

Abstract
Chronic pain associated with injury or disease can result from dysfunction of sensory afferents whereby the threshold for activation of pain-sensing neurons (nociceptors) is lowered. Neurotrophic factors control nociceptor development and survival, but also induce sensitization through activation of their cognate receptors, attributable, in part, to the modulation of ion channel function. Thermal pain is mediated by channels of the transient receptor potential (TRP) family, including the cold and menthol receptor TRPM8. Although it has been shown that TRPM8 is involved in cold hypersensitivity, the molecular mechanisms underlying this pain modality are unknown. Using microarray analyses to identify mouse genes enriched in TRPM8 neurons, we found that the glial cell line-derived neurotrophic factor (GDNF) family receptor GFRα3 is expressed in a subpopulation of TRPM8 sensory neurons that have the neurochemical profile of cold nociceptors. Moreover, we found that artemin, the specific GFRα3 ligand that evokes heat hyperalgesia, robustly sensitized cold responses in a TRPM8-dependent manner in mice. In contrast, GFRα1 and GFRα2 are not coexpressed with TRPM8 and their respective ligands GDNF and neurturin did not induce cold pain, whereas they did evoke heat hyperalgesia. Nerve growth factor induced mild cold sensitization, consistent with TrkA expression in TRPM8 neurons. However, bradykinin failed to alter cold sensitivity even though its receptor expresses in a subset of TRPM8 neurons. These results show for the first time that only select neurotrophic factors induce cold sensitization through TRPM8 in vivo, unlike the broad range of proalgesic agents capable of promoting heat hyperalgesia.
Introduction
An important feature of the peripheral nervous system is identifying potentially tissue-damaging stimuli. Painful stimuli excite specialized sensory neurons termed nociceptors, whose sensitivity is enhanced during conditions of injury, thereby lowering pain thresholds (Woolf, 2007; Basbaum et al., 2009). As a result, pain intensity in response to noxious stimuli is increased (hyperalgesia), or stimuli that would normally be innocuous become painful (allodynia). Pain is further broken down by modality, including mechanical, heat, and cold hypersensitivity. Cold allodynia is a source of serious discomfort and pain, occurring in conditions such as diabetic neuropathy and complex regional pain syndrome, and is a side-effect of many chemotherapeutics (Tahmoush et al., 2000; Vinik, 2004; Attal et al., 2009). However, the mechanisms for cold hypersensitivity remain unknown.
Investigations into the mechanisms underlying injury-induced hypersensitivity have revealed that specific proalgesic agents are able to directly sensitize nociceptors (Chuang et al., 2001; Ji et al., 2002; Basbaum et al., 2009; Linley et al., 2010). Many are upregulated and released at the site of injury where they sensitize neurons that innervate the region, often by modulating the activity of ion channels (Julius and Basbaum, 2001; Pezet and McMahon, 2006). Among the many classes of proalgesics, neurotrophic factors, including nerve growth factor (NGF) and the glial cell-line derived neurotrophic factor (GDNF) family of ligands (GFLs), have been extensively studied (Woolf et al., 1994; Chuang et al., 2001; Malin et al., 2006; Schmutzler et al., 2011). NGF potentiates heat and capsaicin responses transduced by transient receptor potential vanilloid 1 (TRPV1), involving both direct sensitization of the channel as well as insertion of new channels into the cell membrane (Chuang et al., 2001; Zhuang et al., 2004; Zhang et al., 2005). Although focus has been placed on the role of proalgesics in sensitizing heat and mechanical responses, it is unclear whether they are able to modulate cold responses as well.
Transient receptor potential melastatin 8 (TRPM8) acts as the principal cold sensor in the peripheral nervous system (Bautista et al., 2007; Knowlton et al., 2010), and is activated by temperatures both in the innocuous and noxious range (McKemy et al., 2002). Behavioral assays in mice lacking TRPM8 channels or neurons show that it is not only required for sensing acute cold stimuli, but is also involved in cold hypersensitivity in conditions of inflammation and nerve injury (Bautista et al., 2007; Chung and Caterina, 2007; Knowlton et al., 2013). Here, we investigated whether cold responses were modulated by an array of classical neurotrophic factors and their cellular receptors, finding that the GDNF family receptor α3 (GFRα3) is extensively coexpressed with TRPM8 and that its ligand, artemin, is able to significantly sensitize cold responses in mice in a manner that requires TRPM8 channels. We also observed a TRPM8-dependent increase in cold sensitivity after injection of NGF. Other proalgesics were ineffective in inducing cold pain. This is the first evidence of direct sensitization of cold responses in vivo and suggests that artemin may be the chief agent in the development of cold allodynia in pathological conditions.
Materials and Methods
Microarray and qPCR analysis
All experiments were approved by the University of Southern California Institutional Animal Care and Use Committee and performed in accordance with the recommendations of the International Association for the Study of Pain and with the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals. To identify genes enriched in TRPM8 neurons, total RNA was purified from sensory ganglia collected from control mice and animals depleted of TRPM8 neurons (Trpm8DTR mice; n = 8 mice per group; Knowlton et al., 2013) using RNAlater buffer (Qiagen) then precipitated and purified with the RNeasy Mini kit (Qiagen) with in-column DNA digestion following the manufacturer's instructions. Microarray analyses were performed in triplicate and assayed using the GeneChip Mouse Gene 1.0 ST Arrays by the University of Southern California CHLA Affymetrix MicroArray Core Facility as described previously (Knowlton et al., 2013). For qPCR, RNA was isolated as above from hindpaw skin or sensory ganglia of wild-type or Trpm8−/− mice. The iScript cDNA synthesis kit (Bio-Rad) was used for synthesizing cDNA from purified RNA samples, and qPCR was performed using Ssofast EvaGreen supermix (Bio-Rad) and a Bio-Rad CFX96 detection system. All samples were run in duplicate, and ΔCt values normalized to the reference standard were obtained as follows: ΔCt = Ct (gene of interest) − Ct (GAPDH). Relative changes in expression were calculated using the ΔΔCt method. The primers used are listed below.
GFRα3 FWD: 5′ GTGCAGATCACGCCTGATGG 3′ (362bp) REV: 5′ GCTGCAGTCTCAGAGCAGGG 3′
Ret FWD: 5′ GCGCCCCGAGTGTGAGGAATGTGG 3′ (441bp) REV: 5′ GCTGATGCAATGGGCGGCTTGTGC 3′
TRPM8 (5′) FWD: 5′ GCTGCCTGAAGAGGAAATTG 3′ (600bp) REV: 5′ GCCCAGATGAAGAGAGCTTG 3′
Artemin FWD: 5′ TACTGCATTGTCCCACTGCCTCC 3′ (217bp) REV: 5′ TCGCAGGGTTCTTTCGCTGCACA 3′
GAPDH FWD: 5′ TGTAGACCATGTAGTGAGGTCA 3′ (123bp) REV: 5′ AGGTCGGTGTGAACGGATTTG 3′.
Immunohistochemistry
As previously described (Takashima et al., 2007), immunolabeling was performed on sections of dorsal root ganglia (DRG) and trigeminal ganglia (TG) dissected from adult (10–12 weeks) Trpm8GFP mice (Takashima et al., 2007) or Trpm8−/− mice. Primary antibodies were diluted in a working solution (0.3% Triton X-100 and 1–5% normal donkey or goat serum in PBS, pH 7.4). Primary antibodies and dilutions used are as follows: 1:500 chicken anti-GFP (GFP-1020; Aves Laboratories), 1:200 goat anti-GFRα1, goat anti-GFRα2, and goat anti-GFRα3 (GT15004, GT15005, and GT15123; Neuromics), 1:500 guinea pig anti-CGRP (T-5027; Bachem), 1:500 rabbit anti-peripherin (AB1530; Millipore), 1:500 rabbit anti-NF200 (N4142; Sigma-Aldrich), 1:1000 rabbit anti-TRPV1 (ab31895; Abcam), 1:500 guinea pig anti-TRPV1 (a gift from D. Julius, University of California San Francisco), 1:500 mouse anti-Nav1.8 (75-166; Neuromab), 1:2000 rabbit anti-P2X3 (AB5895; Millipore), 1:200 rabbit anti-bradykinin B2 R (H-50); sc-25671, Santa Cruz Biotechnology), 1:500 rabbit anti-TrkA (sc-118; Santa Cruz Biotechnology), 1:200 rabbit anti-c-Ret (R787; JP18121; IBL International), 1:500 rabbit anti-NCAM (AB5032; Millipore), and 1:500 rabbit anti-PGP9.5 (A01398; Genscript). Sections were incubated with primary antibody solutions at 4°C overnight or for two nights, then washed and incubated with the appropriate Alexa Fluor-conjugated secondary antibody solutions (1:1000 Alexa Fluor 350, Alexa Fluor 488 or Alexa Fluor 594, Invitrogen) at room temperature for 1–2 h. To detect isolectin B4 (IB4) binding, 1:2000 Griffonia simplicifolia isolectin GS-IB4-Alexa 568 (I-21412; Life Technologies) was added to the secondary antibody solutions. Slides were washed and mounted in ProLong Gold reagent (Life Technologies). Digital images were acquired using a Zeiss Axio Imager.M2 with Apotome attachment and Axiovision software, and analyzed using ImageJ. Quantification is reported as percentage overlap between two or three markers ±SE between fields (Takashima et al., 2007, 2010).
Behavioral assays
Hindpaw injections.
Adult (at least 8 weeks of age) wild-type or Trpm8−/− mice of both sexes were used in behavioral assays. Mice were acclimated to the appropriate testing chambers for 40–60 min before the experiments. After baseline testing, mice were lightly anesthetized with isoflurane and administered an intraplantar injection of 20 μl vehicle (saline) or inflammatory mediator. Their cold, heat, or mechanical sensitivity was then tested at the indicated time points. NGF (Life Technologies), GDNF (Sigma-Aldrich), neurturin (NRTN; R&D Systems), and artemin (R&D Systems) were diluted to 10 μg/ml in HBSS (Life Technologies) directly before injection, and bradykinin was diluted to 10 nm in 0.9% saline (Chuang et al., 2001; Malin et al., 2006). All results from behavioral assays are shown as averages ± SEM for each group, and statistical differences were determined using one-way or two-way repeated-measures ANOVA, followed by Bonferroni's and Tukey's HSD post hoc analyses, or Mann–Whitney or Wilcoxon tests as appropriate.
Evaporative cooling assay.
To evaluate cold sensitivity of the hindpaws, the acetone evaporative cooling assay was performed as previously described (Knowlton et al., 2011). Briefly, mice were acclimated for 20 min in an elevated chamber with a mesh floor (IITC), and a syringe with a piece of rubber tubing attached to the end was filled with acetone and the plunger depressed so that a small drop of acetone formed at the tip. The syringe was raised from below, depositing the acetone drop on the paw. Mice were tested with an interstimulation period of 4 min per mouse, alternating paws between stimulations, for a total of three trials per paw for each time point. Responses were video recorded for later quantification by an observer blind to genotype and the solution injected. Behaviors were scored according to the magnitude of the response along the following scale: 0-no response; 1-brief lift, sniff, flick, or startle; 2-jumping, paw shaking; 3-multiple lifts, paw lick; 4-prolonged paw lifting, licking, shaking, or jumping; 5-paw guarding (Colburn et al., 2007; Knowlton et al., 2011, 2013; Brenneis et al., 2013). Artemin-injected wild-type mice and Trpm8−/− mice were tested 1, 3, 9, and 24 h after injection; NGF-, GDNF-, and NRTN-injected mice were tested 1, 2, 3, and 24 h after injection; and bradykinin-injected mice were tested 15 min, 1 h, 2 h, 3 h, and 24 h after injection.
Hargreaves test.
Mice were allowed to acclimate in Plexiglas chambers placed on a glass surface heated to 32°C (Plantar Test Apparatus, IITC) for 40 min. To test hindpaw sensitivity to noxious heat, a radiant heat source was focused on the hindpaw and the time to withdrawal of the paw was measured, with an interstimulation period of 5 min per mouse, alternating paws, for a total of three trials per paw for each time point. Stimulus intensity was set to produce latencies of 8–11 s at baseline. A cutoff point was set at 20 s to avoid tissue damage to the paw. The readings from the three trials were averaged to give the withdrawal latency.
Electronic von Frey.
Stimulation with an electronic von Frey apparatus (IITC) was performed as previously described (Knowlton et al., 2011). Briefly, animals were acclimated to an elevated mesh platform for 20 min. The apparatus was fitted with a semiflexible tip and raised to the plantar surface of the hindpaw. The force at which the mouse removed the paw was measured, and average paw withdraw threshold was measured per foot from five trials per foot, alternating paws, with 4 min between trials.
Results
GFRα3 is expressed in a subset of TRPM8 sensory neurons
Given the role that neurotrophic factors are known to play in nociception, and sensitization in particular (Chuang et al., 2001; Malin et al., 2006; Pezet and McMahon, 2006; Schmutzler et al., 2009), we hypothesized that any neurotrophic factor receptor found to be highly coexpressed with TRPM8 will play a role in cold hypersensitivity. To identify such candidate molecules, we analyzed a dataset from a microarray screen we previously performed on sensory ganglia collected from mice genetically depleted of TRPM8 neurons (Knowlton et al., 2013). Transcripts that showed significantly lower expression levels in TRPM8-neuron ablated compared with wild-type ganglia were considered to be candidate genes enriched in the TRPM8 neuronal population. We found that transcript levels of neurotrophic factors themselves were not different in tissue from ablated versus control animals, but there was a large reduction in expression of the NGF receptor TrkA (79.0 ± 0.01% of control) and the artemin receptor GFRα3 (77.0 ± 0.05%, n = 8 mice of each genotype; data not shown).
We have previously shown that TrkA is expressed in only a subset of TRPM8 neurons (Takashima et al., 2010). However, the direct overlap between GDNF family receptors and TRPM8 is unknown. To examine the expression of these receptors in TRPM8 neurons, we used a previously characterized line of transgenic mice (Trpm8GFP, JAX Stock no. 020650, C57BL/6-Tg(Trpm8-EGFP)1Dmck/J) in which expression of enhanced green fluorescent protein (GFP) is driven by the Trpm8 transcriptional promoter and is a reliable marker for TRPM8 expression in mice (Takashima et al., 2007, 2010; Ramachandran et al., 2013). As predicted from the microarray data, we observed a significant overlap between GFRα3 and GFP expression (Fig. 1A,B), finding that half of TRPM8+ neurons coexpress GFRα3 in sensory ganglia (TG, 52.91 ± 3.10%, n = 741 cells; DRG, 48.29 ± 5.42%, n = 606). The soma areas of double-labeled cells were indicative of small and medium size neurons, with averages of 200.52 ± 11.28 μm2 in TG (n = 115) and 236.37 ± 14.95 μm2 in DRG (n = 84), and a similar area frequency distribution to that of the individual TRPM8 and GFRα3 populations. The two other GFL receptors GFRα1 and GFRα2, however, were not expressed in TRPM8+ neurons (Fig. 1C,D; 0.00%, n = 627 and 667, respectively, for TG; 1.89 ± 1.12% and 0.79 ± 0.47%, n = 467 and 1017, respectively, for DRG), consistent with the observation that their expression levels were unchanged in TRPM8 neuron ablated animals in our microarray analysis.
Figure 1.
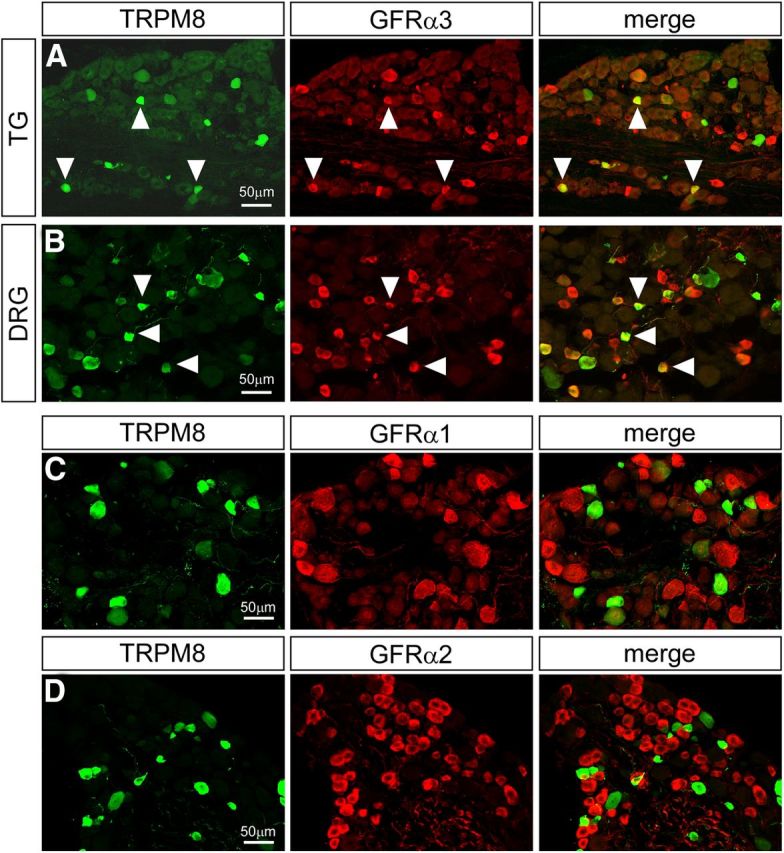
Expression of GFRα receptors in TRPM8 neurons. GFRα3 immunoreactivity (red) was observed in approximately half of TRPM8+ neurons (green, arrowheads) in trigeminal (A) and dorsal root ganglia (B). In contrast, immunoreactivity for neither GFRα1 (C) nor GFRα2 (D) was observed in TRPM8+ neurons in dorsal root ganglia.
TRPM8-GFRα3 coexpressing neurons represent a putative population of thermal nociceptors
Previous immunohistochemical studies have revealed that TRPM8 neurons are a heterogeneous population, expressing a variety of markers, yet rarely overlapping with any given one significantly (Peier et al., 2002; Takashima et al., 2007, 2010). Furthermore, in functional assays, these neurons respond to cold stimuli both in the noxious and innocuous range, in agreement with the observation that TRPM8-null mice are deficient in sensing both noxious and innocuous cold (McKemy et al., 2002; Peier et al., 2002; Bautista et al., 2007; Colburn et al., 2007; Dhaka et al., 2007; Knowlton et al., 2013). Thus, GFRα3 expression in such a large subset of TRPM8 neurons raises the intriguing possibility that it may represent a functionally distinct subpopulation of cold-sensitive afferents, possibly nociceptors involved in mediating cold allodynia. Therefore, we used immunostaining to characterize the TRPM8-GFRα3 population. Consistent with previous reports (Takashima et al., 2007), less than half of TRPM8 neurons express the intermediate filament peripherin (Fig. 2A), a marker of small-diameter, unmyelinated C-fibers (TG, 47.79 ± 2.45%, n = 923; DRG, 34.46 ± 4.03%, n = 398), with an even smaller proportion immunoreactive for NF200 (Fig. 2B), a marker for myelinated Aδ-fibers (TG, 10.36 ± 1.74%, n = 739; DRG, 10.98 ± 1.81%, n = 198). In contrast, TRPM8-GFRα3 coexpressing neurons were largely positive for peripherin (TG, 82.14 ± 3.07%, n = 1067; DRG, 57.00 ± 4.41%, n = 519; Table 1), whereas NF200 expression was negligible in this population (TG, 6.13 ± 2.18%, n = 1064; DRG, 0.00%, n = 269; Table 1). This indicates that GFRα3 is expressed specifically in a subset of TRPM8-expressing C-fibers as opposed to Aδ-fibers (Goldstein et al., 1991).
Figure 2.

DRG neurons coexpressing TRPM8 and GFRα3 are largely peptidergic C-fibers. A, TRPM8 neurons (green) expressing GFRα3 (red) were largely positive for peripherin (blue). B, Few TRPM8 neurons (green) that express GFRα3 (blue) were immunoreactive for NF200 (red). C, TRPM8 neurons (green) do not bind IB4 (red) and therefore do not express GFRα3 (blue). D, In contrast, a large portion of TRPM8 neurons (green) that labeled with GFRα3 (blue) were immunoreactive for CGRP (red). Arrowheads indicate triple-labeled neurons; arrows indicate TRPM8+ cells that are not positive for either marker.
Table 1.
Neurochemical profile of TRPM8-GFRα3 DRG and TG neurons
| Dorsal root ganglia |
Trigeminal ganglia |
|||||||
|---|---|---|---|---|---|---|---|---|
| %TRPM8 | %GFRα3 | %TRPM8/GRFα3 | N | %TRPM8 | %GFRα3 | %TRPM8/GRFα3 | N | |
| Peripherin | 34.5 | 37.1 | 57.0 | 519 | 47.8 | 58.9 | 82.1 | 1067 |
| NF200 | 11.0 | 1.9 | 0.0 | 269 | 10.4 | 6.3 | 6.1 | 1064 |
| CGRP | 36.4 | 43.4 | 57.9 | 821 | 44.3 | 61.3 | 66.2 | 1552 |
| IB4 | 1.9 | 7.1 | 2.9 | 1498 | 1.4 | 11.3 | 9.5 | 541 |
| TRPV1 | 38.5 | 94.1 | 92.1 | 516 | 53.5 | 72.8 | 87.0 | 1159 |
| Nav1.8 | 36.1 | 52.3 | 63.4 | 1215 | 48.9 | 43.7 | 65.5 | 689 |
| TrkA | 50.1 | 48.3 | 67.1 | 1209 | 39.1 | 48.9 | 59.9 | 1660 |
| cRet | 11.3 | 23.4 | 16.5 | 1608 | 8.0 | 15.4 | 8.6 | 1092 |
| B2R | n.d. | n.d. | n.d. | — | 69.4 | 70.2 | 72.1 | 5798 |
Percentages reported indicate the proportion of TRPM8-, GFRα3-, or TRPM8/GFRα3-positive neurons that colabel for the indicated marker.
Sensory neurons can be further classified as peptidergic or nonpeptidergic depending on whether they express calcitonin gene-related peptide (CGRP) or bind the lectin IB4, respectively (Silverman and Kruger, 1988). The number of TRPM8 neurons in DRG and TG that bind IB4 was insignificant (TG, 1.43 ± 1.43%, n = 362; DRG, 1.89 ± 0.61%, n = 1103), and it is therefore unsurprising that very few TRPM8-GFRα3 neurons were found to be IB4 positive as well (TG, 3.57 ± 3.57%, n = 541; DRG, 2.88 ± 1.30%, n = 1498; Fig. 2C; Table 1). In contrast, a substantial percentage of TRPM8 neurons did express CGRP (TG, 44.29 ± 2.72%, n = 1271; DRG, 36.40 ± 4.51%, n = 629), and this number increased when analyzing only the TRPM8-GFRα3 population (TG, 66.15 ± 4.31%, n = 1552; DRG, 57.87 ± 5.65%, n = 821; Fig. 2D; Table 1). Together, these data show that TRPM8-GFRα3 neurons largely correspond to the peptidergic C-fiber subpopulation of TRPM8 neurons, implying that they are involved in nociceptive signaling.
Nociceptors are themselves a diverse population that can be sensitive to a range of stimuli and ligands, and it is therefore of interest to identify which nociceptive markers are expressed in TRPM8-GFRα3 neurons. The noxious heat receptor, TRPV1, is a nociceptor marker and has been shown to extensively colocalize with GFRα3 (Orozco et al., 2001). In agreement with this, although approximately half of TRPM8 neurons were TRPV1-positive (TG, 53.47 ± 3.41%, n = 961; DRG, 38.38 ± 4.84%, n = 500), nearly all TRPM8-GFRα3 cells were immunoreactive for TRPV1 (TG, 86.97 ± 2.87%, n = 1159; DRG, 95.17 ± 2.82%, n = 516; Fig. 3A; Table 1). Thus, TRPM8 neurons that express GFRα3 can also be characterized by their expression of TRPV1, and functionally, would be sensitive to both cooling and noxious heat (McKemy et al., 2002). Another channel believed to be involved in cold sensation is the TTX-resistant voltage-gated sodium channel Nav1.8 (Zimmermann et al., 2007). Evidence suggests that Nav1.8 is not required for sensing innocuous cool, but rather plays a crucial role in mediating cold pain (Abrahamsen et al., 2008). This channel was expressed in a subset of TRPM8 neurons (TG, 48.89 ± 6.92%, n = 494; DRG, 36.09 ± 4.99%, n = 1038), accounting for a majority of the TRPM8-GFRα3 coexpressing population (TG, 65.52 ± 12.07%, n = 689; DRG, 63.36 ± 7.03%, n = 1215; Fig. 3B; Table 1). Although this number is not as striking as for TRPV1, it does indicate that most TRPM8-GFRα3 neurons are equipped with the molecular machinery to mediate thermal pain at both ends of the temperature spectrum.
Figure 3.

DRG neurons coexpressing TRPM8 and GFRα3 are putative cold nociceptors. A, Immunolabeling in sensory ganglia for the noxious heat receptor TRPV1 (red) revealed that GFRα3 (blue)/TRPM8 (green) neurons extensively expressed TRPV1. B, More than half of TRPM8 (green)/GFRα3 (blue) neurons coexpress the voltage-gated sodium channel Nav1.8 (red). C, TrkA immunoreactivity (red) was observed in a large percentage of GFRα3 (blue)/TRPM8 (green) neurons. D, Ret (red) expression does not colocalize with TRPM8 (green) and was absent from GFRα3+ (blue) neurons that express TRPM8. Arrowheads show triple-labeled neurons; arrows indicate TRPM8+ cells that are not positive for either marker.
Next, we labeled for various receptors for proalgesic agents to identify which mechanisms might be involved in modulating TRPM8 neuron activity in addition to GFRα3. We found that over half of TRPM8-GFRα3 neurons were positive for the NGF receptor TrkA (TG, 59.92 ± 5.49%, n = 1660; DRG, 67.05 ± 2.59%, n = 1209; Fig. 3C; Table 1), which, interestingly, is a substantially higher percentage than that of the overall TRPM8 population that labels for TrkA (TG, 39.07 ± 3.56%, n = 1326; DRG, 50.13 ± 1.76%, n = 976; Fig. 3C; Table 1). It is therefore possible that NGF could play a role in modulating the same subset of TRPM8 neurons that express GFRα3. In contrast, the bradykinin receptor B2R, whose expression is widespread in sensory neurons (Wang et al., 2005), was found in a smaller fraction of TRPM8-GFRα3 neurons (46.45 ± 3.98%, n = 5529 TG neurons, data not shown) than of TRPM8 neurons (69.52 ± 1.62%, n = 5798 TG neurons, data not shown). These data suggest that TRPM8 neurons that may be sensitive to bradykinin are distinct from those that are GFRα3 positive, and could represent a functionally distinct subpopulation, consistent with recent evidence suggesting that activation of bradykinin receptors inhibits TRPM8 channel function (Zhang et al., 2012).
We next labeled for the receptor tyrosine kinase Ret, which has long been considered the main coreceptor for GFLs along with their respective GFRα receptors (Takahashi, 2001; Golden et al., 2010). GFRα receptors are extracellular, and dictate specificity of ligand binding, whereas Ret is a transmembrane protein required to transduce the signal intracellularly (Airaksinen and Saarma, 2002). However, consistent with previous studies (Takashima et al., 2010), very few TRPM8 neurons coexpressed Ret (TG, 7.96 ± 2.47%, n = 684; DRG, 11.29 ± 2.25%, n = 1293; Fig. 3D; Table 1), which was also true of the TRPM8-GFRα3 population (TG, 8.57 ± 2.85%, n = 1092; DRG, 16.50 ± 4.45%, n = 1608). The limited expression of Ret in GFRα3 neurons observed here (TG, 15.42 ± 3.56%, n = 989; DRG, 23.39 ± 4.99%; Table 1) is surprising given that the two receptors were previously reported to widely coexpress (Orozco et al., 2001), but is in line with more recent evidence that the overlap is in fact much less extensive (Bennett et al., 2006; Golden et al., 2010). These data imply that in TRPM8-GFRα3 neurons, binding of GFRα3 by its specific ligand artemin must activate an alternative, Ret-independent pathway (Paratcha and Ledda, 2008). There has been increasing evidence for such alternative coreceptors, mainly focusing on the neural cell adhesion molecule (NCAM; Paratcha et al., 2003; Sariola and Saarma, 2003), and we therefore asked whether NCAM is expressed in TRPM8 neurons. We observed a high level of NCAM labeling in TRPM8 (76.17 ± 4.59%, n = 152 TG neurons, data not shown) and TRPM8-GFRα3 neurons (76.35 ± 4.83%, n = 332 TG neurons, data not shown). However, it is worth noting that, given its role as a cell adhesion molecule, NCAM expression is widespread, and it is therefore unsurprising that such a large proportion of TRPM8 and TRPM8-GFRα3 were also NCAM-positive. Nevertheless, NCAM is a potential candidate as a coreceptor for GFRα3 in TRPM8 neurons in the absence of Ret.
Last, the ionotropic purinergic receptor P2X3 is important for inflammatory pain (Vulchanova et al., 1998; North, 2004), and is expressed in small diameter nonpeptidergic nociceptors distinct from those that are CGRP-positive (Vulchanova et al., 1998). Because P2X3 is expressed in a small fraction of TRPM8 neurons (Takashima et al., 2010), and we have demonstrated here that CGRP does not label the entirety of the TRPM8-GFRα3 population, we sought to determine whether the remaining TRPM8-GFRα3 neurons were P2X3-positive. We found similarly low levels of P2X3 labeling in both TRPM8 and TRPM8-GFRα3 cells (9.11 ± 2.52% and 11.50 ± 4.67%, respectively, n = 797 TG neurons; data not shown), confirming that nonpeptidergic neurons represent only a small subset of either of these populations and are not expected to play a large role in signaling cold pain.
Artemin induces TRPM8-dependent cold hypersensitivity in vivo
The neurochemical profile of TRPM8-GFRα3 sensory neurons strongly suggests that they are a subset of cold-sensing neurons involved in signaling thermal pain, possibly both in acute as well as pathological conditions. Next, we sought to determine whether these observations translated in vivo by testing the effect of artemin, which preferentially binds and activates GFRα3 (Jing et al., 1997; Baloh et al., 1998, 2000; Carmillo et al., 2005), on cold sensation in mice. Artemin expression is reported to increase in inflamed skin (Malin et al., 2006), and to confirm these findings we harvested mRNA from inflamed and control wild-type mouse hindpaw skin two days after injection of complete Freund's adjuvant (CFA) and performed qPCR. We observed a 2.45 ± 0.1-fold increase in artemin expression (p < 0.01, n = 3 mice), further supporting the idea that it is involved in pain signaling after injury.
Cold sensitivity was evaluated using the evaporative cooling behavioral assay, in which a droplet of acetone is applied to the hindpaw to cool the skin locally, and responses were scored on a five-point scale based on lifting, flinching, licking, and guarding of the paw (Bautista et al., 2007; Colburn et al., 2007; Knowlton et al., 2011; Brenneis et al., 2013). Using this assay, we measured cold sensitivity after unilateral intraplantar injection of artemin (20 μl of 10 μg/ml) or vehicle (saline) into the mouse hindpaw (Fig. 4A). Cold responses for the paw injected with artemin were significantly larger compared with both the uninjected contralateral paw (p < 0.001, n = 10 mice) and vehicle-injected animals. Response scores in artemin-injected mice were 3.12 ± 0.1 and 3.09 ± 0.1 at 1 and 3 h postinjection, respectively, compared with 2.04 ± 0.2 (p < 0.001, n = 10) and 2.17 ± 0.2 (p < 0.01) in vehicle-injected controls. Responses returned to baseline levels within 24 h, showing that cold sensitization induced by artemin is transient, consistent with similar results obtained for heat hyperalgesia after injection of various neurotrophic factors (Malin et al., 2006).
Figure 4.
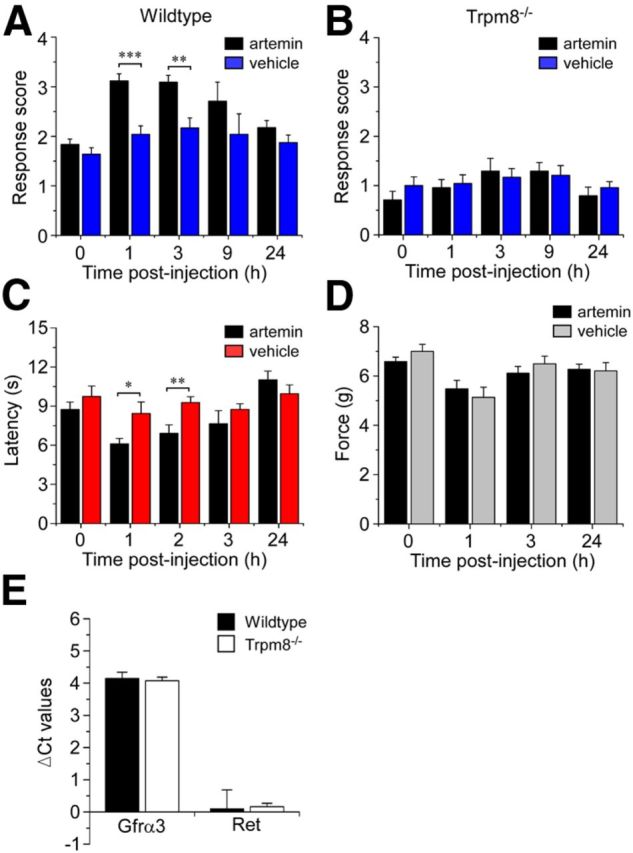
Artemin induces TRPM8-dependent cold hypersensitivity. Cold response scores were assessed before and after injection of artemin or vehicle into the plantar surface of the hindpaw. A, Artemin significantly increased cold behaviors in wild-type mice compared with vehicle-injected controls (***p < 0.001 at 1 h and **p < 0.01 at 3 h, n = 10). B, Artemin-induced cold hypersensitivity was abolished in Trpm8−/− mice, with response scores similar to vehicle-injected animals (p > 0.05, n = 6). C, Artemin induced heat hyperalgesia, with significantly decreased withdrawal latencies 1 and 2 h after injection of artemin compared with controls (*p < 0.05 and **p < 0.01, n = 8). D, There were no differences in paw withdrawal from mechanical stimuli between artemin and vehicle-injected animals (p > 0.05, n = 6). E, Transcript expression levels of Gfrα3 and Ret showed no difference in Trpm8−/− versus wild-type controls as assayed by quantitative PCR. Data expressed as ΔCt values normalized to GAPDH internal control (n = 3; p > 0.05). All data shown as mean ± SEM.
Previous studies indicate that TRPM8 is involved in cold hypersensitivity (Colburn et al., 2007; Knowlton et al., 2011, 2013). To establish whether artemin-induced sensitization to cold relies on TRPM8-dependent or TRPM8-independent mechanisms, we applied the same protocol as described above to mice lacking TRPM8 channels (Trpm8−/− mice; Bautista et al., 2007; Knowlton et al., 2010). As we and others have previously reported, Trpm8−/− mice were significantly less sensitive to cooling than wild-type mice at baseline (Bautista et al., 2007; Knowlton et al., 2011, 2013). In these mice, there was no difference in cold response scores in artemin versus vehicle-injected animals at any of the time points evaluated (Fig. 4B; p > 0.05, n = 6 for each group). These results provide strong evidence that TRPM8 is required for artemin-mediated cold hypersensitivity in vivo, suggesting that artemin, acting through GFRα3, sensitizes TRPM8 neurons.
We next explored the specificity of the effect of artemin on somatosensation by evaluating both heat and mechanical sensitivity in wild-type mice after injection of artemin into the hindpaw. First, we measured paw withdrawal latencies in response to a radiant heat source to test whether artemin modulates sensitivity to noxious heat. In agreement with other reports (Malin et al., 2006; Schmutzler et al., 2009), injection of artemin significantly reduced withdrawal latencies with a rapid time course (Fig. 4C; p < 0.05 and p < 0.01 at 1 and 2 h postinjection, respectively, compared with vehicle-injected controls; n = 8 in each group), which returned to baseline levels within 24 h. Next, we tested the threshold force to paw withdrawal using the electronic von Frey assay. Interestingly, mechanical sensation was unaffected by artemin (Fig. 4D), with withdrawal thresholds in artemin-injected animals remaining at similar levels to control animals after injection (p > 0.05 at all time-points; n = 6 in each group). This finding is in line with the neurochemical profile of GFRα3 neurons, which is more characteristic of thermoreceptors (TrkA- and CGRP-positive) than mechanoreceptors (Basbaum et al., 2009; Cavanaugh et al., 2009). Moreover, the observation that artemin sensitizes heat and cold responses, but does not affect mechanical sensitivity, demonstrates that sensitization can be modality-specific. This opens up the possibility that neurotrophic factors, such as artemin, can act differentially on specific subpopulations of sensory neurons.
One caveat that could account for the lack of artemin-induced cold sensitization in Trpm8−/− mice would be altered expression levels of GFRα3 in these animals. To control for this possibility we compared Gfrα3 transcript levels in dorsal root ganglia harvested from wild-type and Trpm8−/− mice, observing no difference between the two genotypes (Fig. 4E). Furthermore, Ret expression was similarly unchanged in Trpm8−/− mice (Fig. 4E). We also examined the total number of GFRα3+ neurons in the DRGs of wild-type and Trpm8−/− mice to ensure that the phenotype of the former population was not altered by Trpm8 deletion. We found that GFRα3-positive neurons constituted 23.94 ± 1.67% of PGP9.5-labeled neurons in Trpm8−/− ganglia, compared with 21.87 ± 2.51% in wild-type tissue (p > 0.05, n = 1722 and 1453, respectively; data not shown), consistent with previous reports of expression of GFRα3 in sensory ganglia (Naveilhan et al., 1998; Ernsberger, 2008; Wang et al., 2011). In addition, we found that this population was largely TRPV1-positive (90.45 ± 1.36%, n = 1599; data not shown), again showing no difference with wild-type expression patterns (see Table 1).
Specificity of heat and cold sensitization by inflammatory mediators in vivo
Presumably, upon tissue injury, there are a variety of factors at play in sensitizing nociceptors to sensory stimuli (Julius and Basbaum, 2001). Our immunohistochemical data suggest that the TRPM8 population is particularly well disposed to be modulated by artemin, which was confirmed by our observations in vivo (Fig. 4), but it is of interest to establish whether cold sensitization is an effect specific to this neurotrophic factor. We therefore used the evaporative cooling assay to examine a panel of different proalgesics and their effects on cold sensation in vivo. The time course was chosen based on existing data for heat and mechanical sensitization suggesting that single injections are effective within a few hours of administration (Chuang et al., 2001; Malin et al., 2006). The injection of 20 μl of 10 μg/ml GDNF or NRTN, which are the GFLs for GFRα1 and GFRα2, respectively, did not significantly alter cold responses compared with vehicle-injected mice (Fig. 5A,B; p > 0.05, n = 8 mice in each group). This finding is consistent with the fact that neither GFRα1 nor GFRα2 are expressed in TRPM8 neurons (Fig. 1). In contrast, GDNF and NRTN have been reported to sensitize TRPV1 channels and heat-evoked behaviors (Malin et al., 2006; Schmutzler et al., 2009). To investigate whether these growth factors can differentially modulate heat and cold responses, we examined heat sensitivity in mice injected either with GDNF or NRTN. In both cases, withdrawal latencies to noxious heat were significantly reduced within 2 h of injection compared with control animals (Fig. 5C,D; p < 0.01 at 1 h for GDNF and 2 h for NRTN, n = 4 for each group), showing selectivity in the modalities of stimuli modulated by GFLs.
Figure 5.

The GFLs GDNF and NRTN evoke heat hyperalgesia, but not cold hypersensitivity. Cold and heat responses were assessed before and after intraplantar injection of 20 μl of 10 μg/ml growth factors or vehicle (saline). The two GFLs GDNF (A) and NRTN (B) did not significantly change cold responses compared with vehicle-injected controls (p > 0.05, n = 8). In contrast, paw withdrawal latencies to radiant noxious heat were significantly decreased between 1 and 3 h after injection of both GDNF (C) and NRTN (D) compared with vehicle-injected controls (*p < 0.05, **p < 0.01, ***p < 0.001, n = 4). All data shown as mean ± SEM.
Both NGF and bradykinin are well known to induce heat hyperalgesia by potentiating TRPV1 responses (Chuang et al., 2001; Katanosaka et al., 2008), yet there has been no evidence of their effect on TRPM8 in vivo. Thus, we assessed cold responses after injection of NGF (20 μl/0.2 μg), finding that NGF did alter cold sensitivity with a magnitude and time course similar to artemin (Fig. 6A), an effect that required the presence of functional TRPM8 channels (Fig. 6B). This was unsurprising given the established role of NGF as a proalgesic involved in sensitizing heat and mechanical responses (Woolf et al., 1994; Chuang et al., 2001; Pezet and McMahon, 2006). Moreover, its receptor TrkA is expressed in a subset of TRPM8 neurons, particularly those that are also GFRα3-positive and presumably involved in pain signaling. In contrast, injection of the peptide bradykinin (20 μl at 10 nm), which is known to sensitize heat responses (Chuang et al., 2001), did not alter cold sensitivity at any of the evaluated time points (Fig. 6C; p > 0.05, n = 8). Although the bradykinin receptor B2R does colocalize with TRPM8 in sensory neurons, its expression is widespread and is therefore not necessarily an indication as to whether TRPM8 neurons would be modulated by bradykinin. Indeed, recent in vitro data suggest bradykinin activating its receptor may inhibit TRPM8 channel function (Zhang et al., 2012), a finding consistent with our behavioral analysis. Together, these results indicate that, although many proalgesics can induce heat hyperalgesia, only artemin and NGF are able to sensitize cold responses in vivo, and that heat and cold sensitization appear to be regulated by distinct mechanisms.
Figure 6.

Cold sensitivity after injection of NGF and bradykinin in vivo. Intraplantar injection of 20 μl of NGF (10 μg/ml) resulted in increased cold-response scores in the evaporative cooling assay in wild-type (A; *p < 0.05, **p < 0.01, n = 10), but not Trpm8−/− mice (B; p > 0.05, n = 6). C, Injection of bradykinin (10 nm), however, did not affect cold sensitivity at any of the time points evaluated (p > 0.05, n = 8). Data shown as mean ± SEM.
Discussion
There is extensive evidence showing that inflammation and injury result in increased pain due to sensitization of sensory neurons to physical and chemical stimuli (Linley et al., 2010). Many studies have focused on elucidating the mechanisms by which transduction of heat and mechanical stimuli is modulated in pain conditions, such as CFA-induced inflammation, neuropathic pain models, and incisional injury (Davis et al., 2000; Gaus et al., 2003; Spofford and Brennan, 2012). This research has revealed a crucial role for neurotrophic factors in directly potentiating nociceptive signaling. In particular, NGF, bradykinin, and the GFLs GDNF, NRTN, and artemin, have been shown to sensitize TRPV1 responses and are believed to be responsible for heat hyperalgesia induced by inflammation and injury (Davis et al., 2000; Chuang et al., 2001; Zhang et al., 2005; Elitt et al., 2006; Malin et al., 2006; Katanosaka et al., 2008; Schmutzler et al., 2009). However, a third component to this type of pain is manifested as cold hypersensitivity, the mechanisms for which remain largely unexplored. Studies in mice lacking TRPM8 channels show that TRPM8 plays an important role in the development of cold hypersensitivity (Colburn et al., 2007; Knowlton et al., 2011, 2013). In the present study, we used a microarray screen with tissue from mice lacking TRPM8 neurons to identify potential candidates enriched in these cells in an unbiased manner. Our initial focus was on neurotrophic factors and their receptors to investigate the role of TRPM8 in cold hypersensitivity.
Although it was previously reported that a subset of TRPM8 neurons are TrkA-positive (Takashima et al., 2010), it was somewhat surprising that its expression was significantly reduced in ablated tissue, given that TRPM8 is expressed in only ∼10% of TrkA+ neurons (Fig. 3C). More interesting, however, was the finding that gfrα3 transcript levels were substantially reduced in ablated ganglia compared with controls, although expression of other GDNF ligands and receptors was unchanged. GFRα3 is largely restricted to small-diameter sensory neurons, supporting the idea that it could be expressed in TRPM8 neurons involved in signaling cold pain (Orozco et al., 2001). However, this is the first evidence for specific GFRα expression in TRPM8+ neurons. Our data show that GFRα3 is preferentially localized to the subset of TRPM8-positive peptidergic C-fibers that express the nociceptor markers TRPV1, Nav1.8, and TrkA. Cellular recordings have identified two broad cohorts of cold-sensitive TRPM8 neurons, defined by their temperature thresholds for activation: one, which mediates innocuous cool, and a second, which is activated at lower temperatures and represents putative cold nociceptors (Xing et al., 2006; Madrid et al., 2009; Sarria et al., 2012; McKemy, 2013). The latter population is also characterized by its sensitivity to capsaicin, providing further evidence that TRPM8-GFRα3 neurons are cold nociceptors.
This is in contrast to GFRα1 and GFRα2, whose expression overlaps with TRPV1 to a much lesser degree than GFRα3 (Malin et al., 2006) and does not coincide with TRPM8 in naive sensory ganglia. However, in mice in which NRTN was embryonically overexpressed (starting at E10.5) in skin, GFRα2 expression was upregulated and found to coexpress with TRPM8 (Wang et al., 2013). Moreover, TRPM8 transcript expression increased in lumbar DRG, as well as a change in cellular expression in that TRPM8 was observed in nonpeptidergic, IB4-positive neurons, a population that does not express TRPM8 in wild-type animals (Takashima et al., 2007; Wang et al., 2013). Consistent with this change in expression, these mice showed evidence of cold sensitization. How this relates to acute responses to NRTN during injury is unclear, but suggests that long-term exposure to excessive NRTN levels during development alters transcriptional regulation of TRPM8 (Wang et al., 2013). Nonetheless, the high degree of colocalization of TRPV1 in TRPM8-GFRα3 neurons suggests that TRPV1 and capsaicin sensitivity can be used as a marker for TRPM8 neurons that mediate cold pain as opposed to innocuous cold sensation. Moreover, the higher expression of Nav1.8 in TRPM8-GFRα3 neurons compared with the overall TRPM8 population is a further indication that these cells mediate thermal pain. Studies in mice in which Nav1.8 neurons are ablated showed that the channel is specifically required for cold pain, but not innocuous cool sensing (Abrahamsen et al., 2008). This suggests not only that Nav1.8 can be used as a marker for cold nociceptors, but that it could also play a role in TRPM8-GFRα3 neurons in mediating inflammatory cold pain.
GDNF, NRTN, and artemin have all been shown to potentiate TRPV1 responses in vitro, as well as induce heat hyperalgesia in vivo (Malin et al., 2006; Schmutzler et al., 2009). However, the necessity of TRPV1 in GFL-induced thermal hyperalgesia has not been confirmed. Nonetheless, these results add to the broad range of inflammatory mediators, such as NGF and bradykinin, which potentiate responses to heat. Here, we report that of the five tested only artemin and NGF were able to sensitize cold responses in vivo, whereas GDNF, NRTN, and bradykinin had no significant effect on cold sensitivity in mice. These results are consistent with the expression data for each of the molecules' receptors, which indicated that GFRα3 and TrkA were most highly localized to the putative nociceptor subpopulation of TRPM8 neurons. The exception was the bradykinin receptor B2R, which was extensively expressed with TRPM8. In vitro patch clamp and calcium imaging studies investigating the influence of bradykinin on cold responses suggest that it inhibits, rather than potentiates, TRPM8 activity (Linte et al., 2007; Zhang et al., 2012). Although this finding presents evidence for a paradoxical modulation of TRPM8 by different proalgesics, it could be explained by a differential effect of bradykinin and artemin on nonnociceptor versus nociceptor subpopulations of TRPM8 neurons, respectively (McKemy, 2013). Indeed, the bradykinin receptor, although widely expressed, appears to be preferentially localized to TRPM8 neurons distinct from those that express GFRα3. Thus, bradykinin could modulate the putative nonnociceptor population more strongly, inhibiting the signaling of innocuous cool, whereas artemin could specifically sensitize the nociceptor population, increasing pain signaling.
Here we show for the first time that NGF induces cold hypersensitivity in vivo, in line with reports suggesting that NGF increases the cold sensitivity of DRG neurons in vitro (Reid et al., 2002; Babes et al., 2004). However, it is interesting to note that NGF had a milder effect on cold sensitivity in mice compared with artemin, suggesting that although NGF potently affects heat and mechanical responses, its role in cold hypersensitivity is minor. NGF may have an additive effect when acting on TRPM8 neurons in conjunction with artemin, as might be the case with inflammation. In addition, artemin had no effect on mechanical sensitivity, indicating that it specifically modulates thermosensation, which is not the case for NGF (Chuang et al., 2001; Rosenbaum et al., 2004). Furthermore, behavioral studies in mice overexpressing artemin in skin keratinocytes indicated that these animals were more sensitive to noxious heat and cold, but that their mechanical sensitivity was unaffected (Elitt et al., 2006), consistent with our findings.
The sensitization of cold responses by artemin and NGF was abolished in Trpm8−/− mice, similar to results for NGF-induced heat hypersensitivity in TRPV1 knock-out animals (Chuang et al., 2001). Thus, artemin- and NGF-induced sensitization of cold responses requires TRPM8, and could largely account for cold hypersensitivity after inflammation or injury. However, it is worth noting that there is still some degree of sensitization after inflammation in Trpm8−/− mice (Knowlton et al., 2011, 2013), such that the two neurotrophic factors acting on TRPM8 neurons are likely not the only mechanisms for cold hypersensitivity. Other candidate molecules that could be involved in cold hypersensitivity include TRPA1 and Nav1.8, which have both been implicated in mediating cold pain (Obata et al., 2005; Abrahamsen et al., 2008), although recent evidence suggests that artemin inhibits TRPA1 channels (Yoshida et al., 2011). It will be of interest to determine whether artemin sensitizes TRPM8 channels themselves, be it through an intracellular signaling cascade or a more direct interaction, or whether it affects other channels in these cells to cause them to become sensitized. One important consideration in this regard is that Ret, which is considered to be the main coreceptor for GFRα3, is not expressed in the vast majority of TRPM8 neurons (Airaksinen and Saarma, 2002; Takashima et al., 2010). Thus, in TRPM8-GFRα3 neurons, artemin must signal through an alternative coreceptor, such as NCAM, or through another mechanism altogether (Paratcha et al., 2003; Schmutzler et al., 2011). Together, these results have identified TRPM8-GFRα3 neurons as a subpopulation of nociceptors involved in signaling cold pain, which can be specifically sensitized by artemin in a TRPM8-dependent manner. These neurons could serve as a specific target for the development of drugs that aim to alleviate the pain caused by cold allodynia after inflammation or injury.
Footnotes
This study was supported by National Institutes of Health Grants NS078530 and NS071364 (D.D.M.K.).We thank D. Julius for providing the TRPV1 antibody, C. Messinger and Y. Yang for histology and behavior analysis assistance, and members of the McKemy Laboratory for encouragement and guidance.
References
- Abrahamsen B, Zhao J, Asante CO, Cendan CM, Marsh S, Martinez-Barbera JP, Nassar MA, Dickenson AH, Wood JN. The cell and molecular basis of mechanical, cold, and inflammatory pain. Science. 2008;321:702–705. doi: 10.1126/science.1156916. [DOI] [PubMed] [Google Scholar]
- Airaksinen MS, Saarma M. The GDNF family: signalling, biological functions and therapeutic value. Nat Rev Neurosci. 2002;3:383–394. doi: 10.1038/nrn812. [DOI] [PubMed] [Google Scholar]
- Attal N, Bouhassira D, Gautron M, Vaillant JN, Mitry E, Lepère C, Rougier P, Guirimand F. Thermal hyperalgesia as a marker of oxaliplatin neurotoxicity: a prospective quantified sensory assessment study. Pain. 2009;144:245–252. doi: 10.1016/j.pain.2009.03.024. [DOI] [PubMed] [Google Scholar]
- Babes A, Zorzon D, Reid G. Two populations of cold-sensitive neurons in rat dorsal root ganglia and their modulation by nerve growth factor. Eur J Neurosci. 2004;20:2276–2282. doi: 10.1111/j.1460-9568.2004.03695.x. [DOI] [PubMed] [Google Scholar]
- Baloh RH, Tansey MG, Lampe PA, Fahrner TJ, Enomoto H, Simburger KS, Leitner ML, Araki T, Johnson EM, Jr, Milbrandt J. Artemin, a novel member of the GDNF ligand family, supports peripheral and central neurons and signals through the GFRalpha3-RET receptor complex. Neuron. 1998;21:1291–1302. doi: 10.1016/S0896-6273(00)80649-2. [DOI] [PubMed] [Google Scholar]
- Baloh RH, Enomoto H, Johnson EM, Jr, Milbrandt J. The GDNF family ligands and receptors - implications for neural development. Curr Opin Neurobiol. 2000;10:103–110. doi: 10.1016/S0959-4388(99)00048-3. [DOI] [PubMed] [Google Scholar]
- Basbaum AI, Bautista DM, Scherrer G, Julius D. Cellular and molecular mechanisms of pain. Cell. 2009;139:267–284. doi: 10.1016/j.cell.2009.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bautista DM, Siemens J, Glazer JM, Tsuruda PR, Basbaum AI, Stucky CL, Jordt SE, Julius D. The menthol receptor TRPM8 is the principal detector of environmental cold. Nature. 2007;448:204–208. doi: 10.1038/nature05910. [DOI] [PubMed] [Google Scholar]
- Bennett DL, Boucher TJ, Michael GJ, Popat RJ, Malcangio M, Averill SA, Poulsen KT, Priestley JV, Shelton DL, McMahon SB. Artemin has potent neurotrophic actions on injured C-fibres. J Peripher Nerv Syst. 2006;11:330–345. doi: 10.1111/j.1529-8027.2006.00106.x. [DOI] [PubMed] [Google Scholar]
- Brenneis C, Kistner K, Puopolo M, Segal D, Roberson D, Sisignano M, Labocha S, Ferreirós N, Strominger A, Cobos EJ, Ghasemlou N, Geisslinger G, Reeh PW, Bean BP, Woolf CJ. Phenotyping the function of TRPV1-expressing sensory neurons by targeted axonal silencing. J Neurosci. 2013;33:315–326. doi: 10.1523/JNEUROSCI.2804-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmillo P, Dagø L, Day ES, Worley DS, Rossomando A, Walus L, Orozco O, Buckley C, Miller S, Tse A, Cate RL, Rosenblad C, Sah DW, Grønborg M, Whitty A. Glial cell line-derived neurotrophic factor (GDNF) receptor alpha-1 (GFR alpha 1) is highly selective for GDNF versus artemin. Biochemistry. 2005;44:2545–2554. doi: 10.1021/bi049247p. [DOI] [PubMed] [Google Scholar]
- Cavanaugh DJ, Lee H, Lo L, Shields SD, Zylka MJ, Basbaum AI, Anderson DJ. Distinct subsets of unmyelinated primary sensory fibers mediate behavioral responses to noxious thermal and mechanical stimuli. Proc Natl Acad Sci U S A. 2009;106:9075–9080. doi: 10.1073/pnas.0901507106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang HH, Prescott ED, Kong H, Shields S, Jordt SE, Basbaum AI, Chao MV, Julius D. Bradykinin and nerve growth factor release the capsaicin receptor from PtdIns(4,5)P2-mediated inhibition. Nature. 2001;411:957–962. doi: 10.1038/35082088. [DOI] [PubMed] [Google Scholar]
- Chung MK, Caterina MJ. TRP channel knock-out mice lose their cool. Neuron. 2007;54:345–347. doi: 10.1016/j.neuron.2007.04.025. [DOI] [PubMed] [Google Scholar]
- Colburn RW, Lubin ML, Stone DJ, Jr, Wang Y, Lawrence D, D'Andrea MR, Brandt MR, Liu Y, Flores CM, Qin N. Attenuated cold sensitivity in TRPM8 null mice. Neuron. 2007;54:379–386. doi: 10.1016/j.neuron.2007.04.017. [DOI] [PubMed] [Google Scholar]
- Davis JB, Gray J, Gunthorpe MJ, Hatcher JP, Davey PT, Overend P, Harries MH, Latcham J, Clapham C, Atkinson K, Hughes SA, Rance K, Grau E, Harper AJ, Pugh PL, Rogers DC, Bingham S, Randall A, Sheardown SA. Vanilloid receptor-1 is essential for inflammatory thermal hyperalgesia. Nature. 2000;405:183–187. doi: 10.1038/35012076. [DOI] [PubMed] [Google Scholar]
- Dhaka A, Murray AN, Mathur J, Earley TJ, Petrus MJ, Patapoutian A. TRPM8 is required for cold sensation in mice. Neuron. 2007;54:371–378. doi: 10.1016/j.neuron.2007.02.024. [DOI] [PubMed] [Google Scholar]
- Elitt CM, McIlwrath SL, Lawson JJ, Malin SA, Molliver DC, Cornuet PK, Koerber HR, Davis BM, Albers KM. Artemin overexpression in skin enhances expression of TRPV1 and TRPA1 in cutaneous sensory neurons and leads to behavioral sensitivity to heat and cold. J Neurosci. 2006;26:8578–8587. doi: 10.1523/JNEUROSCI.2185-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernsberger U. The role of GDNF family ligand signalling in the differentiation of sympathetic and dorsal root ganglion neurons. Cell Tissue Res. 2008;333:353–371. doi: 10.1007/s00441-008-0634-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaus S, Moriwaki K, Suyama H, Kawamoto M, Yuge O. Capsaicin treatment inhibits osteopenia and heat hyperalgesia induced by chronic constriction injury to the sciatic nerve in rats. Hiroshima J Med Sci. 2003;52:43–51. [PubMed] [Google Scholar]
- Golden JP, Hoshi M, Nassar MA, Enomoto H, Wood JN, Milbrandt J, Gereau RW, 4th, Johnson EM, Jr, Jain S. RET signaling is required for survival and normal function of nonpeptidergic nociceptors. J Neurosci. 2010;30:3983–3994. doi: 10.1523/JNEUROSCI.5930-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein ME, House SB, Gainer H. NF-L and peripherin immunoreactivities define distinct classes of rat sensory ganglion cells. J Neurosci Res. 1991;30:92–104. doi: 10.1002/jnr.490300111. [DOI] [PubMed] [Google Scholar]
- Ji RR, Samad TA, Jin SX, Schmoll R, Woolf CJ. p38 MAPK activation by NGF in primary sensory neurons after inflammation increases TRPV1 levels and maintains heat hyperalgesia. Neuron. 2002;36:57–68. doi: 10.1016/S0896-6273(02)00908-X. [DOI] [PubMed] [Google Scholar]
- Jing S, Yu Y, Fang M, Hu Z, Holst PL, Boone T, Delaney J, Schultz H, Zhou R, Fox GM. GFRalpha-2 and GFRalpha-3 are two new receptors for ligands of the GDNF family. J Biol Chem. 1997;272:33111–33117. doi: 10.1074/jbc.272.52.33111. [DOI] [PubMed] [Google Scholar]
- Julius D, Basbaum AI. Molecular mechanisms of nociception. Nature. 2001;413:203–210. doi: 10.1038/35093019. [DOI] [PubMed] [Google Scholar]
- Katanosaka K, Banik RK, Giron R, Higashi T, Tominaga M, Mizumura K. Contribution of TRPV1 to the bradykinin-evoked nociceptive behavior and excitation of cutaneous sensory neurons. Neurosci Res. 2008;62:168–175. doi: 10.1016/j.neures.2008.08.004. [DOI] [PubMed] [Google Scholar]
- Knowlton WM, Bifolck-Fisher A, Bautista DM, McKemy DD. TRPM8, but not TRPA1, is required for neural and behavioral responses to acute noxious cold temperatures and cold-mimetics in vivo. Pain. 2010;150:340–350. doi: 10.1016/j.pain.2010.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowlton WM, Daniels RL, Palkar R, McCoy DD, McKemy DD. Pharmacological blockade of TRPM8 ion channels alters cold and cold pain responses in mice. PLoS One. 2011;6:e25894. doi: 10.1371/journal.pone.0025894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowlton WM, Palkar R, Lippoldt EK, McCoy DD, Baluch F, Chen J, McKemy DD. A sensory-labeled line for cold: TRPM8-expressing sensory neurons define the cellular basis for cold, cold pain, and cooling-mediated analgesia. J Neurosci. 2013;33:2837–2848. doi: 10.1523/JNEUROSCI.1943-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linley JE, Rose K, Ooi L, Gamper N. Understanding inflammatory pain: ion channels contributing to acute and chronic nociception. Pflugers Arch. 2010;459:657–669. doi: 10.1007/s00424-010-0784-6. [DOI] [PubMed] [Google Scholar]
- Linte RM, Ciobanu C, Reid G, Babes A. Desensitization of cold- and menthol-sensitive rat dorsal root ganglion neurones by inflammatory mediators. Exp Brain Res. 2007;178:89–98. doi: 10.1007/s00221-006-0712-3. [DOI] [PubMed] [Google Scholar]
- Madrid R, de la Peña E, Donovan-Rodriguez T, Belmonte C, Viana F. Variable threshold of trigeminal cold-thermosensitive neurons is determined by a balance between TRPM8 and Kv1 potassium channels. J Neurosci. 2009;29:3120–3131. doi: 10.1523/JNEUROSCI.4778-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malin SA, Molliver DC, Koerber HR, Cornuet P, Frye R, Albers KM, Davis BM. Glial cell line-derived neurotrophic factor family members sensitize nociceptors in vitro and produce thermal hyperalgesia in vivo. J Neurosci. 2006;26:8588–8599. doi: 10.1523/JNEUROSCI.1726-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKemy DD. The molecular and cellular basis of cold sensation. ACS Chem Neurosci. 2013;4:238–247. doi: 10.1021/cn300193h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKemy DD, Neuhausser WM, Julius D. Identification of a cold receptor reveals a general role for TRP channels in thermosensation. Nature. 2002;416:52–58. doi: 10.1038/nature719. [DOI] [PubMed] [Google Scholar]
- Naveilhan P, Baudet C, Mikaels A, Shen L, Westphal H, Ernfors P. Expression and regulation of GFRalpha3, a glial cell line-derived neurotrophic factor family receptor. Proc Natl Acad Sci U S A. 1998;95:1295–1300. doi: 10.1073/pnas.95.3.1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- North RA. P2X3 receptors and peripheral pain mechanisms. J Physiol. 2004;554:301–308. doi: 10.1113/jphysiol.2003.048587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obata K, Katsura H, Mizushima T, Yamanaka H, Kobayashi K, Dai Y, Fukuoka T, Tokunaga A, Tominaga M, Noguchi K. TRPA1 induced in sensory neurons contributes to cold hyperalgesia after inflammation and nerve injury. J Clin Invest. 2005;115:2393–2401. doi: 10.1172/JCI25437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orozco OE, Walus L, Sah DW, Pepinsky RB, Sanicola M. GFRalpha3 is expressed predominantly in nociceptive sensory neurons. Eur J Neurosci. 2001;13:2177–2182. doi: 10.1046/j.0953-816x.2001.01596.x. [DOI] [PubMed] [Google Scholar]
- Paratcha G, Ledda F. GDNF and GFRalpha: a versatile molecular complex for developing neurons. Trends Neurosci. 2008;31:384–391. doi: 10.1016/j.tins.2008.05.003. [DOI] [PubMed] [Google Scholar]
- Paratcha G, Ledda F, Ibáñez CF. The neural cell adhesion molecule NCAM is an alternative signaling receptor for GDNF family ligands. Cell. 2003;113:867–879. doi: 10.1016/S0092-8674(03)00435-5. [DOI] [PubMed] [Google Scholar]
- Peier AM, Moqrich A, Hergarden AC, Reeve AJ, Andersson DA, Story GM, Earley TJ, Dragoni I, McIntyre P, Bevan S, Patapoutian A. A TRP channel that senses cold stimuli and menthol. Cell. 2002;108:705–715. doi: 10.1016/S0092-8674(02)00652-9. [DOI] [PubMed] [Google Scholar]
- Pezet S, McMahon SB. Neurotrophins: mediators and modulators of pain. Annu Rev Neurosci. 2006;29:507–538. doi: 10.1146/annurev.neuro.29.051605.112929. [DOI] [PubMed] [Google Scholar]
- Ramachandran R, Hyun E, Zhao L, Lapointe TK, Chapman K, Hirota CL, Ghosh S, McKemy DD, Vergnolle N, Beck PL, Altier C, Hollenberg MD. TRPM8 activation attenuates inflammatory responses in mouse models of colitis. Proc Natl Acad Sci U S A. 2013;110:7476–7481. doi: 10.1073/pnas.1217431110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid G, Babes A, Pluteanu F. A cold- and menthol-activated current in rat dorsal root ganglion neurones: properties and role in cold transduction. J Physiol. 2002;545:595–614. doi: 10.1113/jphysiol.2002.024331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbaum T, Gordon-Shaag A, Munari M, Gordon SE. Ca2+/calmodulin modulates TRPV1 activation by capsaicin. J Gen Physiol. 2004;123:53–62. doi: 10.1085/jgp.200308906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sariola H, Saarma M. Novel functions and signalling pathways for GDNF. J Cell Sci. 2003;116:3855–3862. doi: 10.1242/jcs.00786. [DOI] [PubMed] [Google Scholar]
- Sarria I, Ling J, Xu GY, Gu JG. Sensory discrimination between innocuous and noxious cold by TRPM8-expressing DRG neurons of rats. Mol Pain. 2012;8:79. doi: 10.1186/1744-8069-8-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmutzler BS, Roy S, Hingtgen CM. Glial cell line-derived neurotrophic factor family ligands enhance capsaicin-stimulated release of calcitonin gene-related peptide from sensory neurons. Neuroscience. 2009;161:148–156. doi: 10.1016/j.neuroscience.2009.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmutzler BS, Roy S, Pittman SK, Meadows RM, Hingtgen CM. Ret-dependent and Ret-independent mechanisms of FGL-induced sensitization. Mol Pain. 2011;7:22. doi: 10.1186/1744-8069-7-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverman JD, Kruger L. Lectin and neuropeptide labeling of separate populations of dorsal root ganglion neurons and associated “nociceptor” thin axons in rat testis and cornea whole-mount preparations. Somatosens Res. 1988;5:259–267. doi: 10.3109/07367228809144630. [DOI] [PubMed] [Google Scholar]
- Spofford CM, Brennan TJ. Gene expression in skin, muscle, and dorsal root ganglion after plantar incision in the rat. Anesthesiology. 2012;117:161–172. doi: 10.1097/ALN.0b013e31825a2a2b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahmoush AJ, Schwartzman RJ, Hopp JL, Grothusen JR. Quantitative sensory studies in complex regional pain syndrome type 1/RSD. Clin J Pain. 2000;16:340–344. doi: 10.1097/00002508-200012000-00011. [DOI] [PubMed] [Google Scholar]
- Takahashi M. The GDNF/RET signaling pathway and human diseases. Cytokine Growth Factor Rev. 2001;12:361–373. doi: 10.1016/S1359-6101(01)00012-0. [DOI] [PubMed] [Google Scholar]
- Takashima Y, Daniels RL, Knowlton W, Teng J, Liman ER, McKemy DD. Diversity in the neural circuitry of cold sensing revealed by genetic axonal labeling of transient receptor potential melastatin 8 neurons. J Neurosci. 2007;27:14147–14157. doi: 10.1523/JNEUROSCI.4578-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takashima Y, Ma L, McKemy DD. The development of peripheral cold neural circuits based on TRPM8 expression. Neuroscience. 2010;169:828–842. doi: 10.1016/j.neuroscience.2010.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinik AI. Advances in diabetes for the millennium: new treatments for diabetic neuropathies. MedGenMed. 2004;6:13. [PMC free article] [PubMed] [Google Scholar]
- Vulchanova L, Riedl MS, Shuster SJ, Stone LS, Hargreaves KM, Buell G, Surprenant A, North RA, Elde R. P2X3 is expressed by DRG neurons that terminate in inner lamina II. Eur J Neurosci. 1998;10:3470–3478. doi: 10.1046/j.1460-9568.1998.00355.x. [DOI] [PubMed] [Google Scholar]
- Wang H, Kohno T, Amaya F, Brenner GJ, Ito N, Allchorne A, Ji RR, Woolf CJ. Bradykinin produces pain hypersensitivity by potentiating spinal cord glutamatergic synaptic transmission. J Neurosci. 2005;25:7986–7992. doi: 10.1523/JNEUROSCI.2393-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Molliver DC, Jing X, Schwartz ES, Yang FC, Samad OA, Ma Q, Davis BM. Phenotypic switching of nonpeptidergic cutaneous sensory neurons following peripheral nerve injury. PLoS One. 2011;6:e28908. doi: 10.1371/journal.pone.0028908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Jing X, DeBerry JJ, Schwartz ES, Molliver DC, Albers KM, Davis BM. Neurturin overexpression in skin enhances expression of TRPM8 in cutaneous sensory neurons and leads to behavioral sensitivity to cool and menthol. J Neurosci. 2013;33:2060–2070. doi: 10.1523/JNEUROSCI.4012-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolf CJ. Central sensitization: uncovering the relation between pain and plasticity. Anesthesiology. 2007;106:864–867. doi: 10.1097/01.anes.0000264769.87038.55. [DOI] [PubMed] [Google Scholar]
- Woolf CJ, Safieh-Garabedian B, Ma QP, Crilly P, Winter J. Nerve growth factor contributes to the generation of inflammatory sensory hypersensitivity. Neuroscience. 1994;62:327–331. doi: 10.1016/0306-4522(94)90366-2. [DOI] [PubMed] [Google Scholar]
- Xing H, Ling J, Chen M, Gu JG. Chemical and cold sensitivity of two distinct populations of TRPM8-expressing somatosensory neurons. J Neurophysiol. 2006;95:1221–1230. doi: 10.1152/jn.01035.2005. [DOI] [PubMed] [Google Scholar]
- Yoshida N, Kobayashi K, Yu L, Wang S, Na R, Yamamoto S, Noguchi K, Dai Y. Inhibition of TRPA1 channel activity in sensory neurons by the glial cell line-derived neurotrophic factor family member, artemin. Mol Pain. 2011;7:41. doi: 10.1186/1744-8069-7-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Huang J, McNaughton PA. NGF rapidly increases membrane expression of TRPV1 heat-gated ion channels. EMBO J. 2005;24:4211–4223. doi: 10.1038/sj.emboj.7600893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Mak S, Li L, Parra A, Denlinger B, Belmonte C, McNaughton PA. Direct inhibition of the cold-activated TRPM8 ion channel by Gαq. Nat Cell Biol. 2012;14:851–858. doi: 10.1038/ncb2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang ZY, Xu H, Clapham DE, Ji RR. Phosphatidylinositol 3-kinase activates ERK in primary sensory neurons and mediates inflammatory heat hyperalgesia through TRPV1 sensitization. J Neurosci. 2004;24:8300–8309. doi: 10.1523/JNEUROSCI.2893-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann K, Leffler A, Babes A, Cendan CM, Carr RW, Kobayashi J, Nau C, Wood JN, Reeh PW. Sensory neuron sodium channel Nav1.8 is essential for pain at low temperatures. Nature. 2007;447:855–858. doi: 10.1038/nature05880. [DOI] [PubMed] [Google Scholar]