) appears to reflect the changes in CC1+ OLs. Overexpression of Sox17 (Trans) first downregulated myelin proteins from P15 to P30, then upregulated them at P60. Western blot values are mean and SEM using mice from at least three independent litters, expressed as fold change over WT. *p < 0.05, **p < 0.01 versus WT, unpaired Student's t test.
Sox17 regulates Olig2+ cells in a biphasic fashion, ultimately expanding the oligodendroglial lineage cell population
To understand the origin of these changes in the WM of the CNP-Sox17 mouse, we sought to characterize the cellular mechanisms that may modulate lineage progression. Olig2 is a widely used marker of the OL lineage (Lu et al., 2002). Signals that regulate the number of cells expressing Olig2 thus bear the potential to impact oligodendrogenesis and maturation. Immunohistochemical analysis of the Olig2+ cell population in the CNP-Sox17 WM revealed a biphasic pattern of modulation by recombinant Sox17 starting from P15. First, Olig2+ cells displayed a decrease at P15 (Fig. 4A,B) that coincided with the decrease in the CC1+ population (Fig. 3A,B). Thereafter, Olig2+ cells were consistently increased over WT levels from P30 through P120 (Fig. 4B). The developmental profile of oligodendroglial lineage cells in the CNP-Sox17 mouse WM is summarized in Figure 4C. Note that NG2 and O4 remain elevated even at P120 (Fig. 4C). This plot indicates that the large increase in Olig2+ cells at P30 consists of NG2+ and O4+ cells. In addition, the changes in the CNP-Sox17 WM appeared to be specific to the OL lineage, since GFAP+ astrocytes remained unaffected (Fig. 4D,E) at both P15 and P60.
Figure 4.

Overexpression of Sox17 modulates the oligodendroglial lineage and not GFAP+ astrocytes. A, Confocal images of Olig2+ cells (red) in corpus callosum. B, Quantitative analysis of Olig2+ cells in corpus callosum. The density of Olig2+ cells is plotted as a function of postnatal age, indicating increased oligodendroglial commitment in Trans beginning at P30. Values represent means ± SEM. *p < 0.05, **p < 0.01 versus WT (unpaired Student's t test). C, Summary of postnatal changes, from P8 through P120, in oligodendroglial lineage cells in the CNP-Sox17 WM illustrated as a line plot. The horizontal line at 1 represents WT levels. At P30, the large increase in Olig2+ cells likely consists primarily of NG2+ and O4+ cells. D, Confocal images of GFAP+ astrocyte cells (red) in corpus callosum. E, Quantitative analysis of GFAP+ cells in corpus callosum. The density of astrocytes is plotted as a function of postnatal age, indicating no significant effect of Sox17 overexpression on astrocyte development. Values represent mean and SEM of all cell counts (B, E) using mice obtained from at least four independent litters.
Sox17 potentiates early postnatal apoptosis in developing WM
The biphasic nature of the changes in the OL lineage may be the result of Sox17-mediated regulation of cell survival, proliferation, or both. To determine changes in cell survival, we examined the levels of cleaved caspase-3, a critical downstream mediator of programmed cell death. Immunohistochemical analysis showed that caspase-3 activity was increased by Sox17 between P8 and P22 (Fig. 5A). This increase in apoptosis was reflected in total cell loss in the WM, as demonstrated by decreased DAPI+ cells at these same time points (Fig. 5B). These reciprocal changes arising from Sox17 overexpression are illustrated in Figure 5C. The results suggest that Sox17-induced death led to decreased size of the Olig2+ population, and may underlie the early hypomyelination observed in the WM of the CNP-Sox17 mouse before P30. To determine the identity of the apoptotic cells, dual-labeling experiments showed localization of cleaved caspase-3 in NG2+, O4+, Olig2+, and CC1+ cells at P8 and P15 (Fig. 5D). The results indicate that increased number of NG2 cells was not accompanied by increased survival of O4 or Olig2 cells at these ages, indicating that apoptosis in the oligodendroglial lineage, beginning with OPCs, likely contributed to the reduction in mature OLs and myelin proteins in the CNP-Sox17 mouse. Analysis of cell proliferation showed that, in agreement with our previous findings, Sox17 induced cell cycle exit, significantly reducing the percentage of Ki67+ cells in WM at both P15 and P30 (Fig. 5E,F). The preferential reduction of Ki67 colocalization in Olig2+ over NG2+ cells (Fig. 5F) suggests an effect on differentiation, because at P30 this occurred in the absence of cell death.
Figure 5.

Overexpression of Sox17 enhances developmental apoptosis of oligodendroglial cells. A, Quantitative analysis of apoptotic cells during postnatal WM development by cleaved caspase-3 immunohistochemistry shows significant increases in density between P8 and P22 in the Trans WM. Cleaved caspase-3+ cells were no longer detected at P30 in either strain. B, Sox17 overexpression downregulates DAPI cells in developing subcortical WM between P8 and P22. C, Combined plots of mean caspase-3 and DAPI+ cell changes during development (data from A and B), expressed as fold over WT, reveal a strongly reciprocal relationship. It is likely that increase of total caspase-3+ cells accounts for the decreases of DAPI+ cells. Note that no further change in cell death occurs at P30. D, Quantitative analysis of doubly labeled caspase-3+ NG2+, caspase-3+ O4+, caspase-3+ CC1+, and caspase-3+ Olig2+ double-positive cells in corpus callosum at the indicated postnatal ages. E, Confocal microscope images of Ki67+ cells (red) at P15 and P30, showing fewer Ki67 cells in Trans. F, Quantitative analysis of Ki67+ cells shows reduced densities of Ki67+ proliferating cells in Trans at both P15 and P30. At P30, doubly labeled Ki67+ NG2+ and Ki67+ Olig2+ cells were further quantified. All mean and SEM shown (A, B, D, F) were obtained using mice from at least three independent litters. *p < 0.05, **p < 0.01 versus WT, unpaired Student's t test.
It is noteworthy that cell apoptosis was also clearly detected in the developing WT WM (Fig. 5A) from P8 through P30. There was a general decline in postnatal apoptosis in the WM toward P30 (Fig. 5A), after which death was no longer detected in either mouse strain by caspase-3 immunolabeling (data not shown). The P30 time point not only coincides with the lack of an effect of Sox17 on total cell numbers labeled by DAPI (Fig. 5B), but also marks a turning point in cellular response when both O4+ (Fig. 2D) and Olig2+ (Fig. 4B) showed the first signs of Sox17-induced increase. These observations of coordinated increases in OPCs and mature OLs suggest an overall enhancement of the lineage caused by Sox17 overexpression, and is likely the result of a process downstream from developmental signals, such as Hedgehog activation or Wnt suppression.
Sox17 modulates β-catenin in biphasic fashion and increases Gli2-expressing cells in developing WM
To determine whether the changes in the numbers of OLs and Olig2+ cells may be related to altered Wnt or Hedgehog signaling, we first analyzed the numbers of cells immunostained with β-catenin and TCF4 (or TCF7L2) in the WM at P15 and P30. Fewer cells positive for β-catenin were found in the CNP-Sox17 mouse WM at P15, but by P30 the density of these cells had increased and exceeded that in the WT (Fig. 6A). Although at P15 the density of TCF4+ cells in both strains was indistinguishable, by P30, as with β-catenin, an increase was clearly observed in the CNP-Sox17 (Fig. 6A). The TCF4+ cells found were mostly nuclear, in agreement with the findings of Fancy et al. (2009). In contrast, the increase in β-catenin consisted of both cytoplasmic/membrane as well as nuclear forms (Fig. 6B), so that all positive cells were included in the analysis. These changes in the numbers of β-catenin and TCF4-expressing cells are shown in Figure 6B. The decrease in β-catenin at P15 would be consistent with our previous observations in culture (Chew et al., 2011), but our finding of increased TCF4+ cells and β-catenin at P30 in the CNP-Sox17 WM (Fig. 6A,B) was unexpected. Using Western blotting, we failed to detect any significant change in the levels of active β-catenin as a result of Sox17 overexpression during postnatal development (P15, P120, data not shown). Nonetheless, since TCF4 is necessary for OL development, and its interaction with β-catenin negatively regulates differentiation (Ye et al., 2009), the Sox17-mediated increase in both TCF4-expressing and β-catenin-expressing cells at P30 suggests activation of the OPC population, which may lead to the delayed appearance of CC1+ OLs.
Figure 6.
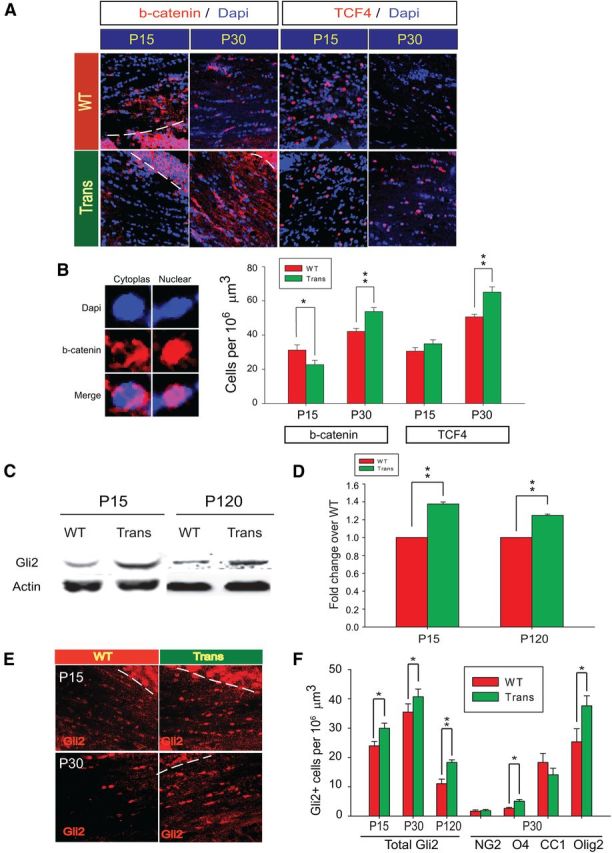
Overexpression of Sox17 in transgenic mice modulates β-catenin and increases Gli2 protein levels. A, Confocal microscope images of β-catenin (b-catenin, red) or TCF4 (red) with DAPI-stained nuclei (blue) at P15 and P30. The dashed lines delimit the regions of high expression beyond the WM (striatal area in P15 WT and gray matter in Trans) that were excluded from the quantitative analysis. Note that the β-catenin signal, where abundant, is cytoplasmic or membranous, whereas TCF4 is predominantly nuclear. B, Magnified examples of cytoplasmic/membrane versus nuclear β-catenin (red) immunostaining in WM cells against nuclear DAPI (blue) counterstain. Quantitative analysis of cell populations expressing β-catenin and TCF4 in WM at P15 and P30. Cell density values are means ± SEM. *p < 0.05, **p < 0.01 versus WT, Student's t test. C, Expression of Gli2 protein in the P15 and P120 corpus callosum as detected by Western blotting. Each lane contained 20 μg of protein. D, Quantitative analysis of Gli2 expression by densitometry performed on multiple Western blots similar to C. Values of Gli2 abundance relative to actin are mean and SEM using mice from at least four independent litters, expressed as fold change over WT. **p < 0.01 versus WT, Student's t test. E, Confocal images of nuclear Gli2 expression in corpus callosum at P15 and P30. DAPI stain was omitted for clarity. F, Quantitative analysis of Gli2-expressing cells in the corpus callosum at P15, P30, and P120. Doubly labeled Gli2 with NG2, O4, CC1, and Olig2 cells at P30 were also quantified. The areas of high Gli2 signal in cortical gray matter areas are bounded by dotted lines in E and excluded from analysis. Values are mean and SEM with 1–2 mice obtained from each of at least four independent litters. *p < 0.05, **p < 0.01 versus WT, Student's t test.
Sonic hedgehog was recently shown to act upstream of Wnt in the expression of TCF4 (Alvarez-Medina et al., 2009), and may therefore be involved in the Sox17-induced changes in TCF4 levels. To test for Hedgehog pathway activation, we analyzed the protein levels of Hedgehog pathway mediators Gli1 and Gli2 in the CNP-Sox17 WM. Gli1 protein could not be detected in our WM samples, but Gli2 showed consistent increases at P15 and P120 by Western blotting (Fig. 6C,D). This increase was due, at least in part, to an increase in the numbers of cells expressing Gli2, as detected by immunohistochemistry (Fig. 6E,F), supporting the notion that Sox17 overexpression enhanced activity of the Hedgehog pathway. At P30 in the absence of developmental apoptosis, we found that the increased Gli2 colocalized primarily with O4 and Olig2 (Fig. 6F) rather than with NG2 or CC1, suggesting increased differentiation. This coincides temporally with the accumulation of O4 and Olig2 cells at P30, consistent with a role for peak Sox17 expression in O4 cells (Sohn et al., 2006). Activation of Hedgehog signaling by Sox17 would thus support an essential contribution to OL generation.
Sox17 prevents myelin protein loss in demyelination injury
Based on our findings of increased OL lineage cells in the adult CNP-Sox17 mouse, we hypothesized that this property of Sox17 may reduce WM damage in an adult injury by enhancing the remyelination potential of OPCs recruited to the lesion site. We chose to test this hypothesis in the focal demyelination model of stereotaxic lysolecithin injections in the P30 corpus callosum. Although such lesions are typically performed on mice at 8 weeks or older when recovery occurs over 10–14 d (Aguirre et al., 2007; Etxeberria et al., 2010; Jablonska et al., 2010), we selected P30 for these studies in the CNP-Sox17 for the following reasons: (1) at P30, CC1+ OLs and myelin protein levels in the CNP-Sox17 mouse are below WT levels (Figs. 3, 4C); (2) developmental apoptosis was no longer detected in either mouse strain at P30; (3) any additive effect between remyelination signals and ectopic Sox17 would be more readily detected or amplified at an age with a robust response (Shen et al., 2008); and (4) at P30 both Olig2+ and O4+ cells begin to increase in the CNP-Sox17. Our results show that MBP immunostaining in both strains had recovered at 7 DPL, but differences were apparent at 3 DPL. At 3 DPL, the ipsilateral lysolecithin-injected site in the WT showed a clearly demarcated region of demyelination, as revealed by MBP immunostaining (Fig. 7A), whereas the damage was far less apparent in the CNP-Sox17 mouse. Compared with Trans, the loss of membrane-associated MBP was visibly greater in the WT at 3 DPL (Fig. 7B); however, by 7 DPL, MBP in both strains was not visibly different from their respective saline-injected, control sides. Figure 7C,D shows that, after normalizing against the levels in the contralateral saline-injected sites for each strain, both MBP and CNP protein levels after lysolecithin injection were still quantitatively lower in the WT at 3 DPL compared with the CNP-Sox17. These data indicate that recombinant Sox17 renders the transgenic WM less vulnerable to the loss of myelin protein by toxin injection.
Figure 7.

WM damage by lysolecithin injection at P30 is attenuated by Sox17 overexpression. A, Epifluorescence images (100× magnification) of MBP immunohistochemistry at 3 DPL showing unilaterally demyelinated lesions in the WT and CNP-Sox17 (Trans) caused by stereotaxic lysolecithin injection into the cingulate WM. Saline was injected into the contralateral cingulate area. Pairs of saline-injected (Sal) and lysolecithin-injected (Lys) images were obtained from brain sections of the same animal. Unlike in the WT, loss of MBP is not apparent in the CNP-Sox17 mouse (Trans). B, Confocal microscope images showing MBP (red) and DAPI (blue) staining at 3 and 7 DPL. Remyelination is observed in lysolecithin sides for both WT and Trans at 7 DPL. C, Western blot showing changes in the myelin-specific proteins MBP and CNP at 3 DPL. D, Quantitative analysis of MBP and CNP following densitometry of Western blots. Values consist of the mean relative abundance of myelin proteins to actin in lysolecithin-injected samples, represented as fold change over respective relative abundance in saline-injected samples. Mean and SEM were obtained using mice from at least four independent litters. *p < 0.05, versus saline-injected samples, paired Student's t test.
Sox17 increases CC1+ OL and Olig2+ cell density in lysolecithin-induced WM lesions
Further immunohistochemistry showed that lysolecithin-induced demyelination resulted in the loss of CC1+ OL in the WT at 3 DPL, whereas no such loss was observed in the CNP-Sox17 (Fig. 8A). Instead, CC1+ signal intensity and cell density in the CNP-Sox17 appeared greater after lysolecithin-induced injury (Fig. 8A). Interestingly, Olig2+ cells were unaffected in the WT, while the transgenic showed a modest but significant increase in Olig2+ cells (Fig. 8A,B). This would support increased CC1+ cells in the CNP-Sox17 (Fig. 8A). Upon quantitation of OPC and OL markers at 3 and 7 DPL, we found that, although both WT and CNP-Sox17 responded to lysolecithin with increased NG2+ OPCs and O4+ premyelinating OLs at 3 DPL (Fig. 8B, left, showing cell numbers), the response in Trans was relatively smaller, reflected as a decline in the ratio (Fig. 8B, right, showing ratios of lysolecithin and saline). Whereas CC1+ cells were lost in the WT at 3 DPL, lysolecithin unexpectedly stimulated a significant increase in the numbers of CC1+ cells in the CNP-Sox17 mouse. However, a modest increase in CC1+ cells was also later observed in the WT at 7 DPL, when no further change was found in the transgenic (Fig. 8B). Similarly, at 3 DPL, Olig2+ cells in the CNP-Sox17 mouse displayed a significant response to injury, whereas this was not observed in the WT until 7 DPL (Fig. 8B, left). These results indicate that Sox17 overexpression both accelerated and enhanced the extent of the Olig2+ cell response to lysolecithin-induced damage, leading to an early increase in CC1+ cells. These observations may be explained by two possible mechanisms regarding WM protection that are not mutually exclusive: (1) damage was more rapidly repaired by enhanced OL maturation between the O4 and CC1 stages, and (2) lysolecithin-induced cell death and subsequent loss was prevented.
Figure 8.
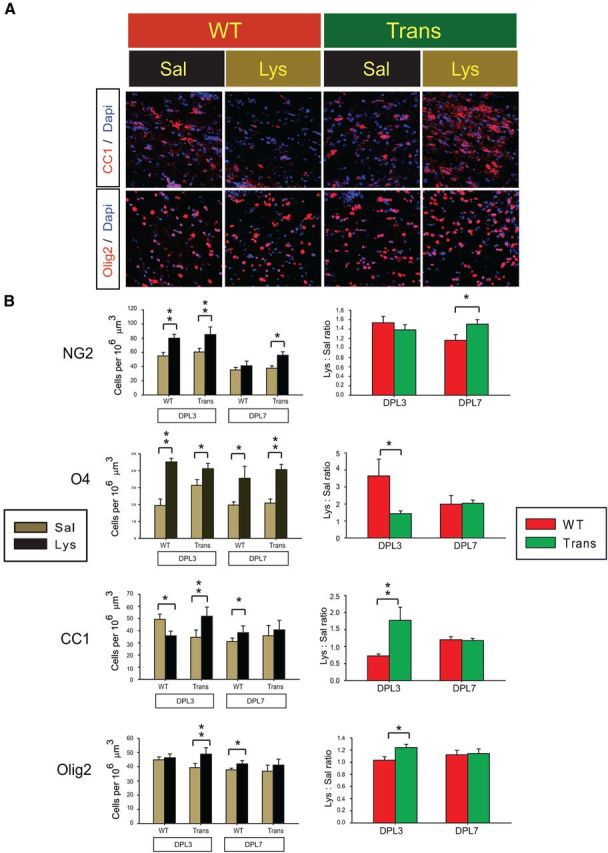
Lysolecithin injection stimulates an increase in OL lineage cells in the CNP-Sox17 mouse. A, Confocal microscope images of CC1+ OLs (top row, red) or Olig2+ cells (bottom row, red), superimposed on DAPI-stained nuclei (blue) in the lesion site at 3 DPL. B, Quantitative analysis of NG2+, O4+, CC1+, Olig2+ cells in the lesion area at 3 and 7 DPL. Left, Cell densities in saline-injected samples (Sal, khaki bars) and lysolecithin-injected samples (Lys, black bars). Right, lysolecithin/saline ratios (Lys:Sal ratios) of NG2+, O4+, CC1+, and Olig2+ cells in Trans (green bars) and WT (red bars). Cell density values are mean and SEM obtained using mice from four independent litters. *p < 0.05, **p < 0.01 versus respective saline-injected samples in left panels (paired Student's t test), or versus WT in right panels (unpaired Student's t test).
When the data are expressed as lysolecithin/saline ratios (Fig. 8B, right), it becomes clear that, shortly after injury, the response of CC1+ and Olig2+ cells in the CNP-Sox17 significantly exceeded that of the WT, whereas changes in NG2+ cells were not different at 3 DPL. Another striking feature of WM injury in the CNP-Sox17 was the increase in NG2+ cells even at 7 DPL (Fig. 8B). This suggests that Sox17 promotes continuous oligodendrogenesis after injury through expanding the OPC population and eventually enhancing their maturation.
Sox17 prevents apoptotic cell death and further change in TCF4+ cells or Gli2 levels after demyelination injury
We determined the extent of apoptotic death at 3 DPL in these lesions and found that lysolecithin-induced increase in cleaved caspase-3 was not observed in lesions of the CNP-Sox17 mouse (Fig. 9A), indicating prevention of apoptosis by Sox17. In addition, Sox17 increased the number of Ki67+ cells in the lysolecithin lesions (Fig. 9B), which was not observed in WT. Wnt signaling has been shown to be upregulated in demyelinating lesions, with increased TCF4 expression among the OPC responses to adult WM injury (Fancy et al., 2009). Based on our previous findings of Wnt/β-catenin antagonism by Sox17 in vitro (Chew et al., 2011), we wanted to determine whether Sox17 regulated components of the canonical Wnt pathway in demyelinated lesions. Consistent with previous reports, we found that the number of cells positive for β-catenin was significantly upregulated in WT (Fig. 9C). A smaller, but nonetheless significant increase was also observed in the CNP-Sox17. However, the injury-induced increase in cells expressing TCF4, which was significant in the WT, was not detected in the CNP-Sox17 transgenic mouse (Fig. 9D). As previously reported (Fancy et al., 2009), TCF4 in the lesions was largely nuclear (Fig. 9D), which is indicative of progenitor status. These results suggest that further increase in TCF4-mediated Wnt responses to injury could be suppressed by Sox17.
Figure 9.

Sox17 overexpression prevents apoptosis in the WM and maintains levels of TCF4 and Gli2 after lysolecithin injection. A, Quantitative analysis of apoptotic cells at 3 DPL as detected by immunostaining for cleaved caspase-3 (Cl. Caspase 3). Lysolecithin increased caspase-3+ cells only in WT WM. **p < 0.01 versus saline, paired Student's t test. B, Quantitative analysis of Ki67+ proliferating cells at 3 DPL. Lysolecithin injection increased cell cycle entry only in Trans. **p < 0.01 versus saline, paired Student's t test. C, Confocal microscope images of β-catenin (b-catenin, red) at 3 DPL, and quantitative analysis of β-catenin+ cells (histograms). The number of β-catenin-expressing cells is elevated by lysolecithin in both mouse strains. *p < 0.05, **p < 0.01 versus saline, paired Student's t test. D, Confocal microscope images of TCF4 (red) at 3 DPL, and quantitative analysis of TCF4+ cells (histograms). The number of TCF4+ cells is elevated by lysolecithin only in WT. **p < 0.01 versus saline, paired Student's t test. E, Western blot of Gli2 protein in the lesion sites at 3 DPL. Fifteen micrograms of total protein was loaded per lane. F, Quantitative analysis of Gli2 protein levels at 3 DPL following densitometric measurement of Western blots. Values consist of mean relative abundance to actin, further expressed as fold change over saline (Lys:Sal). Data are mean ± SEM of at least three independent experiments. **p < 0.01 versus saline, paired Student's t test. G, Fold change of lysolecithin/saline (Lys:Sal) ratio of CNP-Sox17 (Trans) over that of WT, based on data in F. Levels of Gli2 after lysolecithin injection are significantly greater in Trans than WT. **p < 0.01 versus WT, unpaired Student's t test. All mean and SEM of cell count (A–D) and Western blots (F, G) above were obtained using mice from four independent litters.
Since we had found the Olig2+ cell response to be enhanced by Sox17 at 3 DPL (Fig. 8B), we analyzed how Gli2 was regulated in these WM lesions. Figure 9E–G shows that, at 3 DPL, Gli2 protein levels in CNP-Sox17 lesions were not downregulated, whereas a significant decrease was observed in the WT. Together, these results in Figure 9 indicate a preferential activation or maintenance of Hedgehog signaling in the CNP-Sox17 that prevents caspase-mediated apoptosis, whereas it was clearly downregulated in the WT lesions.
Lysolecithin injury induces the antiapoptotic protein Bcl-2 in OLs of CNP-Sox17 mice
We sought to determine whether WM protection in the CNP-Sox17 mouse involved modulation of Gli2-expressing cells and antiapoptotic proteins. In agreement with Western blotting, immunocytochemical analysis shows lower levels of Gli2 immunostaining in the WT after demyelination, whereas no change was apparent in the CNP-Sox17, with the nuclear localization of Gli2 maintained after injury (Fig. 10A). Quantification of cells with Gli2 immunostaining showed a significant decrease only in the WT (Fig. 10B), so that the ratio of Gli2 cells in the lesion to those in the saline-injected site was greater in the CNP-Sox17 mouse (Fig. 10C). Since Hedgehog signaling is important in oligodendroglial survival mechanisms (Chen et al., 2013), we analyzed the lesions for the antiapoptotic protein, Bcl-2, which is involved in Hedgehog-mediated protection (Mazumdar et al., 2011a,b), and which has been identified as a bona fide Gli2 transcriptional target in basal cell carcinoma (Regl et al., 2004). Figure 10D shows that Bcl-2+ cells are more clearly detectable in the CNP-Sox17 than in the WT. Quantitation revealed a significant decrease of Bcl-2-expressing cells in the WT following lysolecithin injury, whereas no decrease was observed in the CNP-Sox17 (Fig. 10E). In this experiment, we also noticed a difference in the total numbers of Bcl-2+ cells between saline-injected controls in both strains, with a significantly higher density in the CNP-Sox17 mouse (Fig. 10E, Sal WT vs Sal Trans; p < 0.001, unpaired student's t test). We then performed dual labeling to determine changes in Bcl-2 expression in CC1+ OLs within the lesions. Figure 10F,G shows that basal densities of Bcl-2+ CC1+ cells were not different between the mouse strains, but after lysolecithin injury, Bcl-2 labeling in CC1+ cells in the CNP-Sox17 was significantly greater than in the WT, presumably due at least in part to the difference in total CC1 cells. Together, these observations indicate that CC1+ cells in the CNP-Sox17 were protected from WM injury through Bcl-2-mediated prevention of apoptosis.
Figure 10.
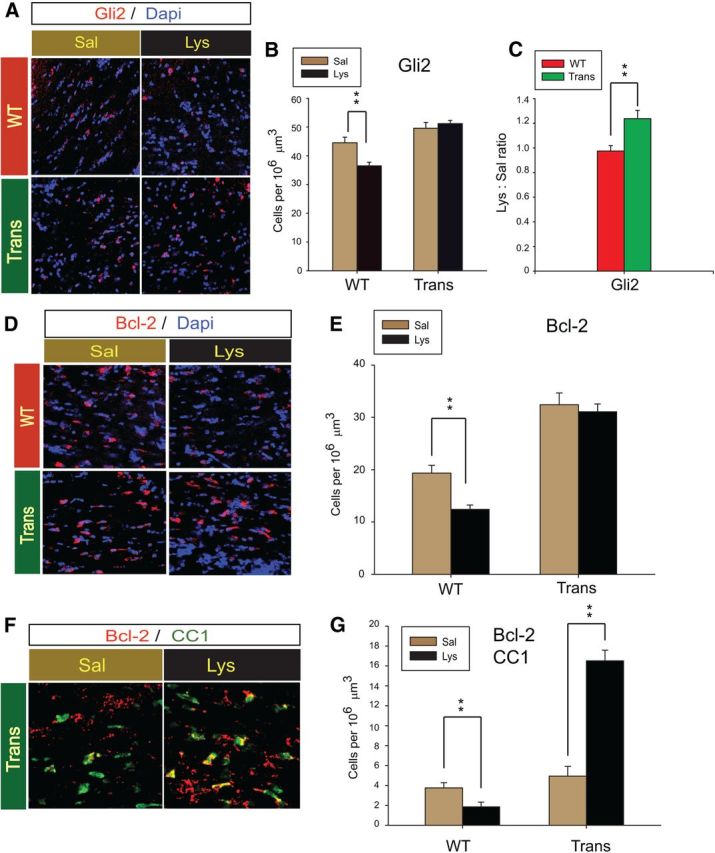
Maintenance of Gli2-expressing cells at 3 DPL is associated with Bcl2-mediated OL survival in the CNP-Sox17 mouse. A, Confocal microscope images of Gli2+ cells (red) in the lesion area at 3 DPL. B, Quantitative analysis of Gli2+ cells at 3 DPL. No change is observed in Trans. Values are means ± SEM of three experiments. **p < 0.01 versus saline, paired Student's t test. C, Plot of lysolecithin/saline (Lys:Sal) ratios for WT and Trans shows increased density of Gli2+ cells after lysolecithin injection. **p < 0.01 versus WT, unpaired Student's t test. D, Confocal microscope images of Bcl-2+ cells (red) at 3 DPL. E, Quantitative analysis of total Bcl-2+ cells at 3 DPL. No change is observed in Trans. Values are mean ± SEM of three experiments. **p < 0.01 versus saline, paired Student's t test. F, Confocal images of Bcl-2+ (red) and CC1+ (green) cells at 3 DPL. Double-positive cells appear yellow. G, Quantitative analysis of Bcl-2-expressing CC1 cells at 3 DPL. **p < 0.01 versus saline, paired Student's t test. All cell counts are mean and SEM using mice obtained from four independent litters.
Discussion
We had previously demonstrated that Sox17 promotes OPC differentiation (Sohn et al., 2006; Chew et al., 2011), and that its suppression of Wnt/β-catenin signaling enhances progenitor cell maturation. Based on these findings, we have now tested our hypothesis that increasing the levels of Sox17 in vivo would promote oligodendroglial lineage progression in either WM development or pathology. We chose the gain-of-function approach in transgenic mice based on the selectivity of the CNP promoter and the proven success of the CNP-driven epidermal growth factor receptor expression in promoting oligodendrogenesis and remyelination (Aguirre et al., 2007).
Sox17 overexpression produced a protracted developmental profile of postnatal lineage progression between P15 and P120, beginning with increased NG2+ OPCs over WT. The initial maturation delay and OPC accumulation coincided with Olig2+ and CC1+ cell loss, while the subsequent recovery of Olig2 and CC1+ overlapped with the decline of apoptosis at ∼P30. This suggests that lineage regulation by Sox17 overexpression is dependent on the dynamics of normal developmental apoptotic events, since apoptosis in both mouse strains was undetectable after P30. The initial increase in apoptosis is reminiscent of studies in embryonic stem cell clones, where Sox17 overexpression caused apoptotic cell death (Serrano et al., 2010). The reason for the biphasic response to Sox17 is presently unknown; a partial explanation includes Sox17-induced reduction of the already declining β-catenin protein levels before P30 (Sohn et al., 2006; Chew et al., 2011). Indeed, β-catenin has been shown to be antiapoptotic in neural progenitor cells, not stem cells (Holowacz et al., 2011), and high levels of β-catenin/TCF4 are implicated in glioma progression via Akt2 expression (Zhang et al., 2011). In myelinating cells, TCF4 was found to be associated with only moderate levels of β-catenin (Fu et al., 2012); this supports differential effects of Wnt activation between OPCs and myelinating cells, where Wnt instead promotes myelin gene expression (Tawk et al., 2011).
Similar to our finding in OPCs, Sox17 overexpression in vivo not only decreased the population of Ki67+ cells in the WM at P15 and P30, indicating increased cell cycle exit, but also decreased β-catenin at P15. At P15, the Hedgehog transcriptional mediator Gli2 was already increased. Wnt and Hedgehog pathways have been described to show reciprocal function (Borycki et al., 2000), with Hedgehog as a negative regulator of β-catenin signaling (Schneider et al., 2010). Our previous findings of Sox17-regulated secreted Frizzled related protein (SFRP) 1/4 expression (Chew et al., 2011), which are Hedgehog targets (He et al., 2006; Katoh and Katoh, 2006), would be consistent with the action of Sox17 as a Hedgehog effector, not only through its production of SFRP, but also in its cell-autonomous destabilization of β-catenin (Sinner et al., 2007; Chew et al., 2011). The effect of Sox17 on cell survival appears to represent a departure from our previous conclusions drawn from purified OPCs. The difference may be explained by the highly mitogenic culture environment with platelet-derived growth factor producing high levels of cellular β-catenin, in which cell survival was unaffected by Sox17 knockdown or overexpression (Sohn et al., 2006).
Through a loss-of-function approach, Gli2 has been shown to be required for oligodendrogenesis in the spinal cord, but not in the forebrain, probably because of redundancy (Qi et al., 2003). In the CNP-Sox17 mouse, elevated Gli2 levels may have mediated the increase in O4+ and Olig2+ cells, leading to growth of the oligodendroglial lineage (Figs. 2–4, 6, 8). Since Olig2 overexpression in neural stem cells produces actively remyelinating OLs in vitro and in remyelination (Copray et al., 2006), and Olig2 is sufficient to initiate the OL differentiation program (Yu et al., 2013), it is possible that the induction of Olig2+ cells by Sox17 promoted the increase in CC1+ cells both during development and after lysolecithin-induced injury.
Oligodendroglial specification, growth, and maturation are dependent on Hedgehog signaling (Poncet et al., 1996; Rowitch et al., 1999; Nery et al., 2001; Wu et al., 2012), and Gli factors transmit graded canonical Hedgehog signals through mechanisms that are not fully understood. Gli2 and Gli1 are both considered activators (Bai and Joyner, 2001), but only Gli2 plays a predominant role as a Hedgehog mediator (Bai et al., 2004; Lei et al., 2004). In support of our observations that Gli1 was not detected in developing CNP-Sox17 transgenic mouse, Gli2, but not Gli1, has been reported to be essential for mediating ectopic Sonic hedgehog signaling (Bai et al., 2002). Although it is presently unknown whether Gli2 induction in the CNP-Sox17 mouse involves increased Hedgehog–Patched receptor interaction, it is likely that Gli2 mediates many of the observed cellular properties that are established effects of Hedgehog signaling.
In addition to changes in Olig2 and Gli2, several other downstream effects of Hedgehog signaling have been observed in the CNP-Sox17 mouse, namely cell proliferation (Kenney and Rowitch, 2000) and survival. However, these were only evident after WM injury at P30. We speculate that the absence of developmental apoptosis after P30 may have contributed to the emergence of Sox17-mediated changes in the adult WM. At 3 DPL, cell apoptosis was decreased and cell cycle entry promoted by Sox17 overexpression; these events likely led to the protection of CC1 cells at 3 DPL and the emergence of NG2+ cells at 7 DPL. It is possible that Sox17 overexpression (1) increased the sensitivity and cellular response to injury-induced signals, and (2) provided protection from injury through increased differentiation and oligodendrogenesis, as well as enhanced antiapoptotic mechanisms. It is also likely that both lysolecithin injury and Sox17 overexpression share the same signaling pathway, since the injury stimulus enhanced cell cycle entry and proliferation but produced no further changes in Hedgehog-regulated targets TCF4, Gli2, or Bcl-2 in the adult CNP-Sox17 mouse (Figs. 9, 10).
Lineage expansion by Sox17 is also observed in non-neural cells. Chronic ectopic Sox17 overexpression was shown to promote self-renewal and eventually leukemogenesis in adult hematopoietic progenitor cells (He et al., 2011), supporting the notion that Sox17 confers fetal identity. While cell fate properties have not been explored in the CNP-Sox17 mouse, our analysis of macroglial cells in the WM indicate that Sox17 did not enhance the generation of astrocytes, and suggests that its effects may be restricted to oligodendroglia. The maintenance of an increased NG2+ OPC population in the developing WM (P15–P60; Fig. 2) suggests the possibility of an increase in lineage commitment rather than local cell proliferation (Fig. 5, Ki67). Alternatively, increased cell survival may contribute to lineage enhancement, both in maturation and resistance to injury. We found that Sox17 overexpression increased the levels of the antiapoptotic proteins Bcl-2 (Fig. 10) and c-FLIP (data not shown) at P30. Both proteins are Hedgehog-Gli targets (Dierks et al., 2007; Kump et al., 2008), which could contribute to the protection of CC1+ OLs from lysolecithin demyelination. Although it is presently unknown how Hedgehog signaling is activated after lysolecithin injection in the CNP-Sox17 mouse, an indirect mechanism is possible, since Sox10 has been shown to regulate Desert hedgehog expression (Küspert et al., 2012).
The interaction between Hedgehog and Wnt signaling is complex (Alvarez-Medina et al., 2008) and intricately interdependent (Lei et al., 2006; Alvarez-Medina et al., 2009). Interestingly, the focal disruption of Wnt signaling within the ventral Hedgehog-defined domain in the neural tube causes ventral progenitor expansion through the regulation of Gli3 (Yu et al., 2008). It is possible that the changes in TCF4/TCF7L2 levels in the CNP-Sox17 may also result from Wnt–Hedgehog interaction. TCF7L2, or TCF4, has been reported to bind β-catenin or histone deacetylase (HDAC) competitively in OL development (Ye et al., 2009). HDAC1 and HDAC2 mediate Sonic hedgehog-induced OL differentiation (Wu et al., 2012), and their disruption of TCF4-β-catenin binding is critical for maturation (Ye et al., 2009). The increase in TCF4 at P30 could have contributed to the delayed appearance of CC1+ OLs. We previously demonstrated that Sox17 forms a complex with β-catenin and TCF4 in samples from postnatal WM (Chew et al., 2011), which declined by P30. Maintenance of the β-catenin/TCF4/Sox17 complex through Sox17 overexpression may be involved in regulating the balance between the emergence of NG2+ OPCs and their maturation to OLs. Although a reliable progenitor marker (Fancy et al., 2009), TCF4 was unexpectedly found in postmitotic progenitors in remyelinating WM lesions (Fu et al., 2012). Our observation of increased TCF4+ cells in the CNP-Sox17 WM at P30, accompanied by increased β-catenin and decreased Ki67+ cells, suggests an expanded pool of premyelinating cells, which also coincides with the Sox17-mediated increase in Olig2+ and O4+ cells. Consistent with this conclusion, analysis of CNPase promoter-driven ablation of Sox17 revealed a significant contribution to the maintenance of the Olig2+ population in the adult WM (L-J Chew, M Catron, E Hong, V Gallo, unpublished observations).
This study represents the first in vivo characterization of Sox17 function in the OL lineage in development and injury, and demonstrates a novel role for Sox17 as a mediator of Hedgehog signaling in promoting OL generation and survival. These findings will provide clues toward further understanding of mechanisms involved in the protection of the lineage and will aid in the design of therapeutic strategies to improve repair in demyelinating disease.
Footnotes
This work was supported by National Institutes of Health (NIH) Grant R01NS056427 (V.G.), National Multiple Sclerosis Society Grant RG3954A1/2 (L.-J.C.), and NIH Intellectual and Developmental Disabilities Research Centers Grant P30HD40677 Imaging Core Resources (V.G.). We thank Timothy Hammond and Drs. Joseph Scafidi, Jonathan Ritter, Peijun Li, and Hui-ling Chen for their patient assistance with surgical lesions, imaging analysis, technical advice, and discussion. We appreciate Dr. Joseph Scafidi's critical reading of the manuscript and helpful comments. We are particularly indebted to Ms. Ying Cheng for valuable assistance in establishing the transgenic mouse colony, and in developing reliable methods for screening and copy number determination.
The authors declare no competing financial interests.
References
- Aguirre A, Gallo V. Postnatal neurogenesis and gliogenesis in the olfactory bulb from NG2-expressing progenitors of the subventricular zone. J Neurosci. 2004;24:10530–10541. doi: 10.1523/JNEUROSCI.3572-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguirre AA, Chittajallu R, Belachew S, Gallo V. NG2-expressing cells in the subventricular zone are type C-like cells and contribute to interneuron generation in the postnatal hippocampus. J Cell Biol. 2004;165:575–589. doi: 10.1083/jcb.200311141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguirre A, Dupree JL, Mangin JM, Gallo V. A functional role for EGFR signalling in myelination and remyelination. Nat Neurosci. 2007;10:990–1002. doi: 10.1038/nn1938. [DOI] [PubMed] [Google Scholar]
- Alvarez-Medina R, Cayuso J, Okubo T, Takada S, Martí E. Wnt canonical pathway restricts graded SHH/Gli patterning activity through the regulation of Gli3 expression. Development. 2008;135:237–247. doi: 10.1242/dev.012054. [DOI] [PubMed] [Google Scholar]
- Alvarez-Medina R, Le Dreau G, Ros M, Martí E. Hedgehog activation is required upstream of Wnt signalling to control neural progenitor proliferation. Development. 2009;136:3301–3309. doi: 10.1242/dev.041772. [DOI] [PubMed] [Google Scholar]
- Bai CB, Joyner AL. Gli1 can rescue the in vivo function of Gli2. Development. 2001;128:5161–5172. doi: 10.1242/dev.128.24.5161. [DOI] [PubMed] [Google Scholar]
- Bai CB, Auerbach W, Lee JS, Stephen D, Joyner AL. Gli2, but not Gli1, is required for initial Shh signaling and ectopic activation of the Shh pathway. Development. 2002;129:4753–4761. doi: 10.1242/dev.129.20.4753. [DOI] [PubMed] [Google Scholar]
- Bai CB, Stephen D, Joyner AL. All mouse ventral spinal cord patterning by Hedgehog is Gli dependent and involves an activator function in Gli3. Dev Cell. 2004;6:103–115. doi: 10.1016/S1534-5807(03)00394-0. [DOI] [PubMed] [Google Scholar]
- Borycki A, Brown AM, Emerson CP., Jr Shh and Wnt signaling pathways converge to control Gli gene activation in avia somites. Development. 2000;127:2075–2087. doi: 10.1242/dev.127.10.2075. [DOI] [PubMed] [Google Scholar]
- Chen HL, Chew LJ, Packer RJ, Gallo V. Modulation of the Wnt/beta-catenin pathway in human oligodendroglioma cells by Sox17 regulates proliferation and differentiation. Cancer Lett. 2013;335:361–371. doi: 10.1016/j.canlet.2013.02.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chew LJ, Gallo V. The yin and yang of Sox proteins: activation and repression in development and disease. J Neurosci Res. 2009;87:3277–3287. doi: 10.1002/jnr.22128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chew LJ, Shen W, Ming X, Senatorov VV, Jr, Chen HL, Cheng Y, Hong E, Knoblach S, Gallo V. SRY-box containing gene 17 regulates the Wnt/beta-catenin signaling pathway in oligodendrocyte progenitor cells. J Neurosci. 2011;31:13921–13935. doi: 10.1523/JNEUROSCI.3343-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copray S, Balasubramaniyan V, Levenga J, de Bruijn J, Liem R, Boddeke E. Olig2 overexpression induces the in vitro differentiation of neural stem cells into mature oligodendrocytes. Stem Cells. 2006;24:1001–1010. doi: 10.1634/stemcells.2005-0239. [DOI] [PubMed] [Google Scholar]
- Dierks C, Grbic J, Zirlik K, Beigi R, Englund NP, Guo GR, Veelken H, Engelhardt M, Mertelsmann R, Kelleher JF, Schultz P, Warmuth M. Essential role of stromally induced hedgehog signaling in B-cell malignancies. Nat Med. 2007;13:944–951. doi: 10.1038/nm1614. [DOI] [PubMed] [Google Scholar]
- Etxeberria A, Mangin JM, Aguirre A, Gallo V. Adult-born SVZ progenitors receive transient synapses during remyelination in corpus callosum. Nat Neurosci. 2010;13:287–289. doi: 10.1038/nn.2500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fancy SP, Baranzini SE, Zhao C, Yuk DI, Irvine KA, Kaing S, Sanai N, Franklin RJ, Rowitch DH. Dysregulation of the Wnt pathway inhibits timely myelination and remyelination in the mammalian CNS. Genes and Development. 2009;23:1571–1585. doi: 10.1101/gad.1806309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu H, Kesari S, Cai J. Tcf7L2 is tightly controlled during myelin formation. Cell Mol Neurobiol. 2012;32:345–352. doi: 10.1007/s10571-011-9778-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gravel M, Di Polo A, Valera PB, Braun PE. Four kilobase sequence of the mouse CNP gene directs spatial and temporal expression of lacZ in transgenic mice. J Neurosci Res. 1998;53:393–404. doi: 10.1002/(SICI)1097-4547(19980815)53:4<393::AID-JNR1>3.3.CO%3B2-Y. [DOI] [PubMed] [Google Scholar]
- Gubbay J, Collignon J, Koopman P, Capel B, Economou A, Münsterberg A, Vivian N, Goodfellow P, Lovell-Badge R. A gene mapping to the sex-determining region of the mouse Y chromosome is a member of a novel family of embryonically expressed genes. Nature. 1990;346:245–250. doi: 10.1038/346245a0. [DOI] [PubMed] [Google Scholar]
- He J, Sheng T, Stelter AA, Li C, Zhang X, Sinha M, Luxon BA, Xie J. Suppressing Wnt signaling by the hedgehog pathway through sFRP1. J Biol Chem. 2006;281:35598–35602. doi: 10.1074/jbc.C600200200. [DOI] [PubMed] [Google Scholar]
- He S, Kim I, Lim MS, Morrison SJ. Sox17 expression confers self-renewal potential and fetal stem cell characteristics upon adult hematopoietic progenitors. Genes Dev. 2011;25:1613–1627. doi: 10.1101/gad.2052911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holowacz T, Huelsken J, Dufort D, van der Kooy D. Neural stem cells are increased after loss of beta-catenin, but neural progenitors undergo cell death. Eur J Neurosci. 2011;33:1366–1375. doi: 10.1111/j.1460-9568.2011.07632.x. [DOI] [PubMed] [Google Scholar]
- Jablonska B, Aguirre A, Raymond M, Szabo G, Kitabatake Y, Sailor KA, Ming GL, Song H, Gallo V. Chordin-induced lineage plasticity of adult SVZ neuroblasts after demyelination. Nat Neurosci. 2010;13:541–550. doi: 10.1038/nn.2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi M, Keith Pittman H, Haisch C, Verbanac K. Real-time PCR to determine transgene copy number and to quantitate the biolocalization of adoptively transferred cells from EGFP-transgenic mice. Biotechniques. 2008;45:247–258. doi: 10.2144/000112913. [DOI] [PubMed] [Google Scholar]
- Kanai Y, Kanai-Azuma M, Noce T, Saido TC, Shiroishi T, Hayashi Y, Yazaki K. Identification of two Sox17 messenger RNA isoforms, with and without the high mobility group box region, and their differential expression in mouse spermatogenesis. J Cell Biol. 1996;133:667–681. doi: 10.1083/jcb.133.3.667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh Y, Katoh M. WNT antagonist, SFRP1, is Hedgehog signaling target. Int J Mol Med. 2006;17:171–175. [PubMed] [Google Scholar]
- Kenney AM, Rowitch DH. Sonic hedgehog promotes G1 cyclin expression and sustained cell cycle progression in mammalian neuronal precursors. Mol Cell Biol. 2000;20:9055–9067. doi: 10.1128/MCB.20.23.9055-9067.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kump E, Ji J, Wernli M, Häusermann P, Erb P. Gli2 upregulates cFlip and renders basal cell carcinoma cells resistant to death ligand-mediated apoptosis. Oncogene. 2008;27:3856–3864. doi: 10.1038/onc.2008.5. [DOI] [PubMed] [Google Scholar]
- Küspert M, Weider M, Müller J, Hermans-Borgmeyer I, Meijer D, Wegner M. Desert hedgehog links transcription factor Sox10 to perineurial development. J Neurosci. 2012;32:5472–5480. doi: 10.1523/JNEUROSCI.5759-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei Q, Zelman AK, Kuang E, Li S, Matise MP. Transduction of graded Hedgehog signaling by a combination of Gli2 and Gli3 activator functions in the developing spinal cord. Development. 2004;131:3593–3604. doi: 10.1242/dev.01230. [DOI] [PubMed] [Google Scholar]
- Lei Q, Jeong Y, Misra K, Li S, Zelman AK, Epstein DJ, Matise MP. Wnt signaling inhibitors regulate the transcriptional response to morphogenetic Shh-Gli signaling in the neural tube. Dev Cell. 2006;11:325–337. doi: 10.1016/j.devcel.2006.06.013. [DOI] [PubMed] [Google Scholar]
- Lu QR, Sun T, Zhu Z, Ma N, Garcia M, Stiles CD, Rowitch DH. Common developmental requirement for Olig function indicates a motor neuron/oligodendrocyte connection. Cell. 2002;109:75–86. doi: 10.1016/S0092-8674(02)00678-5. [DOI] [PubMed] [Google Scholar]
- Mazumdar T, DeVecchio J, Shi T, Jones J, Agyeman A, Houghton JA. Hedgehog signaling drives cellular survival in human colon carcinoma cells. Cancer Res. 2011a;71:1092–1102. doi: 10.1158/0008-5472.CAN-10-2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazumdar T, Devecchio J, Agyeman A, Shi T, Houghton JA. Blocking Hedgehog survival signaling at the level of the GLI genes induces DNA damage and extensive cell death in human colon carcinoma cells. Cancer Res. 2011b;71:5904–5914. doi: 10.1158/0008-5472.CAN-10-4173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nery S, Wichterle H, Fishell G. Sonic hedgehog contributes to oligodendrocyte specification in the mammalian forebrain. Development. 2001;128:527–540. doi: 10.1242/dev.128.4.527. [DOI] [PubMed] [Google Scholar]
- Nicolay DJ, Doucette JR, Nazarali AJ. Transcriptional control of oligodendrogenesis. Glia. 2007;55:1287–1299. doi: 10.1002/glia.20540. [DOI] [PubMed] [Google Scholar]
- Poncet C, Soula C, Trousse F, Kan P, Hirsinger E, Pourquié O, Duprat AM, Cochard P. Induction of oligodendrocyte progenitors in the trunk neural tube by ventralizing signals: effects of notochord and floor plate grafts, and of sonic hedgehog. Mech Dev. 1996;60:13–32. doi: 10.1016/S0925-4773(96)00595-3. [DOI] [PubMed] [Google Scholar]
- Potzner MR, Griffel C, Lütjen-Drecoll E, Bösl MR, Wegner M, Sock E. Prolonged Sox4 expression in oligodendrocytes interferes with normal myelination in the central nervous system. Mol Cell Biol. 2007;27:5316–5326. doi: 10.1128/MCB.00339-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi Y, Tan M, Hui CC, Qiu M. Gli2 is required for normal Shh signaling and oligodendrocyte development in the spinal cord. Mol Cell Neurosci. 2003;23:440–450. doi: 10.1016/S1044-7431(03)00067-8. [DOI] [PubMed] [Google Scholar]
- Regl G, Kasper M, Schnidar H, Eichberger T, Neill GW, Philpott MP, Esterbauer H, Hauser-Kronberger C, Frischauf AM, Aberger F. Activation of the Bcl2 promoter in response to Hedgehog/GLI signal transduction is predominantly mediated by GLI2. Cancer Res. 2004;64:7724–7731. doi: 10.1158/0008-5472.CAN-04-1085. [DOI] [PubMed] [Google Scholar]
- Rowitch DH, S-Jacques B, Lee SM, Flax JD, Snyder EY, McMahon AP. Sonic hedgehog regulates proliferation and inhibits differentiation of CNS precursor cells. J Neurosci. 1999;19:8954–8965. doi: 10.1523/JNEUROSCI.19-20-08954.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider FT, Schänzer A, Czupalla CJ, Thom S, Engels K, Schmidt MH, Plate KH, Liebner S. Sonic hedgehog acts as a negative regulator of beta-catenin signaling in the adult tongue epithelium. Am J Pathol. 2010;177:404–414. doi: 10.2353/ajpath.2010.091079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano AG, Gandillet A, Pearson S, Lacaud G, Kouskoff V. Contrasting effects of Sox17- and Sox18-sustained expression at the onset of blood specification. Blood. 2010;115:3895–3898. doi: 10.1182/blood-2009-10-247395. [DOI] [PubMed] [Google Scholar]
- Shen S, Sandoval J, Swiss VA, Li J, Dupree J, Franklin RJ, Casaccia-Bonnefil P. Age-dependent epigenetic control of differentiation inhibitors is critical for remyelination efficiency. Nat Neurosci. 2008;11:1024–1034. doi: 10.1038/nn.2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinner D, Kordich JJ, Spence JR, Opoka R, Rankin S, Lin SC, Jonatan D, Zorn AM, Wells JM. Sox17 and Sox4 differentially regulate beta-catenin/T-cell factor activity and proliferation of colon carcinoma cells. Mol Cell Biol. 2007;27:7802–7815. doi: 10.1128/MCB.02179-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohn J, Natale J, Chew LJ, Belachew S, Cheng Y, Aguirre A, Lytle J, Nait-Oumesmar B, Kerninon C, Kanai-Azuma M, Kanai Y, Gallo V. Identification of Sox17 as a transcription factor that regulates oligodendrocyte development. J Neurosci. 2006;26:9722–9735. doi: 10.1523/JNEUROSCI.1716-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stolt CC, Wegner M. SoxE function in vertebrate nervous system development. Int J Biochem Cell Biol. 2010;42:437–440. doi: 10.1016/j.biocel.2009.07.014. [DOI] [PubMed] [Google Scholar]
- Stolt CC, Rehberg S, Ader M, Lommes P, Riethmacher D, Schachner M, Bartsch U, Wegner M. Terminal differentiation of myelin-forming oligodendrocytes depends on the transcription factor Sox10. Genes Dev. 2002;16:165–170. doi: 10.1101/gad.215802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stolt CC, Lommes P, Sock E, Chaboissier MC, Schedl A, Wegner M. The Sox9 transcription factor determines glial fate choice in the developing spinal cord. Genes Dev. 2003;17:1677–1689. doi: 10.1101/gad.259003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tawk M, Makoukji J, Belle M, Fonte C, Trousson A, Hawkins T, Li H, Ghandour S, Schumacher M, Massaad C. Wnt/β-catenin signaling is an essential and direct driver of myelin gene expression and myelinogenesis. J Neurosci. 2011;31:3729–3742. doi: 10.1523/JNEUROSCI.4270-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu M, Hernandez M, Shen S, Sabo JK, Kelkar D, Wang J, O'Leary R, Phillips GR, Cate HS, Casaccia P. Differential modulation of the oligodendrocyte transcriptome by sonic hedgehog and bone morphogenetic protein 4 via opposing effects on histone acetylation. J Neurosci. 2012;32:6651–6664. doi: 10.1523/JNEUROSCI.4876-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye F, Chen Y, Hoang T, Montgomery RL, Zhao XH, Bu H, Hu T, Taketo MM, van Es JH, Clevers H, Hsieh J, Bassel-Duby R, Olson EN, Lu QR. HDAC1 and HDAC2 regulate oligodendrocyte differentiation by disrupting the beta-catenin-TCF interaction. Nat Neurosci. 2009;12:829–838. doi: 10.1038/nn.2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan X, Chittajallu R, Belachew S, Anderson S, McBain CJ, Gallo V. Expression of the green fluorescent protein in the oligodendrocyte lineage: a transgenic mouse for developmental and physiological studies. J Neurosci Res. 2002;70:529–545. doi: 10.1002/jnr.10368. [DOI] [PubMed] [Google Scholar]
- Yu W, McDonnell K, Taketo MM, Bai CB. Wnt signaling determines ventral spinal cord cell fates in a time-dependent manner. Development. 2008;135:3687–3696. doi: 10.1242/dev.021899. [DOI] [PubMed] [Google Scholar]
- Yu Y, Chen Y, Kim B, Wang H, Zhao C, He X, Liu L, Liu W, Wu LM, Mao M, Chan JR, Wu J, Lu QR. Olig2 targets chromatin remodelers to enhancers to initiate oligodendrocyte differentiation. Cell. 2013;152:248–261. doi: 10.1016/j.cell.2012.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Huang K, Shi Z, Zou J, Wang Y, Jia Z, Zhang A, Han L, Yue X, Liu N, Jiang T, You Y, Pu P, Kang C. High β-catenin/Tcf-4 activity confers glioma progression via direct regulation of AKT2 gene expression. Neuro Oncol. 2011;13:600–609. doi: 10.1093/neuonc/nor034. [DOI] [PMC free article] [PubMed] [Google Scholar]